

Abstract
Increased levels of the nuclear export protein, exportin 1 (XPO1), were demonstrated in multiple myeloma (MM) patients. Targeting XPO1 with selinexor (the selective inhibitor of nuclear export; SINE compound KPT-330) demonstrates broad antitumor activity also in patient cells resistant to bortezomib; hence, it is a promising target in MM patients. Hypoxia is known to mediate tumor progression and drug resistance (including bortezomib resistance) in MM cells. In this study, we tested the effects of selinexor alone or in combination with bortezomib in normoxia and hypoxia on MM cell survival and apoptosis in vitro and in vivo. In vitro, selinexor alone decreased survival and increased apoptosis, resensitizing MM cells to bortezomib. In vivo, we examined the effects of selinexor alone on tumor initiation and tumor progression, as well as selinexor in combination with bortezomib, on tumor growth in a bortezomib-resistant MM xenograft mouse model. Selinexor, used as a single agent, delayed tumor initiation and tumor progression, prolonging mice survival. In bortezomib-resistant xenografts, selinexor overcame drug resistance, significantly decreasing tumor burden and extending mice survival when combined with bortezomib.
Introduction
Multiple myeloma (MM) is a plasma cell malignancy localized primarily in the bone marrow (BM). Some of the most novel therapies being developed are based on interrupting the interactions between MM cancer cells and the supportive BM microenvironment, which contributes to pathogenesis and facilitates MM progression [1]. The introduction of these therapeutics, including proteasome inhibitors (such as bortezomib, carfilzomib and ixazomib), immunomodulatory drugs, and monoclonal antibodies, substantially improved the survival of MM patients [1], [2], [3]; however, more than 90% of MM patients relapse [1], [4]. Bortezomib is the first-in-class reversible proteasome inhibitor which has been used as the backbone of frontline therapy and relapsed/refractory settings in combination regimen in MM patients; nevertheless, its use is limited due to the development of drug resistance and side effects [5].
Exportin 1 (XPO1), also known as chromosome region maintenance 1, is the main nuclear exporter of leucine-rich proteins through the nuclear pore complex to the cytoplasm, which is driven by guanosine triphosphate hydrolysis [6]. This tightly controlled protein shuttling is deregulated in cancer cells, where some of the XPO1 cargo proteins, such as tumor suppressors (p53 [7], [8], p21, and p27 [9]) and inhibitor of NFκB [10], [11], are abnormally localized in the cytosol and thus cannot properly function in suppressing cancer initiation and progression [9]. Overexpression of XPO1 protein is associated with tumor growth and poor patient survival as demonstrated in solid tumors including lung, pancreatic, cervical, and ovarian cancers [8], [12], [13], [14]; melanoma; glioma; and osteosarcoma [15], [16], as well as in hematologic malignancies including acute myeloid leukemia [17], non-Hodgkin lymphoma [18], and MM [11], [19], [20]. Interestingly, whole-genome sequencing in chronic lymphocytic leukemia patients identified recurring mutations in the highly conserved region of XPO1, suggesting that these oncogenic changes may contribute to the clinical evolution of the disease [21], [22]. Inhibition of the XPO1 protein mediated by selective inhibitor of nuclear export (SINE) compounds, developed by Karyopharm Therapeutics, has shown broad preclinical antitumor activity in many solid and hematologic malignancies independent of p53 status [15], [23], [24], [25]. SINE is currently in several phase I/II/III clinical trials including more than 20 studies for myeloma patients, both newly diagnosed and relapsed/refractory, in combination with different treatments.
Tai et al. have shown previously that XPO1 protein expression is higher in MM cells in newly diagnosed patients as compared to normal plasma cells [20]. Moreover, XPO1 mRNA expression is augmented with MM disease progression and correlates with bone lytic lesions and poor patient survival. It was reported that XPO1 protein plays a crucial role in MM pathophysiology, malignant growth, and cancer cell survival. These findings are based on XPO1 knockdown studies in MM cells which were also mimicked using SINE (KPT-185, KPT-251, KPT-276, and KPT-330). KPT-185 and KPT-330 were the most potent XPO1 inhibitors, which significantly decreased MM cell proliferation, induced cytotoxicity of MM cells, and increased the percentage of apoptotic MM cells that were associated with inhibited export of tumor suppressors such as p53, p27, PP2A, and FOXO3a. In addition, it was shown that KPT-185 and KPT-330 inhibited osteoclastogenesis and bone resorption via reducing NFκB activity; however, they did not affect osteoblastogenesis in vivo [20].
Therapy resistance acquired by MM cells is one of the main problems in MM treatment and tumor recurrence. Interestingly, XPO1 protein and mRNA levels were demonstrated to be increased during MM progression, and MM cells isolated from patients resistant to bortezomib expressed higher XPO1 protein levels [20]. These results suggest that XPO1 could potentially be a biomarker of cancer progression as well as development of drug resistance in MM. Likewise, tumor hypoxia (low oxygenation) is a phenomenon observed in growing malignant tumors, and hypoxia-induced genes have been used in detecting MM tumor growth and drug resistance [26], [27]. We and others have shown that tumor hypoxia develops due to uncontrollable MM cell proliferation and correlates with poor prognosis of cancer patients [28], [29], [30]. Hypoxia confers treatment resistance of cancer cells by regulating processes such as quiescence via cell cycle arrest [31], [32], [33]; inhibiting apoptosis and senescence of cells; controlling autophagy, endoplasmic reticulum (ER) stress, and p53 and mitochondrial activity [26], [32]; and maintaining stem cell–like phenotype through induction of dedifferentiated and immature phenotype of cells [34], [35], [36], [37]. We have previously tested the role of hypoxia in MM cell proliferation in the presence of proteasome inhibitors, where MM cells were cultured in hypoxic and normoxic conditions and subsequently treated with or without bortezomib and carfilzomib. We have shown that, in hypoxic conditions, cancer cells lose their sensitivity to proteasome inhibitors in vitro [27].
In this study, we examined whether inhibiting XPO1 with the oral, slowly reversible inhibitor selinexor resensitized hypoxia-induced drug resistance to bortezomib in MM. Indeed, selinexor reversed the hypoxia-induced resistance to bortezomib in MM cells both in vitro and in vivo, thus providing evidence for clinical trials combining proteasome inhibitors with XPO1 inhibition.
Materials and Methods
Cell Culture
The MM cell lines (MM.1S and MM.1S-GFP-Luc) were a kind gift from Dr. Irene Ghobrial (Dana-Farber Cancer Institute in Boston, MA). Cells were cultured in RPMI-1640 (Corning CellGro, Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Gibco, Life Technologies, Grand Island, NY), 2 mmol/l of L-glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (CellGro, Mediatech, Manassas, VA). Cells were cultured at 37°C and 5% CO2 in a humidified tissue culture incubator (21% O2), in the NuAire water jacket incubator (Plymouth, MN), or in hypoxia (1% O2) using the hypoxic chamber purchased from Coy (Grass Lake, MI).
Selinexor (KPT-330)
Selinexor (KPT-330) was provided by Karyopharm Therapeutics (Newton, MA). For in vitro experiments, selinexor was solubilized in dimethylsulfoxide, aliquoted, and stored at −80°C. For in vivo studies, the selinexor formulated drug product contained ~70% active ingredient, ~15% Plasdone PVP K-29/32, and ~15% Poloxamer Pluronic F-68 (provided by Karyopharm Therapeutics) and was stored at 4°C for 7 days. Plasdone PVP K-29/32 with Poloxamer Pluronic F-68 served as a vehicle in the mice studies.
Effects of Selinexor on MM Cell Survival
Cell survival was assessed using MTT solution (Sigma-Aldrich, St. Louis, MO), followed by absorbance measurement at 570 nm using a spectrophotometer according to the manufacturers' protocol, where the absorbance is proportional to the number of viable cells. The effects of increasing concentrations of selinexor (KPT-330; 0, 100, 250, and 500 nM), bortezomib (Selleck Chem, Houston, TX; 10 nM), and combination treatment (100 nM of KPT-330 and 10 nM of bortezomib) were tested on MM cell survival/cytotoxicity under normoxic and hypoxic conditions for 24 hours. In addition, the effect of selinexor (0, 50, 100, and 250 nM) and bortezomib (0, 1, 5, and 10 nM) was examined on MM.1S cell survival in normoxic conditions by MTT.
Effect of Selinexor on MM Cell Apoptosis
Cell apoptosis was performed using Annexin V–propidium iodide (PI) staining (BD Biosciences, San Jose, CA) according to the manufacturer's protocol. Briefly, MM.1S cells (1 × 106 cell/ml) were cultured with selinexor (0, 100, 250, and 500 nM) with or without bortezomib (10 nM) for 24 hours. Then, cells were washed and resuspended in 1× Annexin binding buffer, followed by Annexin V staining for 15 minutes and PI staining for additional 15 minutes, and analyzed with MACSQuant Flow Cytometer (Miltenyi, San Diego, CA). The results were demonstrated as a frequency (%) of viable (Ann−PI−), early apoptotic (Ann+PI−), and late apoptotic/dead (Ann+PI+) MM cells posttreatment.
Western Blotting
To test cell signaling involved in proliferation, apoptosis, and cell cycle, MM cells were first treated with selinexor (0, 100, 250, and 500 nM) with or without bortezomib (10 nM) in normoxia or hypoxia for 24 hours. Cells were collected, washed with 1× PBS, and lysed for 30 minutes on ice using 1× lysis buffer (Cell Signaling, Danvers, MA). Protein concentration was assessed by Quick Start Bradford dye reagent (BioRad, Hercules, CA). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed using NuPAGE 4% to 12% Bis-Tris gels (Novex, Life Technologies, Grand Island, NY) and transferred to a nitrocellulose membrane using iBlot (Invitrogen, Life Technologies). Membranes were blocked with 5% skim milk in Tris-buffered saline/Tween 20 buffer and incubated with primary antibodies overnight at 4°C for apoptosis signaling (cleaved PARP, cPARP; cleaved caspase-3, cCasp3; cCasp8), proliferation signaling (p-p44/42 MAPK; p-S6R), and cell cycle signaling (p-Rb; CDK-6). α-Tubulin was used as a loading control (antibodies were purchased from Cell Signaling). The membrane was washed with Tris-buffered saline/Tween 20, incubated for 1 hour at room temperature with HRP-conjugated secondary antibody, then washed and developed using Novex ECL Chemiluminescent Substrate Reagent Kit (Invitrogen). The density of bands was quantified using ImageJ Software and normalized to α-Tubulin.
Animal Studies: Tumor Initiation, Tumor Progression, and Survival Study
SCID-beige mice (females, 8 weeks old) were obtained from Charles Rivers Laboratories (Wilmington, MD). Approval for these studies was obtained from the Ethical Committee for Animal Experiments at Washington University in St. Louis School of Medicine.
For the tumor initiation study, 10 mice were injected intravenously (IV) with MM.1S-GFP-Luc cells at the concentration of 3 × 106 cells per mouse and were treated instantaneously with vehicle (n = 5) or selinexor (15 mg/kg, n = 5) administered by oral gavage (PO) three times a week (TIW at day 1, 3, and 5). Mice survival was recorded daily, whereas mice weight and tumor growth using bioluminescent imaging (BLI) were monitored at day 0, 28, and 42.
In the tumor progression study, MM.1S-GFP-Luc cells were injected IV into 16 SCID mice at a concentration of 2 × 106 cells per mouse and allowed to grow for 3 weeks; the tumor growth was determined by BLI. At week 3, mice were randomly allocated into two groups (eight mice per group) which received vehicle (n = 8) or selinexor (15 mg/kg; n = 8). Vehicle and selinexor were administered by oral gavage three times a week (TIW at day 1, 3, and 5). Survival of mice was monitored every day by the investigator, whereas mice weight and tumor growth (BLI) were examined at day 0, 21, and 28.
To test the effect of selinexor on bortezomib-resistant cancer, first we demonstrated development of xenograft mouse model resistant to bortezomib. MM.1S-GFP-Luc cells were injected IV into 16 SCID mice at a concentration of 2 × 106 cells per mouse and allowed to grow for 3 weeks when the tumor burden was determined by BLI. Mice with established MM tumors were pretreated with vehicle (n = 8) or bortezomib (n = 8; 1.0 mg/kg) administered intraperitoneally (IP) twice a week (BIW) for 2 weeks. A treatment with bortezomib showed initial response in the first week and then relapsed in week 2, demonstrated as ROI normalized to tumor size before pretreatment (Supplementary Figure 1).
Supplementary Figure 1.
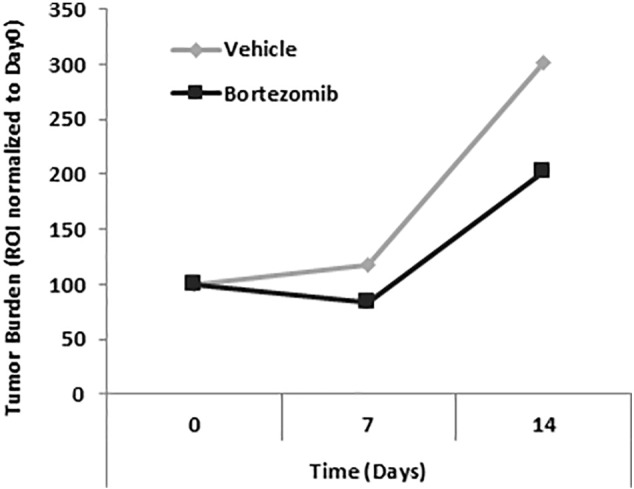
Initial administration of bortezomib induces drug resistance in MM-bearing mice.
Tumor burden of mice with established MM tumors was determined by BLI and shown as a region of interest (ROI) normalized to tumor size before pretreatment. MM-bearing mice were pretreated with vehicle (n = 8) or bortezomib (n = 8; 1.0 mg/kg) administered IP twice a week (BIW) for 2 weeks. A treatment with bortezomib showed initial response in the first week and then relapse in week 2.
Finally, to test the effect of selinexor on bortezomib-resistant cancer, MM.1S-GFP-Luc cells were injected IV into 24 SCID mice at a concentration of 2 × 106 cells per mouse and allowed to grow for 3 weeks, confirmed by BLI. Then mice were pretreated with bortezomib (1.0 mg/kg) for the next 2 weeks to induce resistance to bortezomib, after which the mice were randomly allocated (day 14) into 3 groups as follow: (1) bortezomib (0.5 mg/kg; n = 8) alone, (2) selinexor (10 mg/kg; n = 8) alone, and (3) a combination of selinexor (10 mg/kg) and bortezomib (0.5 mg/kg; n = 8). Bortezomib was injected IP twice a week (BIW; day 1 and day 4), and selinexor was administered by oral gavage three times a week (TIW; day 1, 3, and 5) along with vehicle controls. Survival of mice was monitored every day by the investigator, whereas mice weight and tumor growth (BLI) were examined at day 0, 14, 21, 35, and 42.
Statistical Analysis
The in vitro experiments were performed in quadruples and replicated independently at least three more times. Results are shown as mean ± S.D., and statistical significance was analyzed using Student’s t test. The in vivo data were analyzed using Student’s t test for statistical significance, and results are depicted as mean ± S.E.M. Variation within each group was equally variant and similar between the groups that were statistically compared. Values were considered significantly different for P values less than .05.
Results
Selinexor Inhibits Survival and Induces Apoptosis in a Dose-Dependent Manner in MM.1S Cells
Within 24 hours, in vitro studies showed that selinexor dose-dependently decreased the survival of both normoxic and hypoxic MM.1S cells (Figure 1A), whereas it increased the percentage (%) of apoptotic and dead cells in normoxic and hypoxic cells (Figure 1B). The mechanism of selinexor-induced cell apoptosis was based on its effects on the expression of the cell signaling proteins (cleaved PARP, Casp3, and Casp8), which was regulated in a dose-dependent manner in both conditions. On the contrary, phosphorylation of ERK1/2 and S6R (proteins involved in cell proliferation) was decreased with increasing concentrations of selinexor, as demonstrated by the intensity of the bands (Figure 1Ci) and by quantitative densitometry (Figure 1Cii). The effect of selinexor concentrations was more pronounced in hypoxic than normoxic conditions.
Figure 1.

Selinexor inhibits survival and induces apoptosis in a dose-dependent manner in MM cells.
Cell survival and apoptosis of MM.1S cells treated with increasing concentrations of selinexor for 24 hours in normoxic and hypoxic conditions using (A) a survival/cytotoxic MTT assay, (B) an apoptosis assay based on Annexin V/PI staining, and (Ci) immunoblotting analyses of proteins involved in apoptosis and proliferation, with (Cii) the densitometric analysis from a representative study performed on MM.1S cells.
Selinexor Reverses Hypoxia-Induced Resistance to Bortezomib In Vitro
We have previously demonstrated the development of drug resistance to proteasome inhibitors in hypoxic conditions in MM cell lines [27]. Using the MTT assay, we showed that the combination of selinexor and bortezomib increased the cytotoxic effect of bortezomib and overcame hypoxia-induced resistance to bortezomib (Figure 2A). Similarly, the apoptosis assay showed that the combination of bortezomib and selinexor increased the % of apoptotic and dead cells compared to bortezomib alone, especially in hypoxia (Figure 2B). Mechanistically, the combination of selinexor with bortezomib induced greater cleavage of PARP, Casp3, and Casp8; decreased phosphorylation of Rb; and downregulated the expression of CDK-6 compared to bortezomib alone; there was almost complete abrogation of the latter two proteins, as demonstrated by the intensity of the bands (Figure 2Ci) and by quantitative densitometry (Figure 2Cii). These results imply halted cell cycle and hence reduced cell proliferation. We tested the effect of combination of increasing concentrations of bortezomib and selinexor on MM.1S cell survival, and we found that the combination had an additive effect, and no synergy was observed (Figure 2D).
Figure 2.

Selinexor reverses hypoxia-induced resistance to bortezomib in vitro.
(A) Cell survival of MM.1S cells treated with selinexor (100 nM) in combination with bortezomib (10 nM) for 24 hours in normoxic and hypoxic conditions using a survival/cytotoxic MTT assay; (B) apoptosis assay based on Annexin V/PI staining, and (Ci) immunoblotting analyses of proteins involved in apoptosis and cell cycle, with (Cii) densitometric analysis. (D) The effect of selinexor (0, 50, 100, and 250 nM) in combination with bortezomib (0, 1, 5, and 10 nM) on MM.1S survival using MTT assay.
Selinexor as a Single Agent Delays Tumor Initiation and Improves Mice Survival
Next, we tested the effects of selinexor (15 mg/kg) as a single agent on tumor initiation in vivo. On the first day following MM.1S-GFP-Luc cell injection, we initiated oral gavage treatment of selinexor three times a week (TIW) (see the schematic diagram in Figure 3A). Selinexor delayed tumor initiation, which was detectable in the vehicle-treated group 28 days after treatment (Figure 3B, P = .034). At 42 days after treatment, the tumor burden in the selinexor-treated group was nearly half that of the vehicle-treated group (P = .078) (Figure 3B). Moreover, the vehicle-treated mice deteriorated rapidly and stayed on the study until day 53, whereas mice treated with selinexor stayed on the study for 10 more days until day 63 (P = .007) (Figure 3C).
Figure 3.
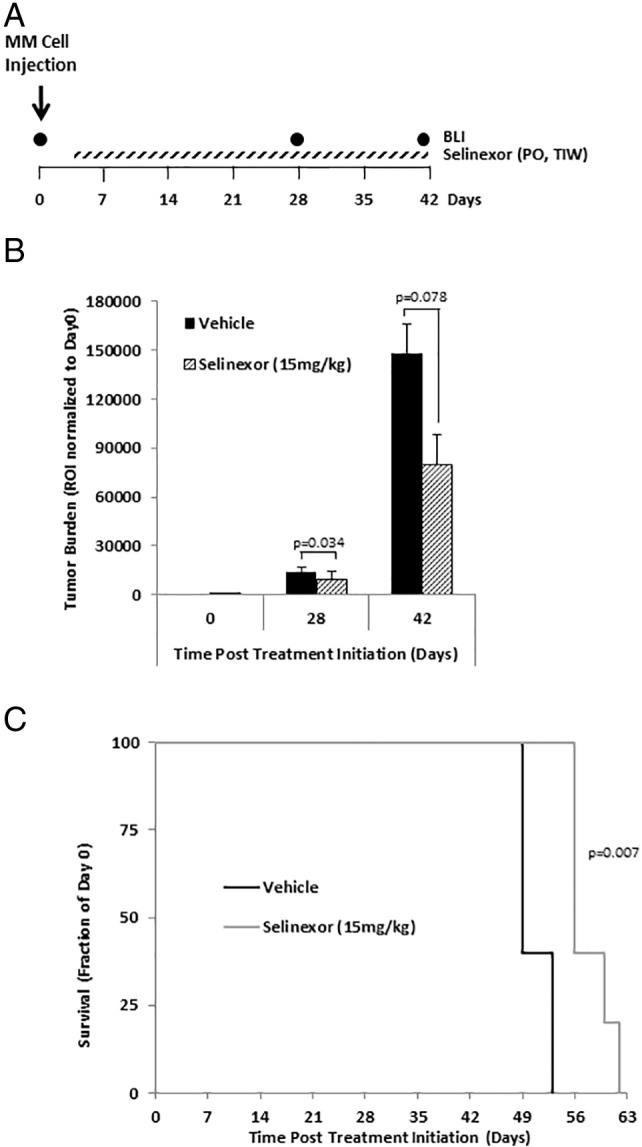
Selinexor as a single agent delays tumor initiation and improves survival in mice.
(A) Schematic diagram of mice treatment regimen in the tumor initiation study where selinexor (15 mg/kg) was given orally (PO) three times a week (TIW) as a single agent the first day following MM.1S-GFP-Luc cell injection. (B) Tumor burden in SCID mice treated with vehicle (n = 5) or selinexor (15 mg/kg administered by oral gavage three times per week, TIW; n = 5) determined by BLI and shown as an ROI normalized to initial tumor size in each group (mean ± S.E.M.). (C) Survival of mice depicted as Kaplan-Meier curve.
Selinexor as a Single Agent Delays Tumor Progression in MM-Bearing Mice
Next, we tested the effects of selinexor (15 mg/kg) as a single agent on tumor progression in MM-bearing mice administered by oral gavage three times a week (TIW). Treatment started 3 weeks post–MM cell injection (see the schematic diagram in Figure 4A). Selinexor significantly delayed tumor progression and reduced tumor burden by approximately half when compared to the vehicle-treated group at 21 and 28 days post–treatment initiation (P = .032) (Figure 4B). Moreover, selinexor-treated mice survived significantly longer than vehicle-treated mice; the vehicle-treated group stayed on the study until 36 days after treatment initiation, whereas the selinexor-treated group stayed on the study until 42 days after treatment initiation (P = .014) (Figure 4C).
Figure 4.
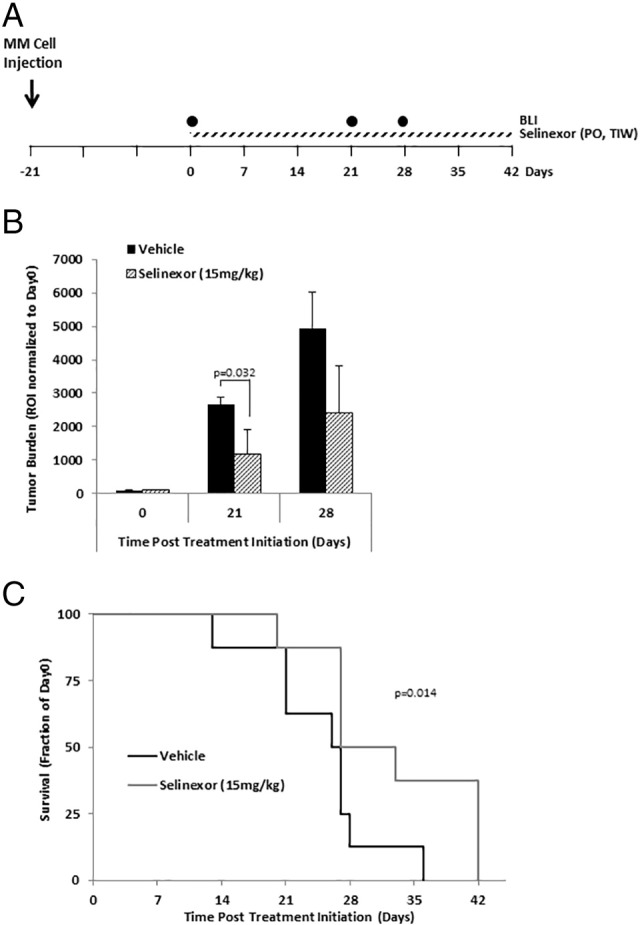
Selinexor as a single agent delays tumor progression in MM-bearing mice.
(A) Schematic diagram of mice treatment regimen in the tumor progression study. (B) Tumor burden in MM-bearing SCID mice treated with vehicle (n = 8) or selinexor (15 mg/kg administered by oral gavage three times per week, TIW; n = 8) determined by BLI and shown as an ROI normalized to initial tumor size in each group (mean ± S.E.M.). (C) Survival of mice depicted as Kaplan-Meier curve.
Combination of Selinexor with Bortezomib Delays Tumor Growth and Improves Survival in Bortezomib-Resistant MM-Bearing Mice
First, we emulated MM disease refractory to bortezomib by demonstrating that the initial administration of bortezomib alone in established MM mouse model caused bortezomib resistance. Three weeks post–MM cell injection to mice, mice were treated with vehicle (n = 8 mice) or bortezomib (n = 8; 1.0 mg/kg). Whereas the MM tumor constantly grew over 2 weeks in the vehicle-treated group, a treatment with bortezomib showed initial response in the first week and then relapse in week 2 (Supplementary Figure 1). We therefore used this mouse model to examine the effect of selinexor, with and without bortezomib, on tumor growth in bortezomib-resistant MM-bearing mice (see the schematic diagram in Figure 5A). Mice with established tumors were first treated with bortezomib IP two times a week (BIW), for 2 weeks, to induce resistance to bortezomib. Mice were then randomized and treated with bortezomib alone, selinexor alone, or the combination of bortezomib and selinexor. At day 35 (21 days after the randomization) and at day 42 (28 days after the randomization), tumor burden in mice treated with the combination of selinexor and bortezomib was 60% smaller than in mice treated with bortezomib alone (P = .012 and .036, respectively) (Figure 5B). Similarly, the survival of mice treated with the combination of selinexor and bortezomib was significantly longer than that of mice treated with bortezomib alone, in which the group treated with bortezomib alone all died at day 50, whereas 50% of the mice in the group treated with the combination of bortezomib and selinexor were still alive at day 52 (P = .005) (Figure 5C).
Figure 5.

Combination of selinexor with bortezomib delays tumor growth and improves survival in bortezomib-resistant MM-bearing mice.
(A) Schematic diagram of mice treatment regimen in the tumor progression study where the MM-bearing mice were pretreated with bortezomib (1.0 mg/kg; administered IP twice a week, BIW) for 2 weeks to develop drug resistance. Mice were then randomized and treated with bortezomib (0.5 mg/kg; n = 8), selinexor (10 mg/kg administered by oral gavage three times per week, TIW; n = 8), or combination treatment (n = 8). (B) Tumor burden was determined by bioluminescence imaging (BLI) and shown as a ROI normalized to tumor size after bortezomib pretreatment (mean ± S.E.M.). (C) Survival of mice depicted as Kaplan-Meier curve.
Discussion
Despite the implementation of novel therapies (which include proteasome inhibitors), the majority of MM patients relapse due to the development of drug resistance [38]. Low oxygenation (hypoxia) in the BM microenvironment was shown to occur beyond physiologic BM conditions during the progression of MM and, together with cellular and acellular components, plays a critical role in MM dissemination, stem cell–like properties, and drug resistance [27], [30], [39]. Our research group reported enhanced MM drug resistance to current therapies, including bortezomib and carfilzomib, in hypoxic MM cells [27], [40]. Therefore, targeting hypoxic cells to abolish the drug-resistant cancer population is the vital goal to prevent recurrence in MM patients.
Increased levels of XPO1, the nuclear exporter of tumor suppressors, were observed in MM. Moreover, XPO1 was shown to be upregulated in MM cells from patients who became resistant to bortezomib, suggesting that XPO1 could be involved in drug resistance [20]. Because hypoxia has been known to be involved in developing treatment resistance, we hypothesized that inhibition of XPO1 using the novel, orally available inhibitor selinexor (KPT-330) will reverse the hypoxia-induced bortezomib resistance in MM cells both in vitro and in vivo.
In vitro, we have demonstrated that selinexor as a single agent increased apoptosis and decreased survival of MM cells in both normoxia and hypoxia in a dose-dependent manner. In addition, combination treatment of selinexor and bortezomib demonstrated an additive effect on cell survival, and selinexor overcame hypoxia-induced drug resistance to bortezomib in cell culture. In vivo, the tumor initiation study inhibiting XPO1 with selinexor as a single agent delayed tumor progression, decreased tumor growth, and extended mice survival, indicating that XPO1 may be involved in tumorigenesis. In the tumor progression study on MM-bearing mice, which exemplified newly diagnosed patients, selinexor used as a single agent significantly reduced tumor burden and prolonged mice survival. These results were in accordance with previously published data showing that XPO1 inhibitors decreased tumor size and increased survival in MM-bearing mice as compared to the vehicle-treated group [11], [20].
Next, we have mimicked MM patients with refractory disease utilizing a xenograft mouse model resistant to bortezomib achieved by pretreating MM-bearing mice with bortezomib, where bortezomib-treated mice initially responded to chemotherapy but eventually relapsed. In this study, we used the same method to develop the bortezomib-resistant mouse model by pretreating mice with established tumors with bortezomib before adding selinexor to the treatment. Hence, this model emulates the refractory stage of the disease. These in vivo results demonstrate that the combination of selinexor with low-dose bortezomib in bortezomib-resistant MM-bearing mice significantly decreased tumor growth and extended mice survival.
We suggest that the mechanism of the combined effect of bortezomib and selinexor is related to protein homeostasis that each of these drugs (and hypoxia) affects. Protein homeostasis is controlled by several mechanisms such as protein synthesis, nuclear-cytoplasm protein trafficking, protein folding in the ER, and protein recycling by degradation in the proteasome [41]. By blocking XPO1 with SINE, the nuclear-cytoplasm trafficking of the growth-promoting proteins and oncogenic proteins is hampered, causing cell death [9], [10]. Hypoxia was shown to induce inhibition of protein synthesis through the mTOR pathway, as well as induction of protein misfolding in the ER due to the lack of recycling of glutathione for production of S-S bonds, ultimately contributing to the ER stress [42], [43]. Bortezomib is a proteasome inhibitor that causes inhibition of protein recycling and accumulation of abnormal and misfolded proteins by preventing their degradation in the proteasome, triggering cell death [5]. Hence, we speculate that subsequently the combination of selinexor with bortezomib, especially in hypoxic conditions, perturbs intracellular protein homeostasis causing protein overload in different cell compartments, induces cell stress, and enhances anticancer activity. A more thorough investigation of the mechanism is warranted.
In conclusion, we report that XPO1 inhibitor selinexor is an effective antitumor agent and can overcome hypoxia-induced drug resistance; hence, it represents a promising novel therapeutic strategy in treating patients with MM. Selinexor decreased survival and increased apoptosis of MM cells as a single agent in vitro and slowed down tumor initiation and tumor progression in the xenograft MM mouse models, significantly prolonging mice survival. In addition, selinexor dramatically reduced tumor growth when combined with low-dose bortezomib in a bortezomib-resistant xenograft model. These results provide further support for current and future clinical trials to treat newly diagnosed as well as relapsed/refractory MM patients with selinexor, especially those who are resistant to bortezomib (and possibly other proteasome inhibitors).
Conflict of Interest
Dr. Azab receives research support from Karyopharm, Verastem, Selexys, Cell Works, Cleave Bioscience, Glycomimetics, Abbvie, and Tioma and is the founder and owner of Targeted Therapeutics LLC and Cellatrix LLC. Yosef Landesman is employed by Karyopharm Therapeutics. Other authors state no conflicts of interest.
The following are the supplementary data related to this article.
Acknowledgement
The study was supported partially by a grant from Karyopharm Therapeutics and partially by the award from the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) and the National Cancer Institute of the NIH under U54CA199092. The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
References
- 1.Nair RR, Gebhard AW, Emmons MF, Hazlehurst LA. Emerging strategies for targeting cell adhesion in multiple myeloma. Adv Pharmacol. 2012;65:143–189. doi: 10.1016/B978-0-12-397927-8.00006-3. [DOI] [PubMed] [Google Scholar]
- 2.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsiades CS, Hayden PJ, Anderson KC, Richardson PG. From the bench to the bedside: emerging new treatments in multiple myeloma. Best Pract Res Clin Haematol. 2007;20:797–816. doi: 10.1016/j.beha.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludwig H, Milosavljevic D, Zojer N, Faint JM, Bradwell AR, Hubl W, Harding SJ. Immunoglobulin heavy/light chain ratios improve paraprotein detection and monitoring, identify residual disease and correlate with survival in multiple myeloma patients. Leukemia. 2013;27:213–219. doi: 10.1038/leu.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120:947–959. doi: 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanai M, Hanashiro K, Kim SH, Hanai S, Boulares AH, Miwa M, Fukasawa K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat Cell Biol. 2007;9:1175–1183. doi: 10.1038/ncb1638. [DOI] [PubMed] [Google Scholar]
- 8.Shao C, Lu C, Chen L, Koty PP, Cobos E, Gao W. p53-Dependent anticancer effects of leptomycin B on lung adenocarcinoma. Cancer Chemother Pharmacol. 2011;67:1369–1380. doi: 10.1007/s00280-010-1434-6. [DOI] [PubMed] [Google Scholar]
- 9.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci U S A. 2000;97:1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turner JG, Kashyap T, Dawson JL, Gomez J, Bauer AA, Grant S, Dai Y, Shain KH, Meads M, Landesman Y. XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IkappaBalpha and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget. 2016;7:78896–78909. doi: 10.18632/oncotarget.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH, Sun YJ. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009;32:E315–E321. [PubMed] [Google Scholar]
- 13.Noske A, Weichert W, Niesporek S, Roske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–1743. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- 14.van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD. The karyopherin proteins, Crm1 and karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009;124:1829–1840. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen P, Sun YJ, Tang LN, Zheng SE. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21:229–235. [PubMed] [Google Scholar]
- 16.Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther. 2016;164:135–143. doi: 10.1016/j.pharmthera.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azmi AS, Al-Katib A, Aboukameel A, McCauley D, Kauffman M, Shacham S, Mohammad RM. Selective inhibitors of nuclear export for the treatment of non-Hodgkin's lymphomas. Haematologica. 2013;98:1098–1106. doi: 10.3324/haematol.2012.074781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, Tiedemann RE, Palmer SE, Garbitt VM, McCauley D. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia. 2013;27:2357–2365. doi: 10.1038/leu.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–165. doi: 10.1038/leu.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, Pekarsky Y, Croce CM. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119:329–331. doi: 10.1182/blood-2011-10-386144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kashyap T, Argueta C, Aboukameel A, Unger TJ, Klebanov B, Mohammad RM, Muqbil I, Azmi AS, Drolen C, Senapedis W. Selinexor, a selective inhibitor of nuclear export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget. 2016;7:78883–78895. doi: 10.18632/oncotarget.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muqbil I, Aboukameel A, Elloul S, Carlson R, Senapedis W, Baloglu E, Kauffman M, Shacham S, Bhutani D, Zonder J. Anti-tumor activity of selective inhibitor of nuclear export (SINE) compounds, is enhanced in non-Hodgkin lymphoma through combination with mTOR inhibitor and dexamethasone. Cancer Lett. 2016 doi: 10.1016/j.canlet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Camacho C, Silvers TR, Razak AR, Gabrail NY, Gerecitano JF, Kalir E, Pereira E, Evans BR, Ramus SJ. Inhibition of the nuclear export receptor Xpo1 as a therapeutic target for platinum resistant ovarian cancer. Clin Cancer Res. 2016;23:1552–1563. doi: 10.1158/1078-0432.CCR-16-1333. [DOI] [PubMed] [Google Scholar]
- 26.Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14:191–201. doi: 10.1016/j.drup.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Muz B, de la Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J. 2014;4:e262–e265. doi: 10.1038/bcj.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asosingh K, De Raeve H, de Ridder M, Storme GA, Willems A, Van Riet I, Van Camp B, Vanderkerken K. Role of the hypoxic bone marrow microenvironment in 5T2MM murine myeloma tumor progression. Haematologica. 2005;90:810–817. [PubMed] [Google Scholar]
- 30.Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–5794. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaupel P, Kelleher DK, Hockel M. Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol. 2001;28:29–35. doi: 10.1016/s0093-7754(01)90210-6. [DOI] [PubMed] [Google Scholar]
- 32.Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26:1818–1830. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 33.Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–2037. [PubMed] [Google Scholar]
- 34.Axelson H, Fredlund E, Ovenberger M, Landberg G, Pahlman S. Hypoxia-induced dedifferentiation of tumor cells—a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005;16:554–563. doi: 10.1016/j.semcdb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diabira S, Morandi X. Gliomagenesis and neural stem cells: key role of hypoxia and concept of tumor "neo-niche". Med Hypotheses. 2008;70:96–104. doi: 10.1016/j.mehy.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Blazek ER, Foutch JL, Maki G. Daoy medulloblastoma cells that express CD133 are radioresistant relative to CD133− cells, and the CD133+ sector is enlarged by hypoxia. Int J Radiat Oncol Biol Phys. 2007;67:1–5. doi: 10.1016/j.ijrobp.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 38.de la Puente P, Muz B, Azab F, Luderer M, Azab AK. Molecularly targeted therapies in multiple myeloma. Leuk Res Treatment. 2014;2014:976567. doi: 10.1155/2014/976567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92. doi: 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muz B, de la Puente P, Azab F, Luderer MJ, King J, Vij R, Azab AK. A CD138-independent strategy to detect minimal residual disease and circulating tumour cells in multiple myeloma. Br J Haematol. 2016;173:70–81. doi: 10.1111/bjh.13927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 42.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]