

Abstract
In mammalian cells, nonhomologous end-joining (NHEJ) repairs DNA double-strand breaks created by ionizing radiation and V(D)J recombination. We have developed a cell-free system capable of processing and joining noncompatible DNA ends. The system had key features of NHEJ in vivo, including dependence on Ku, DNA-PKcs, and XRCC4/Ligase4. The NHEJ reaction had striking properties. Processing of noncompatible ends involved polymerase and nuclease activities that often stabilized the alignment of opposing ends by base pairing. To achieve this, polymerase activity efficiently synthesized DNA across discontinuities in the template strand, and nuclease activity removed a limited number of nucleotides back to regions of microhomology. Processing was suppressed for DNA ends that could be ligated directly, biasing the reaction to preserve DNA sequence and maintain genomic integrity. DNA sequence internal to the ends influenced the spectrum of processing events for noncompatible ends. Furthermore, internal DNA sequence strongly influenced joining efficiency, even in the absence of processing. These results support a model in which DNA-PKcs plays a central role in regulating the processing of ends for NHEJ.
Keywords: DNA end-processing, DNA repair, double-strand break repair, nonhomologous end-joining, V(D)J recombination
Introduction
Nonhomologous end-joining (NHEJ) is important for repairing DNA double-strand breaks produced by ionizing radiation. Furthermore, NHEJ is absolutely required for resolving double-strand breaks during V(D)J recombination, the pathway that generates immunological diversity. While the error-free pathway of homologous recombination restores broken DNA to its original sequence, the error-prone pathway of NHEJ often processes the DNA by adding or deleting nucleotides before joining the ends. Processing occurs after ionizing radiation, which produces chemically modified DNA ends that are not directly ligatable. Furthermore, processing contributes to the immunological diversity generated by V(D)J recombination.
NHEJ in vivo requires Ku, DNA-PKcs, Artemis, and XRCC4/Ligase4. Ku is a heterodimer of 70 and 80 kDa that binds to DNA ends (Smider et al, 1994; Taccioli et al, 1994). DNA-PKcs, the catalytic subunit of DNA-dependent protein kinase, is a 465 kDa ser/thr kinase in the phosphoinositol 3-kinase (PI3K) superfamily (Hartley et al, 1995). DNA-PKcs binds to DNA ends upon recruitment by Ku (Hammarsten and Chu, 1998), mediates synapsis of the ends, and then undergoes activation of its kinase (DeFazio et al, 2002). Kinase activity is required for NHEJ, but its function remains obscure. Artemis exists in a complex with DNA-PKcs and has nuclease activity (Ma et al, 2002). XRCC4/Ligase4 executes the final step of ligation (Grawunder et al, 1997).
Cell-free systems for NHEJ have been reported previously, but each system was unable to reproduce all of the key characteristics observed in vivo. Some systems were independent of DNA-PKcs (Boe et al, 1995; Johnson and Fairman, 1996; Mason et al, 1996). A subsequent end-joining system was only partially dependent on Ku (Feldmann et al, 2000). Extracts from wild-type K1 hamster cells joined compatible and noncompatible DNA ends, but extracts from Ku-deficient xrs6 hamster cells were still capable of joining ends with 14–46% of the efficiency in wild-type K1 extracts. By contrast, intact xrs6 cells have a much more severe defect. They join ends created during V(D)J recombination with less than 0.1% of wild-type efficiency (Taccioli et al, 1993). Another cell-free system yielded circularization and dimerization of linear DNA with noncompatible ends (Pospiech et al, 2001). Circularization was blocked by wortmannin, which inhibits kinases in the PI3K family. However, dimerization, which accounted for most of the joining activity, was unaffected by wortmannin or anti-Ku antibodies. Furthermore, junctions were studied for only one set of noncompatible ends, and deletion of internal DNA sequences was a rare event.
A cell-free system fully dependent on Ku, DNA-PKcs, and XRCC4/Ligase4 has been established, but it was limited to the joining of compatible ends (Baumann and West, 1998). The reaction required inositol hexakisphosphate (Hanakahi et al, 2000) and polynucleotide kinase for ends with 5′-OH groups (Chappell et al, 2002). There are no reports that this system can support processing of other types of noncompatible ends. Thus, a cell-free system for the entire NHEJ reaction has remained elusive.
Here, we report that human cell extract supports NHEJ of noncompatible DNA ends. To observe the processing reactions, we employed a PCR assay that enabled us to recover junctions specific for the noncompatible ends. After discovering that processing included polymerase activity, we were able to achieve high joining efficiencies by including dNTPs in the reaction. Processing also included nucleotide deletion to regions of microhomology, as observed for V(D)J recombination in vivo. Joining was highly dependent on proteins required in vivo: Ku, DNA-PKcs, and XRCC4/Ligase4. Characterization of this system led to new insights into the pathway of NHEJ.
Results
NHEJ is an intermolecular reaction that can be measured by quantitative PCR (qPCR)
We reproduced NHEJ of compatible DNA ends as previously reported (Baumann and West, 1998). A radiolabeled DNA fragment (0.5 μg/ml) was incubated with cell extract and the DNA products were resolved by gel electrophoresis. We used DNA molecules with lengths of 2686 base pairs (bp), similar to the DNA used by Baumann and West, and 820 bp, the same length as the DNA used later in this report. Gel electrophoresis revealed that the extract joined the DNA into linear dimers, trimers, and higher order multimers, but not into circular monomers (data not shown). This result is also present in data from other groups (Baumann and West, 1998; Chen et al, 2001b). By contrast, a parallel incubation of T4 DNA ligase with the 2686 bp DNA generated products that were 75% circular monomers. We concluded that the failure to observe circular monomers in the NHEJ reaction was due to extensive binding of the DNA to proteins in the extract. Therefore, we focused on intermolecular joining in our assay for NHEJ.
To study intermolecular joining, we prepared a series of DNA substrates with different ends. Since we wanted to investigate processing of ends prior to joining, it was critically important to be confident of the sequence at the DNA ends. We carefully prepared DNA substrates free of unwanted DNA contaminants, as described in Materials and methods. Sequencing of 364 junctions from the different DNA ends reported in this paper showed no evidence of contaminating DNA fragments.
To measure the joining efficiency of various noncompatible ends, we developed a PCR assay (Figure 1). Although the PCR primers amplified junctions joined in a specific orientation, restriction enzyme analysis showed that the DNA fragments were joined in all possible orientations. Head-to-head joining of the same fragment to itself could potentially make a spurious contribution to our measurement of joining efficiency. To rule out this possibility, we carried out PCR with a single primer. No product was detected, consistent with the known failure of PCR to amplify long inverted repeats. Therefore, our PCR assay successfully detected joining of two well-defined DNA ends.
Figure 1.
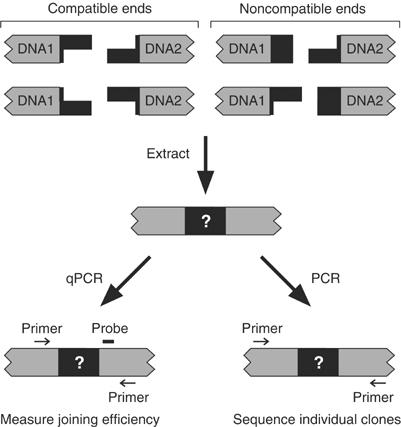
Assay for NHEJ in vitro. DNA substrates were synthesized with various DNA ends attached to the same internal DNA sequences, DNA1 and DNA2. The figure shows substrates with compatible and noncompatible ends. In some cases, we synthesized DNA substrates in which the DNA ends were exchanged with respect to the internal DNA sequences, as illustrated for the noncompatible ends in the figure. Joining efficiency was measured by qPCR. Processing of the ends was analyzed after amplifying DNA junctions by PCR and sequencing individual clones.
Extract supports the processing of noncompatible ends for NHEJ
Cell extract supported joining of both compatible and noncompatible DNA ends (Figure 2A). ATP and Mg2+ were necessary for the joining reaction (data not shown). As a control, T4 ligase joined compatible ends but failed to join noncompatible ends. Thus, the extract actively processed noncompatible ends into a ligatable form.
Figure 2.
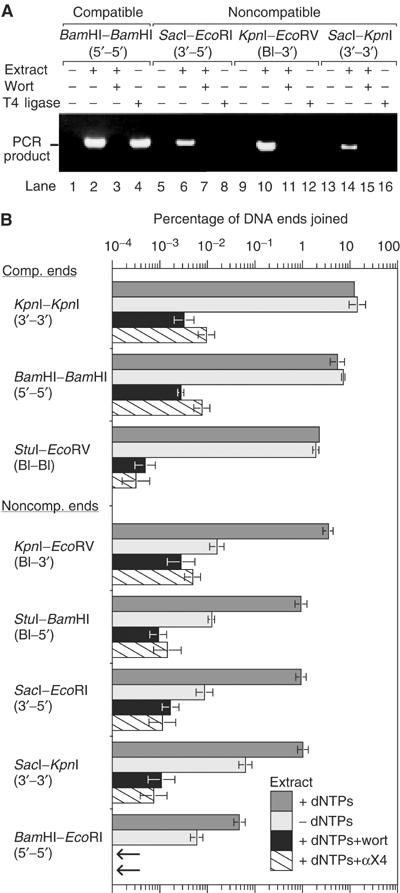
Extract supports NHEJ of noncompatible ends. (A) NHEJ is detectable by a gel-based assay. In lanes 2, 6, 10, and 14, DNA substrates with compatible or noncompatible ends were incubated with extract and dNTPs. The resulting junctions were amplified by 30 cycles of PCR, and the PCR product was resolved by gel electrophoresis. In lanes 3, 7, 11, and 15, the extract was pre-incubated for 30 min with 10 μM wortmannin (wort) to inhibit the kinase activity of DNA-PKcs. In lanes 4, 8, 12, and 16, end-joining was catalyzed by T4 DNA ligase, which joins only compatible DNA ends. (B) The NHEJ reaction requires DNA-PKcs and XRCC4, while dNTPs stimulate joining of noncompatible ends. DNA substrates with compatible or noncompatible ends were incubated with extract (+dNTPs), extract (−dNTPs), extract (+dNTPs) treated with wortmannin, or extract (+dNTPs) immunodepleted with αXRCC4 antibody (2 × 4). Joining efficiency was measured by qPCR and displayed on a log plot to account for the large variation in joining. The small arrows in place of the bottom two bars represent joining efficiency of less than 0.0001%.
To test the dependence of the joining reaction on kinase activity, we added wortmannin to the extract and confirmed the loss of DNA-stimulated kinase activity (data not shown). Wortmannin strongly inhibited the joining of both compatible and noncompatible ends (Figure 2A). Thus, DNA-dependent kinase activity was required for joining both in the presence and absence of processing.
To measure joining efficiency as a percentage of DNA ends joined, we employed a qPCR assay. To characterize the assay, we performed time courses of the end-joining reactions (Supplementary Figure 1). Joining of compatible and noncompatible ends reached asymptotic levels after 90 min. For the rest of our experiments, we chose to measure joining efficiency at this time point rather than an earlier time point because small errors in reaction time did not affect the results. The percentage of DNA ends joined was determined from parallel PCR amplifications of pre-formed DNA junctions (see Materials and methods).
Joining of both compatible and noncompatible ends was inhibited eight- to 20-fold by immunodepletion with antibodies against Ku70, Ku80, or DNA-PKcs (data not shown). This level of inhibition was approximately equal to the degree of immunodepletion that we were able to achieve. Our failure to completely deplete extracts of Ku and DNA-PKcs may be due to the abundance of these proteins in human cells. More significantly, addition of wortmannin or immunodepletion with anti-XRCC4 antibody inhibited joining by at least 500-fold (Figure 2B). Thus, NHEJ in extract was appropriately dependent on Ku, DNA-PKcs, and XRCC4/Ligase4.
NHEJ efficiency depends on the structure of the ends
To determine how the structure of DNA ends influenced joining, we tested DNA with 5′ overhangs, 3′ overhangs, and blunt ends in various combinations (Figure 2B). We will refer to eight structural categories of paired ends. Three categories are ligatable: compatible 3′–3′, 5′–5′, and blunt–blunt ends. Five categories require processing: noncompatible 3′–3′, 5′–5′, 3′–5′, blunt–3′, and blunt–5′ ends. Each of the last five categories includes paired ends attached to DNA1 and DNA2 in either orientation (Figure 1). For example, blunt–3′ ends include EcoRV–SacI, EcoRV–KpnI, and KpnI–EcoRV ends.
Compatible 3′–3′ ends were usually joined most efficiently (12.3% for KpnI–KpnI, 9.0% for SacI–SacI), followed by compatible 5′–5′ ends (5.4% for BamHI–BamHI, 4.0% for EcoRI–EcoRI) and blunt–blunt ends (2.3% for StuI–EcoRV). These joining efficiencies were quite high, since they refer only to DNA ends joined in the orientation specified by the PCR primers. The total percentage of joined ends was significantly higher since the ends were joined in four possible orientations.
Noncompatible ends were joined with varying efficiencies. Blunt–3′ ends (KpnI–EcoRV) were joined with 3.5% efficiency, while noncompatible 5′–5′ ends (BamHI–EcoRI) were joined with only 0.05% efficiency. This suggests that factors involved in processing 5′ ends were limiting in the extract.
Compatible ends are joined without processing
To characterize junctions formed by the NHEJ reaction, we subcloned PCR-amplified DNA sequences on both sides of the junctions and sequenced DNA from individual clones. We sequenced 10 junctions from each of three reactions involving compatible ends, KpnI–KpnI, BamHI–BamHI, and StuI–EcoRV. All 30 junctions were formed without processing (Figure 3A). The precise joining of compatible ends indicated that processing is suppressed when not required.
Figure 3.
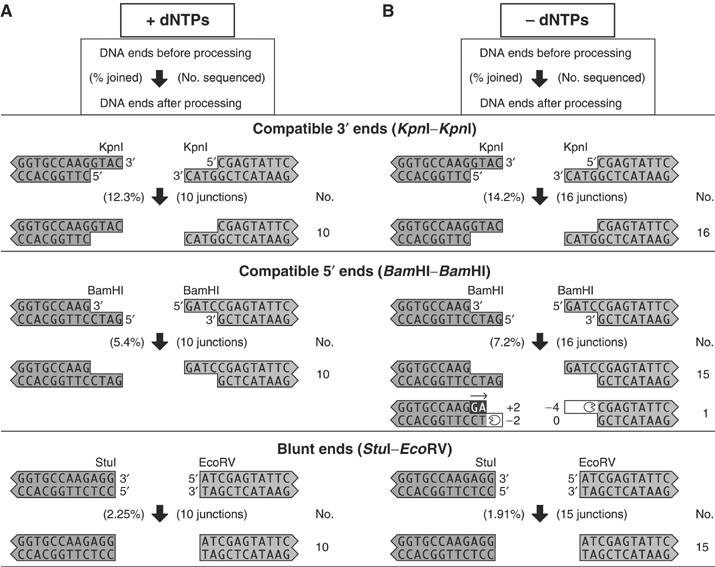
Compatible ends are joined with minimal processing. DNA substrates containing compatible ends were incubated with (A) extract plus added dNTPs, or (B) extract in the absence of added dNTPs. The legends indicate joining efficiency to the left of the arrow, and total number of sequenced junctions to the right of the arrow. The number of times each junction was recovered is shown in the column labeled ‘No'. When the DNA ends are processed, the open-mouthed icon and white background in the DNA indicate nuclease activity, and the arrow and black background indicate polymerase activity. Gray background indicates unprocessed DNA.
Noncompatible ends are processed by DNA synthesis across a discontinuous template strand
When reactions for noncompatible ends were performed with dNTPs added to the extract, the vast majority of junctions showed evidence of templated nucleotide addition (Figure 4). We sequenced 364 junctions and did not detect any mutations from templated addition of a total of 369 nucleotides. Thus, the error rate of the associated polymerase activity was less than 1%.
Figure 4.

In the presence of dNTPs, noncompatible ends are processed largely by templated nucleotide addition. DNA substrates with compatible or noncompatible ends were incubated with extract and dNTPs, and the junctions were sequenced. The processed ends are shown according to the legend in Figure 3. The number of bases of microhomology utilized in the joining reaction is shown in the column labeled ‘MH'. Each section (A–E) depicts junctions from a different structural category of paired ends.
The observed polymerase activity in our extract was not an artifact of the PCR assay. Omitting dNTPs from the reaction inhibited the joining efficiency of noncompatible ends (Figure 2) and suppressed the templated addition of nucleotides in the junctions (see below). Furthermore, adding the DNA polymerase inhibitor dideoxy-NTPs suppressed the joining efficiency of noncompatible ends, while preserving the joining efficiency of compatible ends (data not shown).
Polymerase activity filled in 5′ overhanging ends, as expected. However, polymerase activity also filled in 3′ overhanging ends with high frequency (dotted arrows in Figure 4), a phenomenon previously observed in Xenopus extract (Thode et al, 1990). Indeed, blunt–3′ ends were joined with high efficiency (Figure 2B and Supplementary Figure 3B) and almost always processed by the fill-in of the 3′ end (Figure 4). Thus, a polymerase primed from the blunt end was able to synthesize DNA across a discontinuity in the template strand, perhaps in conjunction with a factor that aligned the ends appropriately. This type of polymerase activity may increase the efficiency of NHEJ by creating base pairing between the opposing ends and thus stabilizing their alignment.
To further study the role of polymerase activity in the NHEJ reaction, we omitted dNTPs from the reaction. The joining efficiency of noncompatible ends was reduced 1.7-fold to over 200-fold, suggesting that polymerase activity made a large contribution to processing of the ends (Figure 2B and Supplementary Figure 3). By contrast, the joining efficiency of compatible ends did not change significantly (Figure 2B and Supplementary Figure 2). This is consistent with the absence of polymerase activity at the junctions of compatible ends (Figure 3).
Noncompatible ends are processed by regulated nuclease activity
To characterize the nuclease activity in NHEJ, we suppressed polymerase activity by omitting dNTPs. In these reactions, addition of wortmannin or immunodepletion of XRCC4 inhibited joining 80- to 5000-fold (data not shown). Thus, joining remained dependent on DNA-PKcs and XRCC4/Ligase4, and reflected bona fide NHEJ.
Joining efficiency was significantly reduced in the absence of added dNTPs (Figure 2). When we sequenced the junctions formed from noncompatible ends, processing was dominated by nuclease activity, which was involved in the formation of 195 of 199 (98%) junctions (Figure 5). Modest polymerase activity was observed, presumably due to nucleotides generated by nuclease activity. For compatible ends, nuclease activity was suppressed, since those ends were almost always joined precisely (Figure 3B).
Figure 5ab.

When dNTPs are omitted, noncompatible ends are processed by deletion and templated addition of nucleotides. DNA substrates with noncompatible ends were incubated with extract in the absence of added dNTPs, and the processed ends are shown according to the legend in Figure 3. Asterisks indicate junctions that utilized microhomology from internal DNA sequences. In these cases, there was ambiguity in the processing events, and we assumed that the microhomology alignment occurred via a 3′ overhang. Each section (A–E) depicts junctions from a different structural category of paired ends.
There were two types of nuclease activity. The first type was specific for single-stranded 5′ or 3′ overhangs. Many of the overhangs were removed precisely to leave a blunt end. This was particularly evident when the opposing end was already blunt. In 15 of 37 junctions from the blunt–3′ ends, EcoRV–KpnI and KpnI–EcoRV, the four terminal nucleotides were removed from the 3′ overhang (Figure 5B). Similarly, in 14 of 24 junctions from the blunt–5′ ends, StuI–BamHI, the four terminal nucleotides were removed from the 5′ overhang (Figure 5C).
Figure 5cde.

When dNTPs are omitted, noncompatible ends are processed by deletion and templated addition of nucleotides. DNA substrates with noncompatible ends were incubated with extract in the absence of added dNTPs, and the processed ends are shown according to the legend in Figure 3. Asterisks indicate junctions that utilized microhomology from internal DNA sequences. In these cases, there was ambiguity in the processing events, and we assumed that the microhomology alignment occurred via a 3′ overhang. Each section (A–E) depicts junctions from a different structural category of paired ends.
The second type of nuclease activity produced deletions that were consistent with alignment of the DNA ends by base pairing in regions of microhomology. Deletions back to one to five bases of microhomology were observed in 148 of 199 (74%) junctions. The deletions were tightly regulated. Only eight junctions (4%) contained deletions larger than 16 nucleotides. Furthermore, all of the larger deletions were more than 150 nucleotides, consistent with the aberrant recruitment of an unregulated, processive nuclease. These large deletions became dominant in the rare junctions sequenced from reactions in which the kinase activity of DNA-PKcs was inhibited by wortmannin (data not shown). This suggests that when DNA-PKcs is kinase-dead, the junctions that were recovered involved backup nucleases that excised large DNA–protein fragments. When DNA-PKcs is active, its kinase activity may regulate the nuclease activity to control the size of deletions.
Junctions utilized microhomology with sequences in 3′ overhangs much more frequently than with sequences in 5′ overhangs. Of the 66 total junctions from 3′–5′ ends, only one utilized microhomology pairing with nucleotides in a 5′ overhang, while 62 utilized a 3′ overhang. Among junctions from the noncompatible blunt–5′ ends, StuI–BamHI, only one of 24 utilized microhomology with the 5′ overhang (Figure 5C), while 15 of 37 junctions from the noncompatible blunt–3′ ends, EcoRV–KpnI and KpnI–EcoRV, utilized microhomology with the 3′ overhang (Figure 5B). Furthermore, 23 of 27 junctions from the noncompatible 3′–3′ ends, SacI–KpnI, utilized microhomology with a 3′ overhang (Figure 5D).
The preference for utilizing 3′ overhangs during NHEJ could be coupled to key enzymatic activities. The DNA-PKcs kinase is activated more efficiently with 3′ overhangs than with 5′ overhangs (Hammarsten et al, 2000). The endonuclease that opens hairpin ends during V(D)J recombination preferentially creates ends with 3′ overhangs (Schlissel, 1998).
Internal DNA sequence influences processing events during NHEJ
We discovered up to 100-fold variations in the efficiency of joining noncompatible ends when different DNA molecules were used. To examine this variation in more detail, we constructed DNA molecules in which the noncompatible ends were exchanged relative to the internal sequences. We shall refer to the reactions for these exchanged noncompatible ends as ‘matched reactions', as illustrated in Figure 1.
Surprisingly, matched reactions often had different joining efficiencies. With dNTPs added to the reaction, EcoRV–BamHI was joined eight-fold more efficiently than BamHI–EcoRV (Supplementary Figure 3). With dNTPs omitted, some matched reactions showed even larger variations in joining efficiency. BamHI–SacI was joined 30-fold more efficiently than SacI–BamHI, and EcoRI–SacI was joined 40-fold more efficiently than SacI–EcoRI. Thus, sequences internal to the DNA ends influenced joining efficiency.
To characterize the role of internal DNA sequence, we sequenced junctions from three pairs of matched reactions (Figure 5A, B and 5E). The matched reactions with noncompatible 5′ ends, BamHI–EcoRI and EcoRI–BamHI, were joined with the same low efficiency of 0.006%. Processing yielded a heterogeneous mixture of junctions, some showing templated addition of nucleotides, but most showing deletions back to regions of microhomology. For another pair of matched reactions, EcoRV–KpnI and KpnI–EcoRV (blunt–3′ ends), we found six-fold differences in joining efficiency of 0.093% and 0.015%, respectively. Part of the difference could arise from polymerase activity across a discontinuous template, which may enhance joining efficiency by creating base pairing to stabilize alignment of the ends. Indeed, polymerase activity was present in 3 of 17 KpnI–EcoRV junctions, but in 11 of 20 junctions from the more efficient EcoRV–KpnI reaction.
The noncompatible 3′–5′ ends, SacI–EcoRI and SacI–BamHI, were joined 40- and 30-fold less efficiently than matched reactions in which the ends were exchanged, EcoRI–SacI and BamHI–SacI (Figure 5E). In the more efficient reactions, all 18 EcoRI–SacI junctions and 14 of 16 BamHI–SacI junctions utilized polymerase activity. Thus, higher joining efficiency was associated with increased polymerase activity, which often created base pairing by synthesis across a discontinuous template. The more efficient reactions also involved microhomology pairing of the terminal two bases of the SacI 3′ overhang with two internal bases near the opposing end, and included nuclease activity directed at only one of the four DNA strands. In the less efficient SacI–EcoRI and SacI–BamHI reactions, microhomology pairing with the terminal two bases of the SacI 3′ overhang was not available for at least 20 bp. Instead, microhomology base pairing was achieved by recruitment of nuclease activity to at least two of the four DNA strands. Thus, the recruitment of nuclease activity appeared to be inefficient, biasing the NHEJ in favor of preserving DNA sequence. These data demonstrate that internal DNA sequence influences the spectrum of processing events for noncompatible ends.
Internal DNA sequence influences joining efficiency in the absence of processing
To further examine the role of internal DNA sequence, we used DNA substrates with five types of blunt DNA ends in 11 separate reactions (Figure 6). As expected, T4 ligase joined all blunt ends with similar efficiency. By contrast, extract joined blunt ends with efficiencies that varied up to 21-fold. For example, SspI–SwaI (2.10%) was joined 19-fold more efficiently than its matched pair of ends, SwaI–SspI (0.11%). Addition of dNTPs did not significantly affect the joining efficiencies of any of these blunt ends (Supplementary Figure 2). Thus, internal DNA sequence strongly influenced the joining efficiency for blunt ends.
Figure 6.
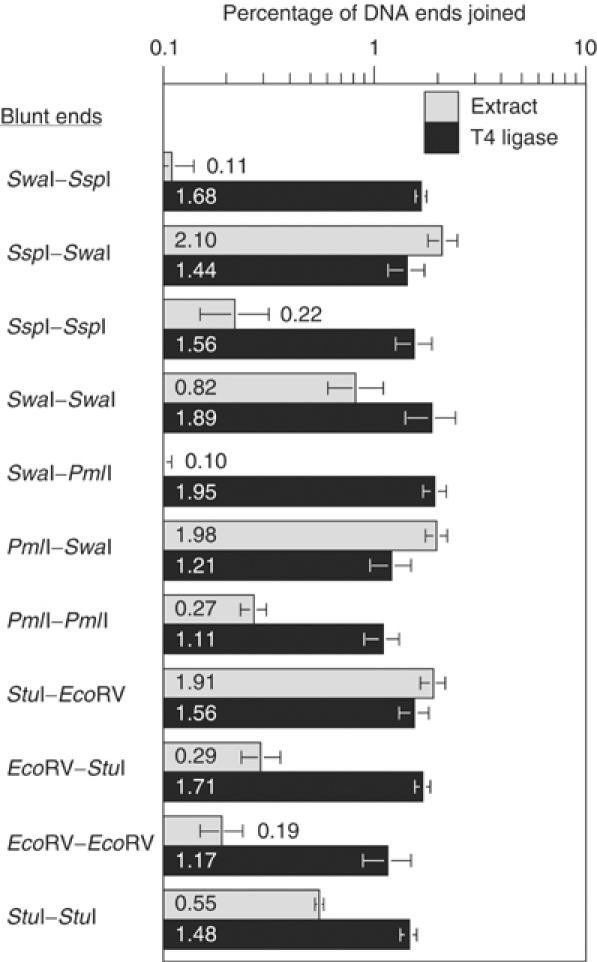
Joining efficiency of blunt DNA ends is influenced by internal DNA sequence. Joining efficiency was measured after incubating DNA substrates with T4 DNA ligase, or after incubating the same DNA substrates with extract. In these experiments, the reactions were performed in the absence of added dNTPs.
We sequenced DNA junctions from four different blunt–blunt reactions that showed large variations in joining efficiency (Supplementary Figure 4). The ends were joined without processing in 49 of 53 sequenced junctions, despite a 19-fold variation in joining efficiency. Thus, we were confronted with the striking result that internal DNA sequence influenced the efficiency of blunt end-joining, even though 95% of the joining occurred without processing.
We considered two possible explanations for how DNA sequence might affect the joining efficiency of blunt ends. First, DNA sequence that permitted microhomology base pairing or the formation of cruciform structures might influence the joining reaction. Second, DNA sequence might affect the interaction of the DNA with DNA-PKcs.
To address the first possibility, we constructed blunt-ended DNA fragments containing up to six bases of microhomology internal to both ends, or containing inverted repeats of up to six bases that would permit formation of a cruciform structure at the ends. Although we observed large variations in the joining efficiency of these blunt ends, there was no correlation with the presence of microhomology or inverted repeats in the paired ends (data not shown).
Thus, we were led to the hypothesis that DNA sequence affects the interaction of the ends with DNA-PKcs. How this interaction might depend on DNA sequence will be explored in Discussion. Furthermore, our hypothesis is supported by recent data that sequence strongly influences kinase activation (Pawelczak et al, 2005).
Discussion
NHEJ in extract has the properties of NHEJ in vivo
It is difficult to study NHEJ in vivo since junctions can be recovered only if the double-strand breaks occur at a defined site. Rouet et al (1994) introduced I–SceI sites into mouse chromosomal DNA and created double-strand breaks by expressing the restriction enzyme I–SceI. The junctions contained various deletions back to short regions of microhomology. A follow-up experiment introduced I–SceI sites into wild-type and Ku-deficient CHO cells (Liang et al, 1996). The site was engineered so that repair of the double-strand break would reconstruct a wild-type neomycin resistance gene if joining occurred after deletion back to a region containing four base pairs of microhomology. Ku-deficient cells were appropriately deficient in rejoining ends by this deletion mechanism. However, characterization of NHEJ in vivo was limited because the double-strand breaks involved only a single sequence with a 3′ overhang. Furthermore, junctions in the Ku-dependent reaction were selected for a single processing event.
NHEJ in vivo has been studied indirectly in the context of V(D)J recombination. The RAG1/RAG2 complex cleaves the chromosome at two recombination signal sequences to create two hairpin-coding ends and two blunt signal ends. The joining of signal ends occurs without processing and requires Ku and XRCC4/Ligase4. The hairpin-coding ends are opened preferentially into 3′ overhangs in vivo by an endonuclease activity (Schlissel, 1998), most likely Artemis (Ma et al, 2002).
The opened coding ends are processed and joined in a reaction requiring Ku, DNA-PKcs, and XRCC4/Ligase4. Processing involves templated nucleotide addition, nucleotide deletion, and utilization of microhomology. A polymerase presumably fills in the 3′ overhangs by synthesizing DNA across a discontinuity in the template strand. In wild-type mice, terminal deoxynucleotidyltransferase (TdT) obscures the utilization of microhomology. However, in TdT knockout mice, 75% of the coding junctions were created by nucleotide deletion back to regions of microhomology within 20 bp of the ends (Gilfillan et al, 1993; Komori et al, 1993).
Data from V(D)J recombination junctions suggest, but cannot define, processing events for double-strand breaks elsewhere in the genome. In particular, breaks produced by RAG1/RAG2 have key differences from breaks induced by ionizing radiation or topoisomerase II inhibitors. First, hairpin ends are created and then opened prior to joining. Second, RAG1/RAG2 remain bound to the DNA in a post-cleavage complex that may influence the NHEJ reaction. Third, it is possible that specialized processing enzymes act at the immunoglobulin locus to promote immunological diversity.
We have shown that cell extract can process and join DNA ends in a reaction that recapitulates NHEJ in vivo. Our in vitro reaction depended on Ku, DNA-PKcs, and XRCC4/Ligase4, as it does in vivo. Polymerase activity was able to jump across a discontinuity in the template strand, as observed for V(D)J recombination in vivo. Nuclease activity deleted nucleotides back to regions of microhomology in 74% of junctions. Deletions were restricted to a region within 20 bp of the DNA ends, which closely mirrored the deletions generated by V(D)J recombination.
Several enzymes are candidates for processing ends during NHEJ
Artemis is a prime candidate for contributing nuclease activity to the NHEJ reaction. Its deficiency leads to the human syndrome of RS-SCID, severe combined immunodeficiency with radiosensitivity (Moshous et al, 2001). As a purified protein, Artemis is a single-strand specific 5′–3′ exonuclease. After phosphorylation by DNA-PKcs, Artemis acquires an endonuclease activity specific for hairpins and 5′ or 3′ overhangs (Ma et al, 2002). Thus, Artemis could be responsible for removing the 5′ and 3′ overhangs in our NHEJ reaction (Figure 5).
The Mre11/Rad50/Nbs1 complex (MRN) is a 3′–5′ exonuclease for blunt and recessed 3′ ends that pauses at sites of microhomology (Paull and Gellert, 2000). MRN also acts as an endonuclease to cleave 3′ overhangs and open DNA hairpins (Paull and Gellert, 1999). Nbs1 and phosphorylated histone H2AX colocalize to sites of V(D)J recombination-induced double-strand breaks (Chen et al, 2000). Furthermore, the equivalent complex in yeast, Mre11/Rad50/Xrs2, is required for NHEJ in vivo (Lewis and Resnick, 2000) and promotes Ku and XRCC4/Ligase4-dependent NHEJ in vitro (Chen et al, 2001a).
The Werner syndrome protein WRN is a RecQ-like ATPase/helicase which catalyzes 3′–5′ exonuclease activity on recessed 3′ ends (Kamath-Loeb et al, 1998; Shen et al, 1998). Ku stimulates WRN exonuclease activity (Cooper et al, 2000). Furthermore, DNA-PKcs phosphorylates WRN in vitro and in vivo (Yannone et al, 2001).
Are WRN and MRN involved in NHEJ? WRN mutant cells are only mildly radiosensitive (Yannone et al, 2001), and have no defect in V(D)J recombination. Patients with Nijmegan breakage syndrome have hypomorphic alleles of NbsI. Their cells are radiosensitive, but show normal V(D)J recombination (Harfst et al, 2000; Yeo et al, 2000). These relatively mild phenotypes could be explained if MRN and WRN contribute redundant nuclease activities to NHEJ. It is notable that the processing of noncompatible ends in our NHEJ system included nuclease activity on recessed ends. This type of processing has not been reported for Artemis, but is consistent with the 3′–5′ exonuclease activities of MRN and WRN.
Which DNA polymerases are involved in NHEJ? DNA polymerase mu is induced and co-localizes with phosphorylated H2AX in nuclear foci after IR (Mahajan et al, 2002). Polymerase mu forms complexes with Ku and XRCC4/Ligase4 on DNA ends and is capable of utilizing microhomology pairing to prime DNA synthesis (Zhang et al, 2001). DNA polymerase lambda can also utilize microhomology pairing at the primer terminus (Bebenek et al, 2003). Extracts supplemented with XRCC4/Ligase4 will join a DNA substrate containing partially complementary 3′ overhangs and a two-base gap, provided that polymerase lambda is present (Lee et al, 2004). This gap-filling NHEJ reaction is only weakly affected by the absence of polymerase mu. Further work is required to determine which polymerases participate in the cell-free NHEJ reaction.
Model for how DNA-PKcs regulates NHEJ to preserve DNA sequence
There is evidence to suggest that NHEJ occurs in an ordered series of steps (Figure 7). Ku binds and protects DNA ends from unregulated nuclease activity. Ku then translocates inward, recruiting DNA-PKcs to the ends (Hammarsten and Chu, 1998; Yoo and Dynan, 1999). DNA-PKcs brings the ends together into a synaptic complex, which promotes activation of the kinase (DeFazio et al, 2002). This sequence of events may ensure that subsequent steps in NHEJ occur only after the DNA ends are protected within a synaptic complex.
Figure 7.
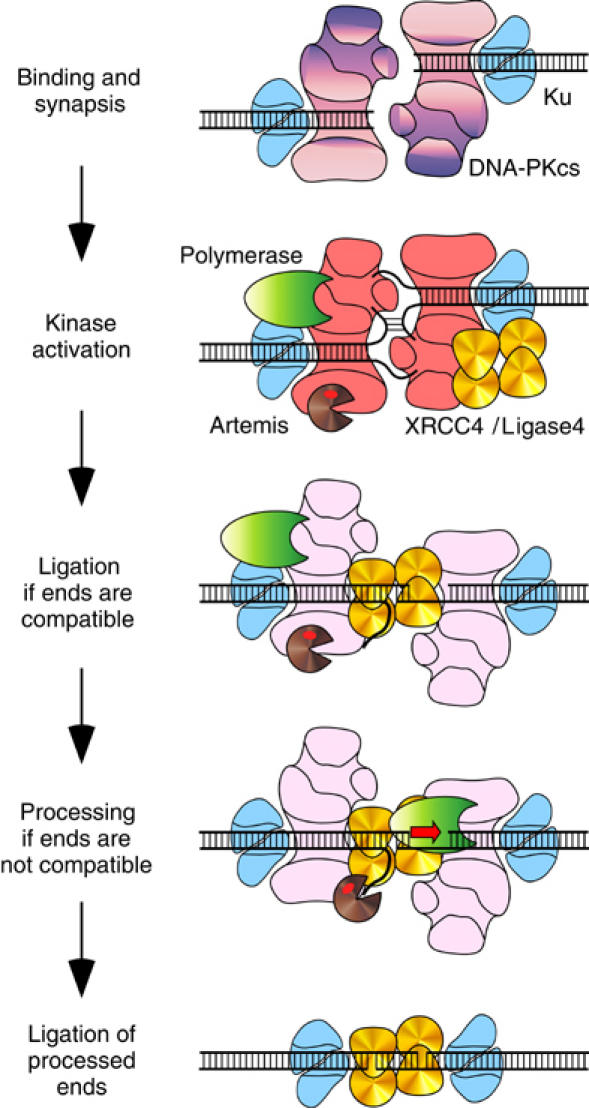
Model for NHEJ. Ku binds to DNA ends and recruits DNA-PKcs, which mediates synapsis of the ends. XRCC4/Ligase4, polymerase, Artemis, and perhaps another nuclease assemble at the synaptic complex. Threading of single-stranded DNA ends into cavities in the DNA-PKcs molecule activates the kinase. DNA-PKcs phosphorylates Artemis, activating its endonuclease activity. DNA-PKcs undergoes autophosphorylation and moves away from the DNA ends. This may provide preferential access to XRCC4/Ligase4. If the ends are compatible, ligation occurs immediately. If the ends are not compatible, XRCC4/Ligase4 remains in the synaptic complex, while polymerase and nuclease activities process the ends. As soon as the ends are processed into a compatible substrate, XRCC4/Ligase4 completes the joining reaction.
DNA-PKcs appears to be activated by a threading mechanism for single-stranded DNA (Hammarsten et al, 2000; Martensson and Hammarsten, 2002). DNA ends with unpaired single strands activate the kinase most efficiently, while DNA ends constructed to prevent threading fail to activate the kinase. Indeed, the DNA-PKcs structure contains an enclosed cavity with openings the size of single-stranded DNA (Leuther et al, 1999).
A threading mechanism may explain why internal sequences influenced joining efficiency, even in the absence of processing. Internal sequences would be exposed when single-stranded DNA is threaded to activate the kinase. Thus, DNA sequence could affect joining efficiency through differences in melting of the ends, formation of secondary structures in the single strands, or in the binding of the single strands to DNA-PKcs.
Upon activation, DNA-PKcs phosphorylates Artemis, itself, and possibly other proteins. Autophosphorylation induces DNA-PKcs to either dissociate from the DNA (Chan and Lees-Miller, 1996) or to move away from the DNA ends (Weterings et al, 2003). The ends then become accessible to XRCC4/Ligase4 and the processing enzymes.
We hypothesize that XRCC4/Ligase4 binds to the DNA ends before the polymerase and nuclease (Figure 7). Perhaps DNA-PKcs remains associated with the synaptic complex in a conformation that grants preferential access to XRCC4/Ligase4. This mechanism would explain why compatible ends are ligated without processing. For noncompatible ends, we further hypothesize that XRCC4/Ligase4 remains in the synaptic complex while the polymerase and nuclease process the DNA ends. Once the ends are compatible, XRCC4/Ligase4 completes the joining reaction. Early recruitment of XRCC4/Ligase4 may suppress the processing of compatible ends and limit processing of noncompatible ends, consistent with our data that NHEJ is biased towards the preservation of DNA sequence.
In conclusion, we have developed a cell-free system that contains all the known enzymatic functions for NHEJ. This is a key step towards identifying the enzymes that process noncompatible ends. Finally, our results provide impetus for developing a cell-free system for V(D)J recombination.
Materials and methods
Extract preparation
Whole cell extract was prepared exactly as described previously (Baumann and West, 1998). Lymphoblastoid cell line GM00558C (Coriell Cell Repositories) was grown in suspension in medium consisting of RPMI 1640, 15% heat-inactivated foetal bovine serum, 2 mM L-glutamine, and 1% penicillin/streptomycin (Gibco). To obtain extracts active for NHEJ, it was necessary to harvest at least eight liters of cells at a density of 8 × 105 cells/ml. The extract was frozen in aliquots, and stored at −80°C. Prior to use, the extract was centrifuged for 2 min in a tabletop centrifuge.
DNA substrates
Plasmid pRL-null (Promega) was the template for PCR amplification of DNA with various restriction enzyme sites at the ends. To avoid the repetitive and palindromic sequences, we used bases 303–1121 from the Renilla luciferase gene as the template for ‘DNA1' and bases 1636–2504 from the amp-r gene as the template for ‘DNA2'. We amplified DNA1 and DNA2 with ends containing cleavage sites for BamHI, EcoRI, KpnI, EcoRV, SacI, StuI, SwaI, SspI, and PmlI using primers listed in the Supplementary data. PCR amplification was performed with Pfu Turbo DNA Polymerase (Stratagene) and PCR products were gel-purified using the QIAquick Gel Extraction Kit (Qiagen). Direct digestion of the PCR products could produce contamination from residual uncut DNA. Therefore, the PCR products were subcloned into the pCR-BluntII-TOPO vector (Invitrogen), released by cleavage with the appropriate restriction endonuclease, and gel-purified. DNA concentrations were determined by spectrophotometer.
End-joining reaction
Reactions were performed in a 20 μl volume containing 50 mM Tris–Cl, pH 8.0, 60 mM KOAc, 0.5 mM MgCl2, 1 mM ATP, 1 mM DTT, 25 μM dNTPs, and 0.1 mg/ml BSA. When indicated, dNTPs were omitted from the reaction mix. DNA fragments (18.4 fmol each) were incubated with 4 μl extract at 37°C for 90 min unless otherwise indicated. Reactions were stopped by adding 2 μl of 0.5 M EDTA, pH 8.0, and DNA was purified on a QIAquick column.
To inhibit the kinase activity of DNA-PKcs, the extract was pre-incubated for 30 min on ice with 10 μM wortmannin. (The wortmannin stock solution was dissolved in DMSO at a concentration high enough to limit the final DMSO concentration in the NHEJ reaction to less than 1%.) For immunodepletion of XRCC4, the extract was pre-incubated at 4°C for 60 min with anti-XRCC4 antibody (Serotec) at a ratio of 1 μl antibody to 15 μl extract. The mixture was added to washed protein A-Sepharose beads (Santa Cruz Biotech), incubated at 4°C for 90 min with rotation, and subjected to centrifugation at 4000 g for 5 min, leaving the supernatant for the NHEJ reaction.
Control reactions with T4 DNA ligase were performed in a 20 μl volume containing T4 DNA ligase buffer. DNA fragments (18.4 fmol each) were incubated with 1 μl of T4 DNA ligase (400 U/μl, New England Biolabs) overnight at room temperature. DNA was purified on a QIAquick column.
PCR amplification and sequencing of DNA junctions
We amplified junctions from the NHEJ reaction using Pfu Turbo HotStart DNA Polymerase (Stratagene). The PCR primers (5′-GAACCATTCAAAGAGAAAGGTG and 5′-GGGAAGCTAGAGTAAGTAG) were located 182 and 583 bases from the ends of DNA1 and DNA2, respectively, to permit detection of deletions. PCR products were purified using the QIAquick PCR purification kit. To visualize the PCR products, DNA was resolved by gel electrophoresis in 1% agarose and stained with ethidium bromide. To analyze individual DNA junctions, we subcloned the PCR products into the pCR-BluntII-TOPO vector. Because the junctions were heterogeneous, we sequenced plasmid DNA from several independent clones. PFU introduces approximately one mutation per 5000 bases, so PCR artifacts made no significant contribution to the junction sequences.
Measurement of joining efficiency by qPCR
The qPCR assay was performed with the Abi Prism 7900 machine (Applied Biosystems). Reactions were carried out in a volume of 20 μl containing 2 μl purified end-joined DNA, 10 μl of 2 × TaqMan PCR Mastermix (Applied Biosystems), 200 nM TaqMan probe (5′FAM-TGTAACCCACTCGTGCACCCAACTGAT-3′TAMRA), and 900 nM of each PCR primer (5′-AGGTGGTAAACCTGACGTTGTACA and 5′-CGCTGTTGAGATCCAGTTCG). Primer and probe sequences were chosen using the recommended Primer Express software. These primer termini were located 109 bases away from the 3′ end of DNA1 and 132 bases away from the 5′ end of DNA2. The probe terminus was located 104 bases away from the 5′ end of DNA2. Although these sequences were closer to the DNA ends than the primers used to sequence the joining junctions, they were still over 100 bases from the ends. Only eight of 284 junctions contained deletions greater than 20 bases, and the vast majority of the junctions in our experiments would have been detected by our qPCR assay.
Thermal cycling was initiated by heating the sample at 50°C for 2 min, 95°C for 10 min, and then treating the sample with denaturation at 95°C for 15 s followed by annealing and extension at 60°C for 1 min for 50 cycles. These conditions were designed to minimize the effect of deletions on the extent of amplification.
To determine the percentage of DNA ends joined, we used pre-formed DNA junctions to establish standards. We tested six preparations of DNA junctions: the dominant nonprocessed junction from compatible BamHI–BamHI ends; the most common junction (−dNTPs) from noncompatible BamHI–KpnI ends; the most common junction (−dNTPs) from noncompatible StuI–KpnI ends; and the three mixtures of junctions obtained from each of these reactions. For each preparation, 18.4 fmol of junction DNA defined 100% joining, matching the amount of each DNA substrate added to the NHEJ reaction. Serial two-fold dilutions of junction DNA established standards down to 0.000006% joining. Each dilution was subjected to the same procedure used for the experimental samples, including passage over a Qiaquick column. Amplification by qPCR produced a standard curve of percentage of DNA ends joined versus the number of cycles needed to synthesize a threshold amount of DNA. The standard curves for the six preparations were completely superimposable. In this assay, the values for ‘percentage of DNA ends joined' refer only to those junctions formed between the DNA ends specified by the qPCR assay. The total percentage of joined ends was higher, since the ends were joined in four possible orientations.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Lisa DeFazio, Sunny Kim, and Tom Tan for helpful discussions, and Steve Sha for assistance in sequencing the junctions. This work was supported by a predoctoral fellowship from the Howard Hughes Medical Institute to JB and NIH grant RO1 GM58120 to GC.
References
- Baumann P, West SC (1998) DNA end-joining catalyzed by human cell-free extracts. Proc Natl Acad Sci USA 95: 14066–14070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K, Garcia-Diaz M, Blanco L, Kunkel TA (2003) The frameshift infidelity of human DNA polymerase lambda. Implications for function. J Biol Chem 278: 34685–34690 [DOI] [PubMed] [Google Scholar]
- Boe SO, Sodroski J, Helland DE, Farnet CM (1995) DNA end-joining in extracts from human cells. Biochem Biophys Res Commun 215: 987–993 [DOI] [PubMed] [Google Scholar]
- Chan DW, Lees-Miller SP (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem 271: 8936–8941 [DOI] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J 21: 2827–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, Nussenzweig A (2000) Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science 290: 1962–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE (2001a) Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell 8: 1105–1115 [DOI] [PubMed] [Google Scholar]
- Chen S, Inamdar KV, Pfeiffer P, Feldmann E, Hannah MF, Yu Y, Lee JW, Zhou T, Lees-Miller SP, Povirk LF (2001b) Accurate in vitro end joining of a DNA double strand break with partially cohesive 3′-overhangs and 3′-phosphoglycolate termini: effect of Ku on repair fidelity. J Biol Chem 276: 24323–24330 [DOI] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA (2000) Ku complex interacts with and stimulates the Werner protein. Genes Dev 14: 907–912 [PMC free article] [PubMed] [Google Scholar]
- DeFazio LG, Stansel RM, Griffith JD, Chu G (2002) Synapsis of DNA ends by the DNA-dependent protein kinase. EMBO J 21: 3192–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann E, Schmiemann V, Goedecke W, Reichenberger S, Pfeiffer P (2000) DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res 28: 2585–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D (1993) Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science 261: 1175–1178 [DOI] [PubMed] [Google Scholar]
- Grawunder U, Wilm M, Xiantuo W, Kulezla P, Wilson TE, Mann M, Lieber MR (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388: 492–494 [DOI] [PubMed] [Google Scholar]
- Hammarsten O, Chu G (1998) DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc Natl Acad Sci USA 95: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O, DeFazio L, Chu G (2000) Activation of DNA-dependent protein kinase by single-stranded DNA ends. J Biol Chem 275: 1541–1550 [DOI] [PubMed] [Google Scholar]
- Hanakahi LA, Bartlet-Jones M, Chappell C, Pappin D, West SC (2000) Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 102: 721–729 [DOI] [PubMed] [Google Scholar]
- Harfst E, Cooper S, Neubauer S, Distel L, Grawunder U (2000) Normal V(D)J recombination in cells from patients with Nijmegen breakage syndrome. Mol Immunol 37: 915–929 [DOI] [PubMed] [Google Scholar]
- Hartley KO, Gell D, Smith GCM, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP (1995) DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82: 849–856 [DOI] [PubMed] [Google Scholar]
- Johnson AP, Fairman MP (1996) The identification and characterization of mammalian proteins involved in the rejoining of DNA double-strand breaks in vitro. Mutat Res 364: 103–116 [DOI] [PubMed] [Google Scholar]
- Kamath-Loeb AS, Shen JC, Loeb LA, Fry M (1998) Werner syndrome protein. II. Characterization of the integral 3′ → 5′ DNA exonuclease. J Biol Chem 273: 34145–34150 [DOI] [PubMed] [Google Scholar]
- Komori T, Okada A, Stewart V, Alt F (1993) Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science 261: 1171–1175 [DOI] [PubMed] [Google Scholar]
- Lee JW, Blanco L, Zhou T, Garcia-Diaz M, Bebenek K, Kunkel TA, Wang Z, Povirk LF (2004) Implication of DNA polymerase lambda in alignment-based gap filling for nonhomologous DNA end joining in human nuclear extracts. J Biol Chem 279: 805–811 [DOI] [PubMed] [Google Scholar]
- Leuther KK, Hammarsten O, Kornberg RD, Chu G (1999) Structure of DNA-dependent protein kinase: implications for its regulation by DNA. EMBO J 18: 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LK, Resnick MA (2000) Tying up loose ends: nonhomologous end-joining in Saccharomyces cerevisiae. Mutat Res 451: 71–89 [DOI] [PubMed] [Google Scholar]
- Liang F, Romanienko P, Weaver D, Jeggo P, Jasin M (1996) Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci USA 93: 8929–8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108: 781–794 [DOI] [PubMed] [Google Scholar]
- Mahajan KN, Nick McElhinny SA, Mitchell BS, Ramsden DA (2002) Association of DNA polymerase mu (pol mu) with Ku and ligase IV: role for pol mu in end-joining double-strand break repair. Mol Cell Biol 22: 5194–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson S, Hammarsten O (2002) DNA-dependent protein kinase catalytic subunit: structural requirements for kinase activation by DNA ends. J Biol Chem 277: 3020–3029 [DOI] [PubMed] [Google Scholar]
- Mason RM, Thacker J, Fairman MP (1996) The joining of non-complementary DNA double-strand breaks by mammalian extracts. Nucleic Acids Res 24: 4946–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP (2001) Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105: 177–186 [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev 13: 1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M (2000) A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci USA 97: 6409–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelczak KS, Andrews BJ, Turchi JJ (2005) Differential activation of DNA-PK based on DNA strand orientation and sequence bias. Nucleic Acids Res 33: 152–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospiech H, Rytkonen AK, Syvaoja JE (2001) The role of DNA polymerase activity in human non-homologous end joining. Nucleic Acids Res 29: 3277–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M (1994) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14: 8096–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlissel M (1998) Structure of nonhairpin coding-end DNA breaks in cells undergoing V(D)J recombination. Mol Cell Biol 18: 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JC, Gray MD, Oshima J, Kamath-Loeb AS, Fry M, Loeb LA (1998) Werner syndrome protein. I. DNA helicase and DNA exonuclease reside on the same polypeptide. J Biol Chem 273: 34139–34144 [DOI] [PubMed] [Google Scholar]
- Smider V, Rathmell WK, Lieber M, Chu G (1994) Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science 266: 288–291 [DOI] [PubMed] [Google Scholar]
- Taccioli G, Gottlieb T, Blunt T, Priestly A, Demengeot J, Mizuta R, Lehmann A, Alt F, Jackson S, Jeggo P (1994) Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science 265: 1442–1445 [DOI] [PubMed] [Google Scholar]
- Taccioli G, Rathbun G, Oltz E, Stamato T, Jeggo P, Alt F (1993) Impairment of V(D)J recombination in double-strand break repair mutants. Science 260: 207–210 [DOI] [PubMed] [Google Scholar]
- Thode S, Schafer A, Pfeiffer P, Vielmetter W (1990) A novel pathway of DNA end-to-end joining. Cell 60: 921–928 [DOI] [PubMed] [Google Scholar]
- Weterings E, Verkaik NS, Bruggenwirth HT, Hoeijmakers JH, van Gent DC (2003) The role of DNA dependent protein kinase in synapsis of DNA ends. Nucleic Acids Res 31: 7238–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ (2001) Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem 276: 38242–38248 [DOI] [PubMed] [Google Scholar]
- Yeo TC, Xia D, Hassouneh S, Yang XO, Sabath DE, Sperling K, Gatti RA, Concannon P, Willerford DM (2000) V(D)J rearrangement in Nijmegen breakage syndrome. Mol Immunol 37: 1131–1139 [DOI] [PubMed] [Google Scholar]
- Yoo S, Dynan WS (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res 27: 4679–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wu X, Yuan F, Xie Z, Wang Z (2001) Highly frequent frameshift DNA synthesis by human DNA polymerase mu. Mol Cell Biol 21: 7995–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information