

Abstract
The endoplasmic reticulum (ER) and mitochondria form contacts that support communication between these two organelles, including synthesis and transfer of lipids, and the exchange of calcium, which regulates ER chaperones, mitochondrial ATP production, and apoptosis. Despite the fundamental roles for ER–mitochondria contacts, little is known about the molecules that regulate them. Here we report the identification of a multifunctional sorting protein, PACS-2, that integrates ER–mitochondria communication, ER homeostasis, and apoptosis. PACS-2 controls the apposition of mitochondria with the ER, as depletion of PACS-2 causes BAP31-dependent mitochondria fragmentation and uncoupling from the ER. PACS-2 also controls formation of ER lipid-synthesizing centers found on mitochondria-associated membranes and ER homeostasis. However, in response to apoptotic inducers, PACS-2 translocates Bid to mitochondria, which initiates a sequence of events including the formation of mitochondrial truncated Bid, the release of cytochrome c, and the activation of caspase-3, thereby causing cell death. Together, our results identify PACS-2 as a novel sorting protein that links the ER–mitochondria axis to ER homeostasis and the control of cell fate, and provide new insights into Bid action.
Keywords: apoptosis, Bid, endoplasmic reticulum, mitochondria, PACS-2
Introduction
The endoplasmic reticulum (ER) controls multiple cellular processes including translocation of soluble and membrane proteins into the secretory pathway, detoxification of metabolites, and biosynthesis of lipids. The ER also serves as the principal internal store of calcium ions that mediate signaling, ATP production, and apoptosis (Voeltz et al, 2002). Extensive biochemical and genetic studies have revealed that communication between the ER, Golgi, and endosome/lysosomes is controlled largely by vesicular traffic mediated by components of the COPII, COPI, and clathrin-based sorting machinery (Bonifacino and Lippincott-Schwartz, 2003). However, high-resolution 3D electron tomography reveals that the expansive, reticulated ER forms close contacts with each of these secretory pathway compartments, and with the mitochondria (Marsh et al, 2001). Indeed, as much as 20% of the mitochondrial surface is in direct contact with the ER, underscoring the dynamic and highly regulated communication between the ER and mitochondria (Rizzuto et al, 1998). The close contacts formed between the ER and mitochondria have led to the model that ER–mitochondria communication may occur by direct transfer rather than vesicular traffic. In support of this model, biochemical studies reveal that the ER also communicates with mitochondria through mitochondria-associated membranes (MAMs), which are ER-contiguous membranes that contain multiple phospholipid- and glycosphingolipid-synthesizing enzymes, including fatty acid CoA ligase 4 (FACL4) and phosphatidylserine synthase-1 (PSS-1), and support direct transfer of lipids between the ER and mitochondria (Piccini et al, 1998; Stone and Vance, 2000).
In addition to supporting lipid transfer, the apposed ER and mitochondria also exchange calcium ions, which regulate processes ranging from ER chaperone-assisted folding of newly synthesized proteins to the regulation of mitochondria-localized dehydrogenases involved in ATP-producing Krebs cycle reactions, and the activation of calcium-dependent enzymes that execute cell death programs (Berridge, 2002). Immunocytochemical studies show that regions of the ER apposed to mitochondria are enriched with IP3 receptors, identifying these zones as ‘hotspots' of calcium transfer from the ER to the mitochondria (Rizzuto et al, 1998). Interference with calcium homeostasis or calcium communication between the ER and mitochondria, for instance, by treatment of cells with thapsigargin, which blocks uptake of calcium by the ER (Hajnoczky et al, 2000), causes a malfunction of ER-localized protein folding, leading to an accumulation of unfolded proteins. As a consequence, ER-localized enzymes that catalyze oxidative protein folding malfunction and unfolded proteins accumulate. This stress induces an unfolded protein response (UPR), which coordinates the suppression of general protein synthesis, with the increased expression of ER chaperones in order to re-establish ER homeostasis. However, if ER homeostasis fails to be re-established, the UPR triggers apoptosis (Rutkowski and Kaufman, 2004).
Apoptosis is executed by caspases, which catalyze the systematic dissolution of structural components, resulting in cell death (Boatright and Salvesen, 2003). The induction of apoptosis is often triggered by initiator caspases, among them caspase-8. Apoptotic signals leading to caspase-8 activation are defined as either extrinsic apoptotic pathways, which are initiated by the binding of Fas ligand or TNF-α to death receptors on the cell surface, or as intrinsic apoptotic pathways, cued by intracellular signals. The subsequent fission of mitochondria marks an early and key step in apoptotic programs. Caspase-8 promotes mitochondria fission by cleaving the ER cargo receptor BAP31 to form p20, which induces Drp1/Dlp1, a mitochondria-localized dynamin, to fissure mitochondria (Breckenridge et al, 2003). Mitochondria fragmentation promotes the recruitment and activation of proapoptotic molecules that cause mitochondria permeabilization, thereby activating distal steps in the apoptotic program (Karbowski and Youle, 2003).
Mitochondria permeabilization is regulated by a balance between the activities of Bcl-2 proteins, which include antiapoptotic members such as Bcl-2 and Bcl-xL, or proapoptotic members such as Bid, Bak, and Bax (Sharpe et al, 2004). Bid is a requisite component of both intrinsic and extrinsic apoptotic pathways. Apoptotic signals induce Bid dephosphorylation, which results in Bid cleavage by caspase-8 to form the potently apoptotic, truncated Bid (tBid) (Gross et al, 1999; Desagher et al, 2001). Myristoylation of tBid has been proposed to act as a ‘switch' to target it to mitochondria (Zha et al, 2000), where the tBid BH3 domain interacts with Bak and Bax on the outer mitochondria membrane to form pores that release cytochrome c into the cytosol (Wei et al, 2001). The released cytochrome c activates caspase-3, an executioner caspase that controls the distal stages of the apoptotic program (Tewari et al, 1995). However, recent studies show that full-length Bid can translocate to the mitochondria, and subsequently lead to membrane permeabilization and cytochrome c release (Tafani et al, 2002; Degli Esposti et al, 2003; Sarig et al, 2003). Together with other studies reporting activated caspase-8 in the cytosol (Micheau and Tschopp, 2003) as well as on the mitochondria (Chandra et al, 2004), these studies suggest that Bid cleavage is not required for mitochondria targeting and that tBid formation may occur subsequent to the targeting of full-length Bid to mitochondria.
Previously, we identified a sorting protein, PACS-1, which binds to cargo molecules in a phosphorylation-state-dependent manner and directs their transport from endosomes to the trans-Golgi network (TGN) (Wan et al, 1998; Crump et al, 2001; Scott et al, 2003). Here we report the identification of PACS-2, a multifunctional sorting protein that controls the ER–mitochondria axis, including the apposition of mitochondria with the ER and ER homeostasis. In addition, we show that, following induction of apoptosis, PACS-2 binds to dephosphorylated Bid and is required to traffic full-length Bid to the mitochondria, where Bid is subsequently cleaved to tBid, leading to the release of cytochrome c, the activation of caspase-3, and cell death.
Results
Identification of PACS-2
We previously identified a sorting connector, PACS-1, that localizes membrane proteins to the TGN by directing their retrieval from endosomal compartments (Wan et al, 1998; Crump et al, 2001; Scott et al, 2003). PACS-1 binds to protein kinase CK2 phosphorylatable acidic-cluster sorting motifs on membrane cargo and links them to the clathrin adaptors AP-1 and AP-3. EST database searches, however, revealed the presence of a second PACS gene. Therefore, we screened a human brain cortex library with DNA probes corresponding to the ESTs and obtained a full-length cDNA encoding a novel PACS family member: PACS-2 (Figure 1A). Sequence alignment showed that the predicted 889 aa PACS-2 protein shares 54% overall sequence identity with the 963 aa human PACS-1, and shares 81% sequence identity with PACS-1 in the 140 aa cargo/adaptor-binding region (FBR).
Figure 1.
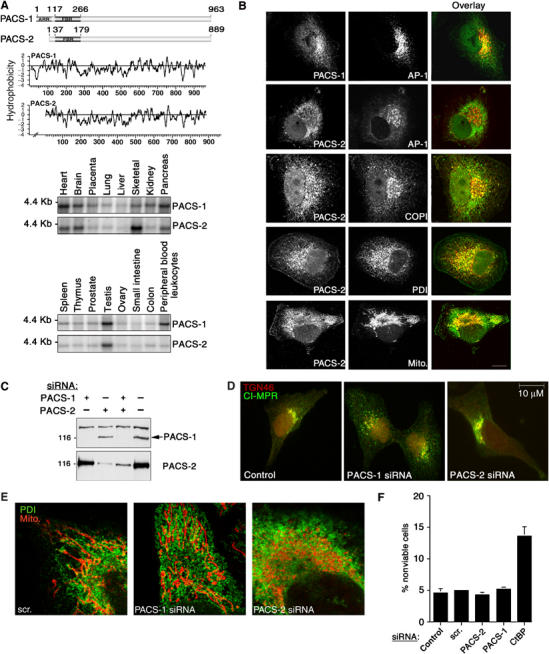
Identification and characterization of PACS-2, a sorting protein found on the ER and mitochondria. (A) Top: Schematic and Kyte–Doolittle hydrophobicity plot of the human PACS-1 and PACS-2 proteins. FBR, cargo/adaptor-binding region; ARR, atrophin-1-related region. Radiation hybrid and genome database analyses mapped the PACS-1 gene to chromosome 11q13.1 (Genbank AY320283) and the PACS-2 gene to chromosome 14q32.33 (Genbank AY320284). Bottom: Northern blot analysis of the tissue distribution for PACS-1 and PACS-2 transcripts. (B) Confocal immunofluorescence of endogenous PACS-1 and PACS-2 in A7 cells. PACS-1/PACS-2 were visualized with Alexa488 (green) and markers were visualized with Alexa546 or mitotracker (red). Scale bar, 10 μm. (C) A7 cells transfected with PACS-1 or PACS-2 siRNAs were analyzed by Western blot 48 h post-transfection. (D) A7 cells were transfected or not with PACS-1 or PACS-2 siRNAs. After 48 h, cells were processed for immunofluorescence microscopy using anti-CI-MPR (green) and anti-TGN46 (red). (E) A7 cells were transfected with control (scrambled), PACS-1, or PACS-2 siRNAs for 48 h and processed for confocal immunofluorescence localization of mitochondria (mitotracker, red) and ER (PDI, green). (F) A7 cells were transfected with the corresponding siRNAs and assayed for cell death by Annexin V/propidium iodide staining and FACS analysis. Treatment of cells with the proapoptotic CtBP siRNA served as a positive control (Zhang et al, 2003). Error for all graphs=s.d.
RNA hybridization and immunofluorescence studies suggested that PACS-1 and PACS-2 have distinct roles. Northern blot analyses showed that the 4.4 kb PACS-1 and the major 3.8 kb PACS-2 transcripts are broadly expressed, with greatest levels in the heart, brain, pancreas, and testis (Figure 1A). PACS-1 is selectively enriched in peripheral blood leukocytes, whereas PACS-2 is selectively enriched in skeletal muscle. Moreover, PACS-1 and PACS-2 show distinct intracellular staining patterns: PACS-1 localized largely to the paranuclear region, where its punctate staining pattern overlapped with that of the AP-1 adaptor, whereas PACS-2 showed a diffuse staining pattern that co-localized largely with the ER chaperone protein disulfide isomerase (PDI) and COPI coatomer, but not AP-1 (Figure 1B). Additional analyses showed a limited overlap of PACS-2 staining with mitochondria.
PACS-2 controls ER–mitochondria contacts
To identify the role of PACS-2 in vivo, we transfected cells with siRNAs that specifically depleted PACS-1 or PACS-2 following a 2-day treatment (Figure 1C). In agreement with our earlier studies, we found that depletion of PACS-1, but not PACS-2, caused the cation-independent mannose-6-phosphate receptor (CI-MPR), a PACS-1 cargo protein (Wan et al, 1998), to mislocalize from the TGN and accumulate in an endosome population (Figure 1D). Surprisingly, we found that depletion of PACS-2, but not PACS-1, caused extensive mitochondrial fragmentation, and appeared to uncouple the fragmented mitochondria from the ER (Figure 1E). To ensure that the uncoupling of the mitochondria from the ER was not a result of PACS-2 siRNA toxicity, we determined by annexin V/propidium iodide staining that siRNA depletion of either PACS-1 or PACS-2 had negligible effects on cell viability or protein synthesis (Figure 1F and data not shown).
To more rigorously determine the effect of PACS-2 depletion on ER/mitochondria, we performed electron microscopic analyses. In PACS-2-depleted cells, we observed an ∼2-fold increase in the area of cytosol containing long ER tubules devoid of associated mitochondria, as the mitochondria were found concentrated in the paranuclear region (Figure 2A and B). Despite the extensive fragmentation, and in agreement with the toxicity analysis (Figure 1F), mitochondria integrity was not disrupted in the PACS-2-depleted cells as determined by loading of tetramethyl rhodamine (TMRM), which requires an intact membrane potential (ΨM, Figure 2C).
Figure 2.
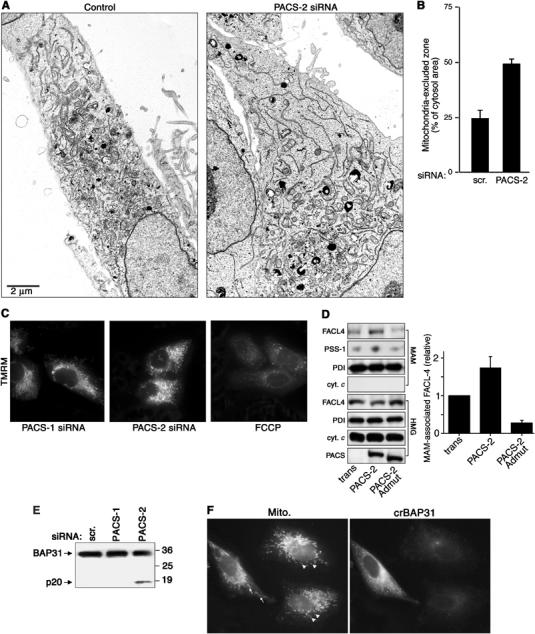
PACS-2 depletion disrupts mitochondrial structure. (A) A7 cells were transfected or not with the PACS-2 siRNA and processed for electron microscopy. Magnification, × 4500. (B) The extent of uncoupling of mitochondria from the ER of either control cells or PACS-2-depleted cells was quantified using morphometric analysis (see Materials and methods). (C) Cells were incubated with 100 nM TMRM for 30 min before microscopic analysis. Control cells were treated with 10 μM FCCP for 30 min, which uncouples the ΨM and blocks TMRM loading. (D) MAMs were isolated from crude homogenates (HMG) of control cells (trans) and PACS-2- and PACS-2Admut-expressing cells by Percoll gradient fractionation, and identified by Western blot using an anti-PSS-1 Ab, which is specific for the MAM fraction (Stone and Vance, 2000). The effect of PACS-2 or PACS-2Admut on the localization of MAM-associated FACL4 was determined by Western blot. Right: Quantitation of MAM-associated FACL4 (n=3). (E) Lysates from control and siRNA-transfected A7 cells were analyzed by Western blot using anti-BAP31. (F) A7 cells were transfected with crBAP31-flag and subsequently transfected with the PACS-2 siRNA for 48 h. Cells were then processed for immunofluorescence with anti-Flag mAb to detect crBAP31-expressing cells (right panel) and mitotracker (left panel). Arrows, rod-like mitochondria. Arrowheads, fragmented mitochondria.
Our discovery that the close apposition of mitochondria with the ER requires PACS-2 prompted us to determine whether localization of lipid biosynthetic enzymes to MAMs is also dependent on PACS-2. PACS-2 overexpression increased the amount of FACL4 associated with isolated MAM fractions (Figure 2D). By contrast, expression of the dominant-negative PACS-2Admut (Köttgen et al, 2005) decreased the amount of FACL4 and PSS-1 present in the MAM fraction. Together, these results further support a key role for PACS-2 in maintaining the ER–mitochondria axis.
Our results are in agreement with recent studies suggesting that proteins involved in ER trafficking may also have essential roles in maintaining mitochondria shape and the apposition of mitochondria against the ER, as shown by inactivating yeast COPI or by the caspase-8-catalyzed cleavage of mammalian BAP31 to form p20, either of which induces mitochondria fragmentation similar to that which occurs in PACS-2-depleted cells (Prinz et al, 2000; Breckenridge et al, 2003). Therefore, we tested the possibility that PACS-2 depletion induced the cleavage of BAP31 to p20 and found that depletion of PACS-2 but not PACS-1 induced cleavage of BAP31 to p20, similar to the process observed during apoptosis (Figure 2E). By contrast, transient expression of a caspase-resistant BAP31 molecule, crBAP31 (Nguyen et al, 2000), blocked the PACS-2 siRNA-induced mitochondria fragmentation, demonstrating that these changes in mitochondrial shape following PACS-2 depletion result from cleavage of BAP31 to p20 (Figure 2F).
PACS-2 mediates ER homeostasis
In addition to promoting lipid transfer between the ER and mitochondria, the juxtaposition of mitochondria against the ER also promotes transfer of ATP to the ER for chaperone-mediated protein folding and allows for calcium-mediated communication between the two organelles (Hajnoczky et al, 2000). Thus, our finding that PACS-2 depletion induced mitochondria fragmentation and uncoupled this organelle from the ER raised the possibility that, in addition to mediating MAM formation, PACS-2 might also influence ER folding and calcium homeostasis. To test this possibility, we first determined whether PACS-2 depletion affected the levels of BiP, an ER chaperone upregulated by ER stressors to maintain efficient folding and export of newly synthesized proteins (Rutkowski and Kaufman, 2004). PACS-2 siRNA induced an ∼2-fold increase in BiP levels, similar to the increase elicited by the potent UPR inducers dithiothreitol (DTT) and thapsigargin (Figure 3A). Second, we examined whether the PACS-2 siRNA-mediated BiP induction was a direct response to the BAP31/p20-mediated uncoupling of ER from mitochondria. We found that stably transfected crBAP31, which blocked the p20-mediated uncoupling of mitochondria from the ER (Figure 2F), also blocked BiP induction (Figure 3B), demonstrating that the mitochondria fragmentation and BiP induction in PACS-2-depleted cells resulted directly from the cleavage of BAP31 to p20. Third, we measured the ability of histamine to elicit IP3 receptor-mediated release of ER calcium in control- or PACS-2-depleted cells loaded with the calcium sensor Fura-2. We found that histamine elicited a two-fold greater increase in calcium released from the ER into the cytosol of PACS-2-depleted cells compared to PACS-1-depleted or control cells (Figure 3C). Together, these results suggest that the PACS-2 siRNA-mediated uncoupling of the ER from the mitochondria was compensated for by increased levels of the ER protein folding machinery and calcium to re-establish ER homeostasis.
Figure 3.
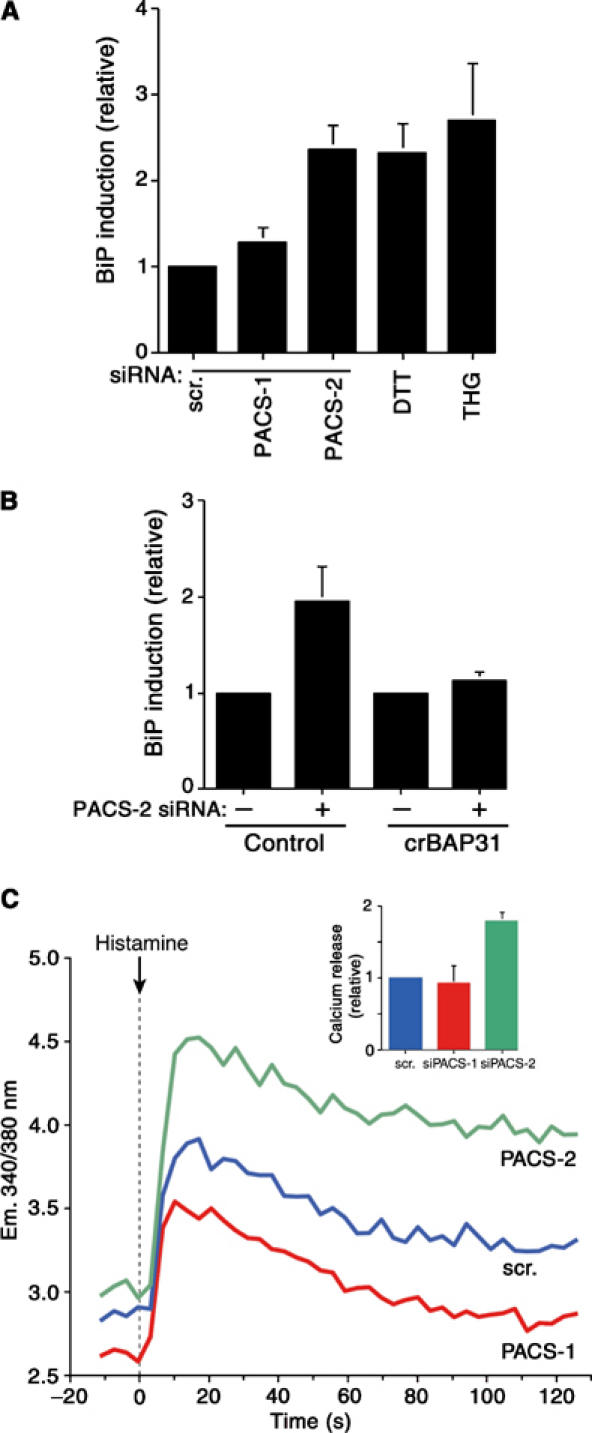
PACS-2 depletion disrupts ER homeostasis. (A) siRNA-treated A7 cells were lysed 48 h post-transfection and the amount of BiP was determined by Western blot. BiP amounts in cells treated with 1 mM DTT or 5 mM thapsigargin (THG) for 16 h served as positive controls. All values are normalized to control cells transfected with scrambled siRNA. (B) Control and HeLa(KB)-crBAP31 cells were transfected or not with PACS-2 siRNA and analyzed for BiP expression by Western blot. (C) A7 cells depleted of PACS-1 or PACS-2 and control cells (transfected with scrambled siRNA) were loaded with Fura-2 and treated with histamine to stimulate calcium release from the ER through IP3R. Inset: Relative amounts of histamine-releasable ER calcium (n=3).
PACS-2 depletion blocks apoptotic programs
In addition to causing mitochondrial fragmentation, p20 also induces apoptotic cell death (Breckenridge et al, 2003). Thus, our paradoxical finding that PACS-2 depletion induced both ER stress (Figure 3) and the p20-mediated mitochondrial fragmentation (Figure 2), but not apoptosis (Figure 1), suggested that PACS-2 is somehow required for p20-mediated apoptotic induction. To test this possibility, we treated cells depleted of PACS-1 or PACS-2 with 1.2 μM staurosporine (STS) for an additional 24 h and quantified the amount of apoptotic and necrotic cells (Figure 4A). STS induced death in ∼95% of the control and PACS-1-depleted cells. By contrast, PACS-2 depletion conferred a marked resistance to STS, increasing the percentage of viable cells by nearly four-fold. The antiapoptotic effects of the PACS-2 siRNA were corroborated by analyzing the STS-mediated caspase cleavage of poly-ADP-ribose polymerase (PARP) to generate p85 (Figure 4B). STS treatment of control cells showed a rapid onset of PARP cleavage beginning at 4 h following the STS addition, with nearly complete cleavage occurring after 6 h incubation, whereas PACS-2 showed no detectable processing at 6 h incubation. Significantly, this delay in PARP cleavage was identical to that observed in cells pretreated with the caspase inhibitor zVAD-fmk, suggesting that PACS-2 depletion inhibits executioner caspase-mediated proteolysis. Similar results were obtained when cells were treated with thapsigargin or Fas Ab, demonstrating the importance of PACS-2 for both the intrinsic and extrinsic apoptotic pathways (data not shown).
Figure 4.
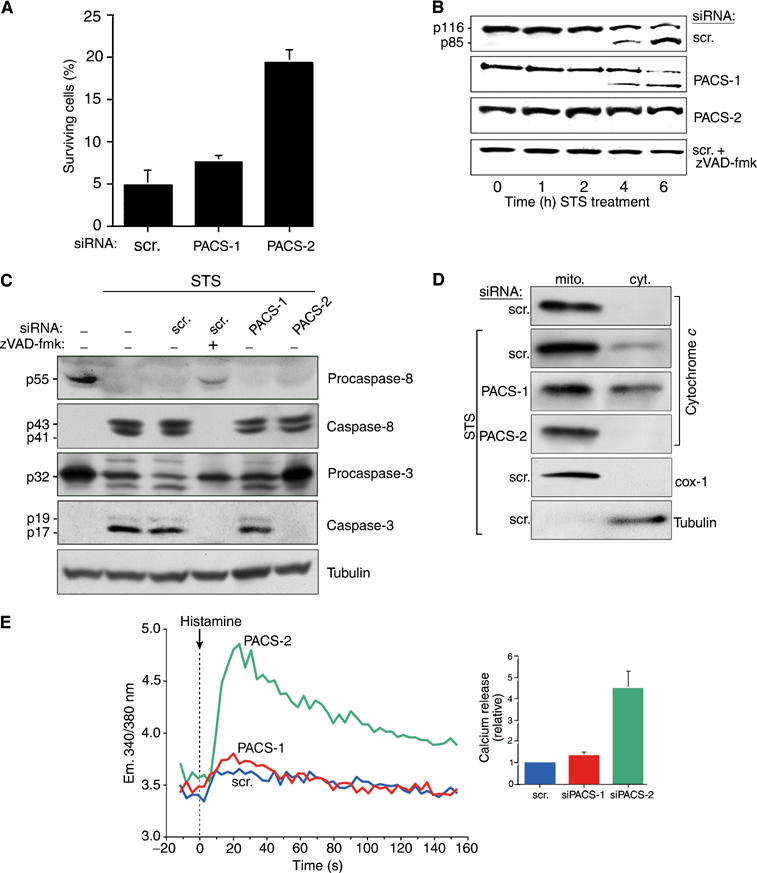
STS-induced cell death depends on PACS-2. (A) A7 cells transfected with PACS-1, PACS-2 or scrambled siRNAs for 48 h were incubated with 1.2 μM STS for an additional 24 h, and the percentage of viable cells was quantified by FACS analysis using anti-annexinV/propidium iodide staining. (B) A7 cells were transfected with PACS-specific or scrambled siRNAs, or treated with oligofectamine alone (control). Apoptosis was induced with 1.2 μM STS for 0, 1, 2, 4, and 6 h in the absence or presence of 10 μM zVAD-fmk and lysates were analyzed by Western blot to detect PARP cleavage to the p85 product. (C) A7 cells were transfected with siRNAs as indicated, treated with 1.2 μM STS for 4 h in the absence or presence of 10 μM zVAD-fmk, harvested, and pro- and activated caspase-8 and -3 or α-tubulin (loading control) were analyzed by Western blot. (D) Post-nuclear supernatants from A7 cells treated with 1.2 μM STS for 4 h were resolved by sedimentation into the cytosol and mitochondria-containing heavy membrane fractions, followed by Western blot with anti-cytochrome c, anti-cytochrome c oxidase I (mitochondria marker) or anti-α-tubulin (cytosol marker). (E) A7 cells depleted of PACS-1 or PACS-2 and control cells (transfected with scrambled siRNA) were incubated with 1.2 μM STS for 4 h and then loaded with Fura-2. Following histamine stimulation, the calcium release was monitored as in Figure 3C.
We next examined whether PACS-2 depletion blocks the STS-mediated activation of caspase-3, an executioner caspase that cleaves PARP to p85. We found that STS elicited caspase-3 activation in control and PACS-1-depleted cells, whereas PACS-2 depletion blocked STS-mediated caspase-3 activation, similar to that observed in cells treated with zVAD-fmk (Figure 4C). We next determined whether PACS-2 depletion affects caspase-8 activation, an initiator caspase that is cleaved following STS treatment (Joshi and Sahni, 2003). We found that STS stimulated the zVAD-fmk-sensitive activation of caspase-8 irrespective of the presence or absence of either PACS-1 or PACS-2 (Figure 4C). Together, these results suggested that the ability of STS to efficiently activate caspase-8, but not caspase-3, following PACS-2 depletion may be due to the inability of PACS-2-depleted cells to stimulate release of cytochrome c from the mitochondria in response to an apoptotic signal. To test this possibility, membrane compartments of cells treated with STS were fractionated and analyzed for the release of cytochrome c. We found a marked translocation of cytochrome c from the mitochondrial, heavy membrane fraction to the cytosol by 4 h following STS addition (Figure 4D). Depletion of PACS-2, however, blocked cytochrome c release under these conditions, thus explaining the failure of caspase-3 to become activated in PACS-2-depleted cells. In addition to activating caspase-3, released cytochrome c increases conductance through the IP3 receptor, leading to a massive release of ER calcium, to amplify the apoptotic pathway (Boehning et al, 2003). Accordingly, we found that the PACS-2 depletion, which blocks cytochrome c release, severely mitigated the ability of STS to invoke depletion of ER calcium stores compared to either a control treatment or PACS-1 depletion (Figure 4E). Together, these results indicate that PACS-2 depletion blocks apoptosis, at least in part, by inhibiting the release of cytochrome c from the mitochondria, thereby blocking both caspase-3 activation and the apoptotic release of ER calcium. Thus, in addition to controlling the ER–mitochondria axis, PACS-2 appears to play an essential role in connecting mitochondria to cell death programs.
Apoptotic programs induce PACS-2 to target Bid to mitochondria
Despite the importance of PACS-2 for the induction of apoptosis, our results did not explain how this ER sorting protein promotes cytochrome c release from the mitochondria in response to apoptotic inducers. We thus investigated the localization of PACS-2 itself during the onset of apoptosis. Surprisingly, we found that apoptotic inducers stimulated the rapid redistribution of PACS-2 from the ER to the mitochondria. In control cells the PACS-2 staining pattern showed a pronounced overlap with the ER marker PDI, and a more limited co-localization with mitochondria (Figure 5A, top, see also Figure 1B). However, treatment of the cells with 1.2 μM STS for 1 h stimulated a dramatic redistribution of PACS-2 staining, revealing a pronounced co-localization with the mitochondria (Figure 5A, bottom). Similar results were obtained by treatment of cells with tunicamycin, or Fas Ab (Figure 6C, bottom, and data not shown). Lastly, we conducted membrane fractionation of cells to determine biochemically whether apoptotic inducers redistributed PACS-2 from ER to the mitochondria. In agreement with the morphological study, apoptotic inducers including STS and tunicamycin stimulated transfer of PACS-2 from cytosolic and ER-enriched light membrane fractions to mitochondria-containing heavy membrane fractions (Figure 5B).
Figure 5.
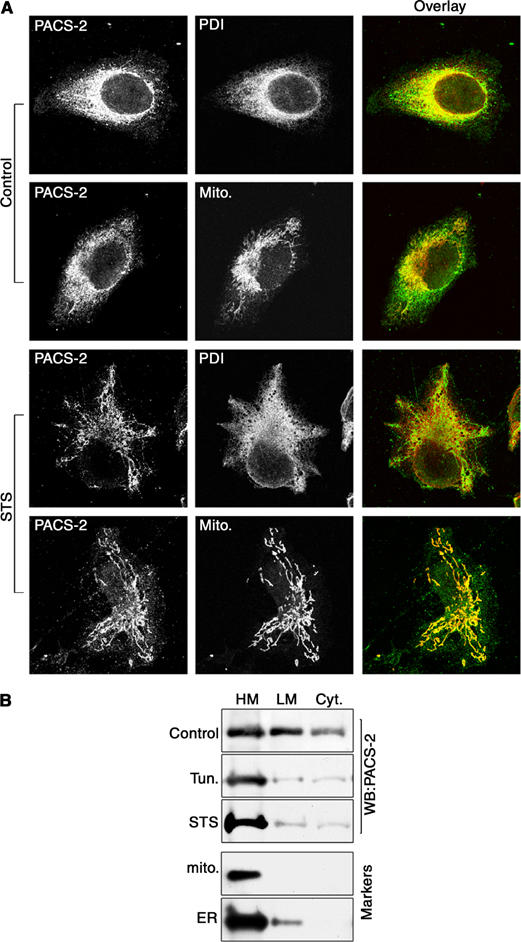
Apoptosis induction redirects PACS-2 onto mitochondria. (A) A7 cells were either untreated (control) or treated with 1.2 μM STS for 1 h and incubated with primary antibodies and species-specific secondary antibodies to detect PACS-2 (green) and PDI (red) or stained with mitotracker (red). (B) Control and tunicamycin (Tun) or STS-treated cells were homogenized and post-nuclear supernatants were resolved by sedimentation into the cytosol, light and heavy (mitochondria fraction) membranes, followed by Western blot with anti-PACS-2, anticytochrome c oxidase I (mitochondria) or anti-BiP (ER).
Figure 6.
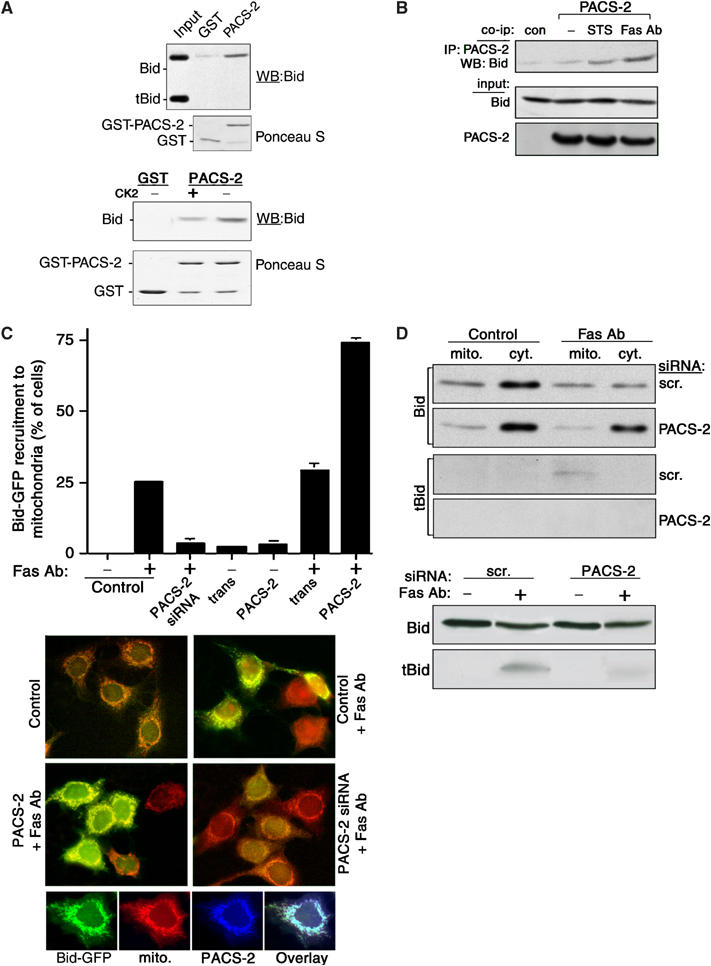
Death signals stimulate PACS-2-mediated trafficking of Bid to mitochondria. (A) Top: GST-PACS-2FBR was incubated with His6-Bid and tBid and bound Bid molecules were detected by Western blot using anti-Bid antibodies. Bottom: GST-PACS-2FBR was incubated with either His6-Bid or CK2-phosphorylated His6-Bid and bound His6-Bid was detected by Western blot using anti-Bid antibodies. (B) Control HeLa cells or cells expressing PACS-2-ha were treated or not for 2 h with 1.2 μM STS or with 0.1 μg/ml Fas Ab and 5 μg/ml cycloheximide. PACS-2-ha was immunoprecipitated from cell lysates and co-immunoprecipitating Bid was detected by Western blot using an anti-Bid Ab. (C) MCF-7:Bid-GFP cells were either transfected with the PACS-2 siRNA for 48 h or infected with Ad:trans (trans, expressing the tet transactivator) alone or together with Ad:PACS-2 for 24 h and then treated with Fas Ab (1 μg/ml) and cycloheximide (20 μg/ml) for 2 h, followed by a 30-min loading with mitotracker. Fixed cells were scored for Fas Ab-mediated recruitment of Bid-GFP to the mitochondria. Top: bar graph normalized to the control (Oligofectamine). Middle: fluorescence microscopy showing Bid-GFP and mitotracker (red) staining under the various conditions. Large fields of the coverslips containing 150–450 cells were scored for translocation of GFP-Bid to the mitochondria. Bottom: Fas Ab stimulates PACS-2 and Bid to translocate to the mitochondria. MCF-7:Bid-GFP treated with Fas Ab for 2 h were processed for immunofluorescence microscopy. (D) Top: HeLa cells transfected with control (scr.) or PACS-2 siRNAs were treated or not with Fas Ab (1 μg/ml) and cycloheximide (20 μg/ml) for 2 h. Bid and tBid in the mitochondria and cytosolic fractions (top panels) or from the total lysates (bottom panels) were detected by Western blot.
The pronounced redistribution of PACS-2 from ER/cytosol to the mitochondria by apoptotic inducers, together with the requirement for PACS-2 to promote apoptosis (Figure 4) and the established role of the PACS proteins in trafficking cargo molecules to cellular compartments, raised the possibility that apoptotic inducers may direct PACS-2 to recruit one or more proapoptotic factors to the mitochondria. One candidate proapoptotic factor is the BH3-only Bcl-2 family member Bid, which is trafficked to the mitochondria, where its caspase-8 cleavage product tBid promotes release of cytochrome c (Gross et al, 1999). Like many PACS cargo proteins, Bid contains an acidic cluster and is phosphorylated by protein kinases CK1 and CK2 (Desagher et al, 2001). To determine whether PACS-2 binds to Bid, tBid, or both proteins, we conducted in vitro binding assays. GST-PACS-2FBR, which contains the PACS-2 cargo-binding region (Figure 1A), bound to Bid but not to caspase-8-generated tBid, indicating that the intact Bid acidic region is required for PACS-2 binding (Figure 6A, top). Next, we determined whether the phosphorylation state of Bid influences the ability of Bid to bind to GST-PACS-2FBR. We found that PACS-2 bound preferentially to nonphosphorylated Bid, suggesting that, during the onset of apoptosis, dephosphorylation of Bid promotes binding to PACS-2 (Figure 6A, bottom). Next, we conducted co-immunoprecipitation studies to determine whether PACS-2 associates with Bid in vivo, and found that treatment of cells with STS or Fas Ab increased the association of PACS-2 with Bid (Figure 6B). Consequently, to determine whether PACS-2 recruits Bid to the mitochondria, MCF7 cells stably expressing Bid-GFP were transfected with PACS-2 siRNA or infected with an adenovirus expressing PACS-2 and then treated or not with cycloheximide and Fas Ab to stimulate Bid-GFP recruitment to mitochondria (Zha et al, 2000). In control cells, Fas Ab or thapsigargin markedly increased the recruitment of Bid-GFP and PACS-2 to the mitochondria (Figure 6C and data not shown). However, depletion of PACS-2 blocked Bid-GFP recruitment, supporting a role for PACS-2 in the Fas Ab- and thapsigargin-stimulated translocation of Bid to mitochondria. In support of the siRNA studies, we found that overexpression of PACS-2 in Fas Ab-treated cells stimulated Bid-GFP recruitment to the mitochondria by nearly two-fold. By contrast, overexpression of PACS-2 in control cells failed to stimulate Bid-GFP translocation to the mitochondria (Figure 6C), demonstrating that apoptotic inducers control the PACS-2-dependent trafficking of Bid to the mitochondria.
Lastly, we conducted biochemical fractionation studies to determine whether Bid translocation to the mitochondria and formation of tBid require PACS-2. In control cells, Fas Ab stimulated the translocation of full-length Bid and the formation of tBid on mitochondria. However, in PACS-2-depleted cells, Fas Ab failed to induce Bid translocation and tBid formation on mitochondria. Analysis of total cell extracts showed that formation of tBid was markedly reduced in PACS-2-depleted cells demonstrating that PACS-2, which binds Bid but not tBid, is required for formation of tBid in Fas Ab-treated cells (Figure 6D). Together, these data suggest that, in response to apoptotic signals, PACS-2 recruits full-length Bid to the mitochondria, where Bid is subsequently cleaved to form tBid to stimulate cytochrome c release, activate executioner caspases, and commit cells to death.
Discussion
We report that PACS-2 is a multifunctional sorting protein that controls the ER–mitochondria axis and the role of this axis in cellular homeostasis and apoptosis. Our results show that PACS-2 is required for the intimate association of mitochondria with the ER: absence of PACS-2 induces the caspase-dependent cleavage of BAP31 to yield the proapoptotic fragment p20, causing mitochondria to fragment and uncouple from the ER. This structural uncoupling also disrupts MAMs and induces ER stress, which is compensated for by increased levels of BiP and ER calcium. Moreover, when either cellular or ER homeostasis is compromised, or when cell death programs are initiated, PACS-2 is redirected to translocate the apoptosis-inducing protein Bid onto the mitochondria. This PACS-2-dependent trafficking of full-length Bid to the mitochondria promotes cell death by inducing formation of tBid and release of cytochrome c, which subsequently results in the depletion of releasable ER calcium, the activation of caspase-3, and the cleavage of caspase-3 substrates.
The requirement of PACS-2 for the apposition of rod-like mitochondria to the ER suggests that PACS-2 has an essential role in ER–mitochondria communication, and influences the dynamic mitochondria fusion/fission events that are coupled with mitochondria homeostasis and intermitochondria communication (Szabadkai et al, 2004). We determined that mitochondria fragmentation in PACS-2-depleted cells requires the caspase-generated p20 fragment of BAP31, which promotes mitochondria fission by regulating Drp1/Dlp1 (Breckenridge et al, 2003), a mitochondria-localized dynamin-related GTPase. Thus, our results indicate that PACS-2 depletion induced mitochondria fragmentation by promoting mitochondria fission rather than by inhibiting fusion. Our demonstration that PACS-2Admut, which fails to bind COPI (Köttgen et al, 2005), or siRNA depletion of either PACS-2 or COPI (data not shown), uncouples the fragmented mitochondria from the ER, suggests that PACS-2/COPI trafficking is essential for maintaining the ER–mitochondria axis. Our results are consistent with studies in yeast which show that inactivation of COPI similarly uncouples the ER from mitochondria (Prinz et al, 2000).
We extended our morphological studies with biochemical analyses to show that PACS-2 mediates the levels of MAM-associated FACL4, which converts fatty acids to fatty acyl-CoA esters used in the formation of complex lipids (Piccini et al, 1998), and PSS-1, which exchanges the head group of phosphatidylcholine with serine (Figure 2). MAMs are ER-contiguous membranes that, in addition to FACL4 and PSS-1, contain multiple phospholipid- and glycosphingolipid-synthesizing enzymes and support the direct transfer of phospholipids from the ER to the mitochondria (Vance, 2003). Our work identifies PACS-2 as the first cellular trafficking protein that regulates MAM formation. However, we do not know whether the reduced levels of MAM-associated FACL4 and PSS-1 in cells expressing dominant-negative PACS-2 result from mistargeting of FACL4 and PSS-1, or whether the MAM itself is redistributed as a result of the uncoupling of the fragmented mitochondria from the ER. Interestingly, we found that the loss of MAM markers in PACS-2-depleted cells was coupled with a pronounced extension of the tubulated, peripheral ER (Figure 2). It is possible that interference with PACS-2 causes the MAM to redistribute into the ER, thereby expanding the ER membrane fraction while reducing the amount of membrane associated with mitochondria. Consistent with this possibility, expression of a dominant-negative Drp1 promotes mitochondria fusion, together with a corresponding reduction in the amount of ER membranes (Pitts et al, 1999). Alternatively, the extended tubulation of the peripheral ER observed by electron microscopy in PACS-2-depleted cells may reflect a requirement of PACS-2 for the function of ER modeling proteins (Nakajima et al, 2004). In addition to contacting the mitochondria, high-resolution 3D electron tomography reveals that the ER also forms close contacts with the trans-Golgi and endosomal compartments (Marsh et al, 2001). Whether there are additional roles for PACS-2 in interorganelle communication remains to be tested, but such an expanded role may explain the high level of PACS-2 expression in skeletal muscle (Figure 1), in which the sarcoplasmic reticulum is apposed with the plasma membrane.
In addition to mediating the ER–mitochondria axis, we found that PACS-2 has a profound role on ER homeostasis as PACS-2 depletion induces a UPR (Figure 3). The UPR is an integrated response to a variety of ER-targeted stressors, which coordinates a repression of cellular protein synthesis by phosphorylating eIF-2α, with an induction of ER chaperone expression to facilitate folding of secreted and membrane proteins (Rutkowski and Kaufman, 2004). We found that PACS-2 depletion leads to a transient increase in phosphorylated eIF-2α, after which ER homeostasis is re-established with a normal rate of protein synthesis by 2 days following treatment (data not shown). The re-establishment of ER homeostasis coincides with increased levels of BiP and ER calcium (Figure 3). Whereas the importance of the ER-trafficking machinery on ER homeostasis is well established (Belden and Barlowe, 2001), the influence of mitochondrial structure on ER homeostasis is unclear. Thus, our finding that the UPR induced by PACS-2 depletion could be prevented by expression of a caspase-resistant crBAP31 demonstrates that this UPR is a direct result of the p20-mediated uncoupling of the fragmented mitochondria from the ER (Figure 4). Several factors may contribute to the control of ER homeostasis by the apposed mitochondria. For example, mitochondria provide ATP to the ER for oxidative protein folding, which is blocked by hypoxic stress (Koumenis et al, 2002). In addition, the increased levels of calcium-binding ER chaperones require a commensurate increase in ER calcium (Koch, 1990), consistent with our finding that PACS-2 depletion increases by ∼2-fold the amount of histamine-releasable ER calcium compared to control cells (Figure 3C).
To what extent the higher ER calcium release in PACS-2-depleted cells is due to promoting ER chaperone activity versus an inability to efficiently transfer calcium to the mitochondria requires further investigation and may provide new insight into ER–mitochondria communication. For example, in addition to transferring lipid intermediates, MAMs may participate in calcium transfer between the ER and mitochondria. In support of this possibility, both IP3Rs and ryanodine receptors possess potential PACS-2-binding sites (Köttgen et al, 2005) and may be associated with MAMs (Hajnoczky et al, 2000). Thus, disruption of PACS-2 may cause mislocalization of IP3Rs, resulting in reduced calcium transfer from the ER to mitochondria. Recent studies show that the mere relocalization of mitochondria is not sufficient to affect ER calcium levels (Varadi et al, 2004), but nonetheless disrupts calcium communication within the mitochondrial system (Szabadkai et al, 2004). Thus, the observed increase in ER calcium in PACS-2-depleted cells cannot be explained by mitochondria fragmentation alone, but rather also by changes in the ER to maintain ER homeostasis.
Ectopic expression of p20 is sufficient to commit cells to apoptosis (Breckenridge et al, 2003). The p20/Drp1-mediated mitochondrial fragmentation primes this organelle for cytochrome c release by promoting the recruitment of Bax/Bak oligomers (Karbowski and Youle, 2003). Thus, we were surprised to find that PACS-2-depleted cells were not only viable but also resistant to apoptosis (Figure 4), despite the p20-mediated mitochondria fragmentation (Figure 1). This block in the apoptotic pathway caused by depletion of PACS-2 led us to determine that apoptotic inducers signal PACS-2 to recruit Bid to the mitochondria (Figure 6), which is required for the induction of cell death. In addition, our finding that apoptotic inducers failed to exhaust ER calcium in PACS-2 siRNA-treated cells (Figure 4) agrees with recent studies showing that cytochrome c released into the cytosol binds to the IP3 receptor to induce release of ER calcium necessary to fully activate cell death proteases (Boehning et al, 2003). Together, our findings suggest that, in response to intrinsic and extrinsic apoptotic inducers, ER/cytosolic PACS-2 collaborates with BAP31/p20 to coordinate cell death programs.
Our demonstration that apoptotic inducers promote PACS-2 to bind and translocate full-length Bid to mitochondria (Figures 5 and 6) provides new insight into the integration of the ER–mitochondria axis with cell death programs. While full-length Bid can associate with mitochondria and induce the release of cytochrome c, the caspase-8-generated cleavage product tBid possesses a much higher affinity for mitochondria, where it accumulates during apoptosis and can more efficiently release cytochrome c (Gross et al, 1999). N-terminal myristoylation further increases tBid apoptotic activity, supporting the model that this lipid addition acts as a molecular ‘switch' that targets tBid to the mitochondria during apoptosis (Zha et al, 2000). Our results, however, cast new insight into the apoptosis-induced targeting of Bid to mitochondria. We found that PACS-2 depletion blocks the apoptotic translocation of Bid-GFP to the mitochondria, whereas overexpression of PACS-2 enhances this sorting step (Figure 6). The PACS-2-dependent translocation of Bid to the mitochondria appears to occur prior to caspase cleavage of Bid as PACS-2 depletion prevents both the Fas Ab-induced formation of tBid and the accumulation of tBid on mitochondria, but has no effect on cellular caspase-8 activation (Figures 4 and 6). Moreover, we found that PACS-2 binds selectively to full-length Bid but not tBid, and that apoptotic inducers promote the association of full-length Bid with PACS-2. These findings suggest that PACS-2 first targets full-length Bid to the mitochondria, where Bid is subsequently cleaved to tBid by cytosolic- or mitochondria-localized caspase-8 (Micheau and Tschopp, 2003; Chandra et al, 2004). Our determination that phosphorylation of Bid by CK2 inhibits binding to PACS-2 suggests that Bid phosphorylation has two antiapoptotic roles; the prevention of binding to PACS-2, which blocks translocation to the mitochondria, and the masking of the caspase-8 cleavage site to produce tBid (Desagher et al, 2001). With our discovery of PACS-2 and the role of this novel sorting protein in controlling the ER–mitochondria axis and apoptosis, we propose a revised model of Bid action. In one leg, apoptotic inducers redirect PACS-2 from maintaining the ER–mitochondria axis, which promotes the BAP31/p20-mediated fragmentation of mitochondria that uncouple from the ER. The fragmentation may ‘prime' mitochondria by enhancing Bak/Bax recruitment. In the second leg, full-length Bid becomes dephosphorylated, thereby enabling it to bind PACS-2. PACS-2 then targets full-length dephosphorylated Bid to the fragmented mitochondria, where Bid can be subsequently cleaved to tBid by mitochondria-localized caspase-8. The cleaved, myristoylated tBid may then combine with Bak/Bax to permeabilize mitochondria, release cytochrome c, and commit cells to death. Whether PACS-2 specifically translocates Bid to the mitochondria or is involved in the translocation of other proapoptotic proteins that contain potential PACS-2-binding sites (e.g. Bak, Bad, Bim, and Bik) warrants future investigation.
In an accompanying paper, we show that PACS-2 is a COPI connector that controls the ER localization of polycystin-2 and likely many other ER localized membrane proteins that contain PACS-2-binding sites (Köttgen et al, 2005). We show here that the role of PACS-2 extends beyond ER trafficking, as it controls ER homeostasis and the ER–mitochondria axis. Our finding that PACS-2Admut, which fails to bind COPI (Köttgen et al, 2005), disrupts MAMs and the apposition of mitochondria with the ER (Figure 2 and data not shown), suggests that PACS-2 must bind COPI to maintain the dynamic communication between the ER and mitochondria. In addition, we show that in response to apoptotic inducers PACS-2 is required to translocate Bid to the mitochondria to control cell death. Interestingly, genomic analyses show that the PACS-2 gene is localized near the telomere on chromosome 14q32:33, a locus susceptible to chromosomal translocation and loss of heterozygosity in B-cell lymphomas and colorectal cancer. Moreover, and in accord with our discovery that PACS-2 is a proapoptotic protein, the PACS-2 gene is mutated in up to 40% of colorectal cancers (G Anderson, personal communication). Based on our findings, the loss of PACS-2 would likely block apoptosis and may provide an opportunity for subsequent genomic insults that lead to cell immortalization and cancer.
Materials and methods
Antibodies and reagents
Reagents were from Sigma except where stated. PACS-1 and PACS-2 antisera (Pocono Farms), antibodies against δ-adaptin (M Robinson), BAP31 (G Shore), CI-MPR (S Pfeffer), PDI (R Sitia), cytochrome oxidase I (Molecular Probes), α-tubulin (Calbiochem), thioredoxin (TRX; Invitrogen), γ-adaptin (Sigma), β-COP (MaD, Abcam), TGN46 (Serotec), BiP (BD Biosciences), Myc (Santa Cruz), HA (Covance), FLAG (Kodak), GFP (Clontech), Fas (Beckman), PARP, cytochrome c (Apotech), His6 (Qiagen), FACL4 (Abgent), PSS1 (O Kuge) and Bid, pro/activated caspase-3 and -8 (Cell Signaling), Alexa (546 and 488)-conjugated secondary antibodies (Molecular Probes), and HRP-conjugated secondary antibodies (Southern Biotech) were provided as indicated. Cell lines and plasmids were received as indicated: HeLa KB/crBAP31 (G Shore), MCF-7:GFP-Bid (S Korsmeyer), pET15b-Bid (H Lu), and pFLAG-crBAP31 (G Shore).
cDNA cloning, expression vectors, cell lines and virus construction
The PACS-2 cDNA was isolated by screening lambda ZAPII-hMC human cortex library (Stratagene) with a probe to EST R50031. TRX-PACS-2 FBR (PACS-2 residues 37–179) was expressed using pET32 (Novagen). MCF7:Bid-GFP, HeLa, and A7 melanoma were cultured as described (Zha et al, 2000; Crump et al, 2001). Adenovirus (Ad) recombinants (tet-off) expressing human PACS-2-ha (wild type and Admut (E89TDLALTF96 → Ala8), which blocks binding of PACS-2 to COPI (Köttgen et al, 2005)) were generated as described (Blagoveshchenskaya et al, 2002). Adenovirus infections were performed for 24 h.
RNA hybridization
Northern hybridization was performed using human multiple tissue blots (Clontech, Palo Alto, CA). cDNAs for PACS-1, PACS-2, and 1B15 (loading control, not shown) were used to generate random-primed 32P-probes. Hybridization was carried out according to the manufacturer's instructions and signals detected by Phosphor-Imager.
Immunofluorescence, electron microscopy, and FACS
Conventional immunofluorescence microscopy was performed as described previously (Wan et al, 1998; Crump et al, 2001). Confocal microscopy used an Olympus Fluo-View FV300 confocal laser-scanning microscope. For the TMRM loading, A7 cells transfected with siRNAs were incubated with 100 nM TMRM (Molecular Probes) for 30 min and then live cells were imaged. Electron microscopy was performed as described (Arvidson et al, 2003). Mitochondrial exclusion zones in electron micrographs were quantified using NIH Image v1.63. Exclusion zones were defined as the total cytoplasmic area minus the area of cytoplasm containing >95% of mitochondria. Using a blind assay, two independent sets of data were recorded for each micrograph (n=7 per condition, P<0.0001). FACS was performed with a FACSCalibur (Becton Dickinson) and using the Annexin V/PI apoptosis staining kit (Oncogene/EMDbiosciences) according to the manufacturer's instructions.
siRNAs
siRNAs specific for human PACS-1 (AACUCAGUGGUCAUCGCUGUG and AAUUCUUCGCUCCAACGAGAU), PACS-2 (AACACGCCCGUGCCCAUGAAC and AAGAGGGAAGGCAACAAGCUU), CtBP (AAGGGAGGACCUGGAGAAGUU), and a nonspecific scrambled control (Dharmacon) were transfected using Oligofectamine (Invitrogen). Cells were analyzed 2–3 days post-transfection.
Bid translocation, Bid binding to PACS-2, and Bid co-immunoprecipitation
MCF-7:Bid-GFP cells expressing Bid-GFP were transfected with PACS-2 or PACS-1 siRNAs for 48 h, or infected with Ad:PACS-2 or Ad:PACS-1 for 24 h. Apoptosis was induced with Fas Ab and cycloheximide as described (Zha et al, 2000). For Bid-binding assays, recombinant Bid (30 μg) was cleaved with 300 U caspase-8 (Calbiochem) for 18 h at 30°C in cleavage buffer (50 mM HEPES, 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 10% glycerol, 0.1% CHAPS, pH 7.4) to generate an equal mixture of Bid and tBid. This mixture was incubated at 4°C for 4 h with 3 μg GST-PACS-2FBR or GST alone, captured with glutathione-agarose and analyzed by Western blot. For the Bid co-immunoprecipitation, cells was infected with Ad:PACS-2 and treated for 2 h with either Fas Ab (1 μg/ml, Beckman)/cycloheximide (2 μg/ml) or 1.2 μM STS. Cells were lysed with m-RIPA (1% NP40, 1% deoxycholine, 150 mM NaCl, 50 mM Tris, pH 8.0, and protease inhibitors), immunoprecipitated with mAb HA.11, and processed for Western blot using an anti-Bid antibody.
MAM isolation, mitochondria fractionation, cytochrome c release, and Bid translocation
MAMs were isolated as described (Stone and Vance, 2000), except that cells were broken with a ball-bearing homogenizer (18 μm clearance). Mitochondria fractionation was performed as described (Kataoka et al, 2001). For cytochrome c release and Bid translocation, A7 or HeLa cells were washed with cold PBS, scraped into 300 μl mitochondrial fractionation buffer (250 mM sucrose, 10 mM Tris–HCl at pH 7.5, 1 mM EGTA, and protease inhibitors) and lysed with 12 passes through a 27 G needle. The post-nuclear supernatant was sedimented at 15 000 g for 15 min to separate mitochondria and cytosol, followed by Western blot with anticytochrome c, anti-Bid, anticytochrome c oxidase I (mitochondria), or antitubulin (cytosol).
Fura-2 measurements
A7 cells treated or not with 1.2 μM STS were incubated with 2 μM Fura-2 (Molecular Probes) for 45 min and then in normal medium for another 30 min. Cells were then trypsinized, washed, and resuspended in 1.5 ml Tyrode's buffer (10 mM glucose, 1 mM MgCl2, 1 mM CaCl2, pH 7.4) and monitored for light emission at 510 nm after excitation at 340 and 380 nm using a Cary Eclipse Fluorescence spectrophotometer. The emission ratio between the two emissions is directly proportional to the cytosolic [Ca2+].
Acknowledgments
We thank J Karpen, S Kaech, G Medigeshi, E Barklis, M Webb, and J Larson for help with experiments and M Forte, J Larson and G Scott for helpful discussions and careful reading of the manuscript. We thank H Lu, O Kuge, G Shore, M Robinson, S Korsmeyer, E Chevet, and R Sitia for reagents. We are supported by fellowships from EMBO and the Swiss National Fund (TS), HFSP (ADB), Association pour la Recherche contre le Cancer (SFF), The Wellcome Trust (CMC), and NIH grants AI49793, AI48585, and DK37274 (GT).
References
- Arvidson B, Seeds J, Webb M, Finlay L, Barklis E (2003) Analysis of the retrovirus capsid interdomain linker region. Virology 308: 166–177 [DOI] [PubMed] [Google Scholar]
- Belden WJ, Barlowe C (2001) Deletion of yeast p24 genes activates the unfolded protein response. Mol Biol Cell 12: 957–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32: 235–249 [DOI] [PubMed] [Google Scholar]
- Blagoveshchenskaya AD, Thomas L, Feliciangeli SF, Hung CH, Thomas G (2002) HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell 111: 853–866 [DOI] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS (2003) Mechanisms of caspase activation. Curr Opin Cell Biol 15: 725–731 [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol 5: 1051–1061 [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Lippincott-Schwartz J (2003) Coat proteins: shaping membrane transport. Nat Rev Mol Cell Biol 4: 409–414 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Choy G, Deng X, Bhatia B, Daniel P, Tang DG (2004) Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion–endoplasmic reticulum cross talk in etoposide-induced cell death. Mol Cell Biol 24: 6592–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump CM, Xiang Y, Thomas L, Gu F, Austin C, Tooze SA, Thomas G (2001) PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J 20: 2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti MD, Cristea IM, Gaskell SJ, Nakao Y, Dive C (2003) Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ 10: 1300–1309 [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, Journot L, Antonsson B, Martinou JC (2001) Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol Cell 8: 601–611 [DOI] [PubMed] [Google Scholar]
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem 274: 1156–1163 [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P (2000) The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J Physiol 529 (Part 1): 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SG, Sahni SK (2003) Immunofluorescent detection of activation of initiator caspases-8 and -9 during pharmacologically induced apoptosis of cultured HeLa and endothelial cells. Histochem Cell Biol 119: 463–468 [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10: 870–880 [DOI] [PubMed] [Google Scholar]
- Kataoka T, Holler N, Micheau O, Martinon F, Tinel A, Hofmann K, Tschopp J (2001) Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J Biol Chem 276: 19548–19554 [DOI] [PubMed] [Google Scholar]
- Koch GL (1990) The endoplasmic reticulum and calcium storage. BioEssays 12: 527–531 [DOI] [PubMed] [Google Scholar]
- Köttgen M, Benzing T, Simmen T, Tauber R, Buchholz B, Feliciangeli S, Huber TB, Schermer B, Kramer-Zucker A, Höpker K, Simmen KC, Tschucke CC, Sandford R, Kim E, Thomas G, Walz G (2005) Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J, [E-pub ahead of print: 3rd February 2005. doi:10.1038/sj.emboj.7600566] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 22: 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Mastronarde DN, Buttle KF, Howell KE, McIntosh JR (2001) Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc Natl Acad Sci USA 98: 2399–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190 [DOI] [PubMed] [Google Scholar]
- Nakajima K, Hirose H, Taniguchi M, Kurashina H, Arasaki K, Nagahama M, Tani K, Yamamoto A, Tagaya M (2004) Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. Embo J 23: 3216–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Breckenridge DG, Ducret A, Shore GC (2000) Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol Cell Biol 20: 6731–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, Zollo M, Borsani G, Ballabio A, Renieri A (1998) FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics 47: 350–358 [DOI] [PubMed] [Google Scholar]
- Pitts KR, Yoon Y, Krueger EW, McNiven MA (1999) The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol Biol Cell 10: 4403–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA, Grzyb L, Veenhuis M, Kahana JA, Silver PA, Rapoport TA (2000) Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Biol 150: 461–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14: 20–28 [DOI] [PubMed] [Google Scholar]
- Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW, Gross A (2003) BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem 278: 10707–10715 [DOI] [PubMed] [Google Scholar]
- Scott GK, Gu F, Crump CM, Thomas L, Wan L, Xiang Y, Thomas G (2003) The phosphorylation state of an autoregulatory domain controls PACS-1-directed protein traffic. EMBO J 22: 6234–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe JC, Arnoult D, Youle RJ (2004) Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 1644: 107–113 [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275: 34534–34540 [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16: 59–68 [DOI] [PubMed] [Google Scholar]
- Tafani M, Karpinich NO, Hurster KA, Pastorino JG, Schneider T, Russo MA, Farber JL (2002) Cytochrome c release upon Fas receptor activation depends on translocation of full-length bid and the induction of the mitochondrial permeability transition. J Biol Chem 277: 10073–10082 [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81: 801–809 [DOI] [PubMed] [Google Scholar]
- Vance JE (2003) Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res Mol Biol 75: 69–111 [DOI] [PubMed] [Google Scholar]
- Varadi A, Cirulli V, Rutter GA (2004) Mitochondrial localization as a determinant of capacitative Ca(2+) entry in HeLa cells. Cell Calcium 36: 499–508 [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, Rapoport TA (2002) Structural organization of the endoplasmic reticulum. EMBO Rep 3: 944–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, Thomas G (1998) PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94: 205–216 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH (2003) Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 115: 177–186 [DOI] [PubMed] [Google Scholar]