

Abstract
ADP-glucose pyrophosphorylase catalyzes the first committed and rate-limiting step in starch biosynthesis in plants and glycogen biosynthesis in bacteria. It is the enzymatic site for regulation of storage polysaccharide accumulation in plants and bacteria, being allosterically activated or inhibited by metabolites of energy flux. We report the first atomic resolution structure of ADP-glucose pyrophosphorylase. Crystals of potato tuber ADP-glucose pyrophosphorylase α subunit were grown in high concentrations of sulfate, resulting in the sulfate-bound, allosterically inhibited form of the enzyme. The N-terminal catalytic domain resembles a dinucleotide-binding Rossmann fold and the C-terminal domain adopts a left-handed parallel β helix that is involved in cooperative allosteric regulation and a unique oligomerization. We also report structures of the enzyme in complex with ATP and ADP-glucose. Communication between the regulator-binding sites and the active site is both subtle and complex and involves several distinct regions of the enzyme including the N-terminus, the glucose-1-phosphate-binding site, and the ATP-binding site. These structures provide insights into the mechanism for catalysis and allosteric regulation of the enzyme.
Keywords: ADP-glucose, pyrophosphorylase, starch biosynthesis
Introduction
The α-1,4-polyglucans (starch and glycogen) are the main repositories of carbon and energy reserves for many organisms. Biosynthesis of polyglucans involves the use of an activated form of glucose: ADP-glucose (ADP-Glc) in photosynthetic eukaryotes and bacteria, and UDP-glucose (UDP-Glc) in mammals, fungi, and eukaryotic heterotrophic microorganisms (Sivak and Preiss, 1998). In plants and bacteria, the main regulatory step of the pathway is ADP-Glc synthesis catalyzed by ADP-glucose pyrophosphorylase (EC 2.7.7.27; ADP-Glc PPase) (Preiss and Sivak, 1998a, 1998b; Okita et al, 1990). The enzymatic reaction requires a divalent metal ion, Mg2+, and is freely reversible in vitro, with equilibrium close to 1. However, the hydrolysis of inorganic pyrophosphate by inorganic pyrophosphatase and the use of sugar nucleotide for polysaccharide synthesis cause the ADP-Glc synthesis reaction to be essentially irreversible in vivo (Preiss et al, 1966; Iglesias and Preiss, 1992). The reaction is sequential, with ATP and Mg2+ binding first, followed by glucose-1-phosphate (G1P; Haugen and Preiss, 1979).
Most of the ADP-Glc PPases so far characterized are allosterically regulated by small effector molecules. In general, ADP-Glc PPase activators are key metabolites that indicate high carbon and energy content within the cell, while inhibitors of the enzyme indicate low metabolic energy levels (Preiss and Sivak, 1998a). For example, many bacterial enzymes are activated by intermediates of glycolysis or the Entner Doudoroff pathway (e.g., pyruvate, fructose-6-phosphate (F6P), fructose-1,6-bisphosphate (FBP)) and inhibited by AMP (Preiss, 1984). The enzymes from blue-green algae and higher plants are activated by 3-phosphoglycerate (3-PGA) and inhibited by Pi, key intermediates in CO2 assimilation by the C3 pathway. In vivo and in situ experiments show that activation by 3-PGA and inhibition by Pi observed in vitro are also physiologically important for starch synthesis in higher plants, showing a direct correlation between the concentration of 3-PGA and starch accumulation and an inverse correlation between Pi concentration and the starch content (Preiss, 1982a, 1982b, 1988). In Chlamydomonas reinhardtii, starch-deficient mutants have been isolated and one class of mutants was shown to have an ADP-Glc PPase that could not be activated by 3-PGA (Ball et al, 1991). In Arabidopsis thaliana, a mutant lacking both subunits of ADP-Glc PPase accumulated only 2% of the starch seen in normal plant (Lin et al, 1988). These data indicate that starch synthesis is almost completely dependent on the synthesis of ADP-Glc by ADP-Glc PPase.
ADP-Glc PPases from all the eukaryotes characterized are composed of α and β subunits (also referred to as small and large subunits, respectively) to form an α2β2 heterotetramer (Okita et al, 1990; Preiss and Sivak, 1998a, 1998b). The α subunit of the higher plant ADP-Glc PPase is highly conserved (85–95% identity), whereas the β subunit is less conserved (50–60% identity; Nakata et al, 1991). In potato tuber ADP-Glc PPase, α and β subunits share 53% identity (Smith-White and Preiss, 1992) (Figure 1).
Figure 1.
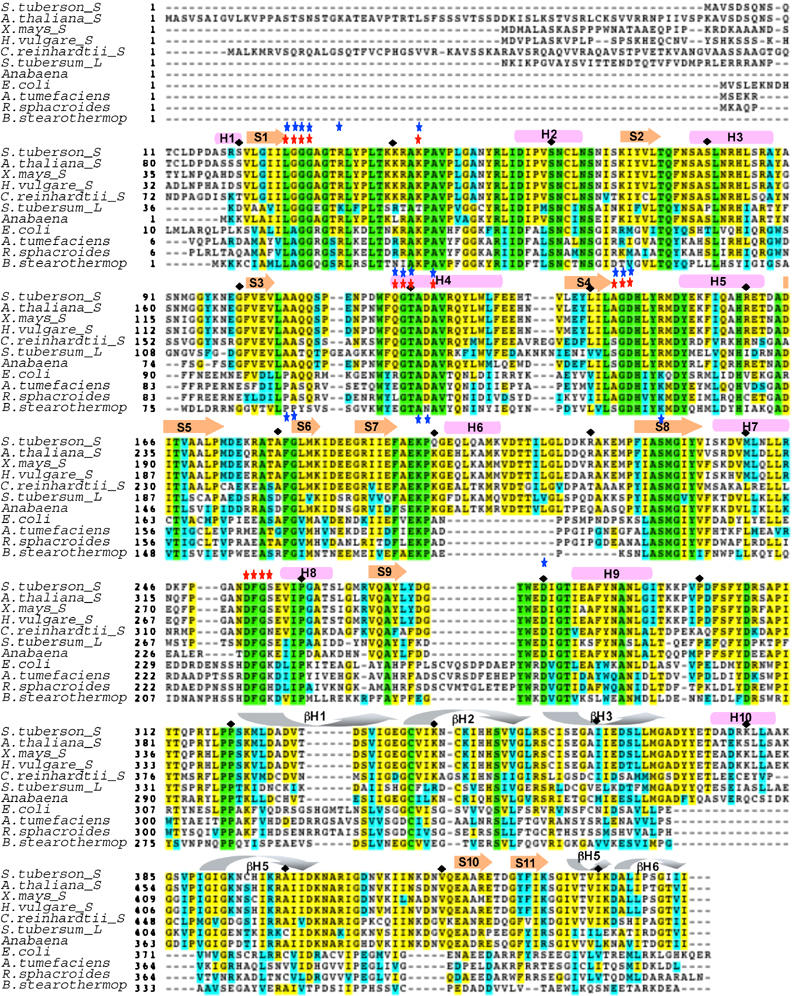
Sequence alignment of ADP-Glc PPase from different species. Secondary structure of the potato tuber enzyme is shown above sequence. Cylinders, helices; straight block arrows, beta strands; curved block arrows, turns in the beta helix domain. Blue stars, residues interacting with ADP-Glc; red stars, residues interacting with ATP. Green residues are identical in all sequences, yellow residues are identical in most sequences, cyan residues are homologous in most sequences. Sequences abbreviations are: S.tuberosom_S, potato tuber α subunit; A.thaliana_S, Arabidopsis thaliana α subunit; Z.mays_S, maize α subunit; H.vulgare_S, Hydra vulgaris α subunit; C.reinhardtii_S: Chlamydomonas reinhardtii α subunit; S.tuberosom_L, potato tuber β subunit; A.tumefaciens, Agrobacterium tumefaciens; R.spheroides, Rhodobacter spheroides; B.stearothermop, Bacillus stearothermophilus. See online version for color.
The ADP-Glc PPase from potato tuber has been isolated and extensively characterized (Ballicora et al, 1995a). Ballicora et al (1995a) demonstrated that the α subunit of potato tuber ADP-Glc PPase expressed as a homotetramer is highly active in high concentrations of the activator 3-PGA, whereas the β subunit could not be expressed in an active form, confirming that α is the catalytic subunit while β is the modulatory subunit. The α4 enzyme has a lower apparent affinity (A0.5=2.4 mM) for the activator 3-PGA than the heterotetramer (A0.5=0.16 mM), and is also more sensitive to the inhibitor Pi (I0.5=0.08 mM in the presence of 3 mM 3-PGA) than the heterotetramer (I0.5=0.63 mM) (Ballicora et al, 1995a).
We report here the crystal structure of potato tuber α4 ADP-Glc PPase determined to 2.1 Å resolution. This is the first atomic resolution structure of ADP-Glc PPase and presents a conformation of this allosteric enzyme in its inhibited state. We also report the structures of the enzyme in complex with ATP and ADP-Glc determined to 2.6 and 2.2 Å, respectively. Based on these structures, we describe the structural basis for the allosteric regulation of the ADP-Glc PPase as well as insights into the enzyme's catalytic mechanism.
Results
Monomer
The potato tuber ADP-Glc PPase α subunit monomer is composed of an N-terminal catalytic domain and a C-terminal β-helix domain (Figure 2A). The overall fold of the catalytic domain shares strong similarity with the three pyrophosphorylases whose structures have been elucidated, namely N-acetylglucosamine 1-phosphate uridylyltransferase (GlmU) (Brown et al, 1999; Kostrewa et al, 2001; Olsen and Roderick, 2001; Sulzenbacher et al, 2001) and two glucose-1-phosphate thymidylyltransferases (RmlA and RffH) (Blankenfeldt et al, 2000; Sivaraman et al, 2002), although the primary sequences for the three distinct enzymes have very low similarities (RmlA and RffH share 68% sequence identity). The catalytic domain is composed of a mostly parallel but mixed seven-stranded β sheet covered by α helices, a fold reminiscent of the dinucleotide-binding Rossmann fold (Rossmann et al, 1975). The catalytic domain makes strong hydrophobic interactions with the C-terminal β-helix domain via an α helix that encompasses residues 285–297. The catalytic domain is connected to the C-terminal β-helix domain by a long loop containing residues 300–320. This loop makes numerous interactions with the equivalent region of another subunit.
Figure 2.
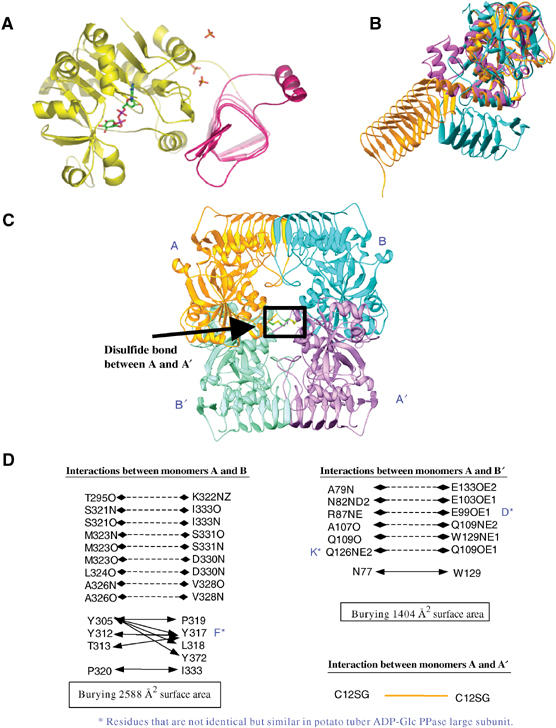
(A) ADP-Glc PPase monomer. Yellow, catalytic domain; pink, β-helix domain; ADP-Glc and sulfates are shown in atom type: carbon, green; oxygen, red; nitrogen, blue; phosphorous, magenta; sulfate, orange. (B) Overlay of ADP-Glc PPase (cyan), RmlA (r.m.s.d. 1.9 Å) (magenta), and GlmU (gold) (r.m.s.d. 2.5 Å). (C) ADP-Glc PPase tetramer. The disulfide bond between A and A′ is boxed. (D) Interactions between monomers in the tetramer.
The C-terminal domain comprising residues 321–451 adopts a left-handed β-helix fold composed of six complete or partial coils with two insertions, one of which encompasses residues 368–390 and the other encompasses residues 401–431. This type of left-handed β-helix domain fold has been found in the structures of several proteins (Raetz and Roderick, 1995; Kisker et al, 1996; Beaman et al, 1997; Brown et al, 1999; Kanamaru et al, 2002) but the orientation and length of the β-helix domain is completely different in the two structures (Figure 2B). Functionally, the other β-helix domains are either acetyltransferases, succinyltransferases, or a ‘membrane-puncturing device' in the case of the gp5 protein (Kanamaru et al, 2002). However, in ADP-Glc PPase, the β-helix domain is involved in cooperative allosteric regulation with the N-terminal catalytic region, interacts with the N-terminus within each monomer, and contributes to the unique oligomerization unprecedented in other structures.
Tetramer
The potato tuber ADP-Glc PPase α4 homotetramer has approximate 222 symmetry with dimensions of ∼80 × 90 × 110 Å3; it can be viewed as a dimer of dimers with each monomer labeled A, A′, B, and B′ (Figure 2C). Monomers A and B interact predominantly by an end-to-end stacking of their β-helix domains, although there is also a significant interface between the linker loop connecting the N-terminal domain and C-terminal domain (residues 300–320) (Figure 2D). This interface buries 2544 Å2 of surface area. The catalytic domains of the A and B′ (and B and A′) subunits also make an extensive interface. Several hydrogen bond and hydrophobic interactions stabilize this interface burying a surface area of 1400 Å2. All of the residues defining the dimerization interfaces are identical or similar in the β subunit, suggesting a similar dimerization interface between α and β subunits in the heterotetrameric enzyme (Figure 2C). Cys12 of monomer A and the equivalent cysteine residue of monomer A′ make a disulfide bond, as do equivalent cysteines of monomers B and B′, and this is the only interaction made between A and A′ (B and B′). The intersubunit disulfide bond between α subunits is preserved in the heterotetramer; however, there is no disulfide bond between β subunits as Cys12 is not conserved. This disulfide bond establishes the relative position of α subunits in the heterotetramer to be like A and A′ in the α4 homotetramer structure.
Sulfate binding
The electron density map for potato tuber α4 ADP-Glc PPase shows features with 20σ peak heights and tetrahedral shape. The size and shape together with the high sulfate concentration (150 mM) in the crystallization solution suggest that these are sulfate ions bound tightly within the enzyme. Two of these sulfates (sulfates 1 and 2) bind within 7.5 Å of each other in a crevice located between the N- and C-terminal domains of the enzyme (Figure 2A) and a third sulfate binds between two subunits of the enzyme (sulfate 3) (Figure 3A). Sulfate is an inhibitor of the potato tuber ADP-Glc PPase α subunit homotetramer with I0.5=2.8 mM in the presence of 6 mM 3-PGA. The enzyme has been shown to be completely inactive in 30 mM (Figure 3B). All three structures contain 12 sulfates within a tetramer in the asymmetric unit (three per subunit), and are all, therefore, representative of the inhibited conformation of the enzyme. In addition, a sulfate ion (sulfate 4) is bound in the active site of the A and A′ subunits in the ATP-bound α4 ADP-Glc PPase structure (Figure 4A).
Figure 3.
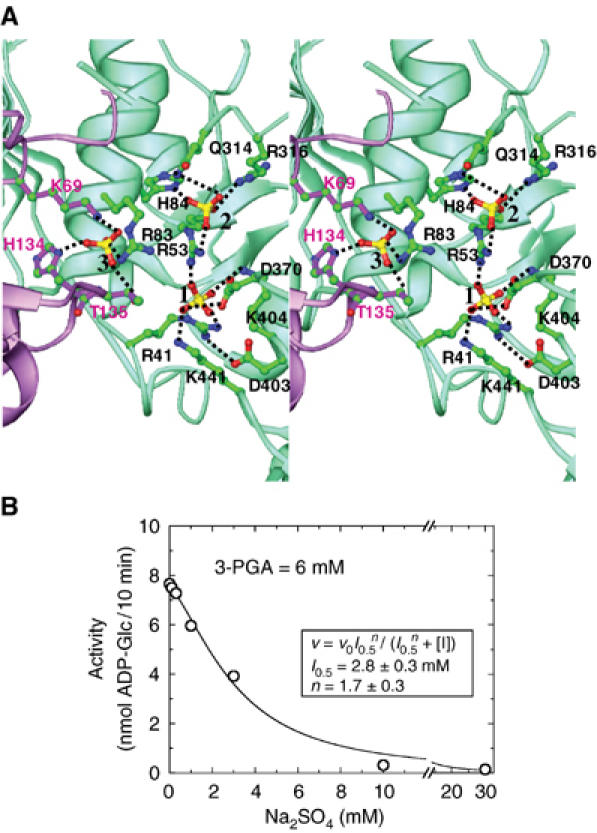
(A) ADP-Glc PPase A subunit sulfate-binding region. A subunit, green ribbon; B subunit, purple ribbon; sulfates and interacting residues are colored by atom type. (B) Inhibition of potato tuber ADP-Glc PPase small subunit by sulfate.
Figure 4.
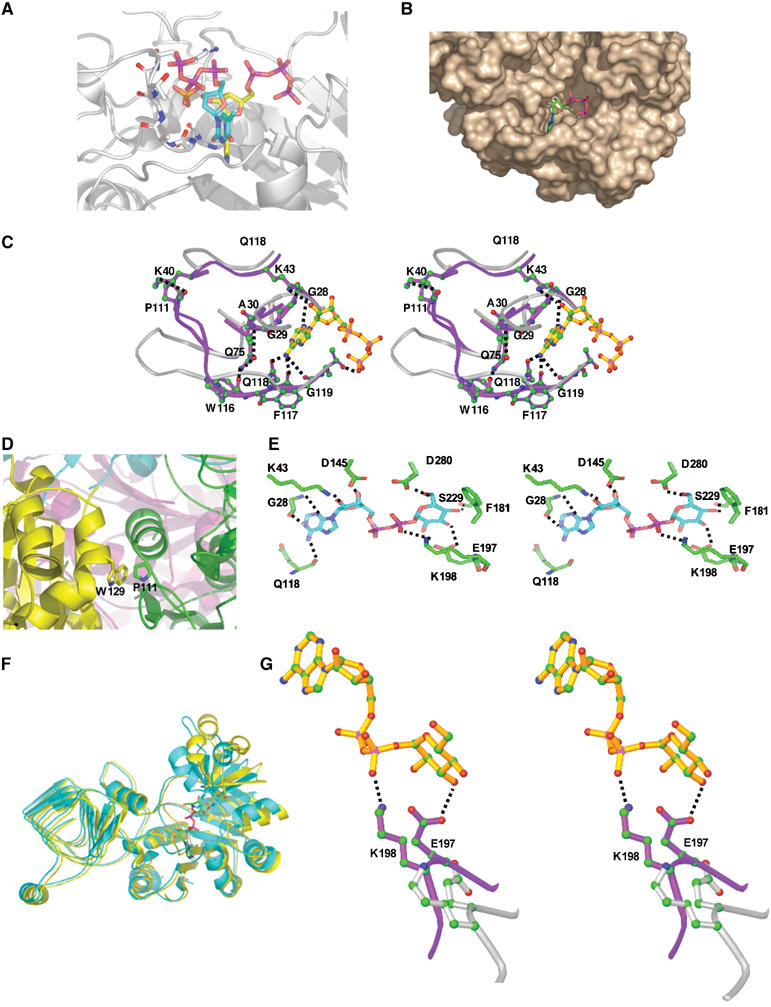
(A) Overlay of ATP in ADP-Glc PPase with TTP in RmlA. Yellow, carbon atoms of ATP from ATP-bound A subunit; cyan, carbon atoms of TTP from RmlA structure. All others are colored by atom type. (B) Surface of ADP-Glc PPase A subunit with bound ATP. (C) Local changes about the adenine occur when ATP binds. Overlay of the A subunit ATP-bound (lavender ribbons) and unbound (gray ribbons) active sites is shown. Mobile loops and residues of the ATP-bound structure are shown in green. Residues that cause the motion to be correlated are also shown (colored by atom type). (D) Intersubunit interaction between P111 and W129. Yellow, B subunit; green, A′ subunit. ATP binding causes a conformational change in P111 transducing motion of the region around W129. (E) Hydrogen bond interactions between ADP-Glc and the ADP-Glc PPase B subunit. Protein carbon bonds are colored green, ADP-Glc carbon bonds are magenta, and all other bonds are colored by atom type as above. (F) Large motion of the 168–231 B subunit subdomain. Cyan, ADP-Glc-bound B subunit; yellow, ATP-bound B subunit. (G) The large domain movement brings E197 and K198 in contact with ADP-Glc. Magenta bonds, ADP-Glc-bound B subunit; gray bonds, ATP-bound B subunit. Atoms are colored by type.
Sulfate 1 makes hydrogen bond interactions with R41, R53, K404, and K441 with D403 making a butressesing salt bridge interaction with R41. All of these residues are conserved in virtually all plant ADP-Glc PPase α subunits, and four of the five (all but K441) are strongly conserved in the bacterial ADP-Glc PPases as well (Figure 1). However, two of these residues are not conserved in the potato tuber β subunit. R41 is a Thr and K441 is a Glu in the β subunit (Figure 1). Given that these are not conservative mutations and a change of charge occurs at K441, this suggests that the potato tuber β subunit binds an anion poorly if at all at this site. Consistent with this conclusion, mutation of K404 or K441 to either Ala or Glu in the α subunit affects both activator and inhibitor allosteric effectiveness, but mutation of the β subunit K417 to Glu or Ala (equivalent to K404 in the α subunit) had no effect on the allosteric effectiveness of either 3-PGA or Pi (Ballicora et al, 1998). However, D413 in the potato tuber enzyme β subunit (D403 in the α subunit) was identified as important for activation by 3-PGA (Greene et al, 1996b), indicating that the region around the sulfate 1-binding site in the β subunit does play a role in the allosteric activation of the enzyme. Taken together, these studies indicate that the activator 3-PGA binds at or near the inhibitor-binding site defined in our structure by sulfate 1.
Sulfate 2 makes interactions with surrounding residues, R53, R83, H84, Q314, and R316 (Figure 3A). All of these residues are conserved in both subunits of all plant ADP-Glc PPases, indicating that the sulfate 2-binding site is used in both subunits of the enzyme. In fact, R53, R83, and H84 are strongly conserved in bacterial ADP-Glc PPases as well, indicating that this anion-binding site is important for all ADP-Glc PPases. Site-directed mutagenesis studies have shown that H83 of the Escherichia coli enzyme (H84 in potato tuber enzyme α subunit) is involved in activator binding (Hill and Preiss, 1998). In addition, mutations of R294 in the Anabaena enzyme (R316 in the potato tuber enzyme α subunit) lowered the apparent affinity for Pi more than 100-fold (Sheng and Preiss, 1997) and also resulted in changes in inhibitor selectivity (Frueauf et al, 2002). Taken together, these studies confirm the importance of this sulfate-binding site in the allosteric regulation of the enzyme, indicate that both 3-PGA and Pi may also bind near the sulfate 2-binding site and suggest that the β subunit may also bind allosteric regulators at this site. Sulfate 3 is located between two subunits (A and B′ or A′ and B). This sulfate makes interaction with R83 of one subunit and K69, H134, and T135 of the other subunit (Figure 3A). Two of these residues (K69 and R83) are conserved in both subunits of all plant ADP-Glc PPases, H134 is conserved in all α subunits, and T135 is conservatively changed to Asn in virtually all α subunits. Although none of these residues have been studied biochemically, the strong conservation of all four of these residues suggests that this site is also allosterically significant.
ATP binding
The active site of ADP-Glc PPase consists of a long cleft bordered at each end with a sugar- or adenine-binding pocket, has approximate dimensions of ∼20 Å × 20 Å × 15 Å (Figure 4B), and is lined by a number of residues that are conserved in related nucleotidyltransferases. Only the A and A′ subunits were bound with ATP. The entire molecule of ATP is ordered in the active site, although the conformation of the phosphates is completely different from that seen in other sugar-nucleotide PPase/NTP complexes (Figure 4A). In fact, the β- and γ-phosphates partially occupy the glucose-binding site, proving that the triphosphate conformation is incompetent for catalysis. A sulfate ion occupies the position in the active site bound by the TTP triphosphate in the TTP-bound RmlA structure (Figure 4A) (Blankenfeldt et al, 2000). When the enzyme is bound with ATP, both A and A′ subunits undergo almost identical conformational changes relative to the apo structure to accommodate ATP binding. Several regions move significantly: the GGXGXRL loop region from residue 27 to 34 conserved in most pyrophosphorylases, another loop region from residue 106 to 119, and residues K40, R41, Q75, and F76 (Figure 4C). Both loop regions make direct interactions with the adenine portion of the nucleotide. Specifically, the main-chain nitrogen of G28 makes a hydrogen bond with the adenine N3; several hydrophobic interactions are established between the adenine ring and L26 and G29; the side chain of Q118 makes a hydrogen bond with the adenine N6 and the main-chain oxygen of Q118 makes a hydrogen bond with N6. These residues all undergo correlated conformational changes upon ATP binding. Interactions of Q75 with both G30 and W116, and interactions of the K40 side chain with P111, couple the motion of the Q75, G30, and 106–119 regions (Figure 4C).
Furthermore, ATP binding in the A and A′ subunits drives conformational change in the B and B′ subunits as P111 of A and A′ is packed snugly against W129 in B′ and B, respectively (Figure 4D). Motion of P111 accompanying ATP binding leads directly to motion of W129 and in fact the entire region from 165 to 231 in the B/B′ subunits.
ADP-Glc binding
Subunits A, A′, and B bind ADP-Glc in the ADP-Glc PPase/ADP-Glc complex. Subunits A and A′ bind ADP-Glc identically and ADP-Glc binding produces identical conformational changes in A and A′ as occurs when ATP binds. The adenine and ribose of ADP-Glc in A and A′ are positioned identically to those of ATP and the interactions between the enzyme and ADP-Glc are also identical to those seen in the ADP-Glc PPase/ATP complex. No electron density is seen for the glucose moiety of ADP-Glc in the A and A′ active sites, indicating it to be disordered. This indicates that the conformational changes seen in A and A′ upon ATP or ADP-Glc binding are due almost exclusively to the adenine and ribose moieties. Both phosphates are also ordered, but are different from the phosphate positions in the other structures. In contrast, the entire ADP-Glc is well ordered in the active site of subunit B. The adenine and ribose positions are very similar to that seen in the A and A′ subunits although the region 112–117, which undergoes conformational change upon ATP or ADP-Glc binding in A and A′, is disordered in B both with and without ADP-Glc in the active site and it is disordered in B′ as well. The two phosphates and glucose moieties are well ordered in the B active site and adopt positions and conformations similar to that seen in other sugar nucleotide PPase complex structures. There are several direct interactions between the enzyme and the glucose moiety of ADP-Glc. These include hydrogen bonds between E197, S229, D280, and the glucose moiety (Figure 4E). In addition, K198 makes a salt bridge with the glucose phosphate.
The B subunit undergoes a very large subdomain movement in response to ADP-Glc binding. While the unbound and ADP-Glc-bound B monomers are similar in this region, the 168–231 region in B is shifted dramatically when ATP is bound in the A and A′ monomers (Figure 4F). When all 12 subunits from the three tetramer structures are overlaid, although the other 10 subunits (A, A′, and B′ in three structures plus B in the ATP-bound B structure for a total of 10 subunits) display some motion in this region, the Cα positions all fall within 2 Å of each other. On the other hand, both the ADP-Glc-bound and apo B subunits are shifted by as much as 6 Å from any of the other 10 subunits in this region. Two residues, E197 and K198, are critical for binding the glucose and phosphate moiety of ADP-Glc; K198 has been characterized as a G1P-binding residue by site-directed mutagenesis (Fu et al, 1998b) and both are part of a motif present in many sugar-nucleotide pyrophosphorylases. These two residues are shifted out of the binding pocket in the B subunit of the ATP-bound structure, while they are pulled more inward in the unbound B subunit and are pulled in significantly in the ADP-Glc-bound molecule (Figure 4G). Among all 12 subunits, only the ADP-Glc-bound B subunit has these two residues positioned properly for interaction with the glucose moiety. In all other subunits, these two residues are positioned too far out of the active site to make direct interaction with the glucose and phosphate moieties of the substrate.
Discussion
Variations in subunit substrate and product affinity
A puzzling result of our work is the apparent difference in binding affinity between subunits that are identical in sequence in α4 ADP-Glc PPase. While only the A and A′ subunits display affinity for ATP, ADP-Glc binds to A, A′, and B subunits, although the glucose moiety is apparently disordered in the A and A′ subunits. This binding pattern agrees well with binding studies of the E. coli ADP-Glc PPase, which also contains four identical subunits. While only two molecules of ATP bind to the tetramer, about three molecules of ADP-Glc bind (Haugen and Preiss, 1979). It would seem that the A and A′ subunits have higher affinity for the adenine and adenine sugar moieties, while the B subunit has higher affinity for the glucose moiety than A and A′. Inspection of the interactions of adenine with A and A′ versus B supports this conclusion. While the position of the adenine moiety is very similar in A/A′ versus B, conformational differences in the 108–118 mobile loop lead to three hydrogen bonds between adenine and the A/A′ subunit: the side chain of Q118 makes a 2.8 Å hydrogen bond with N6, the main-chain O of Q118 makes a 2.7 Å hydrogen bond again with N6, and the main-chain carbonyl of G119 makes a 3.1 Å hydrogen bond also with N6. The distances between N6 and these residues are all greater than 3.2 Å in the ADP-Glc-bound B subunit. In addition, the region between residues 112–117 is disordered in the ADP-Glc-bound B subunit, leaving the edge of the adenine ring partially exposed, while this region is well ordered in the A/A′ subunit, better sequestering the adenine. To understand the reason for the difference in conformation of the 108–118 region, we overlaid the ATP-bound A subunit onto the B subunit of the ADP-Glc-bound tetramer and found that there are significant collisions between residues 109–111 of the overlaid A and W129 of the adjacent A′ subunit, indicating that the 108–118 region of the B subunit cannot assume the conformation seen in the ATP-bound A subunit due to inter-subunit clashes with A′. The conclusion to be drawn from this is that the tetramer enforces nonidentical conformations in the subunits. While the conformations of A and A′ change but are always identical to one another, the B and B′ subunits must adopt different conformations to accommodate them. We expect that this asymmetry will be even more pronounced in the structure of the α2β2 heterotetramer, where the subunits are not identical. While the catalytic α subunits will probably behave similar to the A and A′ subunits in the α4 structure, the β subunits will adopt different conformations enforced by a combination of inter-subunit interactions and differences in amino-acid sequence. Indeed, the region in the β subunit equivalent to residues 112–116 in the α subunit is completely different and contains a two-amino-acid insertion in this region (Figure 1).
What is more difficult to explain is the apparent higher affinity of the B subunit for the glucose moiety. One possible explanation is that the glucose moiety has somehow been cleaved in the A and A′ subunits. We regard this possibility as unlikely since the B subunit shows essentially 100% occupancy for the entire ADP-Glc molecule, indicating that significant quantities of ADP-Glc remain intact during the soaking experiment. We conclude that the B subunit has significantly higher affinity for ADP-Glc and ascribe this increased affinity to the interaction of E197 and K198 with ADP-Glc. These interactions require a significant shift of the 168–231 subdomain in the B subunit, which does not occur in A/A′.
Implication for catalysis
Although we do not have a metal ion bound in the active site of α4 ADP-Glc PPase in any of our structures, the structures of two pyrophosphorylases, GlmU and RffH, have a metal bound in their active sites (Olsen and Roderick, 2001; Sivaraman et al, 2002). A single metal ion (a Co2+ ion in GlmU or am Mg2+ ion in another GlmU structure and in the RffH structure) was located in an almost identical location in the two enzymes when the active sites are structurally aligned. In GlmU, the metal ion is chelated to two conserved residues (D105 and D227) and to the two phosphates of the product UDP-N-acetyl glucosamine. In RffH, the Mg2+ is also bound to two conserved carboxylate residues (D223 and D108) and to the α-phosphate of dTTP. When the ADP-Glc PPase active site is aligned with the active sites of these enzymes, two acidic residues, D145 and D280, are spatially close to the metal chelating residues in RffH and GlmU, and are absolutely conserved in all ADP-Glc PPases (Figure 1). Mutation of D145 to Asn in the potato tuber enzyme and of the equivalent D142 in the E. coli enzyme results in a reduction in catalytic activity by four orders of magnitude (Hill et al, 1991; Frueauf et al, 2001). Taken together, we conclude that the metal-mediated catalytic mechanism proposed for RffH and GlmU is also used by ADP-Glc PPase. We also conclude that the metal ion is chelated by the residues equivalent to D145 and D280 in all ADP-Glc PPases and that the mutational sensitivity of D145 is due to the requirement for metal ion in the reaction. The absolute requirement for a metal ion has been biochemically demonstrated for ADP-Glc PPase from several organisms (Iglesias and Preiss, 1992; Ballicora et al, 2003, 2004).
Structural data from GlmU, RmlA, and RffH have shed considerable light on the mechanism of sugar nucleotide pyrophosphorylases, and the similarity between the active site of α4 ADP-Glc PPase and these enzymes indicates a similar mechanism for all of these enzymes. The structure of the RffH/dTTP complex is particularly informative because it identifies the GXGXRL loop, which is strongly conserved in all of these enzymes, to be the site of ATP triphosphate binding and shows that the residue equivalent to R33 in the α subunit of ADP-Glc PPase is critical for triphosphate binding (Figure 4). Conformational change of this loop will therefore have profound effects on the activity of the enzyme. In addition to R33, D145, D280, R43, E197, and K198 (potato tuber α4 numbering) are also conserved in the other sugar nucleotide pyrophosphorylases of known structures, are in similar locations in the active sites, and make similar interactions with the sugar nucleotide.
Since in our structure the β-phosphate of ATP partially occupies the glucose-binding site (Figure 5A), representing a conformation incompetent for catalysis, we modeled ATP and G1P into the active site of ADP-Glc PPase using the RffH and RmlA structures (Blankenfeldt et al, 2000; Sivaraman et al, 2002) as a guide (Figure 5B). The β- and γ-phosphates of ATP bind to the conserved loop around R33, folding back over the nucleotide and leaving the space opposite the pyrophosphate entity free for G1P binding. The location of the phosphate group in G1P and that of the α-phosphate of ATP are close to the positions in the B subunit ADP-Glc complex. K198 is an absolutely conserved amino acid in ADP-Glc PPases and in other sugar nucleotidyltransferases. K198 stabilizes the negative charge on the phosphate of G1P in this model, increasing the nucleophilicity of the O1P atom. Electrostatic repulsions between the negative charges at the phosphate of G1P and the phosphates of ATP are compensated for by chelation between the phosphates of ATP, G1P, and Mg2+. A number of conserved basic side chains also surround the phosphates at the active site (R33, K43, K198). Additional counterbalancing charges come from the N-terminal dipole of the helical turn at R33 and the main-chain amide nitrogen pocket formed by residues G27 to T32. In fact, R33 makes a close hydrogen bond with a phosphate oxygen of ADP-Glc in the ADP-Glc-bound structure. In the mutagenesis studies of GlmU (Brown et al, 1999), mutation of R15 (equivalent to R33 of ADP-Glc PPase) reduced kcat almost 6000-fold, while KM was doubled, confirming that this residue has an important role in orienting and charge compensation of the pyrophosphate. The P36 within this flexible loop adopts a cis-peptide bond. The loop is located at the interface between the N-terminal catalytic domain and the C-terminal β-helix domain within the immediate vicinity of the regulator-binding site, suggesting a possible route for crosstalk between the active site and the allosteric regulation site.
Figure 5.
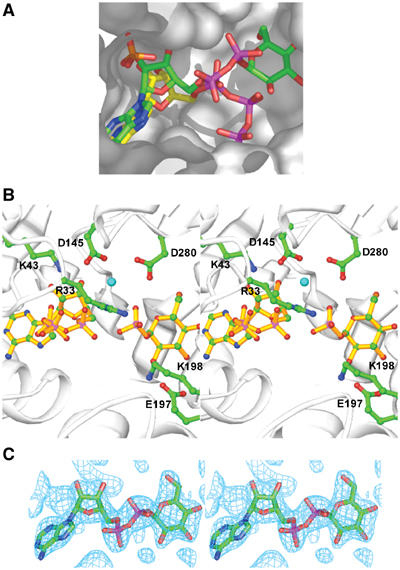
(A) The β-phosphate of ATP collides with the glucose-binding site. The ATP-bound A subunit and the ADP-Glc-bound B subunit are overlayed. The sulfate ion bound in the active site in the ATP-bound A subunit is also shown. ATP carbon bonds are yellow and all other bonds are colored by atom type; ADP-Glc carbon bonds are green and all other bonds are colored by atom type. (B) Modeled ATP and G1P in the ADP-Glc PPase active site. (C) Sigma-A-weighted 2Fo−Fc map contoured at 1.2σ around ADP-Glc in the ADP-Glc-bound B subunit.
While most of the residues in the active site are conserved in the catalytically inactive β subunit, there are two changes: R33 is a lysine and K43 is a threonine. Since K43 interacts with ADP-Glc and R33 is critical for proper ATP triphosphate binding, both are likely to be important in deactivating the β subunit.
Allosteric regulation
Our structure has identified three allosteric inhibitor-binding sites, sulfate 1, sulfate 2, and sulfate 3, two of which (sulfate 1 and sulfate 2) have been shown by numerous biochemical experiments to be involved in the allosteric regulation of the enzyme. The high sequence homology of the residues in the sulfate 3-binding site suggests that it too may represent an important allosteric binding site. The major question is how these allosteric binding sites communicate with the active site to affect catalytic activity. Part of the answer to this question comes from careful study of the conformational changes that occur in the GXGXXG loop encompassing amino acids 27–34 upon active site binding of either ATP or ADP-Glc. This loop has a similar conformation in all unbound subunits where the loop is flipped into the active site. A hydrogen bond between K43 and the main-chain oxygen of either G29 or A30 contributes to the stability of this conformation. Upon binding, however, the loop is forced to move out of the active site to accommodate ATP or ADP-Glc binding and this hydrogen bond is severed (Figure 4C). Given that K41 contacts sulfate 1 in the allosteric binding site, conformational change of K41 upon replacement of the inhibitor sulfate or phosphate with the activator 3-PGA could lead to conformational change of K43, destabilizing the catalytically incompetent flipped in conformation of this loop. K43 is also pointing directly into the active site and makes a hydrogen bond with the adenine sugar in the ADP-Glc-bound B subunit (Figure 4E). Therefore, conformational change of K43 will also directly affect the active site. Conformational change of this loop could also come directly from movement of the K41 region, since it is only six amino acids away from the 27–34 loop. As discussed above, the motion of the 27–34 loop is correlated to the motion of the 108–116 loop, which also undergoes conformational change upon ATP binding (Figure 4C). Conformational change in the K41 region upon activator binding could therefore be correlated with conformational change of the 108–116 loop, preorganizing the active site for ATP binding. In support of this model, the P52L potato tuber β subunit mutant (equivalent to P36 in the α subunit) is substantially less sensitive to 3-PGA activation (Greene et al, 1996a). P36 is a cis proline in the present structure and mutation of this residue is likely to significantly alter communication between the allosteric binding site and the 27–33 GXGXGRL loop.
Several pieces of evidence indicate that inter-subunit motion may also play a role in the allosteric regulation of the enzyme. Removal of the C12 disulfide bond either by mutation or by reduction results in an enzyme that is nearly constitutively active (Fu et al, 1998a; Ballicora et al, 2000), indicating that the inter-subunit interaction between α subunits is an important part of the allosteric mechanism. Release of this constraint could result in more flexible inter-subunit motion. Sulfate 3, which bridges two subunits, may also inhibit inter-subunit motion resulting in a more inhibited enzyme. The binding of ADP-Glc to the B subunit and concomitant motion of the 168–231 subdomain required for the interaction of E197 and K198 with ADP-Glc results in differences in the subunit interface. Inter-subunit motion may therefore drive the motion of this subdomain upon activation preorganizing the active site for binding of glucose phosphate.
The complete mechanism of the allosteric regulation by ADP-Glc PPase cannot be deduced until the conformation of the enzyme in its activated state is elucidated. Nonetheless, the current structural results strongly support previous data on the allosteric regulation of this enzyme and provide some insights into how the binding of allosteric effectors could affect catalysis.
Materials and methods
Protein expression, purification, and crystallization
Potato tuber ADP-Glc PPase α subunit was expressed and purified as described previously (Iglesias et al, 1993; Ballicora et al, 1995a). Protein was crystallized in a condition similar to that reported previously (Binderup et al, 2000). Briefly, crystals were grown from 14% (w/v) PEG MME 2000, 0.15 M (NH4)2SO4, and 0.1 M Na acetate pH 4.5 at 4°C. The space group is P21 with unit cell dimensions a=79.5 Å, b=137.6 Å, c=91.6 Å, and β=112.5°. The mercury derivative crystal used in phasing was obtained by soaking crystals in 1 mM p-chloromercurybenzene sulfonic acid (PCMBS) for 8 h. Crystals were cryoprotected with the mother liquor supplemented with 20% (w/v) glycerol before being flash-frozen for data collection. The ADP-Glc PPase/ATP complex was obtained after soaking apo crystals in the crystal mother liquor supplemented with 5 mM ATP and 10 mM Mg2+. The ADP-Glc PPase/ADP-Glc complex was obtained after soaking apo ADP-Glc PPase crystals in the crystal mother liquor supplemented with 6 mM ADP-Glc.
Data collection and processing
Experimental phases were obtained from a multiwavelength anomalous diffraction (MAD) (Hendrickson, 1991) experiment. Diffraction data (Table I) were collected at BM-19 of the Structural Biology Center (SBC) at the Advanced Photon Source (APS), Argonne National Laboratory. Three data sets were collected around the mercury LIII edge: the peak at 12.308 keV, the inflection point at 12.285 keV, and the high-energy remote at 12.486 keV. The highest resolution data used in the final structure refinement were collected to 2.1 Å at the IMCA-CAT ID-17 beamline at APS. Data for the ADP-Glc PPase/ATP complex and ADP-Glc PPase/ADP-Glc complex crystals were collected at the COM-CAT 32-ID beamline at APS. All data were collected at 100 K, and were reduced and scaled with the HKL 2000 program suite (Otwinowski and Minor, 1996). Data collection statistics are given in Table I.
Table 1.
Data collection and refinement statistics
| Hg-derivative 1 |
|||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
f″ max |
f′ min |
Remote |
Hg-derivative 2b |
ATP-bound |
ADP-Glc-bound |
|||||||||||||||||||||
| Data collectiona | |||||||||||||||||||||||||||
| Beamline | SBC-19BM | SBC-19BM | SBC-19BM | IMCA-17ID | COM-CAT | COM-CAT | |||||||||||||||||||||
| Wavelength (Å) | 1.00739 | 1.00922 | 0.99298 | 1.0087 | 0.99997 | 0.99997 | |||||||||||||||||||||
| Resolution (Å) | 2.8 (2.9–2.8) | 2.69 (2.8–2.69) | 3.04 (3.2–3.04) | 2.1 (2.18–2.1) | 2.6 (2.69–2.6) | 2.3 (2.38–2.3) | |||||||||||||||||||||
| Total observations | 199 596 | 240 609 | 163 501 | 383 291 | 235 693 | 315 737 | |||||||||||||||||||||
| Unique observations | 45 320 | 56 100 | 49 216 | 104 502 | 56 970 | 78 436 | |||||||||||||||||||||
| a (Å) | 79.5 | 79.5 | 79.5 | 79.3 | 79.6 | 79.9 | |||||||||||||||||||||
| b (Å) | 137.6 | 137.6 | 137.6 | 137.3 | 143.0 | 138.0 | |||||||||||||||||||||
| c (Å) | 91.6 | 91.6 | 91.6 | 91.5 | 90.4 | 90.8 | |||||||||||||||||||||
| β (deg) | 112.5 | 112.5 | 112.5 | 112.6 | 112.5 | 112.9 | |||||||||||||||||||||
| Rsym (%) | 8.2 (27.1) | 11.3 (55) | 14 (40.8) | 7.1 (43.7) | 11.2 (49.6) | 8.5 (57.2) | |||||||||||||||||||||
| I/σ(I) | 9.8 (4.0) | 10.5 (1.9) | 5.8 (1.9) | 15.5 (1.9) | 12.3 (2.3) | 18.0 (2.5) | |||||||||||||||||||||
| Completeness (%) | 99.6 (96.3) | 99.8 (99.7) | 96.0 (98.2) | 99.9 (99.5) | 98.8 (97.8) | 99.7 (99.9) | |||||||||||||||||||||
| Refinement | |||||||||||||||||||||||||||
| Resolution range (Å) | 20–2.1 (2.2–2.1) | 20–2.6 (2.72–2.6) | 20–2.3 (2.4–2.3) | ||||||||||||||||||||||||
| No. of reflections | 99 259 | 43 529 | 66 782 | ||||||||||||||||||||||||
| R factord (%) | 17.2 (21.7) | 17.4 (29.4) | 18.3 (25.7) | ||||||||||||||||||||||||
| Rfree (%) | 22.9 (24.7) | 25.2 (35.7) | 24.1 (29.3) | ||||||||||||||||||||||||
| Number of ligands | |||||||||||||||||||||||||||
| PCMBS | 4 | 0 | 0 | ||||||||||||||||||||||||
| Sulfate ions | 12 | 14 | 16 | ||||||||||||||||||||||||
| ATP | 0 | 2 | 0 | ||||||||||||||||||||||||
| ADP-Glc | 0 | 0 | 3 | ||||||||||||||||||||||||
| Residues included in refinement | A: 10–26, 33–451; B: 11–26, 32–112, 117–451; A′: 10–28, 32–451; B′: 11–26, 32–111, 117–451 | A: 10–451; B: 11–113, 118–451; A′: 10–451; B′: 13–112, 119–451 | A: 11–451; B: 12–113, 117–451; A′: 11–451; B′: 12–112, 118–451 | ||||||||||||||||||||||||
| Number of non-hydrogen atoms | 14 740 | 13 768 | 14 017 | ||||||||||||||||||||||||
| Protein | 13 409 | 13 383 | 13 374 | ||||||||||||||||||||||||
| Solvent | 1227 | 259 | 471 | ||||||||||||||||||||||||
| Ligands | 104 | 126 | 172 | ||||||||||||||||||||||||
| R.m.s.d. of bonds (Å) | 0.012 | 0.069 | 0.0073 | ||||||||||||||||||||||||
| R.m.s.d. of angles (deg) | 1.71 | 1.34 | 1.4 | ||||||||||||||||||||||||
| Average B factor (all atoms) (Å2) | 35.7 | 44.5 | 43.5 | ||||||||||||||||||||||||
| Average B factor (ATP, ADP-Glc) |
|
|
|
|
62.8 |
54.6 |
|||||||||||||||||||||
| aValues in parentheses are for the highest resolution shell. | bThe highest resolution data, used for phase extension and final refinement. | cRsym=∑∣I−〈I〉∣∑I, where I is intensity and 〈I〉 is the average I for all equivalent reflections. | dR factor=∑∣F(obs)–F(calc)∣/∑F(obs); 10% of the data that were excluded from the refinement were used to calculate Rfree. | ||||||||||||||||||||||||
Structure determination and refinement
The structure was solved by the MAD method (Hendrickson, 1991). Four Hg atoms were located in anomalous difference Patterson maps using the program SOLVE (Terwilliger and Berendzen, 1999). The figure of merit was 0.49 for data within the resolution range 20–3.3 Å and 0.25 for the highest resolution bin (3.41–3.30 Å). After density modification using RESOLVE (Terwilliger, 2000), the figure of merit was 0.61 for the data within the resolution range 20–3.3 Å and 0.35 for the highest resolution bin (3.41–3.30 Å). The resulting electron density map was partially interpretable, especially the C-terminal β-helix domain. Poly-alanine chains were built in these regions and were used to obtain the non-crystallographic symmetry (NCS) matrix for averaging. Four-fold NCS averaging and solvent flattening were performed using the program DM (Cowtan, 1994). The resulting phase set was transformed to the highest resolution data collected on a mercury derivative crystal soaked with PCMBS, and extended to 2.1 Å with DM (Cowtan, 1994). The electron density map was of good quality and allowed for assignments of side chains of the β-helix domain as well as main chains of the catalytic domain. The NCS matrix and envelope were gradually improved. All of the manual building and refitting were carried out using the program O (Jones et al, 1991). The structure was refined using CNS (Brunger et al, 1998). Strict NCS restraints were applied during the earlier stages of the refinement, and were removed at later stages. The final refinement R factor is 16.2% and Rfree is 22.7%; the model has good stereochemistry and no energetically unfavorable main-chain torsion angles as evaluated by PROCHECK (Laskowski et al, 1993). Each monomer has one PCMBS compound covalently attached to a cysteine residue (C354); the structure also contains 12 sulfate ions (three per monomer) and 1227 solvent molecules. The structures of ADP-Glc PPase in complex with ATP and ADP-Glc were determined by difference Fourier synthesis. Model building for these two complex structures was carried out using the program O (Jones et al, 1991) and TURBO-FRODO (Roussel and Cambillau, 1991) and refinement was carried out with CNS (Brunger et al, 1998). The R factor and Rfree for the ATP-bound structure are 18.1 and 25.6%, respectively, and for the ADP-Glc-bound structure they are 18.3 and 24.1%, respectively (Table I; Figure 5C shows the electron density map around ADP-Glc).
Enzyme inhibition assay
The activity was assayed by the method of Yep et al (2004) after 10 min of incubation at 37°C. The reaction mixture contained 50 mM HEPES (pH 8.0), 8 mM MgCl2, 3 mM dithiothreitol, 0.5 mM G1P (∼1000 c.p.m./nmol), 2 mM ATP, 0.0015 U/ml pyrophosphatase, 0.1 mg/ml bovine serum albumin, and 33 ng of purified potato tuber α subunit in a total volume of 0.2 ml. Concentrations of 3-PGA and Na2SO4 are as indicated. Points are the average of duplicates. Fitting to the Hill plot was performed as described previously (Yep et al, 2004).
Acknowledgments
We are grateful to beamline staff at SBC-CAT 19-BM, IMCA-CAT 17-ID, and COM-CAT 32-ID at the APS, Argonne National Laboratory. Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-ENG-38. This research was supported by DOE research grant DE-FG02-93ER20121 (JP) and by NSF research grant MCB-9982536 (JHG).
References
- Ball S, Marianne T, Dirick L, Fresnoy M, Delrue B, Decq A (1991) A Chlamydomonas reinhardtii low starch mutant is defective for 3-phosphoglycerate activation and orthophosphate inhibition of ADP-glucose pyrophosphorylase. Planta 185: 17–26 [DOI] [PubMed] [Google Scholar]
- Ballicora MA, Frueauf JB, Fu Y, Schurmann P, Preiss J (2000) Activation of the potato tuber ADP-glucose pyrophosphorylase by thioredoxin. J Biol Chem 275: 1315–1320 [DOI] [PubMed] [Google Scholar]
- Ballicora MA, Fu Y, Nesbitt NM, Preiss J (1998) ADP-glucose pyrophosphorylase from potato tubers. Site-directed mutagenesis studies of the regulatory sites. Plant Physiol 118: 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballicora MA, Iglesias AA, Preiss J (2003) ADP-glucose pyrophosphorylase, a regulatory enzyme for bacterial glycogen synthesis. Microbiol Mol Biol Rev 67: 213–225, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballicora MA, Iglesias AA, Preiss J (2004) ADP-glucose pyrophosphorylase: a regulatory enzyme for plant starch synthesis. Photosynth Res 79: 1–24 [DOI] [PubMed] [Google Scholar]
- Ballicora MA, Laughlin MJ, Fu Y, Okita TW, Barry GF, Preiss J (1995a) Adenosine 5′-diphosphate-glucose pyrophosphorylase from potato tuber. Significance of the N terminus of the small subunit for catalytic properties and heat stability. Plant Physiol 109: 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaman TW, Binder DA, Blanchard JS, Roderick SL (1997) Three-dimensional structure of tetrahydrodipicolinate N-succinyltransferase. Biochemistry 36: 489–494 [DOI] [PubMed] [Google Scholar]
- Binderup K, Watanabe L, Polikarpov I, Preiss J, Arni RK (2000) Crystallization and preliminary X-ray diffraction analysis of the catalytic subunit of ADP-glucose pyrophosphorylase from potato tuber. Acta Crystallogr D 56 (Part 2): 192–194 [DOI] [PubMed] [Google Scholar]
- Blankenfeldt W, Asuncion M, Lam JS, Naismith JH (2000) The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J 19: 6652–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K, Pompeo F, Dixon S, Mengin-Lecreulx D, Cambillau C, Bourne Y (1999) Crystal structure of the bifunctional N-acetylglucosamine 1-phosphate uridyltransferase from Escherichia coli: a paradigm for the related pyrophosphorylase superfamily. EMBO J 18: 4096–4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Cowtan K (1994) DM, an automated procedure for density modification and phase improvement. Joint CCP4 and ESF-EACBM Newsl Protein Crystallogr 31: 34–38 [Google Scholar]
- Frueauf JB, Ballicora MA, Preiss J (2001) Aspartate residue 142 is important for catalysis by ADP-glucose pyrophosphorylase from Escherichia coli. J Biol Chem 276: 46319–46325 [DOI] [PubMed] [Google Scholar]
- Frueauf JB, Ballicora MA, Preiss J (2002) Alteration of inhibitor selectivity by site-directed mutagenesis of Arg(294) in the ADP-glucose pyrophosphorylase from Anabaena PCC 7120. Arch Biochem Biophys 400: 208–214 [DOI] [PubMed] [Google Scholar]
- Fu Y, Ballicora MA, Leykam JF, Preiss J (1998a) Mechanism of reductive activation of potato tuber ADP-glucose pyrophosphorylase. J Biol Chem 273: 25045–25052 [DOI] [PubMed] [Google Scholar]
- Fu Y, Ballicora MA, Preiss J (1998b) Mutagenesis of the glucose-1-phosphate-binding site of potato tuber ADP-glucose pyrophosphorylase. Plant Physiol 117: 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene TW, Chantler SE, Kahn ML, Barry GF, Preiss J, Okita TW (1996a) Mutagenesis of the potato ADPglucose pyrophosphorylase and characterization of an allosteric mutant defective in 3-phosphoglycerate activation. Proc Natl Acad Sci USA 93: 1509–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene TW, Woodbury RL, Okita TW (1996b) Aspartic acid 413 is important for the normal allosteric functioning of ADP-glucose pyrophosphorylase. Plant Physiol 112: 1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen TH, Preiss J (1979) Biosynthesis of bacterial glycogen. The nature of the binding of substrates and effectors to ADP-glucose synthase. J Biol Chem 254: 127–136 [PubMed] [Google Scholar]
- Hendrickson WA (1991) Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254: 51–58 [DOI] [PubMed] [Google Scholar]
- Hill MA, Kaufmann K, Otero J, Preiss J (1991) Biosynthesis of bacterial glycogen. Mutagenesis of a catalytic site residue of ADP-glucose pyrophosphorylase from Escherichia coli. J Biol Chem 266: 12455–12460 [PubMed] [Google Scholar]
- Hill MA, Preiss J (1998) Functional analysis of conserved histidines in ADP-glucose pyrophosphorylase from Escherichia coli. Biochem Biophys Res Commun 244: 573–577 [DOI] [PubMed] [Google Scholar]
- Iglesias AA, Barry GF, Meyer C, Bloksberg L, Nakata PA, Greene T, Laughlin MJ, Okita TW, Kishore GM, Preiss J (1993) Expression of the potato tuber ADP-glucose pyrophosphorylase in Escherichia coli. J Biol Chem 268: 1081–1086 [PubMed] [Google Scholar]
- Iglesias AA, Preiss J (1992) Bacterial glycogen and plant starch biosynthesis. Biochem Educ 20: 196–203 [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Kanamaru S, Leiman PG, Kostyuchenko VA, Chipman PR, Mesyanzhinov VV, Arisaka F, Rossmann MG (2002) Structure of the cell-puncturing device of bacteriophage T4. Nature 415: 553–557 [DOI] [PubMed] [Google Scholar]
- Kisker C, Schindelin H, Alber BE, Ferry JG, Rees DC (1996) A left-hand beta-helix revealed by the crystal structure of a carbonic anhydrase from the archaeon Methanosarcina thermophila. EMBO J 15: 2323–2330 [PMC free article] [PubMed] [Google Scholar]
- Kostrewa D, D'Arcy A, Takacs B, Kamber M (2001) Crystal structures of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase, GlmU, in apo form at 2.33 Å resolution and in complex with UDP-N-acetylglucosamine and Mg(2+) at 1.96 Å resolution. J Mol Biol 305: 279–289 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thronton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lin TP, Caspar T, Somerville C, Preiss J (1988) Isolation and characterization of a starchless mutant of Arabidopsis thaliana L. Henyh lacking ADP-glucose pyrophosphorylase activity. Plant Physiol 86: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata PA, Greene TW, Anderson JM, Smith-White BJ, Okita TW, Preiss J (1991) Comparison of the primary sequences of two potato tuber ADP-glucose pyrophosphorylase subunits. Plant Mol Biol 17: 1089–1093 [DOI] [PubMed] [Google Scholar]
- Okita TW, Nakata PA, Anderson JM, Sowokinos J, Morell M, Preiss J (1990) The subunit structure of potato tuber ADP-glucose pyrophosphorylase. Plant Physiol 93: 785–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen LR, Roderick SL (2001) Structure of the Escherichia coli GlmU pyrophosphorylase and acetyltransferase active sites. Biochemistry 40: 1913–1921 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1996) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Preiss J (1982a) Biosynthesis of starch and its regulation. In Encyclopedia of Plant Physiology: Plant Carbohydrates, Tanner W, Loewus F (eds) Vol. 13A, pp 397–417. Berlin, Germany: Springer-Verlag [Google Scholar]
- Preiss J (1982b) Regulation of the biosynthesis and degradation of starch. Annu Rev Plant Physiol 54: 431–454 [Google Scholar]
- Preiss J (1984) Bacterial glycogen synthesis and its regulation. Annu Rev Microbiol 38: 419–458 [DOI] [PubMed] [Google Scholar]
- Preiss J (1988) Biosynthesis of starch and its regulation. In The Biochemistry of Plants, Preiss J (ed) pp 181–254. New York, NY: Academic Press [Google Scholar]
- Preiss J, Shen L, Greenberg E, Gentner N (1966) Biosynthesis of bacterial glycogen. IV. Activation and inhibition of the adenosine diphosphate glucose pyrophosphorylase of Escherichia coli B″. Biochemistry 5: 1833–1845 [DOI] [PubMed] [Google Scholar]
- Preiss J, Sivak MN (1998a) Biochemistry, molecular biology and regulation of starch synthesis. Genet Eng (NY) 20: 177–223 [DOI] [PubMed] [Google Scholar]
- Preiss J, Sivak MN (1998b) Starch and glycogen biosynthesis. In Comprehensive Natural Products Chemistry, Pinto BM (ed) Vol. 3, pp 441–495. Oxford, UK: Pergamon Press [Google Scholar]
- Raetz CR, Roderick SL (1995) A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science 270: 997–1000 [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Liljas A, Branden CI, Bansazak LJ (1975) Evolutionary and structural relationships among dehydrogenases. In The Enzymes, Boyer IPD (ed) Vol. 11, pp 61–102. New York, NY: Academic Press [Google Scholar]
- Roussel A, Cambillau C (1991) The TURBO-FRODO Graphics Package, Silicon Graphics Corp. [Google Scholar]
- Sheng J, Preiss J (1997) Arginine294 is essential for the inhibition of Anabaena PCC 7120 ADP-glucose pyrophosphorylase by phosphate. Biochemistry 36: 13077–13084 [DOI] [PubMed] [Google Scholar]
- Sivak MN, Preiss J (1998) Starch: basic science to biotechnology. In Advances in Food and Nutrition Research, Taylor SL (ed) Vol. 41, pp 1–199. San Diego, CA: Academic Press [Google Scholar]
- Sivaraman J, Sauve V, Matte A, Cygler M (2002) Crystal structure of Escherichia coli glucose-1-phosphate thymidylyltransferase (RffH) complexed with dTTP and Mg2+. J Biol Chem 277: 44214–44219 [DOI] [PubMed] [Google Scholar]
- Smith-White BJ, Preiss J (1992) Comparison of proteins of ADP-glucose pyrophosphorylase from diverse sources. J Mol Evol 34: 449–464 [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G, Gal L, Peneff C, Fassy F, Bourne Y (2001) Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase bound to acetyl-coenzyme A reveals a novel active site architecture. J Biol Chem 276: 11844–11851 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56 (Part 8): 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55 (Part 4): 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yep A, Bejar CM, Ballicora MA, Dubay JR, Iglesias AA, Preiss J (2004) An assay for adenosine 5′-diphosphate (ADP)-glucose pyrophosphorylase that measures the synthesis of radioactive ADP-glucose with glycogen synthase. Anal Biochem 324: 52–59 [DOI] [PubMed] [Google Scholar]