

Abstract
Rv1900c, a Mycobacterium tuberculosis adenylyl cyclase, is composed of an N-terminal α/β-hydrolase domain and a C-terminal cyclase homology domain. It has an unusual 7% guanylyl cyclase side-activity. A canonical substrate-defining lysine and a catalytic asparagine indispensable for mammalian adenylyl cyclase activity correspond to N342 and H402 in Rv1900c. Mutagenic analysis indicates that these residues are dispensable for activity of Rv1900c. Structures of the cyclase homology domain, solved to 2.4 Å both with and without an ATP analog, form isologous, but asymmetric homodimers. The noncanonical N342 and H402 do not interact with the substrate. Subunits of the unliganded open dimer move substantially upon binding substrate, forming a closed dimer similar to the mammalian cyclase heterodimers, in which one interfacial active site is occupied and the quasi-dyad-related active site is occluded. This asymmetry indicates that both active sites cannot simultaneously be catalytically active. Such a mechanism of half-of-sites-reactivity suggests that mammalian heterodimeric adenylyl cyclases may have evolved from gene duplication of a primitive prokaryote-type cyclase, followed by loss of function in one active site.
Keywords: adenylyl cyclase, crystal structure, half-of-sites-reactivity, Mycobacterium tuberculosis, homodimer
Introduction
Worldwide, Mycobacterium tuberculosis is the leading cause of death from a single infectious agent. M. tuberculosis invades phagocytic mammalian cells such as phagocytes, monocytes and macrophages, thus evading the host's immune system. Although its ability to survive within infected cells must depend on extensive crosscommunication between host and pathogen, signal transduction pathways of M. tuberculosis such as the cyclic nucleotide system have not yet been extensively studied.
Cyclic nucleotides regulate diverse cellular processes (Cooper, 2003). In prokaryotes, cyclic nucleotides play key roles in diverse processes such as adaptation to nutrients, response to osmotic stress and acidification and pathogenicity (Leppla, 1982; Süsstrunk et al, 1998; Yahr et al, 1998; Ladant and Ullmann, 1999; Kimura et al, 2002).
Currently, the catalytic domains of nucleotidyl cyclases, enzymes that catalyze the synthesis of cAMP and cGMP, are grouped into five classes based on sequence similarity (Barzu and Danchin, 1994; Cotta et al, 1998; Sismeiro et al., 1998). Most of them, including mammalian adenylyl cyclases and guanylyl cyclases (GCs), contain class III cyclase homology domains (CHDs), which are further subdivided into four subclasses (IIIa–IIId; Linder and Schultz, 2003). Most mammalian ACs are monomeric integral membrane proteins that possess two complementary, although nonidentical, CHDs (Sunahara et al, 1996; Taussig and Zimmermann, 1998), while prokaryotes and lower eukaryotes produce both soluble and membrane-bound nucleotidyl cyclases of variant domain compositions that usually contain only one CHD (Linder and Schultz, 2003) and are catalytically active as homodimers (Guo et al, 2001; Linder et al, 2002). In eukaryotes, stimuli are usually sensed and transmitted to CHDs by a complex network of regulatory proteins, such as calmodulin, protein kinase C and those involved in the G-protein-coupled receptor (GPCR)–G-protein signal transduction pathways (Cooper, 2003). The GPCR–G-protein pathways are conspicuously absent in prokaryotes. Instead, the prokaryotic CHD isoforms, mostly identified from recently available genome sequences, appear to be directly regulated by a variety of potential regulatory domains attached directly to the CHDs. M. tuberculosis has 15 class III AC genes. Yet, their biological role is unknown. Therefore, characterization of these enzymes is essential for elucidating the role of cyclic nucleotides in M. tuberculosis as well as other prokaryotes.
The structures of the G protein- and forskolin-bound complexes of the N-terminal CHD from type V mammalian AC (VC1) and the C-terminal CHD from type II mammalian AC (IIC2) demonstrated that the catalytic site is located at the nearly symmetric interface between the CHDs (Tesmer et al, 1997). Because the catalytic domains are not identical, mammalian ACs possess only one functional active site (Dessauer and Gilman, 1996; Whisnant et al, 1996; Yan et al, 1996; Tesmer et al, 1997, 1999, 2000). In contrast, homodimeric class III nucleotidyl cyclases were expected to contain two identical active sites. From the structure of the mammalian AC VC1·IIC2 heterodimer, extensive mutational studies and the sequences of CHDs, it was inferred that six residues of the CHD fold are essential for substrate recognition and catalysis (Tesmer et al, 1997, 1999; Linder and Schultz, 2003). In mammalian ACs, these correspond to two Mg2+ (or Mn2+)-coordinating aspartates (Tang et al, 1995; Tesmer et al, 1997, 1999; Tesmer and Sprang, 1998) contributed by C1 subunits. The C2 subunits contribute an asparagine (Tesmer et al, 1997; Yan et al, 1997) involved in substrate orientation, an arginine residue to stabilize the transition state (Tang et al, 1995; Tesmer et al, 1997; Yan et al, 1997) and an adenine-specifying lysine–aspartate couple (Tang et al, 1995; Tesmer et al, 1997; Sunahara et al, 1998). While catalysis in the mycobacterial ACs Rv1625c (Guo et al, 2001) and Rv1264 (Linder et al, 2002) relies on all of the six residues, several other CHDs from M. tuberculosis and other prokaryotes do not have a complete complement of these residues, hence cyclase activity cannot be definitively assigned to these CHDs.
Rv1900c is one of these unconventional cyclase genes of M. tuberculosis. The Rv1900c gene product encodes a two-domain protein, of which the 285-residue N-terminal domain has sequence similarity to α/β-hydrolases (Cole et al, 1998) and the C-terminal residues 291–462 (Figure 1) to class IIIc CHDs (McCue et al, 2000; Linder and Schultz, 2003). The presence of the hydrolase homology domain suggested that Rv1900c might possess lipase activity and the enzyme was named ‘LipJ' (Cole et al, 1998), although consensus residues that confer esterase activity are absent (Shenoy et al, 2004a). The Rv1900c CHD shares less than 15 and 28% sequence identity, respectively, with structurally characterized mammalian AC and trypanosomal CHDs, and contains only four of the six residues required for mammalian AC activity. The substrate-orienting asparagine and adenine-specifying lysine are replaced by H402 and N342, respectively (Figure 1). Thus, characterization of the CHD of Rv1900c was expected to provide key insights into variations in substrate binding and catalytic mechanism among nucleotidyl cyclases.
Figure 1.
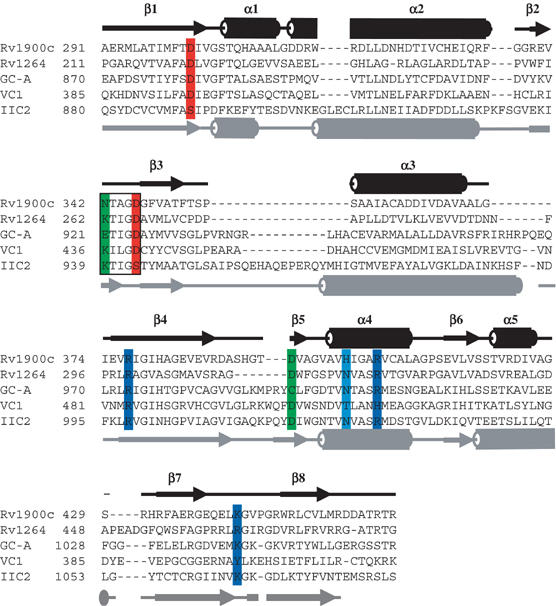
Sequence alignment of selected CHDs. Sequences are as follows: M. tuberculosis Rv1900c (homodimer), M. tuberculosis AC Rv1264 (homodimer), canine AC VC1 (heterodimer), rat AC IIC2 (heterodimer) and human guanylyl cyclase GC-A (homodimer). Amino-acid numbering is left of each block. Secondary structures (cylinders for helices, arrows for β-strands, lines for coils) above the alignment correspond to M. tuberculosis Rv1900c, and those below to rat IIC2 (Tesmer et al, 1997). Eight positions canonically involved in catalysis are shaded. Red: metal-binding aspartates (Rv1900c: D302 and D346); green: substrate specifying (Rv1900c: the unconventional N342 and D395, both dispensable); light blue: transition-state-stabilizing asparagine (Rv1900c: H402, dispensable); dark blue: phosphate coordinating (Rv1900c: R377, R406, K442). Note the distribution of canonical catalytic residues in the VC1·IIC2 heterodimer. The Lys/Glu/polar-X-X-Gly-Asp motif of the β2–β3 loop is boxed.
Here we demonstrate Rv1900c is an active AC with an unusually high GC side-activity. We describe the structures of the Rv1900c CHD, the first of a homodimeric AC, in both an open, unliganded conformation and a closed, substrate-bound conformation. Unlike the mammalian structures (Tesmer et al, 1997, 1999, 2000; Zhang et al, 1997), which were in a forskolin-activated state, the open Rv1900c homodimer structure appears to depict the ground state. By mutagenesis, we confirmed general determinants of substrate specificity in Rv1900c and marked differences of substrate binding by mammalian ACs and noncanonical CHDs. Unexpectedly, although Rv1900c CHDs form homodimers, they are asymmetric in both unliganded and ligand-bound states. These similarities between the homodimeric Rv1900c and heterodimeric mammalian ACs provide a structural and mechanistic basis for the evolution of asymmetry in adenylyl as well as GCs.
Results
Enzymatic activity of Rv1900c
When expressing the Rv1900c holoenzyme in Escherichia coli, only a fraction was produced as a soluble enzyme and purified to homogeneity (see Supplementary data). The specific AC activity was 0.5 μmol/mg/min (Table I). Esterase activity, which was originally anticipated from the sequence of the N-terminal domain, was not detectable using chromogenic substrates (data not shown). The Rv1900c CHD (residues 291–461) and its mutants were expressed as soluble proteins in E. coli and purified to homogeneity (see Supplementary data and Figure 2A). Purified Rv1900c CHD had a specific activity of 1.3 μmol/mg/min using Mn2+·ATP as a substrate (Table I). The enzyme did not accept Mg2+ as a metal cofactor (Table I). The activation energy derived from a linear Arrhenius plot was 73 kJ/mol, with maximal activity at 45°C. Rv1900c CHDs dimerized with a Kdiss of 100 nM, and dimers were detectible as a 46 kDa band even after SDS–PAGE (Figure 2A). Positive cooperativity for cAMP synthesis with respect to ATP (Table I) was consistent with two catalytic sites per functional unit. Like other mycobacterial ACs (Guo et al, 2001; Linder et al, 2002), the specific activity of the Rv1900c CHD depended on protein concentration (Figure 2B), whereas that of a CHD–CHD fusion construct did not (data not shown), confirming that cyclase activity required dimerization. Surprising for an AC, the Rv1900c CHD had considerable GC side-activity with Mn2+·GTP as a substrate, corresponding to 7% of the AC activity (Table I).
Table 1.
Kinetic properties of wild-type and mutant Rv1900c CHD
| Enzyme | Vmax (nmol cAMP/mg/min) | GC (%) | SC50 (μM) | Hill coeff. | Kdiss (nM) |
|---|---|---|---|---|---|
| Holoenzyme | 500 | NM | 590 | 1.4 | 190 |
| CHD | 1300 | 7 | 300 | 1.2 | 100 |
| (CHD)2 | 2000 | 6 | 350 | 1.0 | NA |
| N342A | 700 | 8 | 300 | 1.1 | 80 |
| N342K | 900 | 1 | 420 | 1.4 | 130 |
| N342E | 500 | 30 | 200 | 1.3 | 140 |
| D395A | 1100 | 11 | 80 | 1.3 | <10 |
| N342A/D395A | 900 | 7 | 130 | 1.0 | <10 |
| H402A | 900 | 5 | 240 | 1.3 | 50 |
| H402N | 1200 | 2 | 400 | 1.3 | 50 |
| R406A | 110 | 2 | 250 | 1.7 | ND |
| R377A | 500 | 1 | 530 | 1.5 | 160 |
| K442A | 1000 | 1 | 600 | 1.4 | 40 |
| E340A | 2000 | 5 | 180 | 1.3 | <10 |
| V341A | 800 | 5 | 200 | 1.6 | 40 |
| T343A | 1400 | 5 | 180 | 1.6 | 30 |
| F348A | 1400 | 7 | 250 | 1.5 | 50 |
| GC: relative guanylyl cyclase side-activity; SC50: substrate concentration at half-maximal velocity; Kdiss: dissociation constant of the dimer; (CHD)2: two CHDs linked by a tetradecapeptide; NA: not applicable; ND: not detectable; NM: not measured. Kinetic parameters were measured using 100 nM CHD. All experiments to determine kinetic constants were conducted at least twice with duplicate samples. Variation among individual determinations is typically within 10%. Mean values are reported. | |||||
Figure 2.
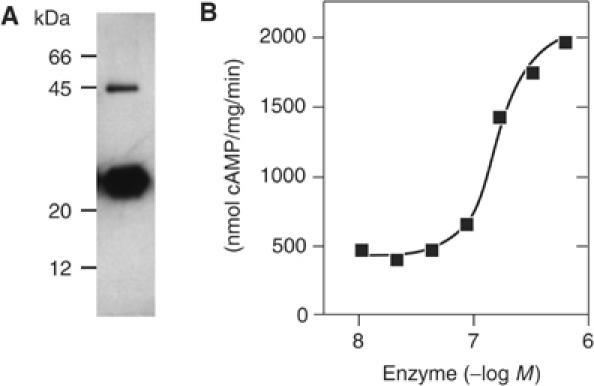
(A) Western blot of Rv1900c CHD probed with an affinity-purified polyclonal antibody against the CHD. (B) CHD specific activity increases four-fold with protein concentration.
Tertiary structures of Rv1900c CHDs
The structures of the Rv1900c CHD (Figure 3) and that of the complex with Mn2+ and the nonreactive ATP analog α,β-methyleneadenosine 5′-triphosphate (AMPCPP) were determined to 2.3 and 2.4 Å resolution, respectively (Table II). The refined model (Table III) of each structure consisted of four Rv1900c monomers per asymmetric unit, arranged as two dimers, AB and CD. The unliganded Rv1900c CHD model (Figures 3 and 4A) included residues 291–457 for all four monomers in the asymmetric unit, and additionally residues 459–462 for monomers B and C. The AMPCPP-bound structure included residues 291–456 for all monomers, except residues 391–393, which were disordered in monomers B and C. A single AMPCPP molecule and one Mn2+ ion were bound per dimer. For both unliganded and AMPCPP-bound structures, subunits within dimers are similar, with root mean square deviations (r.m.s.d.) between corresponding Cα atoms <0.9 Å, whereas corresponding subunits in the two dimers of the asymmetric unit are nearly identical, with r.m.s.d.<0.14 Å (see Supplementary data). Structural differences are confined to flexible loops and terminal residues. The refined models agreed well with the diffraction data and stereochemical criteria (Table III).
Figure 3.
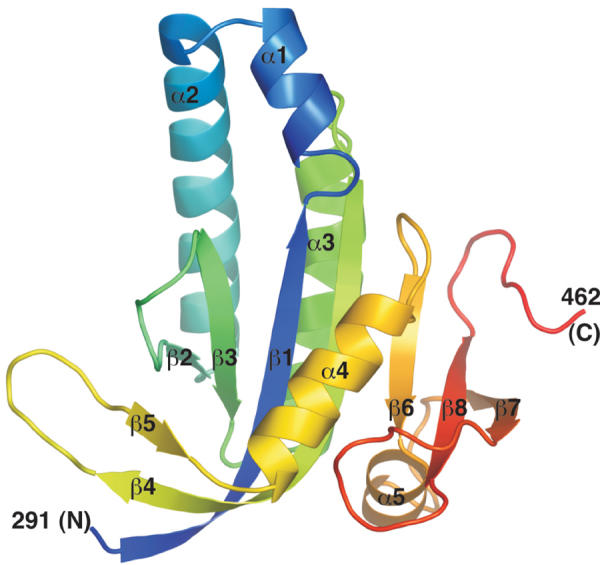
Cartoon of the Rv1900c CHD color-ramped from blue at the N-terminus to red at the C-terminus. Secondary structures are labeled. This figure and other molecular illustrations were made using PYMOL (http://www.pymol.org).
Table 2.
Summary of data collection and phasing statistics
| Apo SeMet MAD data collection |
+AMPCPP |
|||
|---|---|---|---|---|
| λ1 (peak) | λ2 (inflection) | λ3 (remote) | λ (1.0372 Å) | |
| Data | ||||
| Data range (Å) | 50–2.7 | 50–2.3 | 50–2.5 | 50–2.2 |
| Mosaicity | 0.34–0.70 | 0.36–0.73 | 0.35–0.79 | 0.58–0.88 |
| Unique reflections | 33 735 | 53 752 | 43 131 | 29 564 |
| Average multiplicity | 2.58 (2.28) | 2.55 (2.19) | 2.57 (2.54) | 3.8 (3.0) |
| Completeness (%) | 99.8 (99.6) | 99.1 (97.3) | 99.7 (99.7) | 95.3 (75.9) |
| Rsym (%) | 8.4 (32.1) | 8.1 (36.9) | 7.3 (31.4) | 7.1 (29.1) |
| 〈I/σI〉 | 9.2 (1.5) | 12.1 (1.1) | 11.7 (1.1) | 10.4 (2.3) |
| Phasing statistics | ||||
| Phasing power | ||||
| Dispersive—acentric | 0.554 | 0.0 | 1.463 | — |
| Dispersive—centric | 0.497 | 0.0 | 1.161 | — |
| Anomalous—acentric | 1.043 | 1.104 | 0.096 | — |
| Rcullis (%) | ||||
| Dispersive—acentric | 0.820 | 0.0 | 0.689 | — |
| Dispersive—centric | 0.858 | 0.0 | 0.692 | — |
| Anomalous—acentric | 0.814 | 0.807 | 0.978 | — |
| Values in parentheses pertain to the outermost shell of data; Rsym=∑h,i∣Ih,i−〈Ih〉∣/∑h,iIh,i; phasing power=〈∣FH∣〉/r.m.s. lack of closure; Rcullis=r.m.s. lack of closure/r.m.s. isomorphous difference. | ||||
Table 3.
Refinement statistics
| Apo | +AMPCPP | |
|---|---|---|
| Residue range | ||
| Monomer A | 291–457 | 291–456 |
| Monomer B | 291–462 | 291–390, 394–456 |
| Monomer C | 291–462 | 291–390, 394–456 |
| Monomer D | 291–457 | 291–456 |
| AMPCPP molecules | 0 | 2 |
| Metal ions | 0 | 2 |
| Water molecules | 153 | 223 |
| Side chains with two conformers | 6 | 0 |
| Data range (Å) | 50.0–2.3 | 50.0–2.4 |
| Rwork (%) | 19.41 | 23.3 |
| Rfree (%) | 23.11 | 26.2 |
| Average B-values (Å2) | ||
| Main chain | 49.4 | 39.7 |
| Side chain | 60.0 | 46.6 |
| AMPCPP | — | 80.4 |
| Metal | — | 110.4 |
| Water | 48.8 | 48.3 |
| All atoms | 54.1 | 38.5 |
| R.m.s. deviations from target values | ||
| Bond lengths | 0.011 Å | 0.02 Å |
| Bond angles | 1.55° | 2.50° |
| Dihedral angles | 23.9° | 25.1° |
| B-factors between bonded atoms | 6.9 Å2 | 3.4 Å2 |
| Coordinate error (Luzzati, 1952) | 0.26 Å | 0.31 Å |
| Ramachandran outliers (Laskowski et al, 1993) | 0 | 0 |
| R (%): R-factor=∑h∣Fobs∣−∣Fcalc∣∣/∑h∣Fobs∣; test set for Rfree consisted of 4.8% of data. | ||
Figure 4.

Rv1900c CHD dimers. (A) Superimposed open and closed Rv1900c CHD homodimers viewed down the pseudo-dyad dimer axis of the open form. Monomers A and B of the open dimer are rendered in gray and violet, respectively, while those of the closed dimer are rendered sage and salmon, respectively. (B) Closed Rv1900c CHD dimer viewed down the pseudo-dyad dimer axis. Subunits are colored as in (A). The location of the occupied active site is indicated by the bound Mn2+·AMPCPP rendered in atomic detail, and colored by atom type (O red, N blue, P orange and Mn2+ black, and C gray) while the unoccupied, dyad-related site is indicated by an AMPCPP molecule represented as a black stick model. (C) View along the dimer dyad axis showing the dimer active sites. Subunits are colored as in (A). Residues that may play a role in substrate binding or catalysis are rendered in atomic detail and colored by atom type (O red, N blue, P orange and Mn2+ black, and C according to protein subunit). Metal coordination is depicted by solid, black lines while dashed, black lines indicate hydrogen bonds.
The Rv1900c CHD is compact with approximate dimensions of 40 × 40 × 30 Å (Figure 3). Each domain consists of a central seven-stranded β-sheet with strand order β2-β3-β1-β4-β6-β8-β7, with four helices, α1–α3 and α5, on one face of this sheet, and a single helix, α4, and a β-hairpin, formed by COOH-terminal β4 residues and β5, on the opposite face. The Rv1900c CHD is similar in structure to other CHDs, but as a class IIIc CHD it has a shorter dimerization arm than that of mammalian ACs (Tesmer et al, 1997; Linder et al, 2002) and lacks the δ-subdomain of the trypanosomal GRESAG proteins (Bieger and Essen, 2001). Along with palm domains of DNA polymerases, CHDs utilize βαββαβ structural motifs to catalyze the formation of 3′–5′ phosphodiester bonds (Artymiuk et al, 1997). In the Rv1900c CHD, this motif corresponds to β2-α2-β1-β3-α3-β4.
Quaternary structures of Rv1900c CHDs
In crystals of the unliganded as well as the AMPCPP-bound CHDs, the asymmetric unit consists of two nearly identical, pseudosymmetric, isologous homodimers (Figure 4A and B). In addition, each Rv1900c CHD homodimer is asymmetric in that the conformations of quasi-two-fold-related loops and residue side chains at the AB (or DC) subunit interface are different (see Supplementary data). Further discussion is focused on the dimer formed by subunits A and B, and residues or secondary structural elements are distinguished by the suffix ‘A' or ‘B' to denote subunit-specific features. The unliganded, ‘open', CHD dimer interface involves 22 residues from each subunit in polar or nonpolar interactions that bury approximately 3150 Å2 of accessible surface area. Binding of Mn2+-AMPCPP induces dramatic, rigid-body movements of the subunits (Figure 4A) accompanied by conformational changes and repacking of the β2–β3, β4–β5 and β7–β8 loops. Thus, although monomers from the unliganded and AMPCPP-bound structures superimpose well, the dimers do not (r.m.s.d. of about 3.4 Å over 329 superimposable Cα atoms). Relative to the subunits of the open dimer, subunits of the AMPCPP-bound dimer are rotated by 16.6° and translated by 11.4 Å along the rotation axis. In this ‘closed' conformation, the buried surface area increases from 3150 to 5480 Å and the number of residues from each subunit involved in interfacial contacts increase from 22 to 36–38 residues. AMPCPP buries another 230 Å2. The concentration of positive charges at the subunit interface might destabilize the closed conformation of the CHD dimer in the absence of ligand. The closed homodimer (Figure 4B) resembles the VC1·IIC2 heterodimer (Tesmer et al, 1999) except that the shorter dimerization arms (β4–β5 hairpins) tilt away from the opposing subunit.
Rv1900c CHD active site
Two quasi-symmetrical active sites are located in a groove at the CHD dimer interface (Figure 4C). In the open homodimer, residues implicated in substrate binding and catalysis from each of the two subunits are too far apart for interactions with AMPCPP (Figure 5A). The extensive rigid-body rearrangement of subunits to the closed homodimer conformation is required to enable them to simultaneously interact with substrate (Figure 5B). Upon binding of Mn2+·AMPCPP, the asymmetry of the homodimer increases as the conformations of the β2–β3, β4–β5 and β7–β8 loops of opposing subunits diverge further to accommodate substrate at one catalytic site (compare Figures 4C and 5B), but exclude the adenine moiety of ATP from the other, quasi-dyad-related site (compare Figures 4C and 5C). At the AMPCPP-containing active site, monomer A provides the two invariant aspartates that coordinate Mn2+, while monomer B contributes residues that potentially specify substrate and stabilize the transition state (Figure 5B).
Figure 5.

The CHD active site. All three figures are viewed in similar orientations and depict the three crystallographically captured states of the Rv1900c CHD active site. Atomic details of residues that may play a role in substrate binding or catalysis are shown. Subunits, atoms and bonds are represented as in Figure 3. (A) Detailed view of the ligand-unoccupied active site of the open dimer. Active site residues are loosely and asymmetrically packed. (B) Detailed view of the Mn2+-AMPCPP-occupied active site of the closed dimer. Potential hydrogen bonds to the oxygen bridging the ATP α- and β-phosphates (leaving group), which is a nonscissile carbon in AMPCPP, are indicated by dashed, magenta lines. (C) Detailed view of the ligand-unoccupied active site of the closed dimer. Residues that participate in binding substrate are held in inactive conformations by a network of hydrogen bonds.
Metal coordination and binding of the ATP ribose and phosphate
The conformation of the occupied ribose/phosphate-binding site of the closed Rv1900c CHD homodimer is similar to that in the mammalian VC1·IIC2 heterodimer with appropriately located structural equivalents of all residues involved in binding the ATP phosphate (Figures 1 and 5B; Tesmer et al, 1997, 1999, 2000). The AMPCPP γ-phosphate is positioned over the N-terminus of α1A and is hydrogen bonded to amides of the P-loop (β1A–α1A loop) and to two basic residues, R377A in β4A (equivalent to VC1 R484) and K442B in the β7B–β8B loop (IIC2 K1065). The AMPCPP α-phosphate ion pairs with R406B in α4B (IIC2 R1029). Mutation of either R377 or K442 increases SC50 for ATP, but barely affects Vmax, whereas mutation of R406 substantially reduces Vmax without affecting SC50 (Table I). Evidently, these residues have roles with respect to substrate binding and catalysis, which are similar to their counterparts in mammalian ACs. Histidine 402B (α4B) is the topological equivalent of mammalian AC IIC2 N1025, which is implicated in binding the ATP ribose oxygen and orienting the substrate during catalysis (Tang et al, 1995; Tesmer et al, 1997; Yan et al, 1997). However, H402B is too distant from the AMPCPP ribose oxygen to form a hydrogen bond and does not participate in catalysis as demonstrated by mutagenesis (Table I). Two invariant aspartates in Rv1900c, D302A in β2A and D346A in the β2A–β3A loop, coordinate Mn2+ at positions equivalent to the ‘B' metal site of mammalian ACs where this function is filled by D396 and D440 of VC1 (Tesmer and Sprang, 1998; Tesmer et al, 1999). In addition, D346A is also hydrogen bonded to a non-ester oxygen from the ATP α-phosphate. Mutation of either metal-binding aspartate in Rv1900c abolishes catalytic activity (Table I). Inactive D302A mutants are rescued by complementary R406A mutants (Figure 6), confirming that D302 and R406 from different subunits participate in the same interfacial catalytic site. In addition to D302A and D346A, the octahedral coordination shell of the Mn2+ ion consists of S306A, the backbone carbonyl of I303A, two non-ester oxygens from the AMPCPP α-phosphate, the oxygen bridging the AMPCPP β- and γ-phosphates and a water molecule. The Rv1900c equivalent to the mammalian AC ‘A' metal ion site is occupied either partially by Mn2+ or by a water molecule, and contacts the carboxylate oxygens from D302A and D346A, the ester oxygen of the AMPCPP ribose phosphate and a non-ester oxygen from the α-phosphate. The A site may be incompletely occupied in Rv1900c because the AMPCPP ribose is poorly ordered and appears to adopt the C-2′ endo conformation in which the 3′-OH is inaccessible for metal coordination.
Figure 6.

Titration of 100 nM Rv1900c CHD R406A with inactive Rv1900c CHD D302A reconstitutes wild-type AC activity.
Base specificity in Rv1900c
Rv1900c uses ATP and to a much lesser extent GTP as substrates because its purine-binding site does not provide determinants for specificity. The adenine moiety of AMPCPP is inserted into a pocket formed by F300B (β1B), V549B (β3B) and V601B (α4B) and is stacked against the peptide plane of G345A–D346A (β2A–β3A loop). Rv1900c residues D395B (β4B–β5B hairpin) and N342B (β2B–β3B loop) are topological equivalents of D1018 and K938 in mammalian AC IIC2. The latter residues directly hydrogen bond to adenine N1 and N6 (Tesmer et al, 1997). However, in Rv1900c, neither D395B nor N342B hydrogen bonds to the AMPCPP adenine. Individual alanine replacements of N342 or the neighboring residues E340, V341, T343 and F348 of the β2–β3 loop do not substantially affect cyclase activity, whereas replacement of N432 by a charged lysine or glutamate decreases or increases relative GC activity, respectively (Table I). Similarly, mutation of D395A did not affect cyclase activity (Table I). Even the N342A/D395A double mutant had nearly normal cyclase activity (Table I). Thus, the enzymatic data for the mutant enzymes, together with the crystal structure of Rv1900c, indicate that neither of the latter two canonical residues nor their neighbors in the β2–β3 loop are crucial for activity. However, residues in this loop do appear to have a role in the half-of-sites reactivity of Rv1900c (see below). The limited preference of Rv1900c for ATP over GTP is probably dictated by general size and electrostatic complementarity of the purine-binding pocket rather than by base-specific hydrogen bonds. The absence of base-specific interactions may explain why Rv1900c is not sensitive to P-site inhibitors (data not shown). In this respect, Rv1900c is quite distinct from the mammalian ACs.
Substrate binding in dyad-related active sites of Rv1900c CHD homodimers
The β2–β3 loop at the subunit interface of Rv1900c is the central feature of an ATP-driven structural switch that promotes substrate binding in one catalytic site while occluding the dyad-related site (Figures 4C, 5B and C). A network of hydrogen bonds among D346B, S306B, T307B and H402A pull the β2B–β3B loop toward the unoccupied and away from the occupied ATP-binding sites (Figure 5B and C). Further, hydrogen bonds among N395A, T343B and N342A position N342A within 2.0 Å of the potential purine-binding pocket of the unoccupied site and thereby exclude the purine moiety of ATP (Figure 5C). The same interactions withdraw N342B from the quasi-dyad-related adenine-binding pocket and allow substrate to bind (Figure 5B). Binding of the AMPCPP ribose phosphate and Mn2+ disrupts hydrogen bonds from D346A to H402B and S306A, induces reorientation of D346A and stabilizes the alternative conformation of β2A–β3A.
Discussion
Variant substrate recognition in Rv1900c
This study presents one of several possible variations to the paradigm of catalysis by CHDs that was originally inferred from analyses of mammalian ACs. Rv1900c is the first example of a variant class III adenylyl cyclase catalytic site to be characterized. Coordination of the metal cofactor and binding of ATP phosphate is similar to that of the mammalian ACs. A remarkable feature of Rv1900c activity is its strict requirement for Mn2+. Many bacterial CHDs prefer Mn2+ as a cofactor, possibly because the radius of the metal coordination sphere is too large to ligate the smaller Mg2+ ion (Coudart-Cavalli et al, 1997; Kasahara et al, 2001). However, mammalian ACs and a few bacterial ACs can also use Mg2+, which probably is the physiologically relevant cofactor (Dessauer et al, 1997). Rv1900c and other ACs from M. tuberculosis are among those that require millimolar concentrations of Mn2+ for activity (Guo et al, 2001; Linder et al, 2002, 2004; Shenoy et al, 2005). It is well possible that this is physiologically relevant as the M. tuberculosis genome contains a putative Mn2+ transporter that may be capable of sustaining a sufficiently high concentration of intracellular Mn2+ (Cole et al, 1998; Reddy et al, 2001; Linder et al, 2004).
Our mutational and structural analysis demonstrates that the ribose- and purine-binding subsites of Rv1900c differ from those in mammalian ACs. H402, the Rv1900c equivalent of the mammalian AC asparagine that appears to orient the substrate during catalysis, does not contact the substrate analog (Tesmer et al, 1997; Yan et al, 1997). However, this does not appear to have a major impact on the affinity of Rv1900c for ATP or its rate of catalysis, which are comparable to those for unactivated mammalian ACs (Dessauer et al, 1997).
Analyses of nucleotidyl cyclases from different organisms have established that specificity for ATP in ACs is very stringent compared to that for GTP in GCs (Beuve et al, 1993; Coudart-Cavalli et al, 1997; Sunahara et al, 1998; Linder et al, 2000; Guo et al, 2001; Kasahara et al, 2001; Weber et al, 2004). Indeed, Rv1900c is the first example of an AC with significant GC side-activity. In mammalian AC heterodimers, a lysine and an aspartate specify ATP by hydrogen bonds to the adenine N1 and N6, and also sterically prevent GTP binding (Tesmer et al, 1997). Structure-based sequence comparisons with ACs indicated that in GCs these residues are substituted by a glutamate and a cysteine, respectively, which were proposed to interact with the guanine N2 and O6 (Sunahara et al, 1998; Tucker et al, 1998). Rv1900c may be able to use GTP as substrate simply because N342 and D395 do not specify adenine as do their lysine and aspartate counterparts in mammalian AC. Accordingly, mutations of these residues did not dramatically alter activity. In contrast, similar mutations in other adenylyl cyclases completely abolish enzyme activity (Kasahara et al, 2001; Shenoy et al, 2003) and the implementation of the lysine–aspartate couple in GCs switches specificity from GTP to ATP (Sunahara et al, 1998; Tucker et al, 1998; Linder et al, 2000). Therefore, mutational analysis of Rv1900c and other ACs indicates that the canonical lysine–aspartate couple specific to most class III ACs are not the sole determinants of nucleotide specificity, and general determinants such as electrostatic gradients, shape complementarity and size constraints of the purine-binding pocket also play important roles in dictating nucleotide specificity. It is likely therefore that several of the recently identified CHDs that show deviations in the canonical residues required for catalysis by the mammalian ACs (Linder and Schultz, 2003) are also functional cyclases like Rv1900c, but will display differences in substrate specificity, affinity, rate of catalysis and perhaps in mechanism.
Cooperativity in the catalytic cycle
Although CHD homodimers contain two chemically identical and catalytically competent active sites, the structural snapshots of unliganded and AMPCPP-bound Rv1900c CHD dimers show that, at any instance, only one active site binds. Catalytic activity of the D302A-R406A complementation mutant, which has only one functional active site, is comparable to that of the wild-type enzyme (Figure 6), supporting the structural observation that only one site of the homodimer is catalytically competent at a time. Mechanisms of half-of-sites reactivity have been previously observed in enzymes such as adenylyl transferases (Izard and Geerlof, 1999), tyrosyl-tRNA synthetase (First and Fersht, 1993) and dehydrogenases (Eby and Kirtly, 1976; Nichols et al, 2004), as well as ligand-binding proteins like the aspartate receptors (Biemann and Koshland, 1994). However, unlike these proteins, where half-of-sites-reactivity results in negative cooperativity, Rv1900c shows slight positive cooperativity for product formation with respect to ATP. This cooperativity may arise from productive ATP binding in one site, and perhaps weak and nonproductive binding of ATP as an allosteric effector in the dyad-related site.
Progress through the Rv1900c catalytic cycle appears to require a flexible dimer interface, with the open homodimer conformation predominating in the absence of ligand. Entry of ATP into one of the active sites of the homodimer triggers a cooperative movement of the subunits relative to each other, forming the closed homodimer. The productive site, and the ATP bound within, must undergo additional conformational changes relative to the complex to permit full occupancy of Mn2+ at the A-site, as required for catalytic activation of the ribosyl 3′-hydroxyl group for in-line nucleophilic attack on the ATP α-phosphate (Tesmer et al, 2000). The pentavalent phosphate transition state or intermediate may be stabilized by R406B and the catalytic Mn2+ ion. As in mammalian ACs, steric conflict with the cyclized nucleotide phosphate may trigger ejection of cAMP and the A-site Mn2+ (Tesmer et al, 2000). This would allow the β2B–β3B loop and Asn342B to revert to the conformation that is nonproductive for substrate binding at the evacuated site. The release of Mn2+·pyrophosphate then may be the rate-limiting step as in the mammalian ACs, prompted by return of the β2A–β3A loop and D346A to ligand-unoccupied conformations (Dessauer et al, 1997). We have no proof that release of Mn2+·pyrophosphate from one catalytic site is a prerequisite for productive substrate binding in the dyad-related site. However, the comparable rates of cAMP synthesis in wild-type Rv1900c and the D302A-R406A complementation couple indicate that initiation of a new catalytic cycle within the same active site is not rate limiting. Thus, it is plausible to assume that immediately after product release, the two sites will be indistinguishable and therefore equally likely to bind substrate.
The regulation of the CHD activity may well be exerted by the uncharacterized, N-terminal Rv1900c α/β-hydrolase domain in response to signals from hitherto unidentified ligands or protein partners. It can be imagined that a domain interaction occurs between surface-accessible elements of the CHD such as α1, the α1–α2 loop, α2 or the dimerization arm and the N-terminal domain. The poor expression of the Rv1900c holoenzyme precluded attempts to obtain the structure of the full AC.
Exploiting asymmetry for CHD regulation
Like the Rv1900c CHD, other homodimeric CHDs may be expected to form asymmetric homodimers as a consequence of the bifunctionality of the β2–β3 loop. This loop contains a conserved Lys/Glu/polar-X-X-Gly-Asp sequence, of which the first residue regulates nucleotide base binding or specificity in one active site, while in the quasi-dyad-related site, the peptide plane of the invariant glycine–aspartate stacks against the nucleotide base and the invariant aspartate also coordinates metal-phosphate. However, spatial constraints appear to prevent this loop from participating in both active sites simultaneously, permitting only one active site to bind substrate productively and enforcing conformational and mechanistic asymmetry upon the homodimer. This asymmetry might allow differential interaction of sensory/regulatory domains or small molecules with the two subunits of CHD homodimers, and provide mechanistic checkpoints for regulation of cAMP synthesis.
The asymmetric structural features observed in the Rv1900c CHD seem to have been amplified in the evolution of other cyclases. For example, the pseudoheterodimeric mammalian ACs appear to have evolved to enhance and optimize the inherent asymmetry of the CHD homodimer, using one binding site for catalysis while the quasi-dyad-related site acquired a regulatory function. The plant diterpene forskolin, a fortuitous activator of all mammalian AC isoforms except type IX (Seamon and Daly, 1981; Tang and Gilman, 1995; Sunahara et al, 1996), binds to this regulatory site. However, it cannot be accommodated in the catalytic site of homodimeric CHDs. In mammalian ACs, structural asymmetry is then further exploited by regulatory proteins such as Gα subunits, which preferentially bind at nonoverlapping sites on either C1 or C2 domains (Tesmer et al, 1997; Dessauer et al, 1998) and probably act by affecting the interplay between open and closed states (Tesmer et al, 1997) or by disrupting nonproductive or inhibitory interdomain contacts.
Materials and methods
Plasmid construction
The gene Rv1900c (GenBank accession number Z97193) was amplified by PCR from M. tuberculosis genomic DNA (gift of Dr Boettger, University of Zürich Medical School), and cloned into pET-16b between 5′ NdeI and 3′ XhoI sites, adding an N-terminal MGH10SSGHIEGRH tag. The isolated Rv1900c CHD, residues 291–462, was cloned into pQE30 between 5′ BamHI and 3′ XmaCI sites, adding an N-terminal MRGSH6GS tag. Point mutations were introduced by standard methods. In the linked dimmer, the connecting sequence was TRAAGGPPAAGGLE. DNA inserts were verified by double-stranded DNA sequencing.
Expression and purification of Rv1900c proteins
Rv1900c CHD and mutants were expressed in E. coli BL21(DE3)[pRep4]. Expression was induced by 60 μM IPTG (4–20 h, 22°C). Bacteria were washed (50 mM Tris–HCl, 1 mM EDTA, pH 8) and stored at −80°C. Cells from a 200–600 ml culture were suspended in 20 ml lysis buffer (50 mM Tris–HCl, 2 mM 3-thioglycerol, pH 8), sonicated for 30 s, and then incubated on ice for 30 min each with 0.2 mg/ml lysozyme and 5 mM MgCl2+10 μg/ml DNAseI. After centrifugation (31 000 g, 30 min), 15 mM imidazole pH 8 and 250 mM NaCl (final concentrations) were added to the supernatant. Protein was equilibrated on ice for 60 min with 250 μl Ni2+-nitrilotriacetic acid (Ni-NTA)-agarose, and then successively washed with 10 ml of lysis buffer containing 15 mM imidazole, 250 mM NaCl and 5 mM MgCl2 and 5 ml of lysis buffer containing 15 mM imidazole and 5 mM MgCl2. The protein was eluted with 0.4 ml of elution buffer (37.5 mM Tris–HCl pH 8, 250 mM imidazole, 2 mM MgCl2, 1.5 mM 3-thioglycerol), and then stored at −20°C in elution buffer containing 20% glycerol.
Expression and purification of the Rv1900c holoenzyme was similar except that E. coli BL21(DE3)[pLysS] was used for expression, a French Press was used for lysis, lysozyme and DNAseI treatments were omitted, the supernatant and wash buffers contained 30 mM imidazole and 150 μl Ni-NTA-agarose was used for purification.
Cyclase assays
AC activity was determined at 37°C for 10 min in 100 μl reactions (Salomon et al, 1974) containing 50 mM TES–NaOH pH 8.1, 22% glycerol, 3 mM MnCl2, 500 μM [α-32P]ATP and 2 mM [2,8-3H]cAMP. Mutant mixtures were incubated on ice for 10 min prior to the start of the reaction. Standard deviations of the data were usually <10%. GC activity was determined identically using GTP as substrate (Klumpp and Schultz, 1982).
Hydrolase assays
Purified holoenzyme was tested for hydrolysis of p-nitrophenyl caproate (4.5 mM), p-nitrophenyl acetate (saturated) and p-nitrophenyl phosphate (1.35 mM). In all, 14–50 μg/ml enzyme was used in a microtiter plate setup and the product p-nitrophenol quantified photometrically at 410 nm for 10–20 min at ambient temperature. Buffers were varied to cover a range of conditions. Esterase assays were performed in 0.09% Triton X-100, 9 mM Tris–HCl pH 7.5–9.0 and 0.9–9 mM CaCl2, and phosphatase assays in 50 mM buffer (pH 4.5–8.0), supplemented with 1 mM MgCl2 and 0.1 mM EGTA. Staphylococcus hyicus lipase and calf intestinal phosphatase were used as controls.
Western blot analysis
Proteins samples blotted onto PVDF membranes after 15% SDS–PAGE were probed with affinity-purified, anti-Rv1900c CHD, rabbit polyclonal antibodies, followed by peroxidase-conjugated goat anti-rabbit IgG secondary antibodies (Dianova). Peroxidase was detected using the ECL-Plus kit (Amersham-Pharmacia).
Modified purification protocol for structural analysis
His-tagged selenomethionyl (SeMet) Rv1900c CHD was expressed in E. coli strain B834(DE3)[pRep4]. Cultures (1 l) grown at 37°C in minimal media containing selenomethionine were induced with 60 μM IPTG for 12 h at 20°C. Purification was as described above. A HiPrep 26/10 desalting column (Amersham-Pharmacia) was used for fast buffer exchange to 20 mM Tris–HCl, pH 7.5, 10% w/v glycerol, 1 mM MgCl2 and 0.05% β-mercaptoethanol. Purified protein was concentrated to 7.5 mg/ml, flash frozen and stored at −180°C.
Crystallization and crystal mounting
SeMet Rv1900c CHD was crystallized at 20°C by hanging-drop vapor diffusion from a 1:1 mixture of protein solution and well solution (100 mM HEPES pH 7.5, 8–16% PEG 1000, 450 mM NaCl). Rod-shaped crystals harvested in 50 μl of well solution were cryoprotected by five successive exchanges of 10 μl of harvesting solution with an equal volume of cryosolution (50% PEG 1000, 100 mM HEPES pH 7.5) every 5 s. The substrate analog AMPCPP was similarly soaked into crystals using cryosolution containing 2 mM AMPCPP and 2 mM Mn2+. The CHD could not be cocrystallized with several potential ligands under similar conditions. Cryoprotected crystals were immediately flash frozen in liquid N2.
Data collection
Data were recorded as 5-s exposures over 0.5° crystal rotation per image on a 3 × 3 mosaic CCD detector, at a crystal to detector distance of 200 mm at the Structural Biology Center beamline 19ID at the Advanced Photon Source, Chicago. MAD data used to solve the structure were collected from a single SeMet Rv1900c crystal as a single 241.5° sweep at each of three wavelengths selected from X-ray fluorescence spectra, the Se K peak (0.979290 Å), inflection point (0.979399 Å) and a remote wavelength (0.956668 Å). Data at each wavelength were collected from areas separated by ∼0.1 mm along the long crystal axis. Data from an AMPCPP-soaked crystal were recorded as a single 200° sweep at 1.0372 Å.
Data were processed using HKL2000 (Otwinowski and Minor, 1997) (Table I). Both crystals belonged to space group P2, with P21221 pseudosymmetry, but were nonisomorphous. Unit cell parameters are a=90.7 Å, b=44.4 Å, c=80.4 Å, β=90.00°, and a=91.1 Å, b=48.9 Å, c=68.2 Å, β=90.00° for the unliganded and AMPCPP-soaked crystals, respectively.
MAD structure determination of the Rv1900c CHD
Eight out of 16 Se sites (figure-of-merit=0.61) were located from unmerged MAD data using SOLVE (Terwilliger and Berendzen, 1999). Refinement of the Se partial structure and MAD phasing were performed using SHARP (Bricogne and Gilmore, 1990) (Table I). The preliminary Cα backbone model for four copies of the Rv1900c polypeptide per asymmetric unit (Vm=2.0 Å3/Da) was built in the initial 3.0-Å map. A total of 10 cycles of phase refinement and extension via four-fold averaging using noncrystallographic symmetry (NCS) operators determined by superposition of secondary structure elements from each subunit, solvent flattening and histogram matching in the program DM (Cowtan, 1994) yielded an interpretable 2.5-Å map from ∣Fobs∣, φbest.
Molecular replacement solution of Rv1900c·AMPCPP complex
Positions and orientations of monomers A, B and D were determined by molecular replacement in EPMR (Kissinger et al, 1999), using a single Rv1900c monomer as a search model (correlation coefficient=0.474). Monomer C was placed, by applying the NCS operator relating subunits A to B, to monomer D.
Model building and refinement
Models for unliganded and AMPCPP-bound structures were built using O (Jones et al, 1991) and refined using CNS (Brunger et al, 1998) (Table II). The models were restrained by four-fold NCS during preliminary refinement cycles, but during later cycles, as it became apparent that pairs of subunits (A and D, and B and C) from different dimers were more similar than subunits within a dimer, the protein core was restrained by four-fold NCS and the remainder by two-fold NCS restraints relating AB to DC. The unliganded and AMPCPP-bound structures are available in the Protein Data Bank with accession codes 1YBT and 1YBU, respectively.
Note added in proof
While this manuscript was under review, Steegborn et al published a homodimeric adenylyl cyclase structure from Spirulina platensis (Steegborn C, Litvin TN, Levi LR, Buck J, Wu H (2005) Bicarbonate activation of adenylyl cyclase via promotion of catalytic active site closure and metal recruitment. Nat Struct Mol Biol 12: 32–37).
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure Legend
Acknowledgments
We thank the staff of the Structural Biology Center beamline 19ID at the Advanced Photon Source, Argonne, Chicago and Dr Christopher Colbert for assistance with X-ray data collection. Financial support was provided NIH Grant DK46371 and Welch Foundation Grant I-1229 and the John W and Rhonda K Pate Professorship in Biochemistry (SRS) and the Deutsche Forschungsgemeinschaft.
References
- Artymiuk PJ, Poirette AR, Rice DW, Willett P (1997) A polymerase I palm in adenylyl cyclase? Nature 388: 33–34 [DOI] [PubMed] [Google Scholar]
- Barzu O, Danchin A (1994) Adenylyl cyclases: a heterogenous class of ATP-utilizing enzymes. Prog Nucleic Acid Res Mol Biol 49: 241–283 [DOI] [PubMed] [Google Scholar]
- Beuve A, Krin E, Danchin A (1993) Rhizobium meliloti adenylate cyclase: probing of a NTP-binding site common to cyclases and cation transporters. C R Acad Sci III 316: 533–539 [PubMed] [Google Scholar]
- Bieger B, Essen LO (2001) Structural analysis of adenylate cyclases from Trypanosoma brucei in their monomeric state. EMBO J 20: 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemann HP, Koshland DEJ (1994) Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-the-sites cooperativity. Biochemistry 33: 629–634 [DOI] [PubMed] [Google Scholar]
- Bricogne G, Gilmore CJ (1990) A multisolution method of phase determination by combined maximization of entropy and likelihood. I. Theory, algorithms and strategy. Acta Crystallogr A 46: 284–297 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, Mclean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrel BG (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 [DOI] [PubMed] [Google Scholar]
- Cooper DMF (2003) Regulation and organization of adenylyl cyclases and cAMP. Biochem J 375: 517–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta MA, Whitehead TR, Wheeler MB (1998) Identification of a novel adenylate cyclase in the ruminal anaerobe, Prevotella ruminicola D31d. FEMS Microbiol Lett 164: 257–260 [DOI] [PubMed] [Google Scholar]
- Coudart-Cavalli MP, Sismeiro O, Danchin A (1997) Bifunctional structure of two adenylyl cyclases from the myxobacterium Stigmatella aurantiaca. Biochimie 79: 757–767 [DOI] [PubMed] [Google Scholar]
- Cowtan KD (1994) ‘DM': An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsl Protein Crystallogr 31: 34–38 [Google Scholar]
- Dessauer CW, Gilman AG (1996) Purification and characterization of a soluble form of mammalian adenylyl cyclase. J Biol Chem 271: 16967–16974 [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Scully TT, Gilman AG (1997) Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem 272: 22272–22277 [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG (1998) Identification of a Giα binding site on type V adenylyl cyclase. J Biol Chem 273: 25831–25839 [DOI] [PubMed] [Google Scholar]
- Eby D, Kirtly ME (1976) Cooperativity and noncooperativity in the binding of NAD analogues to rabbit muscle glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 15: 2168–2171 [DOI] [PubMed] [Google Scholar]
- First EA, Fersht AR (1993) Mutation of lysine 233 to alanine introduces positive cooperativity into tyrosyl-tRNA synthetase. Biochemistry 32: 13651–13657 [DOI] [PubMed] [Google Scholar]
- Guo YL, Seebacher T, Kurz U, Linder JU, Schultz JE (2001) Adenylyl cyclase Rv1625c of Mycobacterium tuberculosis: a progenitor of mammalian adenylyl cyclases. EMBO J 20: 3667–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izard T, Geerlof A (1999) The crystal structure of a novel bacterial adenylyltransferase reveals half-of-sites-reactivity. EMBO J 18: 2021–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of error in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kasahara M, Unno T, Yashiro K, Ohmori M (2001) CyaG, a novel cyanobacterial adenylyl cyclase and a possible ancestor of mammalian guanylyl cyclases. J Biol Chem 276: 10564–10569 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Mishima Y, Nakano H, Takegawa K (2002) An adenylyl cyclase, CyaA, of Myxococcus xanthus functions in signal transduction during osmotic stress. J Bacteriol 184: 3578–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissinger CR, Gehlhaar DK, Fogel DB (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D 55: 484–491 [DOI] [PubMed] [Google Scholar]
- Klumpp S, Schultz JE (1982) Characterization of a Ca2+-dependent guanylate cyclase in the excitable ciliary membrane from Paramecium. Eur J Biochem 124: 317–324 [DOI] [PubMed] [Google Scholar]
- Ladant D, Ullmann A (1999) Bordatella pertussis adenylate cyclase: a toxin with multiple talents. Trends Microbiol 7: 172–176 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Leppla SH (1982) Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA 79: 3162–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder JU, Hammer A, Schultz JE (2004) The effect of HAMP domains on class IIIb adenylyl cyclases from Mycobacterium tuberculosis. Eur J Biochem 271: 2446–2451 [DOI] [PubMed] [Google Scholar]
- Linder JU, Hoffmann T, Kurz U, Schultz JE (2000) A guanylyl cyclase from Paramecium with 22 transmembrane spans. Expression of the catalytic domains and formation of chimeras with the catalytic domains of mammalian adenylyl cyclases. J Biol Chem 275: 11235–11240 [DOI] [PubMed] [Google Scholar]
- Linder JU, Schultz A, Schultz JE (2002) Adenylyl cyclase Rv1264 from Mycobacterium tuberculosis has an autoinhibitory N-terminal domain. J Biol Chem 277: 15271–15276 [DOI] [PubMed] [Google Scholar]
- Linder JU, Schultz JE (2003) The class III adenylyl cyclases: multi-purpose signalling modules. Cell Signal 15: 1081–1089 [DOI] [PubMed] [Google Scholar]
- Luzzati PV (1952) Traitement statistique des erreurs dans la determination des structure cristallines. Acta Crystallogr 5: 802–810 [Google Scholar]
- McCue LA, McDonough KA, Lawrence CE (2000) Functional classification of cNMP-binding proteins and nucleotide cyclases with implications for novel regulatory pathways in Mycobacterium tuberculosis. Genome Res 10: 204–219 [DOI] [PubMed] [Google Scholar]
- Nichols CE, Dhaliwal B, Lockyer M, Hawkins AR, Stammers DK (2004) High-resolution structures reveal details of domain closure and ‘half-of-sites-reactivity' in Escherichia coli aspartate β-semialdehyde dehydrogenase. J Mol Biol 341: 797–806 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Macromolecular Crystallography Part A, Carter JCW, Sweet RM (eds) Vol. 276, pp 307–325. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Reddy SK, Kamireddi M, Dhanireddy K, Young L, Davis A, Reddy PT (2001) Eukaryotic-like adenylyl cyclases in Mycobacterium tuberculosis H37Rv: cloning and characterization. J Biol Chem 276: 35141–35149 [DOI] [PubMed] [Google Scholar]
- Salomon Y, Londos C, Rodbell M (1974) A highly sensitive adenylate cyclase assay. Anal Biochem 58: 541–548 [DOI] [PubMed] [Google Scholar]
- Seamon K, Daly J (1981) Forskolin: a unique diterpene activator of cyclic AMP-generating systems. J Cyclic Nucleotide Res 7: 201–224 [PubMed] [Google Scholar]
- Shenoy AR, Sivakumar K, Krupa A, Srinivasan N, Visweswariah SS (2004a) A survey of nucleotide cyclases in Actinobacteria: unique domain organization and expansion of the class III cyclase family in Mycobacterium tuberculosis. Comp Funct Genom 5: 17–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy AR, Sreenath NP, Mahalingam M, Visweswariah SS (2005) Characterization of phylogenetically distant members of the adenylyl cyclase family from mycobacteria: Rv1647 from M. tuberculosis and its ortholog ML1399 from M. leprae. Biochem J (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy AR, Srinivasan N, Subramaniam M, Visweswariah SS (2003) Mutational analysis of the Mycobacterium tuberculosis Rv1625c adenylyl cyclase: residues that confer nucleotide specificity contribute to dimerization. FEBS Lett 545: 253–259 [DOI] [PubMed] [Google Scholar]
- Sismeiro O, Trotot P, Biville F, Vivares C, Danchin A (1998) Aeromonas hydrophila adenylyl cyclase2: a new class of adenylyl cyclases with thermophilic properties and sequence similarities to proteins from hyperthermophilic archaebacteria. J Bacteriol 180: 3339–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunahara RK, Beuve A, Tesmer JJG, Sprang SR, Garbers DL, Gilman AG (1998) Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J Biol Chem 273: 16332–16338 [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG (1996) Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol 36: 461–480 [DOI] [PubMed] [Google Scholar]
- Süsstrunk U, Pidoux J, Taubert S, Ullmann A, Thompson CJ (1998) Pleiotropic effects of cAMP on germination, antibiotic biosynthesis and morphological development in Streptomyces coelicolor. Mol Microbiol 30: 33–46 [DOI] [PubMed] [Google Scholar]
- Tang W-J, Gilman AG (1995) Construction of a soluble adenylyl cyclase activated by Gsα and forskolin. Science 268: 1769–1772 [DOI] [PubMed] [Google Scholar]
- Tang W-J, Stanzel M, Gilman AG (1995) Truncation and alanine-scanning mutants of type I adenylyl cyclase. Biochemistry 34: 14563–14572 [DOI] [PubMed] [Google Scholar]
- Taussig R, Zimmermann G (1998) Type-specific regulation of mammalian adenylyl cyclases by G protein pathways. Adv Second Messenger Phosphoprotein Res 32: 81–98 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJ, Dessauer CW, Sunahara RK, Murray LD, Johnson RA, Gilman AG, Sprang SR (2000) Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 39: 14464–14471 [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sprang SR (1998) The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr Opin Struct Biol 8: 713–719 [DOI] [PubMed] [Google Scholar]
- Tesmer JJG, Sunahara RK, Gilman AG, Sprang SR (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα·GTPγS. Science 278: 1907–1916 [DOI] [PubMed] [Google Scholar]
- Tesmer JJG, Sunahara RK, Johnson RA, Gilman AG, Sprang SR (1999) Two metal ion catalysis in adenylyl cyclase. Science 285: 756–760 [DOI] [PubMed] [Google Scholar]
- Tucker CL, Hurley JH, Miller TR, Hurley JB (1998) Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc Natl Acad Sci USA 95: 5993–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JH, Vishnyakov A, Hambach K, Schultz A, Schultz JE, Linder JU (2004) Adenylyl cyclases from Plasmodium, Paramecium and Tetrahymena are novel ion channel/enzyme fusion proteins. Cell Signal 16: 115–125 [DOI] [PubMed] [Google Scholar]
- Whisnant RE, Gilman AG, Dessauer CW (1996) Interaction of the two cytosolic domains of mammalian adenylyl cyclase. Proc Natl Acad Sci USA 93: 6621–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci USA 95: 13899–13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S-Z, Hahn D, Huang Z-H, Tang W-J (1996) Two cytoplasmic domains of mammalian adenylyl cyclase form a Gsα- and forskolin-activated enzyme in vitro. J Biol Chem 271: 10941–10945 [DOI] [PubMed] [Google Scholar]
- Yan S-Z, Huang Z-H, Shaw RS, Tang W-J (1997) The conserved asparagine and arginine are essential for catalysis of mammalian adenylyl cyclase. J Biol Chem 272: 12342–12349 [DOI] [PubMed] [Google Scholar]
- Zhang G, Liu Y, Ruoho AE, Hurley JH (1997) Structure of the adenylyl cyclase catalytic core. Nature 386: 247–253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure Legend