

Abstract
Tumors can be depicted as wounds that never heal, and are infiltrated by a large array of inflammatory and immune cells. Tumor‐associated chronic inflammation is a hallmark of cancer that fosters progression to a metastatic stage, as has been extensively reviewed lately. Indeed, inflammatory cells persisting in the tumor establish a cross‐talk with tumor cells that may result in a phenotype switch into tumor‐supporting cells. This has been particularly well described for macrophages and is referred to as tumor‐associated ‘M2’ polarization. Epithelial‐to‐mesenchymal transition (EMT), the embryonic program that loosens cell–cell adherence complexes and endows cells with enhanced migratory and invasive properties, can be co‐opted by cancer cells during metastatic progression. Cancer cells that have undergone EMT are more aggressive, displaying increased invasiveness, stem‐like features, and resistance to apoptosis. EMT programs can also stimulate the production of proinflammatory factors by cancer cells. Conversely, inflammation is a potent inducer of EMT in tumors. Therefore, the two phenomena may sustain each other, in an alliance for metastasis. This is the focus of this review, where the interconnections between EMT programs and cellular and molecular actors of inflammation are described. We also recapitulate data linking the EMT/inflammation axis to metastasis.
Keywords: cancer, chemokines, cytokines, epithelial‐to‐mesenchymal transition, inflammation, metastasis
Abbreviations
- CAFs
cancer‐associated fibroblasts
- CTCs
circulating tumor cells
- DCregs
regulatory dendritic cells
- EMT
epithelial‐to‐mesenchymal transition
- EMT‐TF
EMT transcription factor
- MMPs
matrix metalloproteinases
- TAMs
tumor‐associated macrophages
- Tregs
regulatory T cells
1. Introduction
While acute, transitory inflammation is an essential actor of tissue damage control and repair, tumor‐associated inflammation – which occurs in virtually all tumors – is of a chronic, unresolved type (Pesic and Greten, 2016) that fosters tumor progression. During tumorigenesis, cancer cells, innate immune cells [such as dendritic cells or tumor‐associated macrophages (TAMs)] and activated resident cells [such as cancer‐associated fibroblasts (CAFs) or endothelial cells] produce a variety of cytokines and chemokines in response to the danger signals originating from the tumor. These soluble factors drive the recruitment of massive amounts of additional bone marrow‐derived innate immune cells, which fuel the so‐called cytokine storm (Crusz and Balkwill, 2015). This prolonged reaction favors tumor cell survival and proliferation, immunosuppression (by the inhibition of effector immune cells and the accumulation of myeloid suppressive cells) and angiogenesis (Becht et al., 2016). Promisingly, multiple anti‐inflammatory agents are under development and/or under clinical testing currently in chemoprevention trials (Crusz and Balkwill, 2015).
The understanding that tumor cells are genetically and phenotypically very heterogeneous has further stimulated studies aiming at deciphering how different tumor cell phenotypes may relate to tumor‐associated inflammation. During the metastatic progression of epithelial tumors, tumor cells indeed undergo phenotypic changes, essentially driven by environmental stimuli, allowing the tumor cells to adapt to the different microenvironment encountered (adjacent stroma, blood, or colonized organs). Epithelial‐to‐mesenchymal transition (EMT) appears today as a major actor modulating these phenotypic conversions. Although two recent studies (Fischer et al., 2015; Zheng et al., 2015) have revived the debate about the universal requirement of EMT in the metastasis process, the current dogma is that EMT processes might be involved in the initial steps of the metastatic cascade, including tumor invasion, intravasation, and micrometastases formation. This has been supported by multiple in vitro and in vivo functional data, as well as by correlative data in human samples (Diepenbruck and Christofori, 2016). The acquisition of EMT‐like changes in tumor cells has been extensively studied and implies increased invasive properties, resistance to DNA damage‐ and chemotherapy‐induced apoptosis, immunosuppression, and the acquisition of stem‐like features. EMT transcriptomic signatures are found highly associated with groups of patients with poorer outcome in multiple cancer entities including breast cancer (Jang et al., 2015), colorectal cancer (Roepman et al., 2014), head and neck cancer (Pan et al., 2016), or malignant pleural mesothelioma (de Reynies et al., 2014). EMT‐associated signaling pathways have lately been considered as therapeutic targets, as recently shown in a murine pancreatic cancer model (Subramani et al., 2016). In this study, metastasis was successfully hampered by the use of nimbolide, a drug that, among other effects, reduced EMT via the induction of excessive production of reactive oxygen species (ROS). EMT signaling pathways have also been targeted in breast cancer in vitro models (Yu et al., 2016), or even in clinical settings (Marcucci et al., 2016).
Increasing literature data have thus emphasized a link between cancer‐associated EMT and chronic inflammation. Indeed, inflammatory mediators (soluble factors, oxidative stress, or hypoxia, for example) can foster the acquisition of EMT‐like features in cancer cells (Lopez‐Novoa and Nieto, 2009) (Table 1). Conversely, these cancer cells can produce a higher amount of proinflammatory mediators, such as cytokines, chemokines, and matrix metalloproteinases (MMPs), which fuel the cancer‐related, smoldering inflammation. In this review, we highlight most recent data including in vitro and in vivo models describing the connections between EMT signaling pathways and (i) innate immune cells and (ii) soluble mediators of inflammation (inflammatory cytokines and chemokines). Finally, we present some in vivo functional studies and human correlative data, associating EMT and inflammation with tumor progression and metastasis.
Table 1.
Soluble actors of inflammation and their described effect on EMT activation in cancer cells in vitro
| Soluble factors | Cellular origin | Evidence of EMT pathway activation in cancer cells | Cell line origin | References |
|---|---|---|---|---|
| TNF‐α | Pleiotropic (mostly macrophages) |
Increased invasive properties E‐cadherin drop N‐cadherin and vimentin expression |
Hepatocellular carcinoma | Zhu et al. (2016) |
|
Slug and ZEB1 expression Increased cell migration E‐cadherin drop |
Renal cell carcinoma | Sun et al. (2016) | ||
|
Increased invasion and migration E‐cadherin drop Vimentin and N‐cadherin expression |
Papillary thyroid cancer | Lv et al. (2015) | ||
| Altered mRNA expression of Snail1, ZEB1, E‐cadherin, fibronectin, vimentin, TGM2 | Breast cancer | Cohen et al. (2015), Elghonaimy et al. (2016) | ||
|
E‐cadherin drop Vimentin expression Increased migration |
Colorectal cancer | Bates and Mercurio (2003), Bhat et al. (2016) | ||
| TGF‐β | Ubiquitous |
Elongated cell shape Low E‐to‐N‐cadherin ratio, vimentin expression Smad‐dependent signaling Smad‐independent signaling |
Breast cancer | Johansson et al. (2015), Moustakas and Heldin (2016), Pang et al. (2016) |
|
Increased migration and invasion E‐cadherin drop N‐cadherin expression Snail1, Snail2, and ZEB1 gene expression |
Lung cancer | Wu et al. (2016b) | ||
|
Morphological changes E‐cadherin drop Vimentin expression Gene and protein expression of EPCAM (and stemness markers) Invasion and migration |
Hepatocellular carcinoma | Malfettone et al. (2017) | ||
| IL‐1β | Macrophages, tumor cells |
ZEB1 expression E‐cadherin drop Morphological changes Increased invasiveness |
Colorectal cancer | Li et al. (2012b) |
|
Snail1 and Slug expression Vimentin E‐cadherin drop Increased migration |
Oral cancer (cancer cell lines and dysplastic oral keratinocytes) | Lee et al. (2015) | ||
| IL‐6 | T cells, macrophages, tumor cells |
Morphological changes JAK2/STAT3/Snail1 pathway activation E‐cadherin drop Vimentin expression Increased migration |
Head and neck cancer | Yadav et al. (2011) |
|
JAK2/STAT3/Twist1 activation E‐cadherin drop, Vimentin, N‐cadherin, and fibronectin expression |
Breast cancer | Kim et al. (2016), Sullivan et al. (2009) | ||
|
Altered mRNA expression of vimentin, Snail1, Slug, and ZEB1 Increased invasive properties |
Colorectal cancer | Rokavec et al. (2014) | ||
|
Multiple markers (E‐cadherin, N‐cadherin, Twist, vimentin, MMP9, VEGF, TGF‐β) Increased migratory properties |
NSCLC | Lee et al. (2016) | ||
| IL‐8 | T cells, macrophages, tumor cells |
ZEB1, Snail1, and Slug activation Increased migratory properties |
Thyroid carcinoma | Visciano et al. (2015) |
|
JAK2/STAT3/Snail1 activation E‐cadherin drop N‐cadherin expression Increased migratory properties |
Hepatocellular carcinoma | Fu et al. (2015) | ||
| E‐cadherin gene repression | Nasopharyngeal carcinoma | Zhang et al. (2016e) | ||
|
Morphological chances Increased migratory and invasive properties E‐cadherin drop N‐cadherin, vimentin, fibronectin expression |
Breast cancer | Ji et al. (2016) | ||
|
E‐cadherin drop Vimentin and fibronectin expression Increased invasive properties |
NSCLC | Fernando et al. (2016) | ||
|
E‐cadherin drop Increased migratory properties |
Ovarian cancer | Yin et al. (2015) | ||
| CCL2 | Monocytes, macrophages, dendritic cells |
Only studied in combination with IL‐6 Twist expression Increased migration E‐cadherin drop Vimentin and fibronectin expression |
NSCLC | Chen et al. (2015b) |
|
E‐cadherin drop, increased Snail1 expression STAT3 activation MMP9 expression Increased migration |
Prostate cancer | Izumi et al. (2013) | ||
|
Increased invasion and migration E‐cadherin/vimentin switch, increased N‐cadherin Increased Snail1 and Twist gene expression Increased MMP9 expression |
Bladder cancer | Rao et al. (2016) | ||
| CCL5 | Cancer stem cells, exhausted T cells, adipocytes |
E‐cadherin drop Expression of vimentin, Snail1, Slug Increased invasive properties |
Ovarian cancer | Long et al. (2015) |
|
E‐cadherin drop Expression of vimentin, Snail1 |
Colorectal cancer | Halama et al. (2016) | ||
| Migratory and invasive properties only | Triple‐negative breast cancer | D'Esposito et al. (2016) | ||
| CCL18 | Macrophages, tumor cells | Migratory and invasive properties only | Ovarian cancer | Wang et al. (2016) |
|
E‐cadherin drop, Vimentin expression Migratory and invasive properties |
Breast cancer | Su et al. (2014) | ||
|
Increased invasive properties Snail1 gene expression |
Pancreatic cancer | Meng et al. (2015) | ||
| CCL20 (+IL‐8) | Lymphocytes |
Morphological changes E‐cadherin drop Vimentin expression Increased migration |
Colorectal cancer | Cheng et al. (2014), Kapur et al. (2016) |
| CCL21 | Lymphocytes |
ERK1/2/NF‐κB signaling Increased invasive properties E‐cadherin drop N‐cadherin and MMP9 expression |
Pancreatic cancer | Zhang et al. (2016c) |
| IL‐23 | Th17 lymphocytes |
Wnt/β‐catenin pathway activation, GSK3β stabilization Snail1 and Slug gene expression E‐cadherin/vimentin switch |
Esophageal cancer | Chen et al. (2015) |
| IL‐17 | Th17 lymphocytes |
Snail1 and Slug expression E‐cadherin/vimentin switch |
Prostate cancer | Zhang et al. (2016d) |
|
E‐cadherin/vimentin switch Increased invasive properties |
Gastric cancer | Jiang et al. (2017) |
2. EMT and the cellular actors of inflammation
Inflammatory cells in a tumor can be of multiple origins. These cells comprise activated resident cells, such as endothelial cells or CAFs or resident macrophages (TAMs) or dendritic cells. In addition, the tumor milieu recruits bone marrow‐derived cells, mostly neutrophils, macrophages, and immature, immunosuppressive myeloid cells called myeloid‐derived suppressive cells (MDSCs). In this section, we review the interconnection between EMT and recruitment and/or polarization of such cells.
2.1. EMT and myeloid cells
2.1.1. TAMs
Among immune cells infiltrating solid tumors, TAMs are most likely the most abundant cell type as they make up to 50% of the tumor mass (Solinas et al., 2009). Even though macrophages should be able to kill tumor cells, provided they get the appropriate activation signals, the chronically inflamed/immunosuppressive microenvironment most often polarizes TAMs into tumor‐supporting cells (schematically categorized in ‘M2‐like macrophages’) that promote extracellular matrix remodeling, angiogenesis, immunosuppression, and foster the acquisition of invasive properties by cancer cells by secreting various soluble factors (Becht et al., 2016).
The global numbers of TAMs have been correlated with EMT‐like features in several cancer entities. This has been reported in a cohort of 178 patients with gastric cancer, where CD68‐positive cell density is associated with the expression of EMT features in cancer cells (E‐cadherin loss and vimentin de novo expression). This was also demonstrated in an independent study using another marker of TAMs, CD163. In that study, high intratumoral CD163 expression was found to be correlated with E‐cadherin loss (Yan et al., 2016). Global density of macrophages also relates to EMT features in non‐small‐cell lung cancer carcinoma (NSCLC) (Bonde et al., 2012) or head and neck cancer (Hu et al., 2016).
Spatial association of cancer cells with EMT features and macrophages has also been described in a murine model of terato‐carcinoma. Tumor areas rich in macrophages indeed contain more tumor cells displaying E‐cadherin loss, β‐catenin accumulation and fibronectin expression compared to areas poor in macrophages (Bonde et al., 2012). Similarly, such a spatial clustering has been revealed in a human cohort of hepatocellular carcinoma (Fu et al., 2015).
Functionally, TAMs have been described as potent EMT inducers in numerous independent studies. TAMs accordingly produce multiple growth factors (HGF, EGF, TGF, PDGF, etc.) and inflammatory cytokines (IL‐1β, IL‐6, and TNF‐α) that each can induce EMT in cancer cells (Fig. 1). In vitro data from pancreatic cancer cell lines have demonstrated that co‐culture of cancer cells with M2‐polarized macrophages is able to foster the acquisition of EMT‐like properties in cancer cells, including spindle‐shaped morphology, decreased E‐cadherin and increased vimentin expression, invasive properties, and enhanced production of MMPs (Liu et al., 2013). Mechanistically, the TLR4/IL‐10 axis has been involved in this process and inhibition of either effectively represses EMT induction. M2‐TAM‐induced EMT has also been reported in co‐cultures of mouse cancer cells in a murine model of terato‐carcinoma. Interestingly, depletion of macrophages using clodronate liposomes in these mice decreases the expression of mesenchymal features in primary tumors (Bonde et al., 2012).
Figure 1.
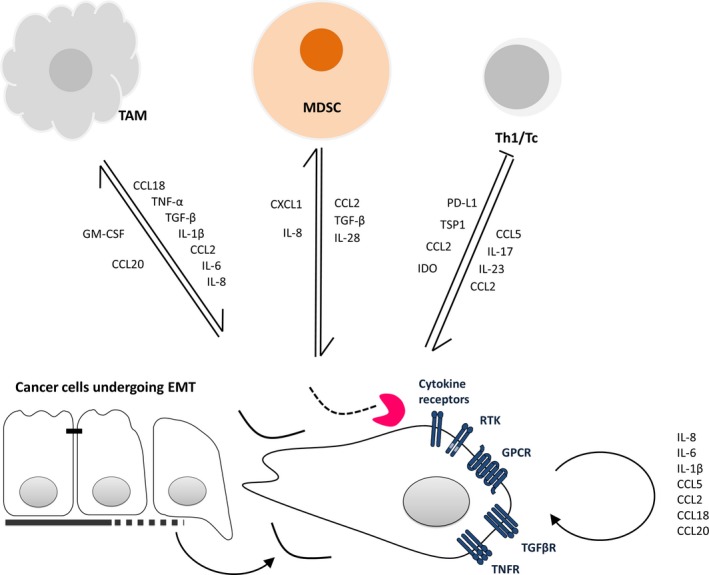
Schematic representation of the soluble factor‐mediated interactions between cancer cells undergoing EMT‐like changes and innate immune cells. RTK, receptor tyrosine kinase; GPCR, G‐protein‐coupled receptor; TAM, tumor‐associated macrophages; MDSCs, myeloid‐derived suppressor cells; Th1/Tc, T helper one cell/cytotoxic T cell. The pink Pacman‐shaped symbol represents some metalloproteinase degrading the extracellular matrix.
In hepatocellular carcinoma, macrophages induce EMT in cancer cells in co‐culture experiments, in an IL‐8‐dependent fashion (Fu et al., 2015) or in a TGF‐β‐dependent fashion (Deng et al., 2016; Fan et al., 2014). In other tumor types such as NSCLC, macrophages are known to induce EMT via EGF (Ravi et al., 2016).
On the other hand, the EMT program in cancer cells might facilitate polarization of TAM into M2‐like, tumor‐supporting cells. Indeed, conditioned media from mesenchymal‐like breast cancer cell lines activate macrophages into a TAM‐like phenotype, including high expression of CD206 (a scavenger receptor also known as macrophage mannose receptor or MMR) and high secretion of CCL17, CCL18, CCL22, and IL‐10 (Su et al., 2014). This effect is mediated by the production of GM‐CSF, which has accordingly been associated with EMT in breast cancer (Su et al., 2014; Suarez‐Carmona et al., 2015). GM‐CSF‐activated macrophages in turn induce EMT in breast cancer cells, in a CCL18‐dependent mechanism (Su et al., 2014), establishing a potential local feedback regulation (Fig. 1).
2.1.2. Other myeloid cells
In a transgenic murine model of melanoma (the RETAAD mice, expressing the activated RET oncogene), MDSCs preferentially infiltrated the primary tumor in comparison with metastases. Mostly recruited following a CXCL5 gradient (whose human orthologues, CXCL1 and IL‐8, are expressed by cancer cells), these immunosuppressive cells could in turn promote EMT‐like changes in cancer cells, via multiple pathways comprising TGF, EGF, and HGF (Fig. 1). Most interestingly, depletion of MDSCs using an anti‐Ly6G antibody led to a decrease in EMT marker expression (namely vimentin and S100A4/FSP1) (Toh et al., 2011).
A couple of years ago, we observed that conditioned media from cancer cells induced for EMT could recruit CD11b+ GR1+ myeloid cells in vivo in a murine model. Similarly, in a cohort of 40 triple‐negative breast cancer samples, tumors infiltrated by high numbers of CD33‐positive myeloid cells were more susceptible to display EMT features (as measured by vimentin expression in above 10% of the tumor cells) (Suarez‐Carmona et al., 2015). Accordingly, in a cohort of 97 high‐grade breast cancer patients, a high extracellular matrix‐related gene expression profile (‘ECM3’, characterized by high SPARC and collagen expression) correlates with a high expression of EMT transcription factor (EMT‐TF) Twist1 and high numbers of infiltrating CD33‐positive myeloid cells (Sangaletti et al., 2016). Whether these myeloid cells are mature (neutrophils) or immature (MDSCs) cells is still unclear, because of a marker overlap both in mice (CD11b‐Ly6G stain neutrophils and their progenitors) and in humans (CD33 stains the whole myeloid lineage) (Ugel et al., 2015). A way to discriminate between immature myeloid cells and neutrophils in human immunohistochemistry studies could be to use a stain for myeloperoxidase (MPO), used in routine to detect finally activated neutrophils, or other markers of differentiated neutrophils, such as CD66b. We mention hereafter two studies in which specifically mature neutrophils were involved in the acquisition of EMT traits by cancer cells. In the first study, the density of infiltrating neutrophils was studied in pancreatic cancer biopsies using a staining for elastase, presumably expressed by differentiated cells (Grosse‐Steffen et al., 2012). As a result, high density of neutrophils was related to the nuclear accumulation of beta‐catenin and ZEB1 in tumors. These findings were not reproducible in hepatocellular carcinoma. In addition, co‐culture of neutrophils induced EMT in cancer cells in vitro. The mechanism involved was a disruption of E‐cadherin‐mediated cell–cell adhesion by neutrophil‐derived elastase (Grosse‐Steffen et al., 2012). In another study, neutrophils were detected using CD66b in lung adenocarcinoma specimens. CD66b is expressed by activated neutrophils and eosinophils. The density of intratumoral CD66b was inversely correlated with E‐cadherin expression (Hu et al., 2015). Consistently with the study mentioned above (Grosse‐Steffen et al., 2012), co‐culture between neutrophils and lung cancer cell lines resulted in EMT trait acquisition and increased migration, in a process requiring TGF‐β this time (Hu et al., 2015).
The important functional role of myeloid cells, particularly immunosuppressive immature myeloid cells (MDSCs), in inducing EMT has been illustrated in an elegant study using high‐grade breast cancers (Sangaletti et al., 2016). Ectopic expression of SPARC in high‐grade breast cancer cells leads to the acquisition of EMT features in vivo but not in vitro, highlighting the role of the microenvironment in this process. Tumors formed by cancer cells ectopically expressing SPARC, in addition to displaying EMT‐like features, produce more metastases and are infiltrated by many myeloid cells (CD11b+GR1+) with strong immunosuppressive functions (as measured by their ability to inhibit anti‐CD3 and anti‐CD28‐induced T‐cell proliferation in vitro). Inhibition of these suppressive functions using zoledronic acid effectively reverses EMT in tumors in vivo. Moreover, zoledronic acid‐treated, SPARC‐expressing tumor cells have a higher mitotic index and are accordingly more sensitive to Doxil, compared to their untreated counterparts (Sangaletti et al., 2016). Supportively, an independent, veterinary group has reported induction of EMT‐like changes in vitro by co‐culturing canine tumor cells with MDSCs. This occurs in an IL‐28/IL‐28R‐dependent way (Mucha et al., 2014) (Fig. 1).
2.2. EMT and adaptive immune cells
Beyond MDSCs, there is a strong connection between EMT and chronic inflammation‐associated immunosuppression in cancer progression (Chen et al., 2014, 2015a). This immunosuppression involves regulatory dendritic cells (DCregs), regulatory T cells (Tregs), and effector T‐cell exhaustion by the expression of immune checkpoints.
In mice bearing NSCLC, the immune checkpoint PD‐L1 is detected on tumor cells expressing the EMT‐TF ZEB1. PD‐L1 binds its receptor PD‐1 at the surface of T cells and this is associated with T‐cell dysfunction, decreased tumor‐infiltrating lymphocyte (TIL) density, and metastasis outburst (Chen et al., 2014). This link between EMT and immunosuppression seems to be conserved in humans as the authors could identify, using two independent gene expression data sets, that EMT gene signature in lung adenocarcinoma is related to the upregulated expression of multiple immune checkpoints (PD‐1/PD‐L1, CTLA‐A, TIM‐3, and others), to high Foxp3‐positive Treg cell density, to immunosuppressive cytokine production (TGF‐β, IL‐10, and IL‐6), and finally, to a strong inflammatory reaction (indeed, CD4+ Treg cells produce high amounts of proinflammatory cytokines such as IL‐10, TNF‐α, or IL‐6) (Chen et al., 2014). This has been recently confirmed in human cancer. Breast cancer cells undergoing EMT indeed express PD‐L1 in a miR‐200‐ and ZEB1‐dependent fashion (Noman et al., 2017).
In mouse melanoma xenografts, Snail1 transfectants (which promote Treg expansion in vitro) recruit both Tregs and DCregs in vivo. The latter would depend on melanoma cell‐derived CCL2 (Kudo‐Saito et al., 2013). Local immunosuppression and resistance to DC immunotherapy is suppressed by intratumoral injection of an anti‐Snail1 siRNA, or by anti‐TSP1 (thrombospondin 1) treatment (Kudo‐Saito et al., 2009), underlining the central role of EMT programs in cancer progression in this model.
Indoleamine 2,3 dioxygenase (IDO) is mainly a tumor cell‐derived, inflammation‐induced, immunosuppressive factor (Muller et al., 2008; Uyttenhove et al., 2003) that promotes tumor progression. Ricciardi et al. (2015) have recently reported a decreased viability and/or proliferation of natural killer (NK) cells and of T cells after co‐culture with cancer cell lines in which EMT had been induced. This effect was triggered by an IFN‐γ‐mediated IDO overexpression in cancer cells and was blocked by co‐culture in the presence of the IDO inhibitor L‐1MT (Ricciardi et al., 2015) (Fig. 1).
Finally, in hepatocellular carcinoma cell lines, hypoxia or HIF‐1α expression triggers EMT‐like changes including E‐cadherin loss and morphological modifications. Simultaneously, HIF‐1α binds to a HRE (hypoxia response element) on the CCL20 promoter and induces CCL20 expression by cancer cells (Ye et al., 2016). Hypoxic tumor cell‐derived CCL20 induced IDO expression in macrophages, which leads to decreased T‐cell proliferation and increased Foxp3+ Treg cell proportions after co‐culture (Ye et al., 2016).
3. EMT and soluble factors of inflammation
Further supporting the relationship between the cellular actors of inflammation and EMT detailed above, extensive literature has also linked EMT and soluble mediators of inflammation. As we review hereafter, several of these soluble mediators, mostly cytokines and chemokines, have been shown to trigger EMT in various cancer cell types. Conversely, EMT‐derived tumor cells have also been shown to overproduce many proinflammatory mediators, thereby establishing a regulatory loop that may contribute to maintain both the EMT phenotype and the proinflammatory context.
It is important to consider that tumor‐associated EMTs rarely involve a complete lineage switching but rather generate intermediate states (hybrid phenotypes) with varying degrees of aggressiveness that distribute along the epithelial‐to‐mesenchymal differentiation spectrum. Correspondingly, there is a multiplicity of EMT molecular repertoires involved in the generation of these hybrid phenotypes. As detailed here under and summarized in Table 1, the extent of EMT induction by specific proinflammatory mediators and the molecular EMT actors affected accordingly vary greatly. If specific responses may be triggered by specific proinflammatory mediators, several EMT‐related changes are commonly induced by several mediators. These common modifications include – but are not limited to – a diminution of E‐cadherin, an induction of vimentin and other mesenchymal markers (FSP1, N‐cadherin, MMP9), an expression of EMT‐TFs, the acquisition of enhanced migratory and invasive properties.
Below, we review in vitro‐based studies that identified relationships and regulatory loops linking EMT and specific well‐known proinflammatory mediators. Animal and clinical studies that functionally involve these EMT/cytokine regulatory loops in the metastatic progression are discussed in a separate chapter (Section 4).
3.1. TGF‐β
TGF‐β is a key factor of cancer‐related inflammation, but its effects on immune cells are very broad. TGF‐β promotes chemotaxis of eosinophils, macrophages, and mast cells. It skews macrophage and neutrophil polarization into tumor‐promoting, immunosuppressive cells. TGF‐β also negatively impacts the antitumor immune response by multiple mechanisms (inhibition of antigen‐presenting functions of DCs, suppression of cytotoxic functions of CD8+ T cells, promotion of development of inflammatory CD4 T cells such as Th17 or Th9). This has been recently reviewed (Chen and ten Dijke, 2016). Finally, in the tumor microenvironment, production of TGF‐β originates from multiple cells such as tumor cells, fibroblasts, macrophages, leukocytes, endothelial cells (Papageorgis and Stylianopoulos, 2015). The role of TGF‐β in cancer metastasis in general, and in cancer EMT in particular, has been massively documented (for a few examples, see Table 1). It has been reviewed, several times, elsewhere (Moustakas and Heldin, 2016; Papageorgis and Stylianopoulos, 2015; Pickup et al., 2013, 2017; Zhang et al., 2016a).
3.2. TNF‐α
Tumor necrosis factor alpha (TNF‐α) was historically so named because it induces hemorrhagic necrosis of tumors. Its dual role has since been studied and it is now widely accepted that chronic exposure to TNF‐α rather promotes tumor cell proliferation, angiogenesis, and dissemination. TNF‐α can control the expression of multiple other cytokines and TNF‐neutralizing agents are used to treat cancer (Crusz and Balkwill, 2015).
The induction of EMT by TNF‐α, particularly in synergy with TGF‐β or other inflammatory factors, has been described (Bates and Mercurio, 2003) (Figure 2 and Table 1). For instance, a mix of TGF‐β, IFN‐α, and TNF‐α can be used to induce EMT‐like changes in human cancer cell lines in vitro (Ricciardi et al., 2015). Treatment of cancer cells with the supernatant from a mixed lymphocyte reaction (MLR) experiment induces a similar phenotype (Ricciardi et al., 2015). In colorectal cancer cell lines, TNF‐α and TGF‐β induce EMT‐like changes either in a NLRP3/Snail1 axis‐dependent way (NLRP3 stands for ‘NOD‐like receptor family, pyrin domain containing 3’) (Song et al., 2016), or via an increase in expression of claudin‐1. In this case, claudin‐1 is delocalized away from the membrane and activates the Src and ERK1/2 MAP kinase pathways (Bhat et al., 2016). TNF‐α‐induced EMT has been reported in cell lines from multiple other cancer entities: hepatocellular carcinoma (Zhu et al., 2016), breast cancer (Cohen et al., 2015), lung cancer (Song et al., 2016), or thyroid cancer (here in combination with IFN‐γ) (Lv et al., 2015) among others. Interestingly, TNF‐α treatment‐induced EMT signaling pathways in renal cell carcinoma cell lines require expression of CXCR2, CXCR3, and their ligands (Sun et al., 2016), highlighting synergy with chemokines.
Figure 2.
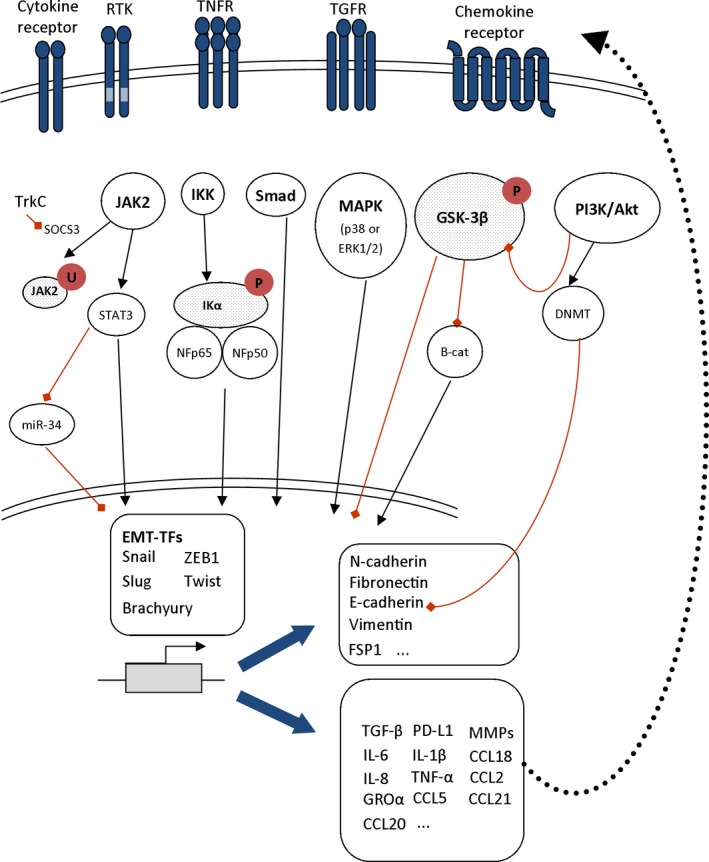
Overview of major signaling cascades leading to EMT program activation and inflammatory target gene activation in cancer cells.
3.3. IL‐1β
Interleukin‐1β (IL‐1β) is a major cytokine involved in cancer‐associated chronic inflammation (Crusz and Balkwill, 2015). A few reports have illustrated a causative link between the IL‐1β/IL‐1R axis and induction of EMT‐like changes in (pre)cancerous cells in vitro (Table 1). Li et al. (2012b) initially showed that IL‐1β treatment promotes EMT in primary colorectal cancer cells via ZEB1 overexpression. IL‐1β also induces EMT in oral squamous cell carcinoma cell lines and in primary dysplastic oral keratinocytes. Interestingly, in these cells, IL‐1β‐induced EMT leads to enhanced secretion of the proinflammatory cytokines IL‐6, IL‐8, and GROα (Lee et al., 2015), thereby initiating a potential autocrine EMT maintenance loop. Finally, in the MCF10A cells, ectopic expression of EMT‐TF Snail1 leads to hyper‐responsiveness to IL‐1β, characterized by high activation of NF‐kB and MAPK pathways and production of IL‐6 and IL‐8 (Lim et al., 2013) (Fig. 2). This is in line with the textbook signaling pathway of IL‐1β, which, upon binding to its IL‐1R (a dimeric cytokine receptor), leads to activation of the NF‐κB pathway and initiates a proinflammatory program in a multitude of cell types.
3.4. IL‐8
Although IL‐8 can be secreted by a variety of stromal cells, EMT pathways induce IL‐8 expression and secretion in tumor cells as shown in human breast cancer cell lines (Brysse et al., 2012; Li et al., 2012a; Lim et al., 2013; Suarez‐Carmona et al., 2015), in immortalized oral keratinocytes (Lyons et al., 2008), in lung cancer cell lines (Fernando et al., 2011), and in colorectal cancer cell lines (Bates et al., 2004) or in colorectal cancer cells freshly isolated from human tumors (Hwang et al., 2011). In this last model, Snail1 has been shown to directly promote IL‐8 transcription upon binding to E3/E4 E‐boxes. Finally, there is a correlation between Snail1 and IL‐8 protein expression in a cohort of 52 human colorectal cancer specimens (Hwang et al., 2011). Other reports have highlighted the functional role of EMT‐TFs Twist (Li et al., 2012a), Brachyury (Fernando et al., 2011), and the Snail family (Suarez‐Carmona et al., 2015) in the regulation of IL‐8 expression in cancer cell lines. Most recently, Lesage et al. (2017) have demonstrated that the molecule ZO‐1, located at the membrane in epithelial cells but transported into the nuclear/cytoplasmic compartment upon EMT‐associated junction disruption, activates the secretion of IL‐8 via the NF‐kB pathway in breast and lung cancer cell lines.
Conversely, IL‐8 can induce EMT (Table 1). IL‐8 can bind to two G‐protein‐coupled receptors, CXCR1/2, and initiate several signaling pathways, such as Ras/Raf/MAPK, PI3K, or JAK/STAT (Fig. 2). IL‐8‐mediated EMT has been demonstrated in ovarian cancer cell lines (Yin et al., 2015), breast (Ji et al., 2016), and lung cancer cells. In these, erlotinib‐induced autocrine IL‐8 production induces EMT via the p38 MAPK kinase pathway (Fig. 2). IL‐8 neutralization effectively restores an epithelial phenotype (Fernando et al., 2016). IL‐8 additionally triggers EMT changes in thyroid cancer cells via the AKT/Slug pathway (Visciano et al., 2015) and in hepatocellular carcinoma, via JAK2/STAT3 activation and Snail1 expression (Fu et al., 2015). Finally, Zhang et al. (2016e) have recently shown that, in nasopharyngeal carcinoma cells, IL‐8 stabilizes the DN methyl‐transferase DNMT1 via AKT, thereby inducing methylation of E‐cadherin promoter, leading to E‐cadherin repression and EMT program activation. Cooperative induction of EMT in colorectal cancer cells has been shown upon IL‐8 and CCL20 treatment, in conditions in which each individual treatment failed to induce EMT. This combined effect involves the activation of the PI3K, Akt, and ERK1/2 pathways (Cheng et al., 2014).
Altogether, the data linking IL‐8 and EMT have established the principle of a mutual loop in which IL‐8 and EMT programs sustain each other in the cancer microenvironment (Long et al., 2016).
3.5. IL‐6
The role of IL‐6 in cancer‐related inflammation has been reviewed elsewhere (Pesic and Greten, 2016). IL‐6, produced both by tumor cells (especially after Ras or p53 mutations) and by reactive stromal cells (Crusz and Balkwill, 2015), promotes both local and systemic inflammation. Elevated circulating levels of IL‐6 are an indicator of a poor outcome in several cancer entities (Crusz and Balkwill, 2015). IL‐6 binds to a dimeric cytokine receptor composed of IL‐6Rα and IL‐6Rβ/gp130. The full receptor is expressed solely by immune cells, while gp130 expression is ubiquitous. In tumors, tumor cells can produce a soluble form of IL‐6Rα which binds to IL‐6, enabling the so‐formed dimer to interact with gp130 at the surface of cancer cells and initiate prosurvival signaling (Naugler and Karin, 2008).
The reciprocal causative link between EMT pathways and IL‐6 has been supported by a number of independent studies (Table 1). Historically, the negative effect of IL‐6 on E‐cadherin expression in breast cancer cell lines was already demonstrated almost 20 years ago (Asgeirsson et al., 1998). Later, Sullivan et al. (2009) demonstrated the induction of EMT phenotypes in estrogen receptor alpha‐positive breast cancer cells. In this work, the ectopic expression of IL‐6 in MCF7 cells activated the JAK2/STAT3 pathway and Twist, leading to the acquisition of mesenchymal markers and properties (Fig. 2). More recently, a role for IL‐6 in inducing and maintaining EMT was shown in other breast cancer cell lines. IL‐6, induced by TrkC (tropomyosin receptor kinase C), triggers EMT and Twist1‐mediated IL‐6 overexpression, initiating a vicious loop securing the EMT phenotype (Kim et al., 2016). IL‐6 also induces EMT in vitro in immortalized epithelial oral cells, in a JAK2/STAT3/Snail1‐dependent way (Yadav et al., 2011), in lung adenocarcinoma cells (in a STAT3‐Snail1‐dependent fashion) (Zhao et al., 2014) and in NSCLC cells (Lee et al., 2016; Shintani et al., 2016). In this latter case, the effects of IL‐6 are more pronounced in the CD133+, stem‐like, subpopulation (Lee et al., 2016). IL‐6 also induces EMT in ovarian cancer cells, similarly via the JAK2/STAT3 signaling pathway (Colomiere et al., 2009) (Fig. 2), in head and neck squamous cell carcinoma cells (Wu et al., 2016a), in gastric cancer cells (Chen et al., 2017), and in uterine cervix cancer cells (in a STAT3‐Slug‐dependent way) (Miao et al., 2014). Finally, an additional pathway has been linked to IL‐6‐mediated EMT in colorectal cancer cells (Rokavec et al., 2014). Phosphorylated STAT3 can directly repress miR‐34 (a miRNA involved in the maintenance of the epithelial phenotype, mostly by inhibiting the EMT‐TF Snail1) by binding on its first intron. miR‐34 is a p53‐regulated target that represses IL‐6Rα (Rokavec et al., 2014).
On the contrary, the induction of EMT in cancer cell lines can foster expression of IL‐6 at quite high levels in breast cancer cell lines via Twist (Sullivan et al., 2009) or Snail1 (Suarez‐Carmona et al., 2015) and in oral keratinocytes via Snail1 (Lyons et al., 2008). The pathways involved in Snail1‐mediated IL‐6 (and IL‐8) mRNA expression have been detailed in the immortalized breast cells MCF10A (Lim et al., 2013) and comprise ERK, STAT2/3, and AKT activation.
3.6. Th17‐related cytokines
Tumor infiltration by the proinflammatory Th17 T cells is associated with increased Th17‐associated cytokines IL‐23, IL‐17, IL‐1β, and IL‐6. In esophageal cancer, IL‐23 expression in the primary tumor relates to the presence of nodal and distance metastases (Chen et al., 2015). In vitro, treatment of esophageal cancer cell lines with recombinant IL‐23 induces EMT‐like changes characterized by loss of E‐cadherin and de novo vimentin expression (Chen et al., 2015). The signaling pathways in these cells comprise activation of the Wnt/β‐catenin pathway, subsequent GSK‐3β stabilization, which finally triggers Slug and Snail1 expression (Fig. 2). Regarding IL‐17, treatment with recombinant IL‐17 was shown to induce EMT‐like changes in three human cell lines (Zhang et al., 2016d) (Table 1). Similarly, treatment of quiescent gastric cancer stem cells with IL‐17 also results in EMT‐like changes including E‐cadherin loss, de novo vimentin acquisition, and increased migratory and invasive properties (Jiang et al., 2017) and associates with STAT3 phosphorylation.
3.7. CCL2
CCL2 [or monocyte chemotactic protein 1 (MCP1)] is produced by several cell types and recruits most immune cells (except from neutrophils and eosinophil) by binding to its receptor CCR2. CCL2 expression in cancer correlates with TAM density (Deshmane et al., 2009).
CCL2 expression leads to EMT in several cancer cell types (Table 1). In lung cancer cell lines, CCL2 and IL‐6 synergistically induce EMT in a Twist/STAT3‐dependent fashion (Chen et al., 2015b) (Fig. 2). CCL2 neutralization also inhibits EMT in prostate cancer cells (Izumi et al., 2013). Finally, in bladder cancer, mast cells induce EMT in cancer cells via stimulation of the ERβ/CCL2/CCR2 axis (Rao et al., 2016).
On the contrary, in mouse melanoma cells, ectopic Snail1 expression leads to EMT and subsequently to CCL2 production (Kudo‐Saito et al., 2013).
3.8. CCL5
CCL5 is a proinflammatory chemokine involved in inflammatory cell recruitment, cancer‐related angiogenesis, and tumor metastasis formation (Aldinucci and Colombatti, 2014). Its receptors, CCR1, CCR3, and mostly CCR5, are expressed at the surface of myeloid cells, T cells, and tumor cells. Recently, CCL5 has emerged as an EMT inducer (Table 1). CCL5 was indeed identified as a T cell‐derived, pro‐EMT factor in human colorectal cancer liver metastasis tissue (Halama et al., 2016). In ovarian cancer models, CCL5 has also been shown to be produced by CD133‐positive stem‐like cancer cells. By binding to CCR1/3/5 on CD133‐negative, non‐stem‐like cells, CCL5 activates NF‐κB and thereby promotes EMT and metastasis (Long et al., 2015). Finally, CCL5 derived from the adipose microenvironment fosters invasive properties of triple‐negative breast cancer cells in vitro (D'Esposito et al., 2016).
3.9. CCL18
CCL18 is a macrophage‐derived, T cell‐attracting chemokine. CCL18 is only expressed in human, not in mice; recent data indicate that it binds to CCR8 (Islam et al., 2013).
A few reports have linked CCL18 to EMT in cancer (Table 1). In pancreatic cancer cells for example, CCL18 is expressed both by tumor cells and by M2‐polarized macrophages. In vitro, recombinant CCL18 induces migration, and invasion, as well as EMT‐TF Snail1 expression in pancreatic cancer cell lines (Meng et al., 2015). In breast cancer cell lines, CCL18 derived from TAMs activates NF‐κB and thereby induces EMT‐like changes including spindle‐shaped morphology, E‐cadherin‐to‐vimentin switch, and increased invasive properties (Su et al., 2014).
Finally in the ovarian cancer cell line Skov3, CCL18 ectopic expression endows tumor cells with enhanced migratory and invasive properties in vitro (although the authors do not investigate the expression of any marker or transcription factor linked to EMT) (Wang et al., 2016).
3.10. CCL20
In human colorectal cancer, CCL20 receptor CCR6 is overexpressed in tumors compared to normal tissue, and expression intensity increases with tumor stage, suggesting a functional role in aggressiveness. Supportively, the authors also show that colorectal cancer cells undergo EMT changes when exposed to CCL20 in vitro. This is inhibited by neutralizing CCR6 (Kapur et al., 2016) (Table 1). Additionally, co‐occurrence of CCL20 expression and EMT has been reported in hepatocellular cancer (Ye et al., 2016), as described in Section 2.2.
3.11. CCL21
In a panel of pancreatic cancer cell lines, CCR7 is found overexpressed specifically in stem‐like CD133‐positive cells (Table 1). Activation of this receptor by CCL21 leads to the acquisition of EMT markers (E‐to‐N‐cadherin switch, MMP9 expression) and increased invasive properties. This involves Erk/NF‐κB signaling (Zhang et al., 2016c).
Expression of the CCL21 receptor CCR7 is also increased in breast cancer cells upon TGF‐β‐induced EMT, which facilitates breast cancer cell migration toward CCL21‐positive, draining lymphatic vessels in mice, in a DC‐like fashion (Pang et al., 2016).
3.12. Reactive oxygen species
During an inflammatory reaction, ROS are mostly produced by activated macrophages or neutrophils. They activate adaptive immune cells and contribute to apoptosis induction. In addition, ROS are major regulators of the production of proinflammatory cytokines and chemokines (such as CXCL12) (Costa et al., 2014). Whether these major actors of inflammation impact the EMT program in cancer cells has been investigated for the past few years.
Nevertheless, the effect of ROS on the induction – or inhibition – of EMT programs in cancer cells is unclear, as opposite effects of ROS on EMT induction in cancer cells have been described. The production of ROS has been shown to inhibit EMT in two prostate cancer cell lines (Das et al., 2014). Similarly, in basal‐like breast carcinoma cells, Snail1‐induced epigenetic silencing of FBP1 (fructose‐1, 6‐bisphosphatase) on the one hand promotes glycolysis and the production of NAPDH (via the pentose phosphate pathway), and on the other hand decreases respiration, leading to the decreased production of ROS, which finally endows tumor cells with cancer stem cell‐ and EMT‐like features (Dong et al., 2013). Conversely, the inhibition of mitochondrial ROS in cervical cancer cells reverses the acquisition of EMT‐like properties (Shagieva et al., 2016). In colorectal cancer cell lines, oxaliplatin‐induced EMT depends on ROS production (Jiao et al., 2016). Accordingly, the drug nimbolide, which has been successfully used to reduce metastasis in a mouse pancreatic cancer model, inhibits EMT by inducing excessive production of ROS (Subramani et al., 2016).
4. EMT, inflammation, and the metastatic progression
There has been a plethora of studies consistently linking chronic inflammation to cancer progression and metastatic outgrowth (Crusz and Balkwill, 2015; Grivennikov and Karin, 2010; Muller et al., 2008; Naugler and Karin, 2008; Pesic and Greten, 2016). Similarly, many publications have illustrated the prometastatic effects of EMT program activation in cancer cells (Bates and Mercurio, 2005; Fassina et al., 2012; Spaderna et al., 2006; Yu et al., 2013). Research data indicating that an alliance, or mutual loop, between EMT‐like cancer cells and established chronic inflammation promotes metastasis in a cooperative fashion are rather scarce. We here focus on these few studies and mention the role of the EMT/inflammation axis in tumorigenesis, invasion, liberation of circulating tumor cells, and metastasis outburst.
The EMT/inflammation axis is already involved in the very first steps of tumor formation. For instance, ectopic expression of Snail1 in colorectal cancer cells induces properties of colonospheres including stem cell properties and IL‐8 expression, leading to increased tumorigenicity in nude mice (Hwang et al., 2011). Similarly, subcutaneous injection of gastric quiescent cancer stem cells to nude mice after 24 h of treatment with IL‐17, which induces EMT‐like changes in vitro, results in the formation of larger xenografts in nude mice (Jiang et al., 2017). Accordingly, in PTENnull MMP7null mice and PTENnull IL‐17RCnull mice, prostate tumors are smaller and a lower proportion of mice develop invasive adenocarcinoma compared to PTEN single‐KO mice. These changes are accompanied by weaker EMT‐like characteristics in cancer cells (Zhang et al., 2016d).
Primary tumors can also become resistant to therapy because of the EMT/inflammation alliance. In lung cancer cells, erlotinib‐induced autocrine IL‐8 production induces EMT and triggers erlotinib resistance via the p38 MAP kinase pathway. IL‐8 neutralization effectively restores both an epithelial phenotype and erlotinib sensitivity (Fernando et al., 2016).
EMT in association with inflammation has also been connected with higher stages of cancer progression in several studies. An important step in metastatic progression of tumors is the liberation and survival of circulating tumor cells. In patients with inflammatory breast cancer, a correlation exists between immune activation and the presence of circulating tumor cells (CTCs) with EMT characteristics (Cohen et al., 2015). Indeed, among 16 tested patients, six patients have high numbers of circulating TNF‐α‐producing T cells. Of these six patients, four had EMT‐like CTCs. In contrast, only one patient with low numbers of TNF‐producing T cells (among 10) has EMT‐like CTCs (Cohen et al., 2015).
Finally, most clinical data regarding the EMT/inflammation axis have highlighted its correlation with metastatic stage and poor outcome. This has been proven to be relevant in multiple cancer entities. In colorectal cancer specimens, concomitant expression of IL‐8 and CCL20 correlates with E‐cadherin loss and with lymph node and liver metastasis (Cheng et al., 2014). CCL20‐IL‐8 double positivity is also linked to a shorter overall survival (OS) and disease‐free survival (DFS) in these patients. Still in colorectal cancer, the expression of IL‐6 and STAT3 and the absence of miR‐34 (altogether constituting an EMT‐maintaining loop) are associated with nodal and distant metastases in a cohort of 425 patients (Rokavec et al., 2014). In breast cancer, the expression of Shohl2 – a transcription factor of the bHLH family which inhibits EMT by repressing IL‐8 expression in vitro (Ji et al., 2016) – is reduced in metastases compared to primary tumors. In mouse xenografts, ectopic expression of Shohl2 inhibits metastasis outgrowth, while Shohl2 knockdown boosts metastasis formation. Finally, Shohl2 and IL‐8 protein expressions are negatively correlated in a cohort of 12 breast cancer samples (Ji et al., 2016). The CCL18/GM‐CSF/EMT loop described above links EMT to macrophages in a prometastatic pathway. CCL18 and GM‐CSF expression are correlated (a) with each other and (b) with EMT‐like features in human breast cancer specimens, and GM‐CSF expression predicts a shorter DFS (Su et al., 2014).
In ovarian carcinoma, CCL5 is produced by ovarian cancer stem‐like cells, where it induces EMT in non‐cancer stem‐like cells. CCL5 and CCR5 expressions are also associated with ovarian cancer metastasis (Long et al., 2015). CCL18, which induces EMT in ovarian cancer cells in vitro, is also associated with high tumor grade and metastasis in patients with ovarian cancer (Wang et al., 2016). High EMT/hypoxia‐associated CCL20 expression has been associated with poor patient DFS and OS in a cohort of 90 hepatocellular carcinomas (Ye et al., 2016). In two independent cohorts of patients with gastric cancer – in which TAM density is associated with the expression of EMT features in cancer cells – both TAM density and EMT features are related to a shorter cumulative survival (Yan et al., 2016; Zhang et al., 2016b).
In addition, mouse models have been extensively used to further dissect the EMT/inflammation/metastasis axis. Using a murine model of breast cancer, De Cock et al. (2016) have elegantly illustrated the functional synergy between inflammation and cancer in the promotion of metastatic outgrowth. They have generated a dormant cell line that is tumorigenic after orthotopic injection, disseminates in the lung but fails to grow there. These cells express low levels of EMT‐TFs Snail1, Twist1, and ZEB1. Interestingly, doxycycline‐inducible Snail1 or ZEB1 ectopic expression increases metastatic outgrowth. Most importantly, lung inflammation, induced by treating the mice with LPS, also leads to a dramatic increase in metastases formation, which can be effectively inhibited by ZEB1 knockdown. This phenomenon occurs in NOD‐SCID mice and is therefore independent of any adaptive immune response, but intriguingly, LPS‐induced metastatic outgrowth is inhibited by neutrophil depletion. So this indicates that EMT and inflammation are mechanistically required to permit metastasis in this model. In a xenograft model of lung adenocarcinoma, injection of cancer cells that stably overexpress IL‐6 leads to the formation of tumors with EMT characteristics (E‐cadherin loss and vimentin expression) and in an increased number of lung metastases (Zhao et al., 2014). In another study, IL‐17‐induced EMT results in increased metastatic numbers in nude mice, in the above‐mentioned gastric cancer xenograft model (Jiang et al., 2017). In murine melanoma RETAAD mice presented above, depletion of MDSCs leads to a decrease in EMT marker expression, impairment of tumor growth, and decreased cancer cell dissemination (Toh et al., 2011). In melanoma xenograft models, Snail1 transfectants (or cells expressing Snail1 following TGF‐β treatment) acquire EMT features and this results in a shorter overall survival of mice (Kudo‐Saito et al., 2009). This aggressiveness relies on local immunosuppression, as characterized by TSP1 expression, impairment of dendritic cells, and recruitment of Tregs.
Finally, mast cells promote metastasis of bladder cancer in mice, in an EMT‐dependent fashion. It has been shown that when mast cells are co‐cultured with bladder cancer cells, CCL2 expression is elevated (after ERβ decrease) and EMT is activated in cancer cells, which express MMP‐9 and display enhanced invasive properties. In mouse xenograft, co‐implantation of mast cells with tumor cells increases metastatic burden. This metastatic advantage is reversed by an ERβ antagonist. So globally, mast cells promote metastasis of bladder cancer in mice in a CCL2/CCR2/EMT/MMP9‐dependent mechanism (Rao et al., 2016).
5. Concluding remarks
Studying EMT and inflammation has been difficult because both phenomena are highly regulated in time and space. All data collected from immunohistochemistry in human specimens are therefore only snapshots that provide associative data. Furthermore, most available models to study EMT in time lines are either cell‐based models (migration, invasiveness in vitro) that fail to reconstitute the complexity in the living environment, or mouse models, in which the study of inflammation, or at least its translation into human data, is complicated by the discrepancies in inflammatory markers between both species.
There is clearly, however, a multifaceted, sustained crosstalk between cancer cells undergoing EMT‐like changes and the cellular actors of inflammation. Tumors are generally described as a wound that never heals, generating a chronic, unresolved inflammatory reaction, which is fueled by a local cytokine storm. EMT programs most certainly play a central role in establishing and maintaining this local cytokine storm. Reciprocally, inflammatory cells present at the tumor site keep producing EMT‐inducing factors. Supportively, tumor cells displaying EMT‐like features are most often encountered at the periphery of tumor islets, at the tumor–stroma interface.
The EMT/inflammation axis definitely fosters aggressiveness of the primary tumors as illustrated in a number of mouse models, and markers of EMT and inflammation are correlated with poor outcome in patient cohorts of multiple cancers. To this day, several anti‐inflammatory agents have been developed and tested for cancer treatment (Crusz and Balkwill, 2015). Conversely, despite a likely implication of EMT in specific steps of the metastatic progression, EMT‐positive tumor cells have not been successfully tackled yet. In this context, we believe that a better understanding of the mechanisms behind the EMT/inflammation alliance might open new perspectives on anti‐inflammatory therapeutic strategies.
References
- Aldinucci D and Colombatti A (2014) The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm 2014, 292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgeirsson KS, Olafsdottir K, Jonasson JG and Ogmundsdottir HM (1998) The effects of IL‐6 on cell adhesion and e‐cadherin expression in breast cancer. Cytokine 10, 720–728. [DOI] [PubMed] [Google Scholar]
- Bates RC, DeLeo MJ 3rd and Mercurio AM (2004) The epithelial‐mesenchymal transition of colon carcinoma involves expression of IL‐8 and CXCR‐1‐mediated chemotaxis. Exp Cell Res 299, 315–324. [DOI] [PubMed] [Google Scholar]
- Bates RC and Mercurio AM (2003) Tumor necrosis factor‐alpha stimulates the epithelial‐to‐mesenchymal transition of human colonic organoids. Mol Biol Cell 14, 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates RC and Mercurio AM (2005) The epithelial‐mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther 4, 365–375. [DOI] [PubMed] [Google Scholar]
- Becht E, Giraldo NA, Germain C, de Reynies A, Laurent‐Puig P, Zucman‐Rossi J, Dieu‐Nosjean MC, Sautes‐Fridman C and Fridman WH (2016) Immune contexture, immunoscore, and malignant cell molecular subgroups for prognostic and theranostic classifications of cancers. Adv Immunol 130, 95–190. [DOI] [PubMed] [Google Scholar]
- Bhat AA, Ahmad R, Uppada SB, Singh AB and Dhawan P (2016) Claudin‐1 promotes TNF‐α‐induced epithelial‐mesenchymal transition and migration in colorectal adenocarcinoma cells. Exp Cell Res 349, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde AK, Tischler V, Kumar S, Soltermann A and Schwendener RA (2012) Intratumoral macrophages contribute to epithelial‐mesenchymal transition in solid tumors. BMC Cancer 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brysse A, Mestdagt M, Polette M, Luczka E, Hunziker W, Noel A, Birembaut P, Foidart JM and Gilles C (2012) Regulation of CXCL8/IL‐8 expression by zonula occludens‐1 in human breast cancer cells. Mol Cancer Res 10, 121–132. [DOI] [PubMed] [Google Scholar]
- Chen W, Gao Q, Han S, Pan F and Fan W (2015b) The CCL2/CCR2 axis enhances IL‐6‐induced epithelial‐mesenchymal transition by cooperatively activating STAT3‐Twist signaling. Tumour Biol 36, 973–981. [DOI] [PubMed] [Google Scholar]
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W et al (2014) Metastasis is regulated via microRNA‐200/ZEB1 axis control of tumour cell PD‐L1 expression and intratumoral immunosuppression. Nat Commun 5, 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Heymach JV, Qin FX‐F and Gibbons DL (2015a) The mutually regulatory loop of epithelial–mesenchymal transition and immunosuppression in cancer progression. OncoImmunology 4, e1002731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li W, Liu S, Su Y, Han G, Xu C, Liu H, Zheng T, Zhou Y and Mao C (2015) Interleukin‐23 promotes the epithelialmesenchymal transition of oesophageal carcinoma cells 11 via the Wnt/b‐catenin pathway. Sci Rep 5, 8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Tang N, Wang C, Xiao L, Yu M, Zhao L, Cai H, Han L, Xie C and Zhang Y (2017) TNF‐α‐inducing protein of Helicobacter pylori induces epithelial‐mesenchymal transition (EMT) in gastric cancer cells through activation of IL‐6/STAT3 signaling pathway. Biochem Biophys Res Comm 484, 311–317. [DOI] [PubMed] [Google Scholar]
- Chen W and ten Dijke P (2016) Immunoregulation by members of the TGFβ superfamily. Nat Rev Immunol 16, 723–740. [DOI] [PubMed] [Google Scholar]
- Cheng XS, Li YF, Tan J, Sun B, Xiao YC, Fang XB, Zhang XF, Li Q, Dong JH, Li M et al (2014) CCL20 and CXCL8 synergize to promote progression and poor survival outcome in patients with colorectal cancer by collaborative induction of the epithelial‐mesenchymal transition. Cancer Lett 348, 77–87. [DOI] [PubMed] [Google Scholar]
- Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, Giordano A, Tin S, Wu Q, Garza RJ et al (2015) Inflammation mediated metastasis: immune induced epithelial‐to‐mesenchymal transition in inflammatory breast cancer cells. PLoS One 10, e0132710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomiere M, Ward AC, Riley C, Trenerry MK, Cameron‐Smith D, Findlay J, Ackland L and Ahmed N (2009) Cross talk of signals between EGFR and IL‐6R through JAK2/STAT3 mediate epithelial‐mesenchymal transition in ovarian carcinomas. Br J Cancer 100, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Scholer‐Dahirel A and Mechta‐Grigoriou A (2014) The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol 25, 23–32. [DOI] [PubMed] [Google Scholar]
- Crusz SM and Balkwill FR (2015) Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 12, 584–596. [DOI] [PubMed] [Google Scholar]
- Das TP, Suman S and Damodaran C (2014) Induction of reactive oxygen species generation inhibits epithelial–mesenchymal transition and promotes growth arrest in prostate cancer cells. Mol Carcinog 53, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock J, Shibue T, Dongre A, Keckesova Z, Reinhardt F and Weinberg RA (2016) Inflammation triggers Zeb1‐dependent escape from tumor latency. Cancer Res 76, 6778–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y‐R, Liu W‐B, Lian Z‐X, Li X and Hou X (2016) Sorafenib inhibits macrophage‐mediated epithelial‐mesenchymal transition in hepatocellular carcinoma. Oncotarget 7, 38292–38305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S and Sawaya BE (2009) Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res 29, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Esposito V, Liguoro D, Abrosio MR, Collina F, Cantile M, Spinelli R, Raciti GA, Miele C, Valentino R, Campiglia P et al (2016) Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 7, 24495–24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepenbruck M and Christofori G (2016) Epithelial–mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol 43, 7–13. [DOI] [PubMed] [Google Scholar]
- Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T et al (2013) Loss of FBP1 by Snail‐mediated repression provides metabolic advantages in basal‐like breast cancer. Cancer Cell 23, 316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elghonaimy EA, Ibrahim SA, Youns A, Hussein Z, Nouh MA, El‐Mamlouk T, El‐Shinawi M and Mostafa Mohamed M (2016) Secretome of tumor‐associated leukocytes augment epithelial‐mesenchymal transition in positive lymph node breast cancer patients via activation of EGFR/Tyr845 and NF‐kappaB/p65 signaling pathway. Tumour Biol 37, 12441–12453. [DOI] [PubMed] [Google Scholar]
- Fan Q‐M, Jing Y‐Y, Yu G‐F, Kou X‐R, Ye F, Gao L, Li R, Zhao Q‐D, Yang Y, Lu Z‐H et al (2014) Tumor‐associated macrophages promote cancer stem cell‐like properties via transforming growth factor‐beta1‐induced epithelial–mesenchymal transition in hepatocellular carcinoma. Cancer Lett 352, 160–168. [DOI] [PubMed] [Google Scholar]
- Fassina A, Cappellesso R, Guzzardo V, Dalla Via L, Piccolo S, Ventura L and Fassan M (2012) Epithelial‐mesenchymal transition in malignant mesothelioma. Modern Pathol 25, 86–99. [DOI] [PubMed] [Google Scholar]
- Fernando RI, Castillo MD, Litzinger M, Hamilton DH and Palena C (2011) IL‐8 signaling plays a critical role in the epithelial‐mesenchymal transition of human carcinoma cells. Can Res 71, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando RI, Hamilton DH, Dominguez C, David JM, McCampbell KK and Palena C (2016) IL‐8 signaling is involved in resistance of lung carcinoma cells to erlotinib. Oncotarget 7, 42031–42044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X‐T, Song K, Zhang Z‐J, Zhou Z‐J, Zhou S‐L, Zhao Y‐M, Xiao Y‐S, Sun Q‐M, Ding Z‐B and Fan J (2015) Macrophage‐secreted IL‐8 induces epithelial‐mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol 46, 587–596. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI and Karin M (2010) Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev 20, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse‐Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Haensch GM and Gaida MM (2012) Epithelial‐to‐mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil‐derived elastase. Clin Dev Immunol 2012, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez‐Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F et al (2016) Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti‐CCR5 therapy in cancer patients. Cancer Cell 29, 587–601. [DOI] [PubMed] [Google Scholar]
- Hu Y, He MY, Zhu LF, Yang CC, Zhou ML, Wang Q, Zhang W, Zheng YY, Wang DM, Xu ZQ et al (2016) Tumor‐associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res 35, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Shen M, Zhang P, Zheng C, Pang Z, Zhu L and Du J (2015) Intratumoral neutrophil granulocytes contribute to epithelial‐mesenchymal transition in lung adenocarcinoma cells. Tumour Biol 36, 7789–7796. [DOI] [PubMed] [Google Scholar]
- Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, Teng HW, Yang SH, Lan YT, Chiou SH et al (2011) SNAIL regulates interleukin‐8 expression, stem cell‐like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 141, 279–291. [DOI] [PubMed] [Google Scholar]
- Islam SA, Ling MF, Leung J, Shreffler WG and Luster AD (2013) Identification of human CCR8 as a CCL18 receptor. J Exp Med 210, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ and Chang C (2013) Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2‐induced STAT3 activation. EMBO Mol Med 5, 1383–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MH, Kim HJ, Kim EJ, Chung YR and Park SY (2015) Expression of epithelial‐mesenchymal transition‐related markers in triple‐negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome. Hum Pathol 46, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Ji S, Zhang W, Zhang X, Hao C, Hao A, Gao Q, Zhang H, Sun J and Hao J (2016) Sohlh2 suppresses epithelial to mesenchymal transition in breast cancer via downregulation of IL‐8. Oncotarget 7, 49411–49424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y‐X, Yang S‐W, Li P‐A, Luo X, Li Z‐Y, Ho Y‐X and Yu P‐W (2017) The promotion of the transformation of quiescent gastric cancer stem cells by IL‐17 and the underlying mechanisms. Oncogene 36, 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Li D‐D, Yang C‐L, Peng R‐Q, Guo Y‐Q, Zhang X‐S and Zhu X‐F (2016) Reactive oxygen species mediate oxaliplatin‐induced epithelial‐mesenchymal transition and invasive potential in colon cancer. Tumor Biology 37, 8413–8423. [DOI] [PubMed] [Google Scholar]
- Johansson J, Tabor V, Wikell A, Jalkanen S and Fuxe J (2015) TGF‐beta1‐induced epithelial‐mesenchymal transition promotes monocyte/macrophage properties in breast cancer cells. Front Oncol 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur N, Mir H, Clark CE, Krishnamurti U, Beech DJ, Lillard JW and Singh S (2016) CCR6 expression in colon cancer is associated with advanced disease and supports epithelial‐to‐mesenchymal transition. Br J Cancer 114, 1343–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Jeong J, Seo J, Kim HS, Kim SJ and Jin W (2016) Dysregulated JAK2 expression by TrkC promotes metastasis potential, and EMT program of metastatic breast cancer. Sci Rep 6, 33899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo‐Saito C, Shirako H, Ohike M, Tsukamoto N and Kawakami Y (2013) CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis 30, 393–405. [DOI] [PubMed] [Google Scholar]
- Kudo‐Saito C, Shirako H, Takeuchi T and Kawakami Y (2009) Cancer metastasis is accelerated through immunosuppression during Snail‐induced EMT of cancer cells. Cancer Cell 15, 195–206. [DOI] [PubMed] [Google Scholar]
- Lee CH, Chang JS, Syu SH, Wong TS, Chan JY, Tang YC, Yang ZP, Yang WC, Chen CT, Lu SC et al (2015) IL‐1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J Cell Physiol 230, 875–884. [DOI] [PubMed] [Google Scholar]
- Lee SO, Yang X, Duan S, Tsai Y, Strojny LR, Keng P and Chen Y (2016) IL‐6 promotes growth and epithelial‐mesenchymal transition of CD133+ cells of non‐small cell lung cancer. Oncotarget 7, 6626–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage J, Suarez‐Carmona M, Neyrinck‐Leglantier D, Grelet S, Blacher S, Hunziker W, Birembaut P, Noel A, Nawrocki‐Raby B, Gilles C et al (2017) Zonula occludens‐1/NF‐kappaB/CXCL8: a new regulatory axis for tumor angiogenesis. FASEB J 31, 1678–1688. [DOI] [PubMed] [Google Scholar]
- Li S, Kendall SE, Raices R, Finlay J, Covarrubias M, Liu Z, Lowe G, Lin YH, Teh YH, Leigh V et al (2012a) TWIST1 associates with NF‐kappaB subunit RELA via carboxyl‐terminal WR domain to promote cell autonomous invasion through IL8 production. BMC Biol 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang L, Pappan L, Galliher‐Beckley A and Shi J (2012b) IL‐1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Becker A, Zimmer A, Lu J, Buettner R and Kirfel J (2013) SNAI1‐mediated epithelial‐mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PLoS One 8, e66558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C‐Y, Xu J‐Y, Shi X‐Y, Huang W, Ruan T‐Y, Xie P and Ding JL (2013) M2‐polarized tumor‐associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL‐10 signaling pathway. Lab Invest 93, 844–854. [DOI] [PubMed] [Google Scholar]
- Long H, Xiang T, Qi W, Li Y, Huang J, Xiz R, Chen J and Zhu B (2015) CD133+ ovarian cancer stem‐like cells promote non‐stem cancer cell metastasis via CCL5 induced epithelial‐mesenchymal transition. Oncotarget 6, 5847–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Ye Y, Zhang L, Liu P, Yu W, Wei F, Ren X and Yu J (2016) IL‐8, a novel messenger to cross‐link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int J Oncol 48, 5–12. [DOI] [PubMed] [Google Scholar]
- Lopez‐Novoa JM and Nieto MA (2009) Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 1, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv N, Gao Y, Guan H, Wu D, Ding S, Teng W and Shan Z (2015) Inflammatory mediators, tumor necrosis factor‐alpha and interferon‐gamma, induce EMT in human PTC cell lines. Oncol Lett 10, 2591–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons JG, Patel V, Roue NC, Fok SY, Soon LL, Halliday GM and Gutkind JS (2008) Snail up‐regulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res 68, 4525–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfettone A, Soukupova J, Bertran E, Crosas‐Molist E, Lastra R, Fernando J, Koudelkova P, Rani B, Fabra Á, Serrano T et al (2017) Transforming growth factor‐β‐induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett 392, 39–50. [DOI] [PubMed] [Google Scholar]
- Marcucci F, Stassi G and De Maria R (2016) Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discovery 15, 311–325. [DOI] [PubMed] [Google Scholar]
- Meng F, Li W, Li C, Gao Z, Guo K and Song S (2015) CCL18 promotes epithelial‐mesenchymal transition, invasion and migration of pancreatic cancer cells in pancreatic ductal adenocarcinoma. Int J Oncol 46, 1109–1120. [DOI] [PubMed] [Google Scholar]
- Miao JW, Liu LJ and Huang J (2014) Interleukin‐6‐induced epithelial‐mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int J Oncol 45, 165–176. [DOI] [PubMed] [Google Scholar]
- Moustakas A and Heldin C‐H (2016) Mechanisms of TGFβ‐induced epithelial‐mesenchymal transition. J Clin Med 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha J, Majchrzak K, Taciak B, Hellmen E and Krol M (2014) MDSCs mediate angiogenesis and predispose canine mammary tumor cells for metastasis via IL‐28/IL‐28RA (IFN‐lambda) signaling. PLoS One 9, e103249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA 3rd, Kahler DJ, Pihkala J, Soler AP, Munn DH et al (2008) Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A 105, 17073–17078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler WE and Karin M (2008) The wolf in sheep's clothing: the role of interleukin‐6 in immunity, inflammation and cancer. Trends Mol Med 14, 109–119. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami‐Chouaib F, Thiery JP and Chouaib S (2017) The immune checkpoint ligand PD‐L1 is upregulated in EMT‐activated human breast cancer cells by a mechanism involving ZEB‐1 and miR‐200. Oncoimmunology 6, e1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Guo X, Yang Z, Chen S, Lei Y, Lin M, Wang L, Feng C and Ke Z (2016) AEG‐1 activates Wnt/PCP signaling to promote metastasis in tongue squamous cell carcinoma. Oncotarget 7, 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang MF, Georgoudaki AM, Lambut L, Johansson J, Tabor V, Hagikura K, Jin Y, Jansson M, Alexander JS, Nelson CM et al (2016) TGF‐[beta]1‐induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21‐mediated chemotaxis. Oncogene 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgis P and Stylianopoulos T (2015) Role of TGFβ in regulation of the tumor microenvironment and drug delivery (Review). Int J Oncol 46, 933–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesic M and Greten FR (2016) Inflammation and cancer: tissue regeneration gone awry. Curr Opin Cell Biol 43, 55–61. [DOI] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S and Moses HL (2013) The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer 13, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup MW, Owens P and Moses HL (2017) TGF‐beta, bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harb Perspect Biol, a022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Q, Chen Y, Yeh C‐R, Ding J, Li L, Chang C and Yeh S (2016) Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 7, 7842–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi J, Elbaz M, Wani NA, Nasser MW and Ganju RK (2016) Cannabinoid receptor‐2 agonist inhibits macrophage induced EMT in non‐small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog 55, 2063–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Reynies A, Jaurand MC, Renier A, Couchy G, Hysi I, Elarouci N, Galateau‐Salle F, Copin MC, Hofman P, Cazes A et al (2014) Molecular classification of malignant pleural mesothelioma: identification of a poor prognosis subgroup linked to the epithelial‐to‐mesenchymal transition. Clin Cancer Res 20, 1323–1334. [DOI] [PubMed] [Google Scholar]
- Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, Bifari F and Krampera M (2015) Epithelial‐to‐mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell‐like immune‐modulatory properties in cancer cells. Br J Cancer 112, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R, Nitsche U et al (2014) Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial‐to‐mesenchymal transition. Int J Cancer 134, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S et al (2014) IL‐6R/STAT3/miR‐34a feedback loop promotes EMT‐mediated colorectal cancer invasion and metastasis. J Clin Investig 124, 1853–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangaletti S, Tripodo C, Santangelo A, Castioni N, Portararo P, Gulino A, Botti L, Parenza M, Cappetti B, Orlandi R et al (2016) Mesenchymal transition of high‐grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep 17, 233–248. [DOI] [PubMed] [Google Scholar]
- Shagieva G, Domnina L, Makarevich O, Chernyak B, Skulachev V and Dugina V (2016) Depletion of mitochondrial reactive oxygen species downregulates epithelial‐to‐mesenchymal transition in cervical cancer cells. Oncotarget 8, 4901–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Fujiwara A, Kimura T, Kawamura T, Funaki S, Minami M and Okumura M (2016) IL‐6 secreted from cancer‐associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial‐mesenchymal transition signaling. J Thorac Oncol 11, 1482–1492. [DOI] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A and Allavena P (2009) Tumor‐associated macrophages (TAM) as major players of the cancer‐related inflammation. J Leukoc Biol 86, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Song J, Feng L, Zhong R, Xia Z, Zhang L, Cui L, Yan H, Jia X and Zhang Z (2016) Icariside II inhibits the EMT of NSCLC cells in inflammatory microenvironment via down‐regulation of Akt/NF‐kappaB signaling pathway. Mol Carcinog 56, 36–48. [DOI] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T and Brabletz T (2006) A transient, EMT‐linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 131, 830–840. [DOI] [PubMed] [Google Scholar]
- Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W et al (2014) A positive feedback loop between mesenchymal‐like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 25, 605–620. [DOI] [PubMed] [Google Scholar]
- Suarez‐Carmona M, Bourcy M, Lesage J, Leroi N, Syne L, Blacher S, Hubert P, Erpicum C, Foidart JM, Delvenne P et al (2015) Soluble factors regulated by epithelial‐mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J Pathol 236, 491–504. [DOI] [PubMed] [Google Scholar]
- Subramani R, Gonzalez E, Arumugam A, Nandy S, Gonzalez V, Medel J, Camacho F, Ortega A, Bonkoungou S, Narayan M et al (2016) Nimbolide inhibits pancreatic cancer growth and metastasis through ROS‐mediated apoptosis and inhibition of epithelial‐to‐mesenchymal transition. Sci Rep 6, 19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM and Hall BM (2009) Interleukin‐6 induces an epithelial‐mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun KH, Sun GH, Wu YC, Ko BJ, Hsu HT and Wu ST (2016) TNF‐alpha augments CXCR2 and CXCR3 to promote progression of renal cell carcinoma. J Cell Mol Med 20, 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, Kato M, Prevost‐Blondel A, Thiery JP and Abastado JP (2011) Mesenchymal transition and dissemination of cancer cells is driven by myeloid‐derived suppressor cells infiltrating the primary tumor. PLoS Biol 9, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugel S, De Sanctis F, Mandruzzato S and Bronte V (2015) Tumor‐induced myeloid deviation: when myeloid‐derived suppressor cells meet tumor‐associated macrophages. J Clin Investig 125, 3365–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T and Van den Eynde BJ (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3‐dioxygenase. Nat Med 9, 1269–1274. [DOI] [PubMed] [Google Scholar]
- Visciano C, Liotti F, Prevete N, Cali G, Franco R, Collina F, de Paulis A, Marone G, Santoro M and Melillo RM (2015) Mast cells induce epithelial‐to‐mesenchymal transition and stem cell features in human thyroid cancer cells through an IL‐8‐Akt‐Slug pathway. Oncogene 34, 5175–5186. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang Y, Yu H, Yin Q, Li M, Shi L, Zhang W, Li D and Li L (2016) CCL18 from tumor‐cells promotes epithelial ovarian cancer metastasis via mTOR signaling pathway. Mol Carcinog 55, 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Cheng J, Sun G, Wu S, Li M, Gao Z, Zhai S, Li P, Su D and Wang X (2016a) p70S6K promotes IL‐6‐induced epithelial‐mesenchymal transition and metastasis of head and neck squamous cell carcinoma. Oncotarget 7, 36539–36550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Tang SJ, Sun GH and Sun KH (2016b) CXCR7 mediates TGFbeta1‐promoted EMT and tumor‐initiating features in lung cancer. Oncogene 35, 2123–2132. [DOI] [PubMed] [Google Scholar]
- Yadav A, Kumar B, Datta J, Teknos TN and Kumar P (2011) IL‐6 promotes head and neck tumor metastasis by inducing epithelial‐mesenchymal transition via the JAK‐STAT3‐SNAIL signaling pathway. Mol Cancer Res 9, 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Zhang J, Li J‐H, Liu X, Wang J‐Z, Qu H‐Y, Wang J‐S and Duan X‐Y (2016) High tumor‐associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial–mesenchymal transition in gastric cancer. Onco Targets Ther 9, 3975–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, Sun X, Li GG, Hu QD, Fu QH et al (2016) Hypoxia‐induced epithelial‐to‐mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res 76, 818–830. [DOI] [PubMed] [Google Scholar]
- Yin J, Zeng F, Wu N, Kang K, Yang Z and Yang H (2015) Interleukin‐8 promotes human ovarian cancer cell migration by epithelial‐mesenchymal transition induction in vitro. Clin Transl Oncol 17, 365–370. [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM et al (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Yan C, Yang X, He S, Liu J, Qin C, Huang C, Lu Y, Tian Z and Jia L (2016) Pharmacoproteomic analysis reveals that metapristone (RU486 metabolite) intervenes E‐cadherin and vimentin to realize cancer metastasis chemoprevention. Sci Rep 6, 22388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu S, Parajuli KR, Zhang W, Zhang K, Mo Z, Liu J, Chen Z, Yang S, Wang AR et al (2016d) Interleukin‐17 promotes prostate cancer via MMP7‐induced epithelial‐to‐mesenchymal transition. Oncogene 36, 687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RL, Peng LX, Yang JP, Zheng LS, Xie P, Wang MY, Huang BJ, Zhao HR, Bao YX and Qian CN (2016e) IL‐8 suppresses E‐cadherin expression in nasopharyngeal carcinoma cells by enhancing E‐cadherin promoter DNA methylation. Int J Oncol 48, 207–214. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tian X‐J and Xing J (2016a) Signal transduction pathways of EMT induced by TGF‐β, SHH, and WNT and their crosstalks. J Clin Med 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang D, Li Y, Liu Y, Xie X, Wu Y, Zhou Y, Ren J, Zhang J, Zhu H et al (2016c) CCL21/CCR7 axis contributed to CD133(+) pancreatic cancer stem‐like cell metastasis via EMT and Erk/NF‐κB pathway. PLoS One 11, e0158529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yan Y, Yang Y, Wang L, Li M, Wang J, Liu X, Duan X and Wang J (2016b) High infiltration of tumor‐associated macrophages influences poor prognosis in human gastric cancer patients, associates with the phenomenon of EMT. Medicine 95, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B and He Y (2014) Metformin inhibits the IL‐6‐induced epithelial‐mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS One 9, e95884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Cheng Y, Guo Y, Chen J, Chen F, Luo R and Li A (2016) Protein kinase D2 contributes to TNF‐alpha‐induced epithelial mesenchymal transition and invasion via the PI3K/GSK‐3beta/beta‐catenin pathway in hepatocellular carcinoma. Oncotarget 7, 5327–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]