

Abstract
GRASP65, a structural protein of the Golgi apparatus, has been linked to the sensing of Golgi structure and the integration of this information with the control of mitotic entry in the form of a Golgi checkpoint. We show that Cdk1–cyclin B is the major kinase phosphorylating GRASP65 in mitosis, and that phosphorylated GRASP65 interacts with the polo box domain of the polo-like kinase Plk1. GRASP65 is phosphorylated in its C-terminal domain at four consensus sites by Cdk1–cyclin B, and mutation of these residues to alanine essentially abolishes both mitotic phosphorylation and Plk1 binding. Expression of the wild-type GRASP65 C-terminus but not the phosphorylation defective mutant in normal rat kidney cells causes a delay but not the block in mitotic entry expected if this were a true cell cycle checkpoint. These findings identify a Plk1-dependent signalling mechanism potentially linking Golgi structure and cell cycle control, but suggest that this may not be a cell cycle checkpoint in the classical sense.
Keywords: docking, Golgi matrix, GRASP65, polo kinase, Rab1
Introduction
Checkpoint control mechanisms are used to ensure the fidelity of a wide variety of cellular processes associated with cell growth and division (Nigg, 2001; Bartek et al, 2004; Hoyt, 2004). The original and perhaps best understood of these mechanisms is the spindle checkpoint necessary for proper chromosome segregation, which keeps the cell cycle arrested in metaphase until all chromosomes have become attached to the mitotic spindle and correctly aligned (Cleveland et al, 2003). More recently, it has been suggested that checkpoint control mechanisms may exist in organelles critical for cell function (Sütterlin et al, 2002), such as the Golgi apparatus, to ensure that like chromosomes they are also correctly inherited by the two daughter cells (Warren, 1993; Warren and Wickner, 1996). Thus, in animal cells, the fragmentation and dispersal of the Golgi apparatus from the pericentriolar region is required for entry into mitosis (Sütterlin et al, 2002). One component of this pathway is GRASP65, originally identified in a screen for proteins active in the establishment of Golgi stacks in vitro and subsequently shown to be an adaptor protein capable of linking Golgi matrix proteins and transmembrane cargo proteins (Barr et al, 1997, 1998, 2001). Antibodies to the C-terminus of GRASP65 or the C-terminal domain itself cause a delay in Golgi fragmentation and subsequent entry into mitosis when microinjected into normal rat kidney (NRK) cells (Sütterlin et al, 2002). These effects were not due to defective DNA replication or activation of the DNA damage checkpoint during mitotic entry, and therefore were interpreted as indicators of a novel checkpoint pathway sensing Golgi integrity (Sütterlin et al, 2002). Additionally, there is evidence that the yeast homologue of GRASP65, Grh1/Ydr517w, plays a role in signalling in mitosis through the spindle checkpoint kinase Mps1 (Norman et al, 1999). What these studies have not addressed is how GRASP65 links Golgi apparatus structure with the regulation of cell cycle entry.
GRASP65 is part of a complex at cis-Golgi membranes with the golgin GM130 that acts as a receptor for the vesicle tethering factor p115 (Barr et al, 1997; Lowe et al, 1998b). During mitosis, GM130 becomes phosphorylated by the Cdk1–cyclin B mitotic kinase and is no longer able to bind to p115 (Lowe et al, 1998b). This contributes to the cessation of vesicle transport and alterations in Golgi structure seen in mitosis. GRASP65 is the major Golgi phosphoprotein in mitosis (Barr et al, 1997), and may be phosphorylated on multiple sites by Cdk1–cyclin B and the polo-like kinase 1 (Plk1) (Lin et al, 2000; Sütterlin et al, 2001; Wang et al, 2003). However, in this case, it is unclear which aspects of GRASP65 function are regulated by phosphorylation. GRASP65 oligomerisation in vitro is negatively regulated by phosphorylation (Wang et al, 2003), and this may relate to its role in Golgi stack formation (Barr et al, 1997). The fact that phosphorylated GRASP65 is still associated with GM130 and present on Golgi membranes (Barr et al, 1997, 1998) suggests an alternative possibility involving the mitotic kinase Plk1.
Plk1 is a key regulator of cell division, and plays a number of roles in regulation of entry into mitosis, mitotic spindle formation, and cytokinesis (Barr et al, 2004). Consistent with these varied functions, it is subject to a complex network of regulation. One aspect of this, controlling both the kinase activity and subcellular localisation of Plk1, is the binding of phosphorylated docking partners to the Plk1 polo box domain (PBD) (Elia et al, 2003a, 2003b). It has recently been shown that another Golgi protein Nir2 binds to Plk1 in mitosis and then plays a role in cytokinesis (Litvak et al, 2004). We therefore asked if GRASP65 is a docking partner for Plk1, and whether or not this could explain the observations that GRASP65 may link Golgi fragmentation with the control of mitotic entry.
Results
Identification of mitotic kinases acting on GRASP65
To identify mitotic kinases acting on GRASP65, cytosolic extracts of HeLa cells arrested in mitosis by nocodazole treatment were fractionated using column chromatography. Two assays were used to screen for kinase activity, either direct incorporation of radioactive phosphate from [γ-32P]ATP into recombinant GRASP65, or upshift of in vitro-translated [35S]methionine-labelled GRASP65. Both methods gave essentially the same results, so only the results of the [γ-32P]ATP phosphorylation screen are shown here (Figure 1). Candidate kinases, Cdk1–cyclin B, Plk1, and Mps1, that have been linked to GRASP65 were followed throughout the purifications. Fractionation of HeLa mitotic extract over Q-sepharose gave a single peak of activity (Figure 1A, QI) that comigrated with Cdk1–cyclin B1, and showed some overlap with the broad peak of Plk1. Mps1 fractionated away from the peak of GRASP65 phosphorylation activity, suggesting that this is not a GRASP65 kinase. The QI pool was then further fractionated by heparin sepharose (Figure 1B), and then Superose 6 size exclusion chromatography (Figure 1C and D). The phosphorylation activity was resolved into one major (Figure 1C, SII) and two minor peaks (Figure 1C, SI and III) on the Superose 6 column. Peak SII corresponds to the Cdk1–cyclin B complex determined by Western blotting (Figure 1D), while the minor SIII peak may be due to a small amount of kinase activity associated with Cdk1 free of cyclin B (note the trailing peak of Cdk1 to low molecular mass fractions 17–19). Peak SI corresponds to the peak of Plk1 (Figure 1D), and like the Cdk1–cyclin B peak, SII causes a partial upshift in GRASP65. A minor pool of kinase activity (Figure 1A, QII) was seen in three independent purifications, and further fractionation on size exclusion columns showed that this has an apparent molecular size of 30 kDa (Figure 1E). This may therefore correspond to a minor pool of Cdk1 that is not complexed with cyclin B. Cdk1–cyclin B is therefore the major mitotic kinase with activity towards GRASP65, while Plk1 contributes a lesser but still significant amount to GRASP65 phosphorylation. Mps1 and Aurora family kinases did not specifically phosphorylate GRASP65 (C Preisinger and FA Barr, data not shown).
Figure 1.
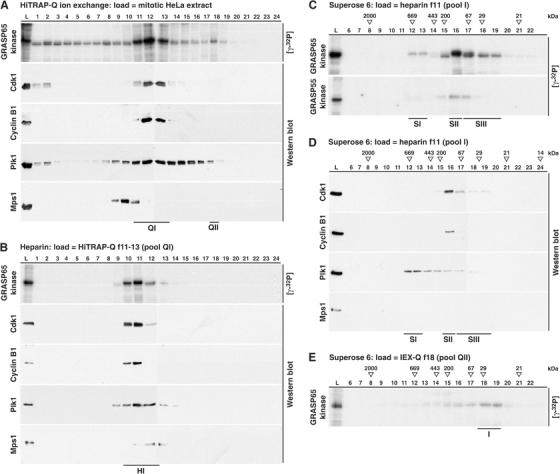
Cdk1–cyclin B is the major GRASP65 kinase in the mitotic HeLa extract. (A) Mitotic HeLa extract was fractionated over HiTRAP-Q ion exchange columns as described in Materials and methods. (B) The QI peak of kinase activity was further fractionated over heparin sepharose to yield a single peak of activity. (C, D) The HI peak was then separated by size exclusion chromatography on a Superose 6 HR10/30 column. This resolved three peaks of GRASP65 kinase activity. Molecular size standards are indicated along the top of the panels. (E) QII, the second peak from the initial ion exchange column, was separated by size exclusion chromatography on a Superose 6 HR10/30 column. This indicates that the kinase activity is associated with a protein of apparent molecular size 30 kDa, possibly Cdk1 free of cyclin B. The load (L) and the numbered fractions were tested for kinase activity towards GRASP65 and GRASP55, and Western blotted for Plk1, Cdk1, cyclin B1, and Mps1 as indicated.
Mapping of GRASP65 phosphorylation sites
Analysis of GRASP65 phosphorylated by mitotic cytosol by mass spectrometry revealed that it was phosphorylated at multiple sites all within the serine- and proline-rich C-terminal domain (summarised in Figure 2A). This is discrete from the first 202 amino acids containing two PDZ-like domains, the second of which forms the binding site for GM130 (Barr et al, 1998). Phosphorylated peptides were identified using MALDI-TOF spectroscopy (Figure 2B), and then subjected to fragmentation to identify the sequence and phosphorylated residues using MS/MS (Figure 2C). In the example shown, a peptide corresponding to amino acids 359–388 of GRASP65 treated with the Plk1 (SI) and Cdk1–cyclin B1 (SII) peaks from the Superose 6 fractionation was analysed (Figure 2B). A single phosphorylated serine corresponding to amino acid 376 was observed only when GRASP65 was treated with Cdk1–cyclin B1 (SII) (Figure 2C). Other results correlating the mapped sites with the biochemical fractionation data are summarised in the table and schematic of GRASP65 (Figure 2A). These sites represent the major sites of mitotic phosphorylation, since when they are mutated to alanine, GRASP65 is no longer phosphorylated or upshifted under mitotic conditions (Figure 3A). Furthermore, mutation of the four consensus Cdk1–cyclin B sites to alanine abolishes phosphorylation by this kinase, indicating that these are the only sites it modifies in GRASP65 (Figure 3B). Phosphospecific antibodies were raised against two of the mitotic Cdk1–cyclin B sites, pT220 and pS376, as well as the interphase and mitotic phosphorylation site pS367 (Figure 3C). All these antibodies are competed by the respective phosphopeptide peptide antigen, and only recognise the protein after treatment with the appropriate interphase or mitotic cytosol (Figure 3C). Treatment of mitotically phosphorylated GRASP65 with phosphatase abolished the reactivity, providing further evidence for their phospho-specificity (Figure 3C). In summary, GRASP65 is phosphorylated at four consensus sites by Cdk1–cyclin B1, and at two other sites by unidentified kinases. One of these sites, serine 367, was also phosphorylated under interphase conditions, suggesting that it is not a site of regulation specific for mitosis.
Figure 2.
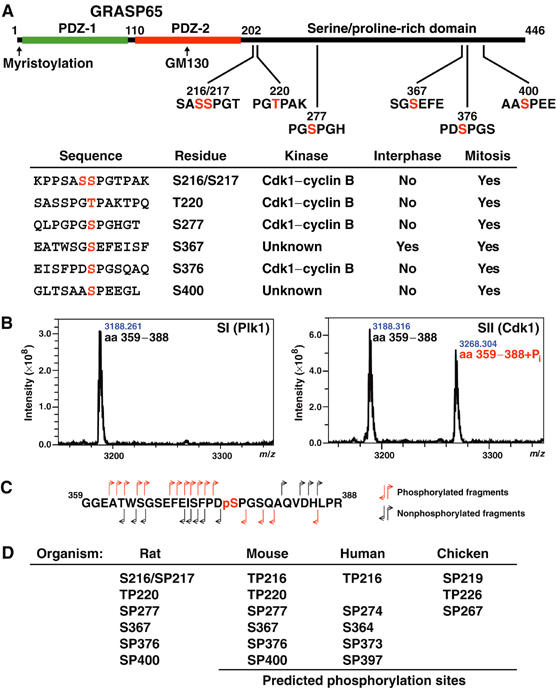
Mapping of phosphorylation sites in GRASP65. (A) Schematic of GRASP65 indicating the two N-terminal PDZ-like domains and GM130 binding region. Phosphorylation sites are shown with the phosphorylated residues numbered and marked in red. The table summarises data on the kinases that phosphorylate these sites and whether or not they are modified in interphase or mitosis. Phosphorylation of S372 was observed under some conditions, but this was substoichiometric and could not be assigned to any kinase with confidence, although it does fall within a Plk1 consensus sequence, and is thus omitted from the table. (B) MALDI-TOF spectra showing a peptide corresponding to GRASP65 amino acids 359–388, phosphorylated with SI and SII from the Superose 6 fractionation of mitotic extract (see Figure 1C). A peak corresponding to the addition of a single phosphate is observed with the Cdk1–cyclin B-containing SII fraction. (C) Fragmentation of the phosphorylated GRASP65359−388 peptide by tandem mass spectrometry gives daughter ions derived from the N- and C-termini of the peptide indicating that serine 376 (pS), shown in red, is the phosphorylated residue. (D) Phosphorylation sites predicted to be equivalent to those identified in rat GRASP65 are shown for mouse, human, and chicken GRASP65.
Figure 3.
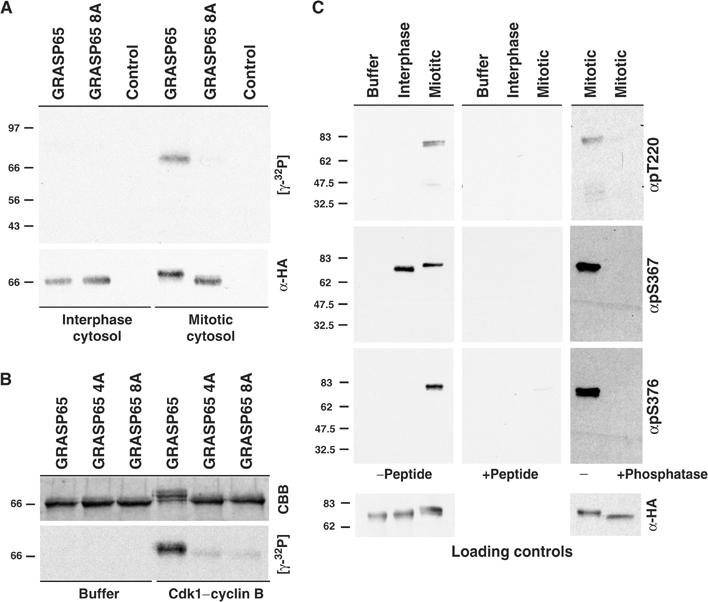
Cdk1–cyclin B is the major GRASP65 kinase in mitotic HeLa extracts. (A) GRASP65 and the GRASP65 8A mutant where the following residues have been mutated to alanine (T220, S216, 217, 277, 367, 372, 376, and 400) were incubated with buffer (control), and interphase and mitotic cytosols. Substrate loading was controlled by Western blotting for the HA epitope tag present on the recombinant GRASP65, and an autoradiograph indicates the amount of [γ-32P] incorporation. (B) GRASP65, GRASP65 8A, and the GRASP65 4A mutants where the following residues have been mutated to alanine (T220, S217, 277, and 376) were incubated with buffer (−), or in the presence (+) of Cdk1–cyclin B. Substrate loading was controlled by Coomassie brilliant blue (CBB) staining, and an autoradiograph indicates the amount of [γ-32P] incorporation. (C) Phospho-antibodies pT220, pS367, and pS376 were used to Western blot recombinant HA-tagged GRASP65 treated with buffer, and interphase and mitotic cytosols. Control experiments were performed with antibodies preincubated with the phosphopeptide antigen to compete specific antibody reactivity (+peptide), and GRASP65 phosphorylated with mitotic cytosol was treated with or without alkaline phosphatase. GRASP65 loading was controlled with the HA epitope antibody.
GRASP65 on Golgi fragments is phosphorylated in mitotic entry
To identify when GRASP65 is phosphorylated, synchronised NRK cells were stained with the GRASP65 pS376 phosphopeptide antibody and antibodies to GRASP65 to mark the Golgi apparatus (Figure 4A). The pS376 antibody gave the strongest staining when Golgi apparatus fragmentation is initiated during prophase and in metaphase when it has become converted into a haze of small vesicles and some larger tubulovesicular remnants (Figure 4A). This correlated with the phosphorylation of GM130 at serine 25 (Figure 4B), which is also phosphorylated by Cdk1–cyclin B during prophase and metaphase (Lowe et al, 2000). Staining decreased through anaphase into telophase, corresponding with the inactivation of Cdk1–cyclin B, and the reassembly of Golgi stacks. Staining was essentially absent from interphase cells in the Golgi region (Figure 4A), although a weak signal over the nucleus was observed in some cells. Therefore, GRASP65, like GM130, is phosphorylated in mitotic entry on Cdk1–cyclin B sites during the period when the Golgi apparatus is fragmented.
Figure 4.
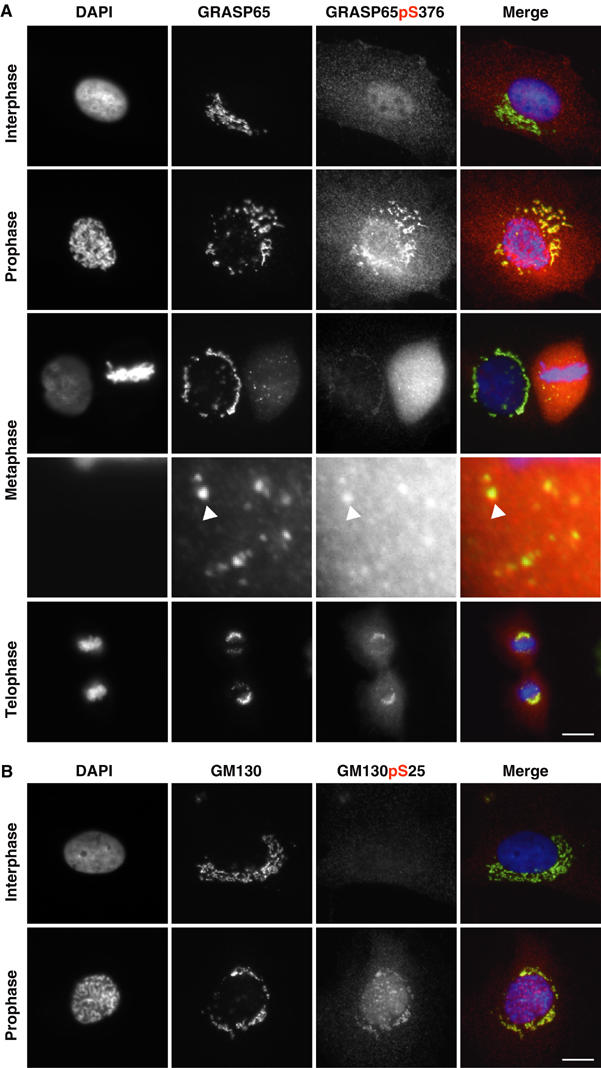
GRASP65 pS376 phosphorylation correlates with entry into mitosis. NRK cells were stained with DAPI to reveal DNA (blue) and antibodies to either (A) GRASP65 (green) and GRASP65 pS376 (red), or (B) GM130 (green) and GM130 phosphoserine 25, pS25 (red). Interphase cells and cells at various stages in mitosis are shown. A 4.6-fold enlargement of a region of the metaphase cells shows the presence of GRASP65 and GRASP65 pS376 staining on mitotic Golgi fragments (arrowhead). The bar indicates 10 μm.
Specific docking of Plk1 to GRASP65 in mitosis
To test if GRASP65 is a specific docking partner for the mitotic kinase Plk1, GRASP65–GM130 and the related GRASP55–Golgin45 complexes were immune precipitated under interphase and mitotic conditions (Figure 5A and B). This approach revealed that Plk1 was co-precipitated with the GRASP65–GM130 complex under mitotic but not interphase conditions (Figure 5B). Plk1 was not present in GRASP55–Golgin45 immune precipitates (Figure 5B), indicating that it specifically interacts with some component of the GRASP65–GM130 complex. To test if Plk1 is directly binding to GRASP65, farwestern blots were performed using wild type and the phosphorylation site mutant of GRASP65 under both interphase and mitotic conditions (Figure 5C). Plk1 bound to wild-type GRASP65 only after phosphorylation with mitotic cytosol or purified Cdk1–cyclin B1 (Figure 5C and D). Under the same conditions, the phosphorylation defective mutant of GRASP65 showed a strongly reduced interaction with Plk1 (Figure 5C and D). No binding of Plk1 to the GRASP65 N-terminal PDZ-like domains, or between the Plk1 kinase domain and GRASP65 was observed (C Preisinger and FA Barr, data not shown). The action of Cdk1–cyclin B therefore creates docking sites for Plk1 on GRASP65, consistent with observations that the PBD of Plk1 is a phosphopeptide binding domain that binds to a subset of sites phosphorylated by Cdk1–cyclin B during mitosis.
Figure 5.
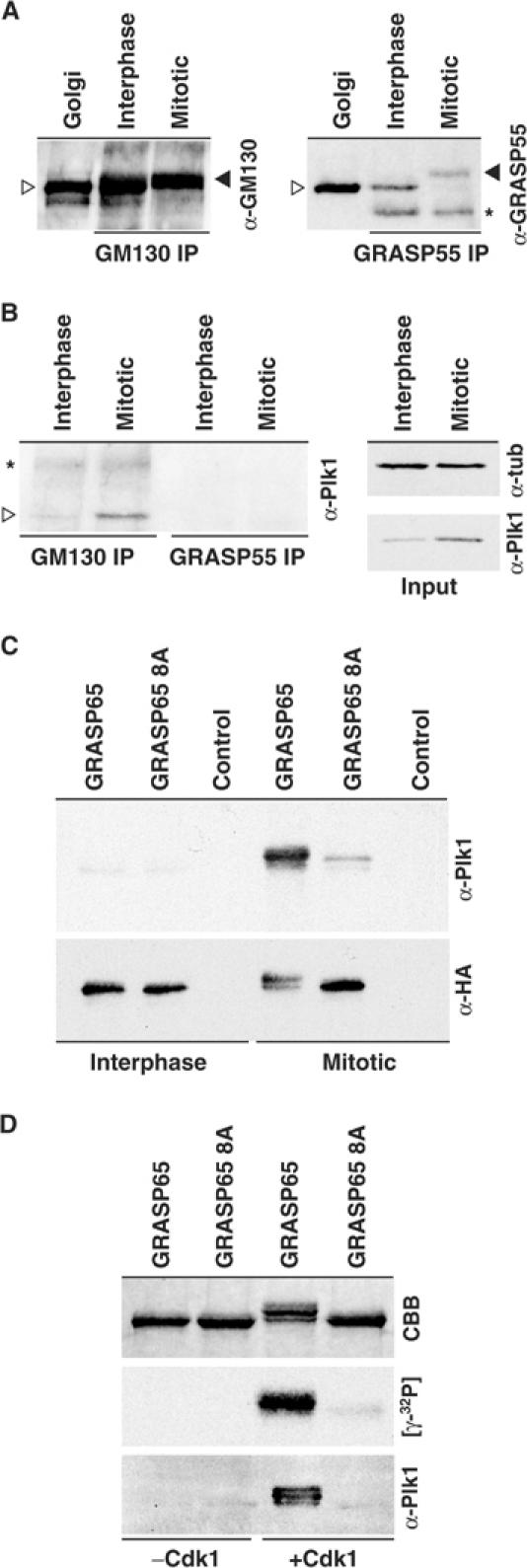
Plk1 interacts with GRASP65 in mitosis. (A) Interphase and mitotic GRASP65–GM130 (GM130 IP) and Golgin45–GRASP55 (GRASP55 IP) complexes were immune precipitated and Western blotted for GM130 and GRASP55, respectively. The asterisk marks a crossreaction with the antibody heavy chain in the GRASP55 blot. (B) Western blot of the GRASP65 and GRASP55 complexes for Plk1. Loading controls for extracts blotted for α-tubulin (α-tub) and Plk1 are shown on the right. Ligand blots were carried out with Plk1 amino acids 305–603 as the probe, and (C) interphase and mitotically or (D) Cdk1–cyclin B phosphorylated GRASP65 and the GRASP65 8A phosphorylation site mutants as the targets.
Mitotic phosphorylation of Rab1 creates a Plk1 docking site
Both the GRASP65–GM130 and GRASP55–Golgin45 complexes have cognate Rab GTP binding proteins, Rab1 and Rab2, respectively. Since Rab1 is phosphorylated during mitosis and the function of this phosphorylation is unclear (Bailly et al, 1991), it seemed possible that it might also be a binding partner for Plk1. To test this possibility, the phosphorylation site on Rab1 was first analysed using mass spectrometry. This revealed that Rab1 is phosphorylated on threonine 195 within a consensus Cdk1–cyclin B site in the variable C-terminal domain (Figure 6A). Mutation of threonine 195 to alanine abolished phosphorylation by mitotic cytosol and purified Cdk1–cyclin B1 (Figure 6B and C), suggesting that Rab1 has a single phosphorylation site recognised by Cdk1–cyclin B. Phosphorylated Rab1 was able to bind to Plk1 (Figure 6B and C), and this interaction was lost if threonine 195 was mutated to alanine. The specificity of this interaction is demonstrated by the fact that GM130, which like Rab1 has a single Cdk1–cyclin B phosphorylation site (Lowe et al, 1998b, 2000), is unable to bind Plk1 even though it is equally phosphorylated (Figure 6D). Farwestern blots showed that Plk1 could bind to phosphorylated GRASP65 and Rab1, but not Rab2 or phosphorylated GRASP55 (Figure 6E and F). Plk1 therefore binds to a specific subset of cis-Golgi proteins phosphorylated by Cdk1–cyclin B during mitosis, including GRASP65 and Rab1.
Figure 6.
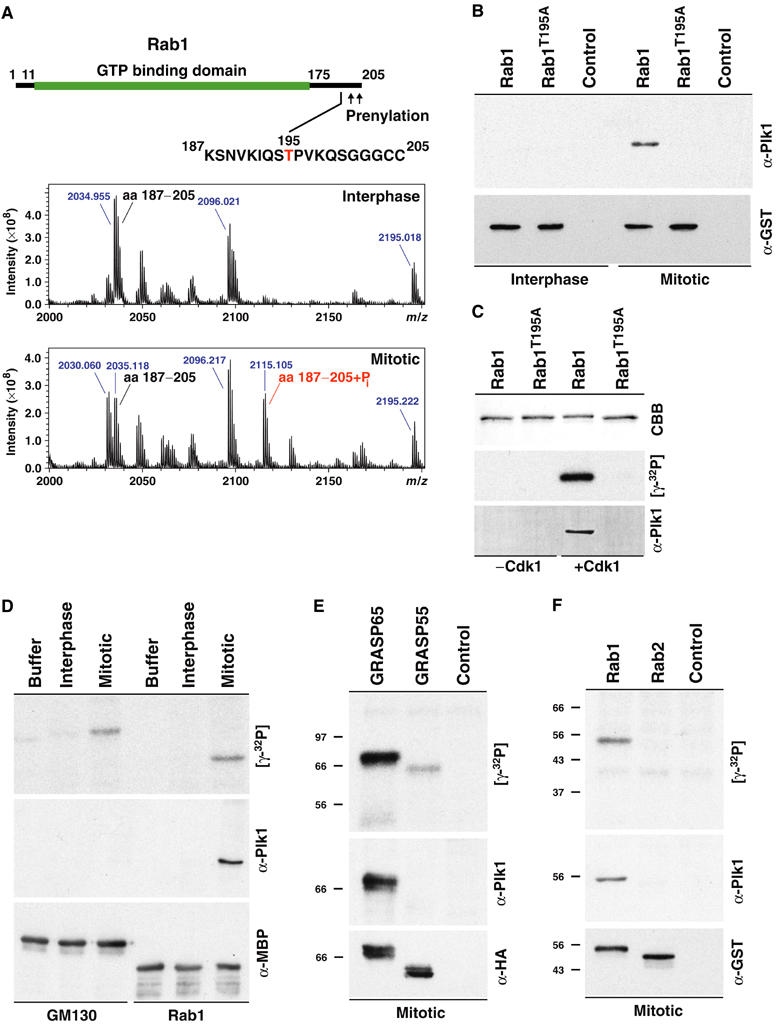
Docking of Plk1 to mitotically phosphorylated cis-Golgi proteins. (A) Schematic of Rab1 indicating the GTP binding domain (green) and prenylation sites. The variable C-terminal region with the phosphorylation site is shown with the phosphorylated residue numbered and marked in red. MALDI-TOF spectra showing a peptide corresponding to Rab1 amino acids 187–205 (m/z 2035.118), after treatment under interphase and mitotic conditions. A peak (m/z 2115.105) corresponding to the addition of a single phosphate is only observed under mitotic conditions. (B) Ligand blots were carried out with Plk1 amino acids 305–603 as the probe, and GST-tagged Rab1 or Rab1 T195A incubated with interphase or mitotic cytosol as the targets. (C) GST-tagged Rab1 or Rab1 T195A were incubated with buffer (−) or Cdk1–cyclin B (+). Samples were analysed by autoradiography ([γ-32P]) or ligand blotting with Plk1. Ligand blots were carried out with Plk1 amino acids 305 to 603 as the probe, and (D) MBP-tagged GM1301−271 or Rab1, (E) HA-tagged GRASP65 or GRASP55, and (F) GST-tagged Rab1 or Rab2 incubated with interphase or mitotic cytosol as the targets.
Golgi fragmentation occurs in the absence of Plk1
Plk1 is known to be required for normal spindle formation and to possibly modulate the rate of entry into mitosis in human cells (Lane and Nigg, 1996; Barr et al, 2004). To assess the requirement for Plk1 in Golgi disassembly during entry into mitosis, HeLa cells were depleted of Plk1 using siRNA. As expected, Plk1 was absent from interphase cells, and ribbon-like perinuclear Golgi morphology was not noticeably altered in interphase cells treated with Plk1 siRNA compared to a lamin A control (Figure 7A and B). In lamin A siRNA cells, the Golgi apparatus, defined by GM130 and GRASP65, fragmented normally in prometaphase cells into a mixture of vesicular clusters and an unresolved haze of small vesicles (Figure 7A). In Plk1-depleted cells, which arrest in a prometaphase-like state with highly condensed chromosomes that fail to attach to the mitotic spindle (Sumara et al, 2002, 2004), there was a pronounced increase in the number of cells with prominent mitotic Golgi clusters defined by GM130 and GRASP65: 89.3±2.2% of Plk1 siRNA cells compared to 12.6±2.5% of lamin A siRNA control cells (Figure 7C). The activity of Plk1 therefore appears to be required for full mitotic fragmentation of the Golgi apparatus, but not the initial breakdown of the ribbon-like perinuclear organisation of Golgi stacks and their initial fragmentation to form mitotic Golgi clusters.
Figure 7.
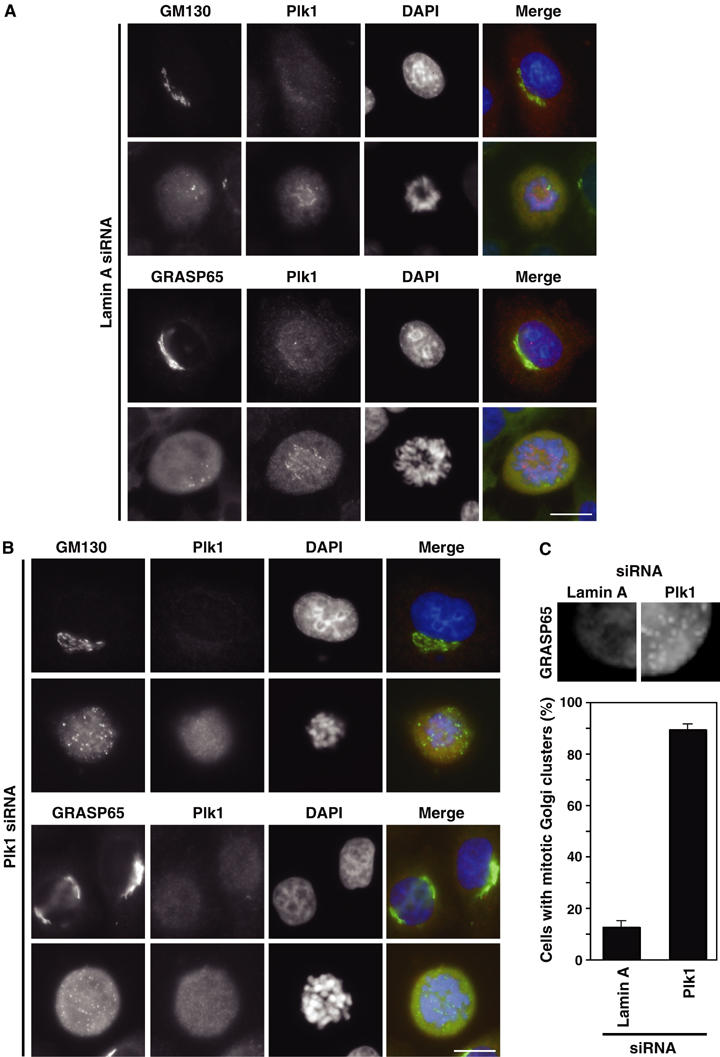
Effect of Plk1 depletion on mitotic Golgi fragmentation. HeLa cells or HeLa cells stably expressing GRASP65-GFP were treated with siRNA duplexes for (A) lamin A and (B) Plk1 for 72 h, and then stained with antibodies to GM130 (green) and Plk1 (red), or stained with an antibody to Plk1 (red) and GRASP65 visualised using GFP (green). DNA was stained with DAPI (blue). Bar indicates 10 μm. (C) Enlarged images showing the increased number of persistent Golgi clusters, marked by GRASP65-GFP, observed in Plk1 siRNA cells. Numbers of mitotic cells containing Golgi clusters were counted for lamin A and Plk1 siRNA and plotted as a bar graph (300 cells per experiment, n=3).
GRASP65 but not a phosphorylation defective mutant blocks entry into mitosis
It has previously been shown that the GRASP65 C-terminal domain, containing the Cdk1–cyclin B phosphorylation sites and docking sites for Plk1, acts as a dominant-negative form of GRASP65 that is postulated to activate a Golgi morphology checkpoint (Sütterlin et al, 2002). This results in a reduced rate of passage of cells through mitosis. To ask if this effect is mediated through Plk1, wild-type GRASP65 C-terminal domain or the phosphorylation and Plk1 binding defective mutants were expressed in NRK cells arrested in S-phase with aphidicolin. After washout of aphidicolin, cells passed synchronously through mitosis, and the number of mitotic cells was counted (Figure 8A). Both control cells and cells expressing the GRASP65 8A mutant C-terminus passed through mitosis with similar kinetics, with the highest mitotic index being observed at 7.5 h after release from the aphidicolin block (Figure 8A). In contrast, cells expressing the wild-type GRASP65 C-terminus showed a delayed entry into and exit from mitosis, with the highest mitotic index being observed between 8 and 8.5 h after release from the aphidicolin block (Figure 8A). Chromosome condensation and spindle formation seem not to be altered, since mitotic spindles appear normal in cells expressing either the wild-type or 8A mutant GRASP65 C-terminus (Figure 8B). Furthermore, Plk1 localised normally to the kinetochores and spindle poles in prometaphase and metaphase cells expressing either the wild-type or 8A mutant GRASP65 C-terminus (Figure 8C). No defects were observed in cytokinesis (C Preisinger and FA Barr, data not shown), suggesting that Plk1 function in this process is also normal. These observations support the idea that the ability of the GRASP65 C-terminus to alter passage through mitosis is a specific effect due to its influence on Plk1 function on mitotic Golgi fragments, and not at other sites of Plk1 action.
Figure 8.
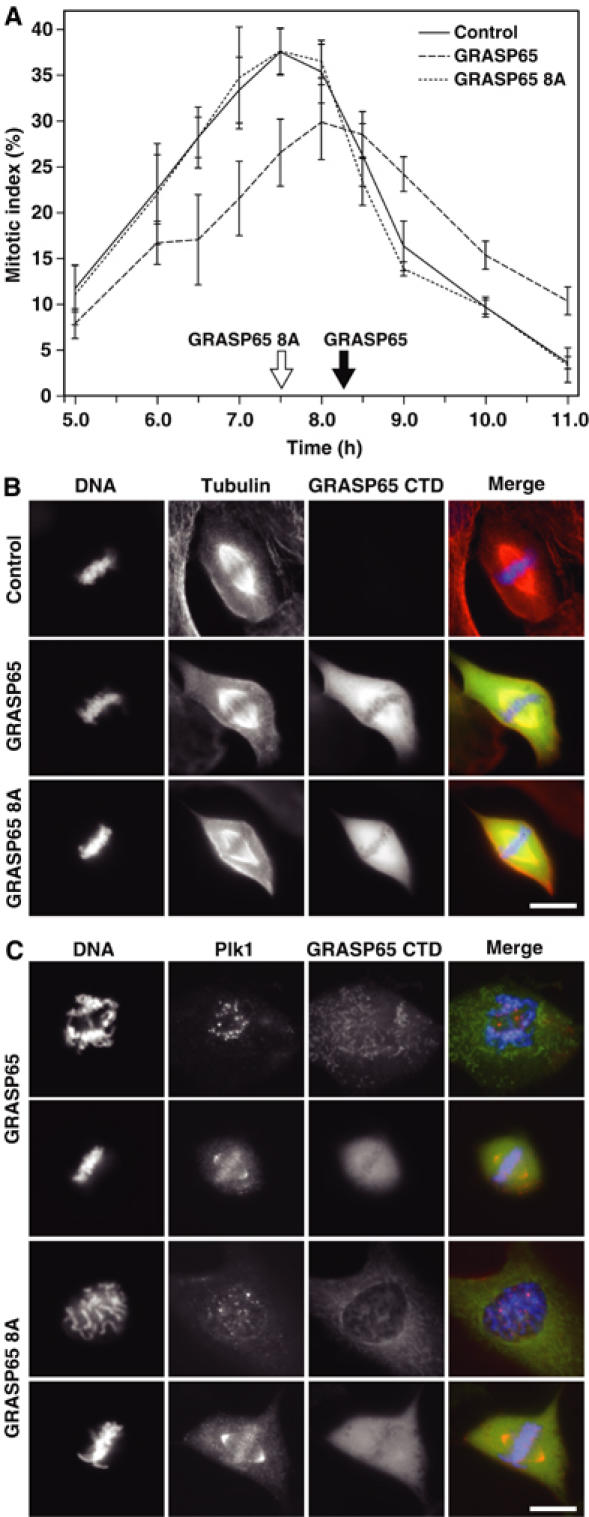
Effect of phosphorylation mutant GRASP65 on mitotic entry. NRK cells expressing the C-terminal domain of wild-type GRASP65 and the phosphorylation defective mutant GRASP65 8A were treated with aphidicolin and then allowed to synchronously passage through mitosis. (A) The mitotic index was counted at the times indicated in the figure after removal of aphidicolin; an average of 150 cells were counted per condition, for three or more experiments. (B, C) The GRASP65 C-terminal domain was visualised by GFP fluorescence, and cells were counterstained with antibodies to α-tubulin or Plk1 (red). DNA was stained with DAPI (blue). Bar indicates 10 μm.
Discussion
While the results presented here suggest a mechanism for the Golgi morphology checkpoint proposed by Sütterlin et al (2002), it remains unclear if this is a checkpoint in the classical sense, or a mitotically active signalling pathway. Checkpoints such as those monitoring bipolar attachment of chromosomes to the mitotic spindle or DNA damage during S-phase are capable of arresting the cell cycle. However, expression of the Plk1 binding GRASP65 C-terminus results in a delay but not a block in passage through mitosis (Sütterlin et al, 2002), suggesting that although the status of the Golgi may be linked to progression through mitosis, this is not in the form of a classical cell cycle checkpoint.
Another issue is what exactly is being sensed and signalled by this pathway? It may be that Golgi integrity per se is being sensed as originally suggested by Sütterlin et al (2002), but a number of other studies have shown that cells with a partially intact Golgi apparatus can actually enter into and passage through mitosis (Altan-Bonnet et al, 2003; Uchiyama et al, 2003). If Golgi integrity is not the important factor, then what is? Recently, it has been shown that the mitotic spindle checkpoint protein ZW10 functions in ER to Golgi traffic in interphase cells (Hirose et al, 2004), and these authors suggested that this might indicate a link between the Golgi checkpoint and ER to Golgi trafficking. In mammalian cells, ER to Golgi transport is blocked during mitosis at the level of vesicle fusion through the action of Cdk1–cyclin B (Lowe et al, 1998a), and significantly both GRASP65 and Rab1 have links to the ER to Golgi vesicle tethering machinery. Rab1 recruits the tethering factor p115 to vesicles (Allan et al, 2000), and GRASP65 is part of a second p115 binding complex containing GM130 on Golgi membranes (Barr et al, 1997; Lowe et al, 1998b). Phosphorylation of GRASP65 and Rab1 by Cdk1–cyclin B may conceivably act as a signal that transport from the ER to the Golgi is blocked, which is then propagated through the recruitment and activation of Plk1 on the surface of Golgi membranes. What the downstream targets for Plk1 are is not known at present, but may include structural proteins of the Golgi as well as cell cycle regulators such as the Cdk1 inhibitory kinase Myt1 (Liu et al, 1997, 1999). Myt1 localises to the ER and Golgi apparatus in animal cells (Liu et al, 1997), and has been reported to be required for complete Golgi fragmentation in Drosophila S2 cells (Cornwell et al, 2002). Myt1 is known to be a substrate for Plk1 (Nakajima et al, 2003), and Plk1 suppresses the activity of Myt1 towards Cdk1, thus promoting Cdk1 activation (Okano-Uchida et al, 2003). Together, these studies suggest a potential link between Cdk1 regulation by Plk1 and Myt1, and hence the control of Golgi fragmentation and mitotic entry.
GRASP65 itself has previously been found to be a Plk1 target (Lin et al, 2000; Sütterlin et al, 2001), and phosphorylation may regulate its oligomeric state (Wang et al, 2003). However, we were unable to identify any specific site stoichiometrically modified by Plk1, and furthermore Plk1 does not appear to be essential for the initial stages of Golgi fragmentation (Figure 7). In the absence of Plk1, Golgi fragmentation is incomplete and an increased number of mitotic Golgi clusters are observed consistent with the idea that Cdk1–cyclin B and Plk1 cooperate with other kinases to achieve full Golgi breakdown (Acharya et al, 1998; Colanzi et al, 2000, 2003). Interestingly, the Cdk1–cyclin B1 sites at amino acids 220, 277, and 376 of GRASP65 also conform to the P-X-S/T-P consensus for mitogen-activated protein kinase kinase/extracellular-activated protein kinase (ERK). GRASP55 is phosphorylated at threonine 225 by ERK, also contained within an overlapping Cdk1–cyclin B/ERK site (Jesch et al, 2001), providing further evidence that multiple kinases cooperate in Golgi fragmentation during mitosis.
These findings suggest a mechanism for ensuring that the Golgi has been fragmented and protein transport stopped, in the form of a signalling pathway involving the docking of Plk1 to phosphorylated GRASP65 and Rab1. This Golgi-associated pool of activated Plk1 could then phosphorylate substrates important for regulating Golgi structure and passage through mitosis. Such a mechanism may be important for ensuring the high fidelity of Golgi partitioning (Shima et al, 1997, 1998), and as a consequence that the two daughter cells inherit the components necessary for establishing a functional Golgi apparatus upon exit from mitosis (Warren, 1993; Warren and Wickner, 1996). The identification and characterisation of these Plk1 substrates, although a far from simple task, will be necessary to test this model.
In summary, the data presented here together with previously published work suggest that the GRASP65–GM130 complex functions as a kinase scaffold on the Golgi apparatus in both interphase and mitosis. In interphase, the Ste20 family kinases YSK1 and MST4 bind to and are activated by GM130, and this is needed for signalling events important for cell polarisation and migration (Preisinger et al, 2004). In mitotic cells, Plk1 docks to phosphorylated GRASP65 and links Golgi function to cell cycle control.
Materials and methods
Reagents and antibodies
Reagents were obtained from Sigma-Aldrich (Germany) unless specified otherwise. Chromatography reagents and [γ-32P]ATP (3000 Ci/mmol and 10 mCi/ml) were obtained from Amersham Biosciences Europe GmbH (Freiburg, Germany). Antibodies were obtained as follows: affinity-purified rabbit antibody against phosphorylated GM130 pS25 (Dr Martin Lowe, Manchester, UK); affinity-purified rabbit antibody against GM130 (Dr Nobohiro Nakamura, Kanazawa, Japan); affinity-purified monoclonal antibody against Mps1 (Professor Erich Nigg, Martinsried, Germany); affinity-purified rabbit antibody against GST (Dr Ulrike Grüneberg, Martinsried, Germany); F-7 mouse monoclonal to the HA epitope (Santa Cruz Biotech). Antibodies specific to the phosphorylated GRASP65 peptide pT220 (CSSPGpTPAKT) were raised in goat, and pS367 (CTWSGpSEFEI) and pS376 (CSFPDpSPGSQ) were raised in rabbits, and then isolated from a protein-A-purified IgG fraction of the serum over the same peptide immobilised on Sulfolink resin according to the manufacturer's instructions (Pierce Biotechnology, PERBIO Science GmbH, Bonn, Germany). Other antibodies used in this study have been described and characterised previously (Barr et al, 1997; Shorter et al, 1999; Short et al, 2001; Neef et al, 2003).
Molecular biology
GRASP65 and Rab1 point mutants were constructed using the Quickchange mutagenesis protocol (Stratagene). DNA oligonucleotides were purchased from Thermo-Hybaid (Germany) and Qiagen GmbH (Hilden, Germany). All constructs were confirmed by DNA sequencing (Medigenomix GmbH, Martinsried, Germany). Mammalian expression constructs were made in pEGFP (Clontech), or pcDNA4TO (Invitrogen) modified to encode EGFP and puromycin resistance. For baculovirus expression, pVL1393 or the pAcSG2 vector (Becton-Dickinson) modified to include the hexahistidine tag from pQE32 (Qiagen) was used. Baculoviruses were produced and proteins expressed in Sf9 cells according to the manufacturer's protocols (Becton-Dickinson). Plk1 and Cdk1–cyclin B complexes were expressed and purified from insect cells as described previously. Bacterial expression was carried out using the MBP vector pMalC2x (New England Biolabs), the His-GST vector pGAT2, and the His-tag vector pQE32 modified to include an HA epitope tag (Qiagen). Inserts encoding GM1301−271, GRASP651−202, GRASP651−252, GRASP651−358, GRASP65 wild type and phosphorylation mutants, GRASP55, Plk1305−603, Rab1 wild type and phosphorylation mutants, and Rab2 were inserted into pGAT2, pMAL, or pQE32 and proteins were expressed in BL21(DE3) or JM109 cells. Proteins were purified over amylose resin (New England Biolabs) or nickel-NTA agarose (Qiagen). Insect cell expressed kinases were desalted into MEB (50 mM Tris–HCl pH 7.3, 50 mM KCl, 10 mM MgCl2, 20 mM β-glycerophosphate, 15 mM EGTA) and other proteins were dialysed into PBS, and then aliquots were snap-frozen in liquid nitrogen for storage at −80°C.
Kinase identification
Cytosolic extracts of interphase and mitotic HeLa cells grown in 1 l spinner flasks were prepared as described previously, and stored frozen at −80°C (Barr et al, 1997). The following buffers were used for column chromatography: dilution buffer (20 mM Tris–HCl pH 7.3), Q50 (50 mM Tris–HCl pH 7.3, 50 mM NaCl, 10 mM MgCl2, 20 mM β-glycerophosphate, 15 mM EGTA), Q1000 (Q50 with 1 M NaCl), H0 (50 mM Tris–HCl pH 7.3, 12.5 mM β-glycerophosphate, 2 mM MgCl2, 2 mM EGTA), and H2000 (H0 with 2 M NaCl). Before use, 2 mM ATP and 1 mM DTT were added to all buffers. A 3 ml Q-sepharose column equilibrated in Q50 was loaded with 25 mg of mitotic HeLa cytosol in MEB at a flow rate of 0.5 ml/min, washed with 5 ml of Q50, and then eluted with a 20 ml linear gradient from 0 to 50% Q1000, followed by a 5 ml step to 100% Q1000. The entire flow through, wash, and fractions of 1 ml were collected throughout the elution and assayed for their ability to phosphorylate GRASP65 and GRASP55. Kinase assays of 12.5 μl were performed using 10 μl of the fraction, 0.1 μl [γ-32P]ATP (3000 Ci/mmol and 10 mCi/ml), and 250 ng substrate for 30 min at 37°C. Reactions were separated on 10% minigels and analysed by autoradiography. The desired fractions from the Q-sepharose column were pooled and diluted to 50 mM NaCl with dilution buffer, and then loaded on to a 1 ml heparin sepharose column at a flow rate of 0.5 ml/min. The column was washed with 5 ml of H0, and then eluted with a 20 ml linear gradient from 0 to 50% H2000, followed by a 5 ml step to 100% H2000. GRASP65 and GRASP55 kinase assays were performed with all fractions. For gel filtration, samples of 1 ml were loaded on to a Superose 6 HR10/30 column at 0.25 ml/min equilibrated in Q50. The column was then eluted with 30 ml of Q50 collecting 1 ml fractions. GRASP65 and GRASP55 kinase assays were performed with all fractions. For Western blot analysis, 10 μl of the column fractions was separated on 10% minigels and probed with antibodies to cell cycle regulatory proteins as indicated in the figures.
Protein phosphorylations and farwestern ligand blots
All cytosols were desalted prior to use over 1 ml Bio-Gel P-6DG Desalting Gel (Bio-Rad) spin columns into MEB (50 mM Tris–HCl pH 7.3, 50 mM KCl, 10 mM MgCl2, 20 mM β-glycerophosphate, 15 mM EGTA, 1 mM DTT, 2 mM ATP). Protein phosphorylation with interphase and mitotic cytosol was performed using 22 pmol of each substrate, 2 mM ATP, 1 mM DTT, and 0.1 μl [γ-32P]ATP (3000 Ci/mmol and 10 mCi/ml) in a total volume of 20 μl MEB containing 1 mg/ml cytosol or 50 ng of active Cdk1–cyclin B1 complex. The reactions were allowed to proceed for 1.5 h (farwestern) or 3 h (autoradiography) at 37°C. Cold phosphorylation for mass spectrometry analysis was carried out with 44 pmol of substrate under the same conditions without the use of labelled ATP. Farwestern ligand blots were performed in TBST (50 mM Tris–HCl pH 7.4, 137 mM NaCl, 0.1% Tween 20, 4% (w/v) skim milk powder). Assays described as above were separated on 7.5% or 10% minigels, Western blotted, and then probed with 1 μg/ml of GST-tagged fragments of Plk1 for 6 h at 4°C. Bound Plk1-PBD was detected using affinity-purified rabbit antibodies to GST or Plk1.
Immune precipitations
Rat liver Golgi membranes were purified and treated with interphase and mitotic cytosols as described previously (Barr et al, 1997). The Golgi membranes were then solubilised in lysis buffer (20 mM HEPES pH 7.2, 150 mM NaCl, 0.2% Triton X-100). For immune precipitations, 2 μg affinity-purified antibody, 20 μl packed protein G-sepharose, and 25 μg Golgi membrane extract in a total volume of 500 μl lysis buffer were incubated for 2 h at 4°C rotating continuously. Beads were washed four times with lysis buffer, 60 μl of reducing sample buffer was added, and then beads were heated at 95°C for 3 min followed by analysis on 7.5% minigels. To detect Plk1 in immune precipitates, biotinylated goat anti-Plk1 antibodies were used for Western blotting and detected with streptavidin-HRP.
Mass spectrometry
Proteins were digested in-gel with sequencing grade porcine trypsin (Promega) or sequencing grade endoproteinase Glu-C (Roche Applied Science) and extracted using a published method (Shevchenko et al, 1996). Extracted peptides were analysed using a MALDI-TOF instrument (Reflex III; Bruker, Germany) and probability database searching (Perkins et al, 1999), or separated by nano-HPLC and analysed by tandem mass spectrometry on a Q-TOF Ultima mass spectrometer (Micromass, Manchester, UK) (Wilm et al, 1996).
Cell culture, RNA interference, and microscopy
HeLa and NRK cells were cultured at 37°C and 5% CO2 in DMEM containing 10% FCS. Transfections were performed with Fugene-6 according to the manufacturer's instructions (Roche Diagnostics). After 24 h, the cells were treated with 2.5 mg/ml aphidicolin for 18 h, released into fresh growth medium, and fixed at the time points indicated in the figures. RNA interference was performed on HeLa cells transfected using oligofectamine (Invitrogen) with duplex RNA (Dharmacon Research Inc.) for 48 h and processed for fluorescence microscopy (Elbashir et al, 2001). Plk1 was targeted with the sequence 5′-CGTGAACCGTTTGGCGGGdTdT-3′, and the lamin A control was described previously (Elbashir et al, 2001). Cells were fixed for 20 min in 3% (w/v) PFA, quenched for 10 min with 50 mM ammonium chloride, and then permeabilised with 0.1% (v/v) Triton X-100 for 5 min. All solutions were made in PBS, and antibody staining was carried out for 60 min using the following dilutions: 1:50 pS25; 1:400 GRASP65 monoclonal; 1:500 rabbit GM130; 1:2000 sheep GM130 on HeLa, 1:5000 on NRK; 1:1000 pS376 (serum and affinity purified); 1:1000 Plk1. Coverslips were mounted in 10% (w/v) Moviol 4-88, 1 μg/ml DAPI, and 25% (w/v) glycerol in PBS. Images were collected using an Axioskop-2 with a × 63 Plan Apochromat oil immersion objective of NA 1.4, standard filter sets (Carl Zeiss GmbH, Germany), and a 1300 × 1030 pixel cooled-CCD camera (model #CCD-1300-Y) with Metavue software (Visitron Systems GmbH, Puchheim, Germany). Images were cropped in Adobe Photoshop 8.0, and then sized and placed in figures using Adobe Illustrator 10.0 (Adobe Systems Inc.).
Acknowledgments
We thank Dr Nobohiro Nakamura and Shin-ichiro Yoshimura (Kanazawa University, Kanazawa, Japan) for discussions and communication of unpublished data, Dr Olaf Stemmann (MPI of Biochemistry, Martinsried, Germany) for Cdk1–cyclin B1 baculovirus constructs, and our colleagues for useful discussions and comments on the manuscript. The Max-Planck Society and EMBO young investigator programme support research in the group of FAB.
References
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V (1998) Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 92: 183–192 [DOI] [PubMed] [Google Scholar]
- Allan BB, Moyer BD, Balch WE (2000) Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science 289: 444–448 [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet N, Phair RD, Polishchuk RS, Weigert R, Lippincott-Schwartz J (2003) A role for Arf1 in mitotic Golgi disassembly, chromosome segregation, and cytokinesis. Proc Natl Acad Sci USA 100: 13314–13319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly E, McCaffrey M, Touchot N, Zahraoui A, Goud B, Bornens M (1991) Phosphorylation of two small GTP-binding proteins of the Rab family by p34cdc2. Nature 350: 715–718 [DOI] [PubMed] [Google Scholar]
- Barr FA, Nakamura N, Warren G (1998) Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J 17: 3258–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Preisinger C, Kopajtich R, Korner R (2001) Golgi matrix proteins interact with p24 cargo receptors and aid their efficient retention in the Golgi apparatus. J Cell Biol 155: 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Puype M, Vandekerckhove J, Warren G (1997) GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 91: 253–262 [DOI] [PubMed] [Google Scholar]
- Barr FA, Sillje HH, Nigg EA (2004) Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5: 429–440 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J (2004) Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 5: 792–804 [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112: 407–421 [DOI] [PubMed] [Google Scholar]
- Colanzi A, Deerinck TJ, Ellisman MH, Malhotra V (2000) A specific activation of the mitogen-activated protein kinase kinase 1 (MEK1) is required for Golgi fragmentation during mitosis. J Cell Biol 149: 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Sütterlin C, Malhotra V (2003) RAF1-activated MEK1 is found on the Golgi apparatus in late prophase and is required for Golgi complex fragmentation in mitosis. J Cell Biol 161: 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell WD, Kaminski PJ, Jackson JR (2002) Identification of Drosophila Myt1 kinase and its role in Golgi during mitosis. Cell Signal 14: 467–476 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Elia AE, Cantley LC, Yaffe MB (2003a) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299: 1228–1231 [DOI] [PubMed] [Google Scholar]
- Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB (2003b) The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115: 83–95 [DOI] [PubMed] [Google Scholar]
- Hirose H, Arasaki K, Dohmae N, Takio K, Hatsuzawa K, Nagahama M, Tani K, Yamamoto A, Tohyama M, Tagaya M (2004) Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi. EMBO J 23: 1267–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA (2004) A new checkpoint takes shape. Nat Cell Biol 6: 801–803 [DOI] [PubMed] [Google Scholar]
- Jesch SA, Lewis TS, Ahn NG, Linstedt AD (2001) Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol Biol Cell 12: 1811–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane HA, Nigg EA (1996) Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 135: 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Madsen ML, Yarm FR, Jang YJ, Liu X, Erikson RL (2000) Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2. Proc Natl Acad Sci USA 97: 12589–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Argov R, Dahan N, Ramachandran S, Amarilio R, Shainskaya A, Lev S (2004) Mitotic phosphorylation of the peripheral Golgi protein Nir2 by Cdk1 provides a docking mechanism for Plk1 and affects cytokinesis completion. Mol Cell 14: 319–330 [DOI] [PubMed] [Google Scholar]
- Liu F, Rothblum-Oviatt C, Ryan CE, Piwnica-Worms H (1999) Overproduction of human Myt1 kinase induces a G2 cell cycle delay by interfering with the intracellular trafficking of Cdc2–cyclin B1 complexes. Mol Cell Biol 19: 5113–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Stanton JJ, Wu Z, Piwnica-Worms H (1997) The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol 17: 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Gonatas NK, Warren G (2000) The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. J Cell Biol 149: 341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Nakamura N, Warren G (1998a) Golgi division and membrane traffic. Trends Cell Biol 8: 40–44 [DOI] [PubMed] [Google Scholar]
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jamsa E, Rahman D, Pappin DJ, Warren G (1998b) Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94: 783–793 [DOI] [PubMed] [Google Scholar]
- Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E (2003) Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem 278: 25277–25280 [DOI] [PubMed] [Google Scholar]
- Neef R, Preisinger C, Sutcliffe J, Kopajtich R, Nigg EA, Mayer TU, Barr FA (2003) Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J Cell Biol 162: 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2: 21–32 [DOI] [PubMed] [Google Scholar]
- Norman TC, Smith DL, Sorger PK, Drees BL, O'Rourke SM, Hughes TR, Roberts CJ, Friend SH, Fields S, Murray AW (1999) Genetic selection of peptide inhibitors of biological pathways. Science 285: 591–595 [DOI] [PubMed] [Google Scholar]
- Okano-Uchida T, Okumura E, Iwashita M, Yoshida H, Tachibana K, Kishimoto T (2003) Distinct regulators for Plk1 activation in starfish meiotic and early embryonic cycles. EMBO J 22: 5633–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567 [DOI] [PubMed] [Google Scholar]
- Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, Kopajtich R, Gettemans J, Barr FA (2004) YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol 164: 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Shima DT, Cabrera-Poch N, Pepperkok R, Warren G (1998) An ordered inheritance strategy for the Golgi apparatus: visualization of mitotic disassembly reveals a role for the mitotic spindle. J Cell Biol 141: 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Haldar K, Pepperkok R, Watson R, Warren G (1997) Partitioning of the Golgi apparatus during mitosis in living HeLa cells. J Cell Biol 137: 1211–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short B, Preisinger C, Korner R, Kopajtich R, Byron O, Barr FA (2001) A GRASP55-rab2 effector complex linking Golgi structure to membrane traffic. J Cell Biol 155: 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Watson R, Giannakou ME, Clarke M, Warren G, Barr FA (1999) GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J 18: 4949–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, De La Torre C, Ellenberg J, Peters JM (2004) Roles of Polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol 14: 1712–1722 [DOI] [PubMed] [Google Scholar]
- Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM (2002) The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol Cell 9: 515–525 [DOI] [PubMed] [Google Scholar]
- Sütterlin C, Hsu P, Mallabiabarrena A, Malhotra V (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109: 359–369 [DOI] [PubMed] [Google Scholar]
- Sütterlin C, Lin CY, Feng Y, Ferris DK, Erikson RL, Malhotra V (2001) Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc Natl Acad Sci USA 98: 9128–9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Lindman M, Jackman M, Kano F, Murata M, Zhang X, Kondo H (2003) The localization and phosphorylation of p47 are important for Golgi disassembly–assembly during the cell cycle. J Cell Biol 161: 1067–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Seemann J, Pypaert M, Shorter J, Warren G (2003) A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J 22: 3279–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren G (1993) Membrane partitioning during cell division. Annu Rev Biochem 62: 323–348 [DOI] [PubMed] [Google Scholar]
- Warren G, Wickner W (1996) Organelle inheritance. Cell 84: 395–400 [DOI] [PubMed] [Google Scholar]
- Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379: 466–469 [DOI] [PubMed] [Google Scholar]