

Abstract
Background
ADPKD is a renal pathology caused by mutations of PKD1 and PKD2 genes, which encode for polycystin-1 (PC1) and polycystin-2 (PC2), respectively. PC1 plays an important role regulating several signal transducers, including cAMP and mTOR, which are involved in abnormal cell proliferation of ADPKD cells leading to the development and expansion of kidney cysts that are a typical hallmark of this disease. Therefore, the inhibition of both pathways could potentiate the reduction of cell proliferation enhancing benefits for ADPKD patients.
Methods
The inhibition of cAMP- and mTOR-related signalling was performed by Cl-IB-MECA, an agonist of A3 receptors, and rapamycin, respectively. Protein kinase activity was evaluated by immunoblot and cell growth was analyzed by direct cell counting.
Results
The activation of A3AR by the specific agonist Cl-IB-MECA causes a marked reduction of CREB, mTOR, and ERK phosphorylation in kidney tissues of Pkd1flox/−: Ksp-Cre polycystic mice and reduces cell growth in ADPKD cell lines, but not affects the kidney weight. The combined sequential treatment with rapamycin and Cl-IB-MECA in ADPKD cells potentiates the reduction of cell proliferation compared with the individual compound by the inhibition of CREB, mTOR, and ERK kinase activity. Conversely, the simultaneous application of these drugs counteracts their effect on cell growth, because the inhibition of ERK kinase activity is lost.
Conclusion
The double treatment with rapamycin and Cl-IB-MECA may have synergistic effects on the inhibition of cell proliferation in ADPKD cells suggesting that combined therapies could improve renal function in ADPKD patients.
Keywords: ADPKD, Sequential double treatment, Cell growth, CREB, mTOR, ERK signalling
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited kidney pathology that accounts for about 10 % of end-stage renal disease (ESRD) and is characterized by the progressive development and expansion of fluid-filled cysts [1]. ADPKD is caused by mutations in either PKD1 or PKD2 genes which encode for polycystin-1 (PC1) or polycystin-2 (PC2), respectively [2]. PC1 and PC2, interacting by each other, form a complex able to regulate different signalling pathways associated with cell proliferation, differentiation, and fluid secretion. Alteration of this complex leads to the dysfunction of a network of pathways, including cAMP, and mTOR signalling that may play an important role in the renal cyst development [2]. Consistently, therapeutic interventions using compounds able to inhibit these pathways have already concluded clinical trials in ADPKD patients [3–6]. However, the treatment with mTOR inhibitors shows limited clinical success, while the use of Tolvaptan that reduces cAMP levels targeting V2 receptors, leads to significant advances in ADPKD kidney clinical picture [7, 8]. Nevertheless, the administration of Tolvaptan causes some side effects, such as polyuria and high activity of hepatic enzymes, increasing the management costs of patients [9]. Therefore, dialysis is still the main therapy for ADPKD patients, but it requires high healthcare and personnel costs and limits the life quality of patients [10]. To improve benefits and reduce healthcare costs, the use of combined compounds able to target different pathways could be an attractive purpose for the treatment of ADPKD [11].
Here, we report that the sequential treatment with rapamycin and Cl-IB-MECA causes a reduction of cell proliferation in cystic cell lines by the inhibition of mTOR, CREB, and ERK signalling. These data suggest that the combination of different compounds could be a useful tool for the treatment of ADPKD patients.
Materials and methods
Media, fetal bovine serum, and plastic material for cell culture were purchased from EuroClone (Italy). Specific mTOR, P-mTOR, CREB, P-CREB, ERK, and P-ERK antibodies were obtained from Cell Signalling Technologies (EuroClone, Italy). β-Actin and A3AR antibodies were acquired from Santa Cruz Technologies (DBA, Italy). Enhanced chemiluminescent substrates for Western blotting and HRP-conjugated goat anti-rabbit and anti-mouse antibodies were purchased from EuroClone (Italy). Rapamycin and Cl-IB-MECA were obtained from Sigma-Aldrich (Italy).
Cell lines and cell culture
Human normal (4/5) and human ADPKD (9.7 and 9.12) kidney epithelial cells as well as mouse PH2 (heterozygous) and PN24 (homozygous) Pkd1 gene knockout cell lines were generated by other laboratories [12, 13]. Cell lines were grown in DMEM 50 % F12 medium supplemented with 10 % FBS. Data shown in this study are in line with the 1975 declaration of Helsinki.
Generation of Pkd1flox/−:Ksp-Cre mice
Pkd1flox/−:Ksp-Cre is a mouse model for ADPKD that develops massive enlarged kidney cysts within few days after birth and was generated by crossing Pkd1flox/flox and Pkd1+/−:Ksp-Cre mice [14]. Pkd1flox/+:Ksp-Cre are heterozygous mice that do not develop disease and were considered as controls. All the mice used in these experiments were in a pure C57/Bl6 genetic background and were maintained in SPF colonies handled by a service company provided at the San Raffaele Scientific Institute. All experiments involving animals were performed under a protocol approved by an institutional ethical committee and, subsequently, by the Italian ministry of health (IACUC number: 548).
Analysis of A3AR expression by immunohistochemistry and saturation binding
Paraffin-embedded tissues, derived from two non-ADPKD adult kidneys with normal histology and from kidneys of six ADPKD patients, were analyzed by immunohistochemistry as already reported [15]. Tissue sections were incubated with 3 % BSA for 15 min at RT and treated with anti-A3AR (1:200) polyclonal antibody at 4 °C overnight. The primary antibody bound was detected by a secondary antibody linked to avidin/biotin/horseradish peroxidase complex (DAKO, Italy) for 30 min at RT and positive regions were visualized by a microscope equipped with CCD camera (Zeiss, Italy) after diaminobenzidine staining. Saturation binding to A3 adenosine receptors was performed using [3H]-MRE 3008F20 in cell membranes derived from renal cells and tissues, according with the previously described methods [16].
Treatment of control and polycystic mice with Cl-IB-MECA
Cl-IB-MECA powder was dissolved in DMSO (0.2 mg/ml) and aliquoted for long-term storage at −80 °C. Cl-IB-MECA (0.2 mg/kg-body) or DMSO as control was first delivered to pups by daily intraperitoneal injection of lactating mothers from birth (P0) for 7 days (P6) and next to control and polycystic neonate mice for further 2 days (P7–P8). After treatment, polycystic and control mice were sacrificed, and kidneys were removed and analyzed.
Western blotting
Total proteins obtained by tissue and cell lysates were electrophoresed and probed with the appropriate antibodies as previously described [17]. Protein band intensity was detected by X-ray film scanning with the Model GS-700 Imaging Densitometer (BIO-RAD, Italy). Relative protein levels were calculated as ratio between bands of interest and β-Actin, while the phosphorylation levels were obtained as ratio between the phosphorylated form of the protein and total protein.
Analysis of cell proliferation
Cells were seeded at low density (5000 cell/ml) in 96-well plates. After 24 h starvation in DMEM/F12 0.4 % BSA, cells treated with rapamycin (500 nM) or Cl-IB-MECA (100 nM) alone or in combination were cultured for 24, 48, and 72 h. After trypan blue staining, cells were directly counted in a Burker chamber [17].
Statistical analysis
Analysis of data was performed using Student’s t test (unpaired analysis). Differences are considered significant at a value of p < 0.05. All data are reported as mean ± standard deviation (SD) of at least 3 independent experiments.
Results and discussion
Cl-IB-MECA reduced CREB, mTOR, and ERK activity in Pkd1flox/−:Ksp-Cre mice
As previously described, adenosine type 3 receptors (A3AR) that have an inhibitory effect on adenylyl cyclase are up-regulated in human ADPKD kidney cells and tissues compared with normal controls [17]. Consistently, A3 receptors positivity detected by immunohistochemistry is stronger in ADPKD kidneys where are confined to flat cells of renal cysts compared with normal kidneys (Fig. 1a). These receptors are also up-regulated in polycystic kidney tissues of Pkd1flox/−:Ksp-Cre mice and PN24 homozygous (Pkd1(−/−)) mouse kidney cystic cells compared with non-polycystic Pkd1flox/+:Ksp-Cre mice and PH2 heterozygous (Pkd1(+/−)) mouse cystic cells, respectively (Fig. 1b–d). The activation of A3 receptors by the specific agonist Cl-IB-MECA causes a reduction of cAMP levels, ERK activity, and cell proliferation in ADPKD cystic cells [17]. Moreover, in these cells also, CREB phosphorylation is increased and the treatment with Cl-IB-MECA decreases its promoter activity [15]. Therefore, it is reasonable to speculate that “in vivo” treatment of polycystic mice with Cl-IB-MECA could decrease the CREB activity. As expected, in basal conditions (vehicle), an higher CREB activation in kidney tissues of Pkd1flox/−:Ksp-Cre polycystic compared with control mice was observed (Fig. 2a). Moreover, the administration of Cl-IB-MECA (0.2 mg/kg) significantly reduces the activity of CREB in Pkd1flox/−:-Ksp-Cre polycystic mice compared with those treated with vehicle (Fig. 2a), but no changes in kidney weight have been observed (Fig. 2b). This finding is not surprising given the aggressive phenotype of this ADPKD model that is characterized by massive renal cystogenesis leading to death in the perinatal life [18]. Moreover, a low “in vivo” efficacy of this compound could be hard to estimate by this ADPKD model.
Fig. 1.
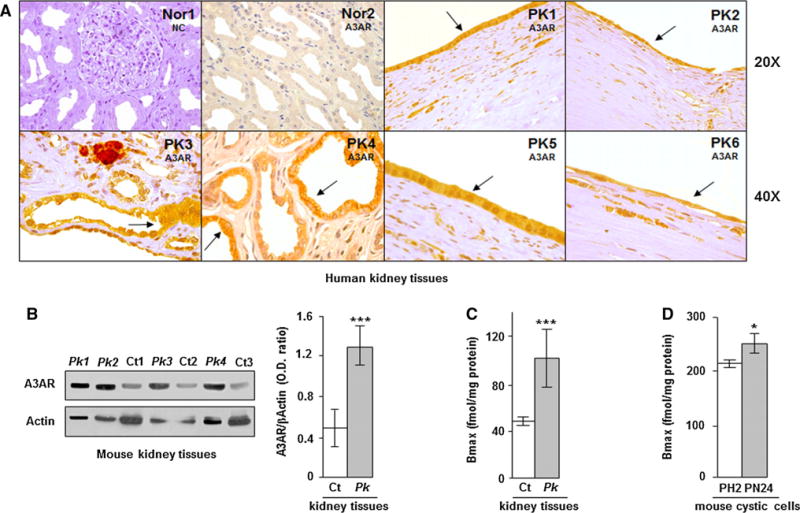
Analysis of A3AR by immunohistochemistry, western blotting, and saturation binding shows an increased expression of these receptors in renal cystic cells and tissues. a A3AR positivity detected by immunohistochemistry in human normal (Nor) and polycystic (PK) kidney tissues is mainly confined to flat cells of kidney cysts (see arrows); nc is the negative control (without primary antibody). b Western blot analysis shows increased levels of A3AR in kidney tissues of Pkd1flox/−:Ksp-Cre polycystic mice (Pk) compared with Pkd1flox/+:Ksp-Cre control mice (Ct). A3AR protein content was calculated as band intensity ratio between A3AR and β-Actin (Pk vs Ct: ***p < 0.001). Adenosine A3 receptors were also measured by saturation binding in cystic (Pk) and control (Ct) mouse kidney tissues (c) as well as in PN24 and PH2 mouse kidney cystic cells (d). A3 receptor density is higher in Pk tissue and PN24 cells than in Ct and PH2 (Pk vs Ct: ***p < 0.001 and PN24 vs PH2: *p < 0.05). Data are expressed as mean ± SD of at least three independent experiments
Fig. 2.
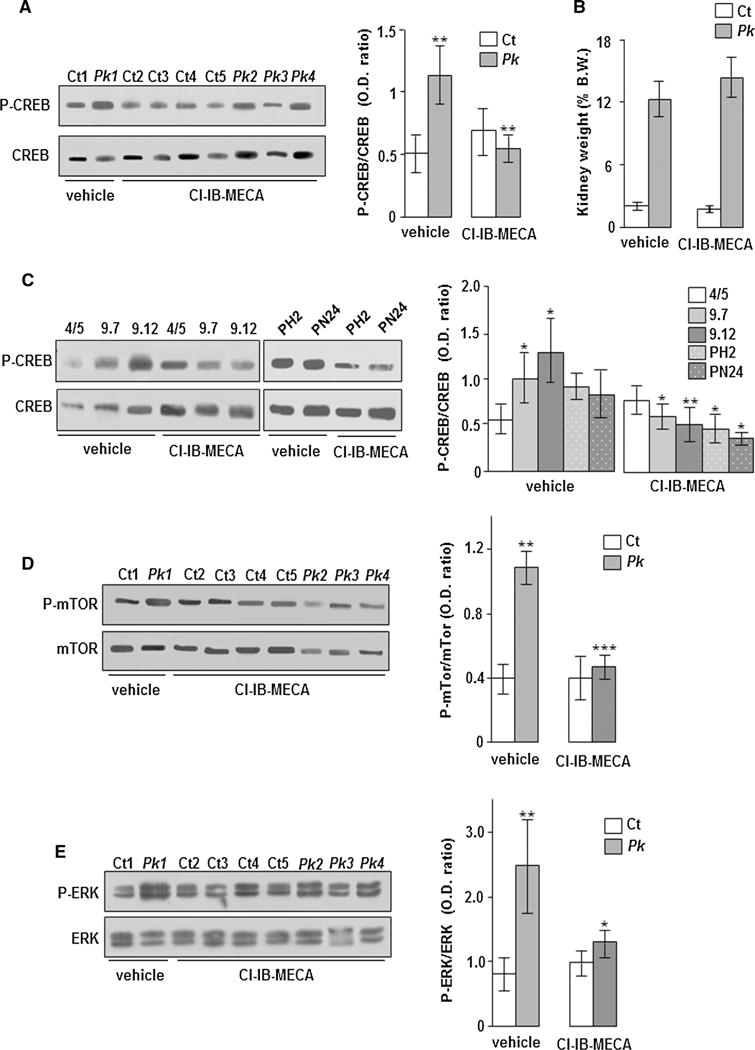
Analysis of CREB, mTOR, and ERK activity in kidney tissuesc and renal cells treated and untreated with Cl-IB-MECA. a In basal conditions (vehicle), CREB phosphorylation is greater in kidney tissues of Pk mice than in Ct mice (Pk mice vs Ct mice: **p < 0.01). The administration of Cl-IB-MECA (0.2 mg/kg) reduces CREB activity in kidney tissues of Pk mice (Pk mice treated with DMSO vs Pk mice treated with Cl-IB-MECA: **p < 0.01). b Cl-IB-MECA administration does not change the kidney weight percentage calculated as ratio between kidney- and total body-weight in both control (Ct) and polycystic (Pk) mice. c 9.7 and 9.12 human kidney cystic cells cultured in the presence of vehicle show an higher activity of CREB than 4/5 control kidney cells (9.7 and 9.12 cells vs 4/5 cells: *p < 0.05). The treatment with 100 nM Cl-IB-MECA causes a reduction of CREB activity in both 9.7 and 9.12 human cystic cells as well as in PH2 and PN24 mouse cystic cells compared with those treated with vehicle (9.7 cells treated with DMSO vs 9.7 cells treated with Cl-IB-MECA: *p < 0.05; 9.12 cells treated with DMSO vs 9.12 cells treated with Cl-IB-MECA: **p < 0.01; PH2 and PN24 cells treated with DMSO vs PH2 and PN24 cells treated with Cl-IB-MECA: *p < 0.05). d In basal conditions (vehicle), the activity of mTOR is higher in kidney tissues of Pk mice than in Ct mice (Pk mice vs Ct mice: **p < 0.01). The treatment with Cl-IB-MECA reduces mTOR phosphorylation in Pk mice compared with those treated with vehicle (Pk mice treated with DMSO vs Pk mice treated with Cl-IB-MECA: ***p < 0.001). e The activity of ERK is increased in kidney tissues of Pk mice compared with Ct mice (Pk mice vs Ct mice: **p < 0.01). The administration of Cl-IB-MECA decreases ERK phosphorylation in Pk mice compared with those treated with vehicle (Pk mice treated with DMSO vs Pk mice treated with Cl-IB-MECA: *p < 0.05). Bars show mean ± SD of at least three different experiments
The reduction of CREB activity by the Cl-IB-MECA treatment has also been observed in both human (9.7 and 9.12) and mouse (PH2 and PN24) kidney cystic cells, but not in human normal kidney cells (4/5) (Fig. 2c).
In different ADPKD mouse models also, mTOR and ERK signalling is inappropriately activated [13, 19, 20]. As expected, an increased mTOR and ERK activity in polycystic kidney tissues of Pkd1flox/−:Ksp-Cre mice compared with controls was found (Fig. 2d, e). Furthermore, an inhibition of both mTOR and ERK signalling pathways after the treatment with Cl-IB-MECA in human ADPKD cystic cells was reported [17]. Consistently, the administration of Cl-IB-MECA induces a marked decrease of mTOR and ERK phosphorylation in Pkd1flox/−:Ksp-Cre polycystic mice compared with those treated with vehicle, whilst any effect in Pkd1flox/+:Ksp-Cre control mice has been observed (Fig. 2d, e). Despite the early lethality of Pkd1flox/−:Ksp-Cre ADPKD model may be a limitation for the pharmacological study of this pathology, the treatment with Cl-IB-MECA causes a significant inhibition of CREB, mTOR, and ERK activity. Further studies using the other ADPKD models will be necessary to evaluate possible “in vivo” benefits of Cl-IB-MECA.
Cl-IB-MECA and rapamycin alone or in sequential combination inhibited cell proliferation in kidney cystic cell lines affecting mTOR, CREB, and ERK activity
Because the treatment with Cl-IB-MECA in the Pkd1flox/−:Ksp-Cre ADPKD model does not improve renal function, we have treated different ADPKD cell lines with both rapamycin and Cl-IB-MECA that target mTOR and cAMP pathways, respectively, to potentiate the inhibition of cell proliferation. As expected, the individual treatment with 500 nM rapamycin or 100 nM Cl-IB-MECA causes a significant inhibition of cell proliferation in both human (9.7 and 9.12) and mouse (PH2 and PN24) kidney cystic cells compared with those treated with vehicle (Fig. 3a, b). However, if the two compounds are simultaneously administrated, no reduction of cell growth in 9.7 and 9.12 as well as in PH2 and PN24 cells was observed (Fig. 3c, d). Conversely, the sequential treatment with these molecules (first rapamycin and subsequently Cl-IB-MECA) after 72 h of culture still reduces cell growth in all cystic cell lines (Fig. 3e, f). Moreover, the sequential application of these drugs significantly potentiates the inhibition of cell proliferation in human (9.7) cells as well as in mouse (PH2 and PN24) cells compared with the individual compound (Fig. 3e, f). Lower dosage as well as the sequential inversion of these compounds causes a milder inhibition of cell growth (data not shown). The simultaneous and sequential treatment with rapamycin and Cl-IB-MECA does not induce the formation of apoptotic nuclei, suggesting that apoptosis is not activated by the combination of these two drugs (supplementary Fig. 1). To clarify the different effects of rapamycin and Cl-IB-MECA on cell proliferation when used in simultaneous or in sequential combination, we have analyzed the activity of mTOR, CREB, and ERK kinases that are inhibited by these compounds [11, 15, 17]. As shown in Fig. 4a, the individual and simultaneous double treatment with rapamycin and Cl-IB-MECA causes a significant reduction in mTOR activity in both 9.7 and 9.12 human cystic cells, while the activity of CREB is inhibited by the individual treatment with Cl-IB-MECA only. ERK phosphorylation is reduced by the individual treatment with both rapamycin and Cl-IB-MECA, but not by the simultaneous application of these two compounds (Fig. 4a). Therefore, the simultaneous application of rapamycin and Cl-IB-MECA reverts the inhibition of CREB and ERK activity induced by Cl-IB-MECA alone in human ADPKD cells. This effect is not an exclusive of these two compounds, because it was also observed after combined treatment with Resveratrol and rapamycin in BV-2 cells activated with LPS [21]. In fact, this double treatment removes the inhibitory effect on both CREB and ERK kinases compared with Resveratrol individually used [21]. On the contrary, the sequential treatment with rapamycin and Cl-IB-MECA causes an overall inhibition of mTOR, CREB, and ERK activity (Fig. 4b). Similar findings are obtained in PH2 and PN24 mouse kidney cystic cells (supplementary Fig. 2). In particular, the reduction of cell proliferation by rapamycin and Cl-IB-MECA treatment (alone or in sequential combination) is always associated with the inhibition of ERK activity (Fig. 4; supplementary Fig. 2), suggesting an important role for these kinases in ADPKD cell growth. Conversely, CREB activity is not inhibited by the individual rapamycin treatment (middle part of Fig. 4a; supplementary Fig. 2a), but this compound causes a decrease of cell proliferation (Fig. 3a, b) likely by the inhibition of mTOR and ERK signalling (Fig. 4a; supplementary Fig. 2a). Thus, the activation of CREB is not sufficient to stimulate cell growth in ADPKD cells. Moreover, the simultaneous combination of rapamycin and Cl-IB-MECA in human 9.7 and 9.12 cystic cells that induces a decrease of mTOR phosphorylation (left part of Fig. 4a) does not affect cell proliferation (Fig. 3c). Therefore, the only reduction of mTOR activity seems unable to slow cell proliferation in human ADPKD cells. Similar results on cell growth were observed in mouse ADPKD cells (Fig. 3d), but differently from human cells, the simultaneous double treatment does not inhibit mTOR activity (supplementary Fig. 2a). However, we cannot exclude that the simultaneous combination of rapamycin and Cl-IB-MECA may also activate/inhibit other pathways affecting cell growth in ADPKD cells.
Fig. 3.
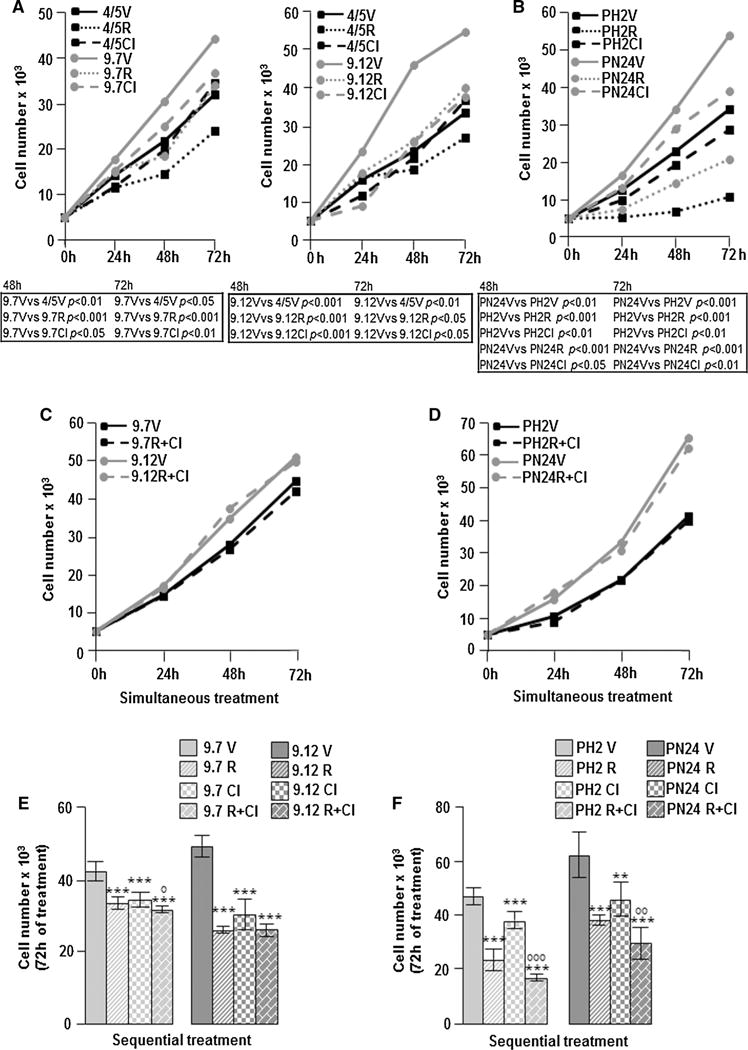
Cell proliferation analysis after treatment with rapamycin and ► Cl-IB-MECA alone or combination. Normal (4/5) and cystic (9.7 and 9.12) human kidney cells (a) as well as cystic (PH2 and PN24) mouse kidney cells (b) were cultured for 24, 48, and 72 h with 500 nM rapamycin (R) and 100 nM Cl-IB-MECA (Cl) alone or with vehicle (V). 9.7 and 9.12 cystic cells as well as mouse Pkd1(−/−) homozygous PN24 cystic cells treated with vehicle show an increased cell growth compared with 4/5 normal cells and mouse Pkd1(+/−) heterozygous PH2 cystic cells, respectively. The treatment with rapamycin and Cl-IB-MECA alone reduces cell growth in both human (9.7 and 9.12) and mouse (PH2 and PN24) cystic cells compared with those treated with vehicle. Statistical significance is shown in tables under the graphs. The simultaneous treatment with 500 nM rapamycin and 100 nM Cl-IB-MECA for 24, 48, and 72 h in 9.7 and 9.12 cystic cells (c) as well as in PH2 and PN24 cystic cells (d) removes the inhibitory effect on cell growth exerted by the individual compound. The sequential treatment before with 500 nM rapamycin for 24 h and after with 100 nM Cl-IB-MECA for a total time of 72 h reduces cell growth in 9.7 and 9.12 human cystic cells (e) as well as in PH2 and PN24 mouse cystic cells (f) compared with those treated with vehicle (9.7 V vs 9.7R, 9.7Cl and 9.7R + Cl: ***p < 0.001; 9.12 V vs 9.12R, 9.12Cl and 9.12R + Cl: ***p < 0.001; PH2 V vs PH2R, PH2Cl and PH2R + Cl: ***p < 0.001; PN24 V vs PN24R and PN24R + Cl: ***p < 0.001; PN24 V vs PN24Cl: **p < 0.01).The sequential treatment potentiates the inhibition of cell growth compared with rapamycin or Cl-IB-MECA alone in human 9.7 cystic cells as well as in mouse PH2 and PN24 cystic cells (9.7R + Cl vs 9.7R or 9.7 Cl: °p < 0.05; PH2R + Cl vs PH2R or PH2Cl: °°°p < 0.001; PN24R + Cl vs PN24R or PN24 Cl: °°p < 0.01). Data are expressed as mean ± SD of at least three different experiments in duplicate
Fig. 4.
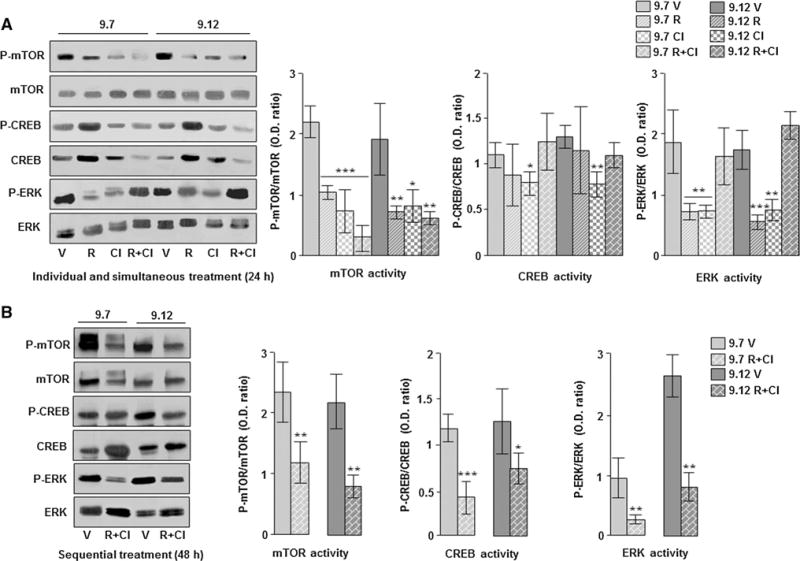
Analysis of mTOR, CREB, and ERK activity in human ADPKD renal cells treated and untreated with rapamycin and Cl-IB-MECA alone or in combination. a 9.7 and 9.12 cystic cells were treated for 24 h with 500 nM rapamycin and 100 nM Cl-IB-MECA alone or in combination and analyzed for the activity of mTOR, CREB, and ERK by western blotting. The treatment with rapamycin and Cl-IB-MECA alone or in simultaneous combination reduces the activity of mTOR in 9.7 and 9.12 cells compared with those treated with vehicle (9.7 V vs 9.7R, 9.7Cl and 9.7R + Cl: ***p < 0.001; 9.12 V vs 9.12R and 9.12R + Cl: **p < 0.01; 9.12 V vs 9.12Cl: *p < 0.05). In 9.7 and 9.12 cells, CREB phosphorylation is reduced by the treatment with Cl-IB-MECA only (9.7 V vs 9.7Cl and 9.12 V vs 9.12Cl: *p < 0.05 and **p < 0.01, respectively). ERK phosphorylation is decreased by treatment with rapamycin and Cl-IB-MECA alone, but not by the simultaneous application of these compounds in 9.7 and 9.12 cells compared with the same cells treated with vehicle (9.7 V vs 9.7R and 9.7Cl: **p < 0.01; 9.12 V vs 9.12R: ***p < 0.001; 9.12 V vs 9.12Cl: **p < 0.01). b 9.7 and 9.12 cells were treated sequentially before with 500 nM rapamycin for 24 h and after with 100 nM Cl-IB-MECA for further 24 h and analyzed by western blotting. This combined treatment causes a reduction of mTOR, CREB, and ERK phosphorylation in 9.7 and 9.12 cells compared with those treated with vehicle (for mTOR: 9.7 V vs 9.7R + Cl and 9.12 V vs 9.12R + Cl: **p < 0.01; for CREB: 9.7 V vs 9.7R + Cl: ***p < 0.001; 9.12 V vs 9.12R + Cl: *p < 0.05; for ERK: 9.7 V vs 9.7R + Cl and 9.12 V vs 9.12R + Cl: **p < 0.01). V vehicle (DMSO), R rapamycin, and Cl Cl-IB-MECA. Data are expressed as mean ± SD of at least three different experiments
Taken together, these findings suggest that the regulation of cell growth in human ADPKD cells may occur in an ERK-dependent manner. Consistently, it is well known that the abnormal ERK activation is associated with the increased cell proliferation of ADPKD cystic cells [22], as well as the inhibition of these kinases ameliorates renal function in mice with polycystic kidney disease [23].
In conclusion, data here reported suggest that the double sequential treatment with rapamycin and Cl-IB-MECA is able to potentiate the inhibition of cell proliferation by the reduction of mTOR, CREB, and ERK signalling in ADPKD cells, suggesting that the use of combined treatments might be more efficient than the individual compound. Consistently, it has been reported that the double treatment with Tolvaptan and Pasireotide, adenylyl cyclase 6 inhibitors, induces an additive beneficial effect on renal function in a hypomorphic Pkd1 model [24]. Moreover, the double treatment with rapamycin and metformin in renal cells is more effective for inhibiting mTOR activity than molecules used alone [25].
Therefore, the combined inhibition of different signalling pathways could open new horizons for the treatment of ADPKD.
Acknowledgments
We greatly thanks Prof. PC Harris, (Mayo Clinic; Rochester, USA) for providing human normal and cystic kidney epithelial cells.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10157-016-1289-1) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors have no conflicts of interest to disclose.
References
- 1.Cornec-Le Gall E, et al. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat. 2014;35:1393–1406. doi: 10.1002/humu.22708. [DOI] [PubMed] [Google Scholar]
- 2.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. 2014;124:2315–24. doi: 10.1172/JCI72272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serra AL, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–9. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 4.Walz G, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–40. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 5.Torres VE, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–18. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caroli A, et al. ALADIN study group: effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013;382:1485–95. doi: 10.1016/S0140-6736(13)61407-5. [DOI] [PubMed] [Google Scholar]
- 7.Riella C, Czarnecki PG, Steinman TI. Therapeutic advances in the treatment of polycystic kidney disease. Nephron Clin Pract. 2014;128:297–302. doi: 10.1159/000368244. [DOI] [PubMed] [Google Scholar]
- 8.Muto S, et al. The effect of tolvaptan on autosomal dominant polycystic kidney disease patients: a subgroup analysis of the Japanese patient subset from TEMPO 3:4 trial. Clin Exp Nephrol. 2015;19:867–77. doi: 10.1007/s10157-015-1086-2. [DOI] [PubMed] [Google Scholar]
- 9.Erickson KF, Chertow GM, Goldhaber-Fiebert JD. Cost-effectiveness of tolvaptan in autosomal dominant polycystic kidney disease. Ann Intern Med. 2013;17:382–9. doi: 10.7326/0003-4819-159-6-201309170-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brunelli SM, et al. End-stage renal disease in autosomal dominant polycystic kidney disease: a comparison of dialysis-related utilization and costs with other chronic kidney diseases. Clinicoecon Outcomes Res. 2015;7:65–72. doi: 10.2147/CEOR.S76269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguiari G, Catizone L, Del Senno L. Multidrug therapy for polycystic kidney disease: a review and perspective. Am J Nephrol. 2013;37:175–82. doi: 10.1159/000346812. [DOI] [PubMed] [Google Scholar]
- 12.Loghman-Adham M, et al. Immortalized epithelial cells from human autosomal polycystic kidney cysts. Am J Physiol Renal Physiol. 2003;285:F397–412. doi: 10.1152/ajprenal.00310.2002. [DOI] [PubMed] [Google Scholar]
- 13.Shibazaki S, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17(11):1505–16. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wodarczyk C, et al. Nephrocystin-1 forms a complex with polycystin-1 via a polyproline motif/SH3 domain interaction and regulates the apoptotic response in mammals. PLoS One. 2010;5:e12719. doi: 10.1371/journal.pone.0012719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguiari G, et al. Polycystin-1 regulates amphiregulin expression through CREB and AP1 signalling: implications in ADPKD cell proliferation. J Mol Med (Berl) 2012;90:1267–82. doi: 10.1007/s00109-012-0902-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varani K, et al. Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:398–406. doi: 10.1164/rccm.200506-869OC. [DOI] [PubMed] [Google Scholar]
- 17.Aguiari G, et al. Deficiency of polycystic kidney disease-1 gene (PKD1) expression increases A(3) adenosine receptors in human renal cells: implications for cAMP-dependent signalling and proliferation of PKD1-mutated cystic cells. Biochim Biophys Acta. 2009;1792:531–40. doi: 10.1016/j.bbadis.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Leuenroth SJ, et al. Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol. 2008;19:1659–62. doi: 10.1681/ASN.2008030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shillingford JM, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466–71. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonhard WN, et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. Am J Physiol Renal Physiol. 2011;300:F1193–202. doi: 10.1152/ajprenal.00419.2010. [DOI] [PubMed] [Google Scholar]
- 21.Zhong LM, et al. Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS One. 2012;7(2):e32195. doi: 10.1371/journal.pone.0032195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi T, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–94. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 23.Omori S, et al. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17(6):1604–14. doi: 10.1681/ASN.2004090800. [DOI] [PubMed] [Google Scholar]
- 24.Hopp K, et al. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J Am Soc Nephrol. 2015;26:39–47. doi: 10.1681/ASN.2013121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mekahli D, et al. Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflugers Arch. 2014;466:1591–604. doi: 10.1007/s00424-013-1394-x. [DOI] [PubMed] [Google Scholar]