

Abstract
It was already in the 18th century when the French surgeon LeDran first noted that breast cancer patients with spread of tumor cells to their axillary lymph nodes had a drastically worse prognosis than patients without spread (LeDran et al., 1752). Since then, metastatic spread of cancer cells to regional lymph nodes has been established as the most important prognostic factor in many types of cancer (Carter et al., 1989; Elston and Ellis, 1991). However, despite its clinical importance, lymph metastasis remains an underexplored area of tumor biology. Fundamental questions, such as when, how, and perhaps most importantly, why tumor cells disseminate through the lymphatic system, remain largely unanswered. Accordingly, no treatment strategies exist that specifically target lymph metastasis. The identification of epithelial‐mesenchymal transition (EMT) as a mechanism, which allows cancer cells to dedifferentiate and acquire enhanced migratory and invasive properties, has been a game changer in cancer research. Conceptually, EMT provides an explanation for why epithelial cancers with poor differentiation status are generally more aggressive and prone to metastasize than more differentiated cancers. Inflammatory cytokines, such as TGF‐β, which are produced and secreted by tumor‐infiltrating immune cells, are potent inducers of EMT. Thus, reactivation of EMT also links cancer‐related inflammation to invasive and metastatic disease. Recently, we found that breast cancer cells undergoing TGF‐β‐induced EMT acquire properties of immune cells allowing them to disseminate in a targeted fashion through the lymphatic system similar to activated dendritic cells during inflammation. Here, we review our current understanding of the mechanisms by which cancer cells spread through the lymphatic system and the links to inflammation and the immune system. We also emphasize how imaging techniques have the potential to further expand our knowledge of the mechanisms of lymph metastasis, and how lymph nodes serve as an interface between cancer and the immune system.
Keywords: chemokines, EMT, immune cell properties, lymph metastasis, targeted migration, TGF‐β
Abbreviations
- APCs
antigen‐presenting immune cells
- CTL
cytotoxic T lymphocytes
- CXADR
coxsackie and adenovirus receptor
- DC
dendritic cells
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- EndoMT
endothelial/mesenchymal transition
- FDCs
follicular dendritic cells
- IVM
intravital two‐photon microscopy
- JAM‐A
junction adhesion molecule A
- LN
lymph node
- MDSC
myeloid‐derived suppressor cells
- PECAM‐1
platelet endothelial cell adhesion molecule 1
- SSM
subcapsular sinus macrophages
- TAMs
tumor‐associated macrophages
- TGF‐β
transforming growth factor beta
- VEGF
vascular endothelial growth factor
1. Introduction
Metastasis is a complex process, during which cancer cells—for not completely known reasons, migrate away from the primary tumor, gain access to the circulation, and subsequently home to distant organs—most frequently the lungs and the liver, where they establish growth. Recent data indicate that the metastatic process is not entirely linked to tumor growth, per se, but is controlled by other factors and can occur from early lesions (Husemann et al., 2008). Although described as a rather inefficient process, the formation of distant metastases is what accounts for the largest numbers of deaths from cancer (Fidler and Balch, 1987; Luzzi et al., 1998). In contrast to locally growing tumors, which generally have better prognosis as they can be removed by surgical resection and/or by irradiation, metastatic cancers are significantly less treatable. Metastatic tumors tend to grow at multiple foci in distant organs and are difficult to remove surgically, or by irradiation. In addition, metastatic cancer cells are often resistant to treatment with chemotherapeutic drugs. This may be explained by the fact that most chemotherapeutic drugs have been developed to specifically target the proliferative capacity of cancer cells rather than their capacity to disseminate and grow at distant sites, which are the characteristic properties of metastatic cancer cells.
Metastatic spread of cancer cells requires dissemination via the systemic circulation. Circulating tumor cells can be found in patients with almost any types of cancer (Allard et al., 2004). However, the subject of how they get access to the circulation is still a matter of debate. Some tumor cells may intravasate into blood vessels within the tumor microenvironment and thereby get direct access to the circulation. Tumor blood vessels induced by overexpression of vascular endothelial growth factor (VEGF) often have discontinuous cell–cell junctions and incomplete coverage of mural cells (Baluk et al., 2005). As such, abnormal blood vessels in tumors have incomplete vascular walls and are leaky and may be used by tumor cells for intravasation. Yet, although angiogenic inhibitors targeting the VEGF pathway are quite efficient in blocking angiogenesis in mouse tumor models, they have shown limited effects on cancer progression and survival in human patients (Vasudev and Reynolds, 2014). In fact, it was shown in 2009 that although angiogenic inhibitors targeting the VEGF pathway block tumor growth in mouse models of pancreatic neuroendocrine carcinoma and glioblastoma, they concomitantly elicit tumor adaptation and progression toward higher malignancy grade (Paez‐Ribes et al., 2009), which was evident by increased lymphatic and distant metastasis.
Spread of cancer cells to regional lymph nodes is the most important prognostic factor in many types of cancer including breast cancer, head and neck cancer, prostate cancer, and colorectal cancer. In line with this, lymphatic invasion may be the predominant method of vascular invasion in breast cancer (Gujam et al., 2014; Mohammed et al., 2007). In head and neck cancers, the formation of distant metastasis was for a long time believed to occur through hematogenous dissemination (Calhoun et al., 1994; Leemans et al., 1993; Leon et al., 2000). However, more recent studies show that distant metastasis without lymph node spread is unusual in oral cell squamous carcinomas, and patients staged N0, N1, N2, and N3 (N0: no spread to lymph nodes; N1: spread to one lymph node; N2: spread to more than one lymph nodes; N3: spread to lymph nodes that are more than 6 cm in size) have a 5‐year survival rate of 63%, 32%, 26%, and 11%, respectively (de Jong et al., 2001). Some tumor cells have been shown to migrate into nondraining lymphatic vessels and form tumors, so‐called skip metastases. The incidence of skip metastases was in 1997 reported in 15.8% of 277 tongue cancer cases (Byers et al., 1997).
Considering the construction of the lymphatic system, it may not be surprising that this system represents a major route for dissemination of tumor cells. In contrast to blood vessels, lymphatic vessels are built for transport of fluid and cells—away from tissues. Blind‐ended lymphatic capillaries are formed by oak leaf‐shaped lymphatic endothelial cells overlapping each other (Oliver and Detmar, 2002). When the interstitial pressure increases in tissues, the space between the endothelial cells widens, which creates the formation of gaps that allow influx of interstitial fluid (Gerli et al., 2000). The creation of this one‐way drainage system, allowing fluid to flow in but not out, was previously interpreted as a defect of endothelial cells lacking intercellular junctions. This theory fell short when gene profiling data demonstrated the expression of endothelial cell–cell adhesion molecules, such as platelet endothelial cell adhesion molecule 1 (PECAM‐1), junction adhesion molecule A (JAM‐A), and occludin (Kriehuber et al., 2001; Podgrabinska et al., 2002). The final solution of how intercellular junctions are organized in lymphatic capillaries came through high‐resolution imaging studies that demonstrated the existence of button‐like junctions allowing gaps to form and support intravasation of fluid and cells while remaining intact (Baluk et al., 2007).
Thus, one reason for why cancer cells may choose lymphatic vessels over blood vessels for intravasation is because lymphatic capillaries are structurally organized to support cellular trafficking away from tissues. Yet, another important question is how cancer cells find their way to lymphatic vessels—do they simply migrate stochastically within the tumor microenvironment until they randomly come in contact with a lymphatic vessel? Or, do mechanisms exist that actively promote targeted migration of cancer cells toward lymphatic vessels?
2. Immune cell trafficking through the lymphatic system
Lymphatic vessels are not only serving as a transport system for fluid and cells but represent an important component of the immune system (Kataru et al., 2014). The lymphatic system functions as an interface between the innate and adaptive immunity, and actively communicates and senses inflammatory stimuli from the periphery (Shields, 2011). The immune response, which is mounted in draining lymph nodes, plays an important role in coordinating the local response toward the cause of the inflammatory response, and promotes tissue‐specific immunity. An essential part of the process of mounting of an adaptive immune response in lymph nodes is the activation and targeted migration of antigen‐presenting immune cells (APCs) from the periphery and through the lymphatic system. Important cells in this respect are dendritic cells (DC) that possess phagocytic and antigen presentation abilities (Randolph et al., 2005). Although DCs are not the only migratory cells, they are at the core of the activation of the adaptive immune system. At steady state, they are also important in promoting peripheral tolerance and thus have a dual role (Steinman and Nussenzweig, 2002). Upon activation, DCs acquire migratory properties characterized by distinct polarization of cellular actin machineries (Vargas et al., 2016). The migratory capacity of DCs is influenced by their subtype and by inflammatory cues (Worbs et al., 2017). Activated DCs in peripheral tissues do not migrate randomly but rather, in a targeted fashion toward lymphatic vessels, and further toward draining lymph nodes. An important driver of the targeted migration of DCs through the lymphatic system is the chemokine CCL21, which is produced and secreted by lymphatic endothelial cells, and its receptor CCR7, which is expressed at the surface of activated DCs. Through their surface expression of CCR7, activated DCs have the capacity to sense and migrate toward lymphatic endothelial cells expressing CCL21.
To generate an adaptive immune response that includes antibody production, antigens also need to reach the follicular dendritic cells (FDCs) that reside in the B‐cell follicles and are important for B‐cell activation. In this respect, a number of mechanisms are involved including active transport by phagocytes to the lymph nodes (Heesters and Carroll, 2016). However, studies have also shown that small soluble antigens can reach lymph nodes even without antigen‐presenting cells (Sixt et al., 2005). Such antigens are processed and transported by subcapsular sinus macrophages (SSM) and B cells to FDCs. The SSM are tissue‐resident macrophages and are positioned so that they can stretch between the lymphatic endothelial cells to capture antigen. Alternatively, antigens may flow into the conduit network, which makes up a tubular system within lymph nodes allowing antigens below 70 kD to enter and be taken up by resident phagocytes and FDCs (Gonzalez et al., 2011). After the B cell has picked up its specific antigen from the FDC, it is processed and presented to T cells activated by DCs. This is a key event and the start of the germinal center reaction, which leads to clonal selection and subsequently specific antibody production. Activated effector lymphocytes can exit the lymph nodes and disseminate into blood vessels (Weisel et al., 2016). These cells can subsequently traffic to the inflamed site to perform cytotoxicity or, in the case of B cells, remain in secondary lymphoid organs to produce antibodies, or migrate to the bone marrow to become long‐lived plasma cells (Roth et al., 2014). Thus, immune cells interact with lymphatic vessels in tissues and lymph nodes. They also secrete factors that may induce remodeling of the lymphatic system through lymphangiogenesis, which is frequently observed in chronic inflammatory and cancer diseases (Kataru et al., 2014). Expansion of the lymphatic vasculature in tumors through lymphangiogenesis is linked to enhanced lymph metastasis (Ji, 2012; Zumsteg and Christofori, 2012).
2.1. EMT in cancer metastasis
Epithelial‐mesenchymal transition is a developmental process whereby epithelial cells transdifferentiate into mesenchymal cells, migrate to other regions of the embryo, and form new cell types (Nieto et al., 2016). Similar to other developmental processes, like angiogenesis and lymphangiogenesis, EMT is silent in normal, healthy tissues. However, reactivation of EMT occurs in pathological conditions including chronic inflammation, fibrosis, wound healing, and cancer (comprehensively reviewed in (Kalluri and Weinberg, 2009; Nieto et al., 2016; Thiery et al., 2009). Epithelial cells undergoing EMT lose epithelial characteristics, such as components of tight and adherens junctions, and apico‐basal cell polarity, and the cytoskeleton is reorganized to support a more migratory state. Loss of E‐cadherin expression, one of the most frequently used markers for EMT, is associated with a more invasive phenotype in many types of human carcinomas (Frixen et al., 1991). The regulation of E‐cadherin has been widely studied, and various transcription factors known for inducing EMT, including Snail1/2, ZEB1/2, and Twist1/2 factors, function as direct or indirect repressors of E‐cadherin (Lamouille et al., 2014; Nieto and Cano, 2012). The expression of these EMT factors is regulated by signaling pathways involved in cellular stemness and transformation, including Ras, Notch, and WNT signaling, and is often associated with poor prognosis and tumor recurrence (Elloul et al., 2005; Fuxe et al., 2010; Martin et al., 2005; Moody et al., 2005). Loss of tight junction proteins including the coxsackie and adenovirus receptor (CXADR) and occludin, are other hallmarks of EMT, and contributes to loss of the epithelial barrier (De Craene and Berx, 2013; Fuxe et al., 2010; Vincent et al., 2009). Increased expression of mesenchymal proteins, such as the intermediate filament protein vimentin, contributes to the enhanced migratory properties of EMT cells (Lamouille et al., 2014).
In addition to being more migratory, EMT cells display other properties associated with cancer progression and metastasis. As such, EMT cells can avoid senescence. Twist factors can disarm oncogene‐induced senescence by inhibiting the tumor suppressor proteins p16 and p21, alongside inducing EMT (Ansieau et al., 2008). Furthermore, early studies provided evidence for the role of EMT in drug resistance (Sommers et al., 1992). High levels of vimentin were detected in adriamycin‐resistant cell lines, where some cells in a heterogenic population possessed typical EMT features. This suggested that EMT cells have advantages in growth capabilities compared to non‐EMT cells after drug treatment. Drug resistance is often accompanied with EMT and has been reported in several cancer types like breast and pancreatic cancer (Singh and Settleman, 2010). High levels of E‐cadherin are associated with sensitivity to epidermal growth factor receptor (EGFR) kinase inhibitors, whereas the opposite is true for cells with low levels of E‐cadherin and a mesenchymal phenotype. Cells expressing EMT markers have also been shown to have a higher resistance to drugs such as oxaliplatin and paclitaxel (Kajiyama et al., 2007; Yang et al., 2006). Recent studies using animal models emphasize the role of EMT as an important mechanism of chemoresistance (Fischer et al., 2015; Shibue and Weinberg, 2017; Zheng et al., 2015).
2.2. Inflammation as an EMT inducer
Recently, much attention has been drawn to the inflammatory milieu within tumors, and infiltrating immune cells have been found to play both anti‐ and protumorigenic roles. Subtypes of tumor‐associated macrophages (TAMs), regulatory T cells, and myeloid‐derived suppressor cells (MDSC) have been identified as promoters of tumor progression and metastasis. These cells produce and secrete cytokines such as transforming growth factor beta (TGF‐β), which may induce EMT, and contribute to an immunosuppressive tumor microenvironment by switching the polarization of immune cells (Fuxe and Karlsson, 2012).
Transforming growth factor beta‐β is frequently overexpressed in cancer and inflammatory tissues (Lamouille et al., 2014; Massague, 2008; Moustakas and Heldin, 2016). The TGF‐β paradox refers to its function both as a tumor suppressor and as a promoter. Normally, TGF‐β controls tissue homeostasis by coordinating apoptosis, and inhibits incipient tumor growth (Massague, 2008). However, during tumor progression, TGF‐β switches from a tumor suppressor to a promoter of EMT and resistance to cell death. Breast cancer cells treated with TGF‐β for several weeks stay in EMT and avoid apoptosis (Gal et al., 2008). TGF‐β signals through a canonical Smad signaling pathway, which involves phosphorylation and activation of receptor‐Smads 2/3, which then interact with Smad4. The Smad complex enters the cell nucleus, where it regulates the transcription of TGF‐β target genes. However, Smads have low affinity for DNA binding and need to interact with cofactors to achieve target gene specificity (Massague, 2008). The identification of Snail1, a master regulator of EMT, as a cofactor for Smad3/4 opened up a new understanding of how TGF‐β cooperates with oncogenic pathways like Ras‐MAPK, Notch, and WNT/β‐catenin to induce EMT (Fuxe et al., 2010; Vincent et al., 2009). Interestingly, TGF‐β can also promote endothelial/mesenchymal transition (EndoMT), which also requires Snail1 and Smad transcription factors (Kokudo et al., 2008; Mihira et al., 2012). EndoMT may contribute to the generation of fibroblasts/myofibroblasts in fibrotic diseases (Piera‐Velazquez et al., 2016), but its role in cancer metastasis is not clear.
2.3. EMT cells hijack the immune system to disseminate through the lymphatic system
It has not been clear what drives cancer cells to disseminate through the lymphatic system, and whether EMT contributes to this process. However, recent data have shed some new light on this question. Our experiments demonstrated that cancer cells that undergo TGF‐β‐induced EMT acquire an enhanced capacity to disseminate through the lymphatic system (Pang et al., 2016). Using a novel three‐dimensional coculture system, in which EMT cells grown on polymeric beads were analyzed for their capacity to migrate toward beads coated with either blood or lymphatic endothelial cells, we found that EMT cells preferentially migrate toward lymphatic compared to blood endothelial cells. These data indicated that lymphatic endothelial cells may produce chemotactic factors that attract EMT cells. Considering the well‐known role of the chemokine receptor CCR7 and its ligand CCL21 in promoting dissemination of DCs through the lymphatic system, we studied whether CCR7/CCL21‐mediated chemotaxis could play a role in targeting EMT cells to lymph nodes. The CCR7 receptor is functioning as an activator of DCs and lymphocytes, and plays an important role for targeted DC migration through the lymphatic system to lymph nodes (Platt and Randolph, 2013). CCR7 has also been shown to mediate lymphatic dissemination of cancer cells (Cunningham et al., 2010; Shields et al., 2007).
In our studies, we found that CCR7 was induced in cancer cells undergoing TGF‐β‐induced EMT (Pang et al., 2016). Further studies revealed that CCR7 mediated lymph node metastasis of EMT cells. The ligand CCL21 is expressed on lymphatic endothelial cells and serves as an important chemoattractant for migrating DCs. We found that CCL21 produced by lymphatic endothelial cells was important for the capacity of EMT cells to migrate toward lymphatic endothelial cells. Moreover, the data showed that TGF‐β induced the expression of CCL21 in lymphatic, but not in blood endothelial cells. Thus, TGF‐β‐induced EMT in cancer cells appears to activate a mechanism used by the immune system to direct migration of DCs through the lymphatic system (Fig. 1). In line with this, others have found that lymphatic endothelial cells may express other chemokines used by tumor cells for lymph node metastasis (Table 1). The CXCR5 receptor and its ligand CXCL13 have been linked to EMT and lymph metastasis in breast cancer (Biswas et al., 2014). The chemokine CXCL12 was found to be upregulated in the lymphatic endothelium of draining lymph nodes, alongside migrating tumor cells expressing the CXCR4 receptor (Hirakawa et al., 2009), and this system is linked to TGF‐β‐induced EMT (Bertran et al., 2009; Taki et al., 2008). Another chemokine, CCL1, was shown to promote lymph node entry of melanoma cells expressing the receptor CCR8, whereas blocking of CCR8 function resulted in decreased lymph node metastasis (Das et al., 2013). Inhibition of CCR8 additionally prevented tumor cells from lymph node entry, creating an arrest of tumor cells in the afferent lymphatic vessels. Together, these results indicate that TGF‐β‐induced EMT provides cancer cells with the capacity to hijack various chemotactic signals and use them for dissemination through the lymphatic system similar to activated DCs during inflammation.
Figure 1.
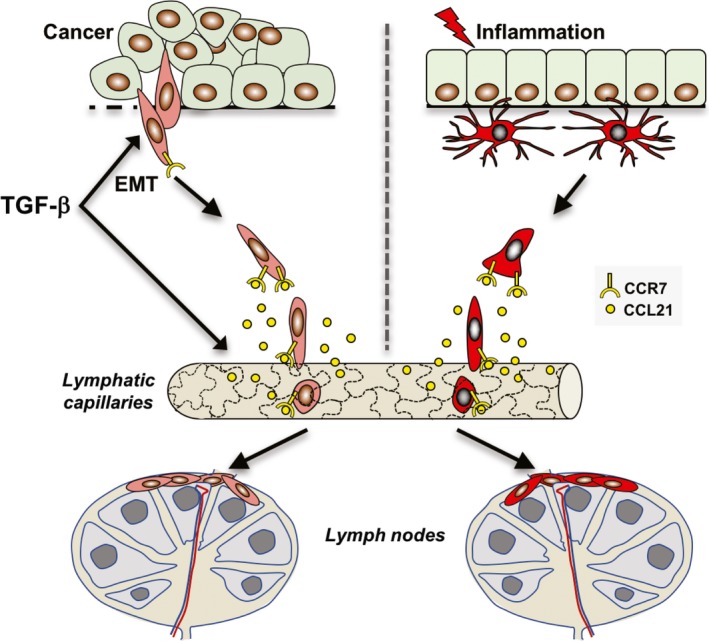
Cancer cells undergoing TGF‐β‐induced EMT acquire properties of immune cells allowing them to disseminate through the lymphatic system similar to DCs during inflammation. Upon activation, DCs acquire cell surface expression of the chemokine receptor CCR7, which allows them to sense and migrate in a targeted fashion toward lymphatic capillaries secreting the CCR7 ligand, CCL21. Endothelial cells of lymphatic capillaries are oak leaf‐shaped and are connected through button‐like junctions, which allows cells to intravasate without junctional disorganization. Subsequently, DCs migrate to lymph nodes where they interact with other cells of the immune system and perform antigen presentation. Recent studies show that similar to activated DCs, cancer cells undergoing TGF‐β‐induced EMT gain the expression of CCR7 and migrate in a targeted fashion through the lymphatic system (Pang et al., 2016). TGF‐β may also enhance the migration of EMT cells toward lymphatic capillaries by inducing the expression of CCL21 in lymphatic endothelial cells. It remains to be determined how EMT cells migrate and interact with the immune system within draining lymph nodes.
Table 1.
Chemokine receptors and ligands linked to TGF‐β‐induced EMT and lymphatic dissemination of cancer cells
Considering the possibility that cancer cells undergoing EMT may acquire additional properties of immune cells, we performed gene profiling analysis of breast cancer cells that had been allowed to adopt an EMT program after treatment with TGF‐β for 2 weeks. Intriguingly, a cluster of genes, which are normally expressed in myeloid type of immune cells including macrophages and DCs, were induced in the EMT cells (Johansson et al., 2015). These genes encode proteins with variable intracellular localization and molecular function, and while some of them previously have been linked to cancer metastasis, others have not. Our conclusion from these studies is that cancer cells undergoing TGF‐β‐induced EMT adopt features of immune cells and that the term ‘mesenchymal’ actually is insufficient to describe the properties of EMT cells. It will be interesting for future studies to further examine to what extent EMT cells make use of immune cell properties to disseminate to lymph nodes and further to distant sites. Intriguingly, the only cells in human bodies that actually share the capacity with prometastatic cancer cells to disseminate through the vascular systems and home to distant organs are immune cells.
3. New technical advances to study metastasis
Sites of regional lymph node spread have been termed ‘bridgeheads’, suggesting that they serve as bridges assisting further spread (Sleeman, 2000). However, the mechanisms by which cancer cells migrate both within lymph nodes and further to other sites remain poorly studied. We foresee that the rapidly evolving field of tumor imaging will lead to better insight into this. Macroscopic imaging techniques such as MRI, CT, and PET are mostly applied to clinical practice and allow the visualization of tumors as well as the evaluation of cancer therapy. Microscopic techniques, instead, allow the characterization of molecular mechanisms that underlie metastasis, and cell–cell interactions in vivo, which are needed to study the dynamic behavior of metastatic cells (Condeelis and Weissleder, 2010).
Advances in photonics have enabled the development of intravital two‐photon microscopy (IVM), a revolutionary technique that allows the study of cell migration in living organisms at cellular and subcellular resolution (Stein and Gonzalez, 2017). The advantage of this technique, compared to classical imaging methods, resides in the use of low‐energy infrared photons that allow the visualization of cells at a greater specimen depth (typically from 100 to 1000 μm), while minimizing tissue photodamage and photobleaching caused by exposure to laser radiation during the image acquisition process (Warren et al., 2003). Moreover, IVM has benefited from the generation of constitutive or inducible fluorescent reporter mouse strains, which have been used for the study of different processes such as the visualization of immune cell interactions (Niesner and Hauser, 2011), the study of the structure and morphology of the lymphatic system (Choi et al., 2011), and the oncogenic transformation and tumor regression in vivo (Ventura et al., 2007). The application of this powerful technology to cancer studies has provided new insights into the spatiotemporal dynamics of tumor progression and the understanding of how the interaction between tumor cells and the stromal compartment influences this process.
However, in order to apply IVM, it is necessary to develop proper microsurgical animal models that combine a minimally invasive surgery, while keeping the organ completely immobilized, which is required for the stable acquisition of micrographs. One of the most commonly used models in IVM is the mouse popliteal lymph node (LN) (Fig. 2) developed by Mempel and colleagues more than a decade ago (Mempel et al., 2004). The selection of this organ as the model to study cell migration is associated with its prominent lymphatic drainage that originates from the mouse footpad (Fig. 2A), and facilitates the direct observation of migratory cells from the injection site to the LN (Fig. 2B; (1) and (2), respectively) (Stein and Gonzalez, 2017). Additionally, the development of transgenic fluorescent reporter mouse strains, such as the Prox1‐GFP (Choi et al., 2011), in which lymphatic endothelial cells are labeled through expression of GFP, has been a valuable tool to study cell migration through the lymphatic system (Fig. 2A).
Figure 2.
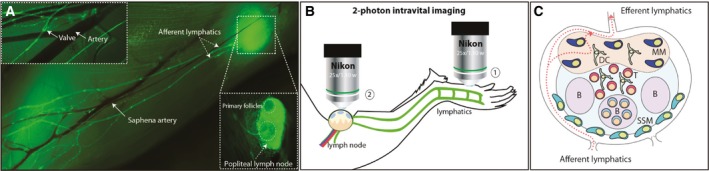
Intravital imaging techniques to study lymphatic dissemination of cancer cells. (A) Photomerge composition of lymphatic and blood circulatory systems of the lower hind limb of a Prox1‐GFP mouse. In the upper box, white arrows indicate a lymphatic valve and the saphena artery. In the main figure, white arrows indicate the afferent vessels, the saphena artery, and the popliteal lymph node (LN). (B) Schematic representation of the surgical intravital two‐photon model in which two different areas of the limb are imaged. On the one hand, the injection site, located in the footpad (1), and on the other hand, the popliteal area, where the arrival of the cancer cells is monitored (2). After administration of anesthesia, the animal is immobilized, the popliteal LN is surgically exposed, and interactions between the cancer and immune cells are recorded using intravital two‐photon microscopy. (C) Schematic representation of the most representative areas and immune cell populations in the popliteal LN. SSM, subcapsular sinus macrophages; MM, medullary macrophages; DC, dendritic cells; B, B‐cell follicle.
Despite being one of the key indicators of tumor aggressiveness, the mechanisms of lymphatic dissemination of tumor cells remain elusive. To date, only relatively few studies have focused on the migration of cancerous cells through the lymphatic system using IVM models. Recently, Das et al. (2013) monitored, using IVM, the metastasis of melanoma cells in the draining LN. The authors observed that the egress of the tumor cells from the afferent lymphatics to the subcapsular sinus area of the LN is an active mechanism directed by the chemokine CCL1. Secretion of the latter by lymphatic endothelial cells at the junction between the afferent lymphatics and the subcapsular sinus allows the entry of CCR8‐expressing tumor cells into the node and their subsequent metastasis.
Other studies have concentrated in the in vivo examination of the encounter of the migratory tumor cell by the immune system. The LN is a highly compartmentalized organ with specific cell groups populating its different areas (Fig. 2C). Among them, subcapsular sinus macrophages (SCS), located next to the afferent lymphatics, act as ‘flypaper’, preventing the dissemination of antigen and pathogens (review in Stein and Gonzalez, 2017). Moalli and colleagues have recently characterized a mechanism by which tumor‐derived antigen (TDA) induces humoral immune responses in tumor‐bearing hosts (Moalli et al., 2015). Using IVM, the authors reported that the capture of TDA was associated with SSM and B cells, and they were both involved in the transport of TDA to the follicular dendritic cell network, located inside the follicle. On the other hand, Junankar et al. (2015) found that the antitumoral action of bisphosphonates, a drug commonly used for the treatment of osteoporosis, was related to the elimination of tumor‐associated macrophages that facilitate tumor progression (Junankar et al., 2015).
Furthermore, other studies focusing on the interactions of effector immune cells with tumor cells in the LN have also been performed using IVM. Deguine and colleagues found that natural killer cells established mainly dynamic contacts with their target cells during tumor regression, while, conversely, cytotoxic T lymphocytes (CTL) formed stable contacts with tumor cells that expressed their cognate antigen (Deguine et al., 2010). Similarly, Boisonnas and colleagues imaged the arresting and killing of tumor cells by adoptively transferring CTL that recognized the expression of cognate antigen. Interestingly, the authors found that CTL infiltrate tumors in depth only when the tumor cells express the cognate CTL antigen. Conversely, the infiltration of CTL was restricted to peripheral regions in tumors that do not express the cognate antigen (Boissonnas et al., 2007).
These studies have demonstrated the power of IVM as a unique technique to elucidate in vivo the mechanisms behind tumor progression. This could also be applied to study the effects on tumors when using checkpoint inhibitors (Sharma and Allison, 2015). Currently, this treatment that targets T cells is changing the way cancer is treated, and there is a drive to enhance the effect of this type of therapy to benefit more patients (Khalil et al., 2016). IVM will be important to evaluate how immunotherapy affects infiltration of CTLs and other immune cells in the TME. Besides activation of T‐cell responses, modulation of other immune cells within the stromal compartment including tumor‐associated macrophages (TAM) is an attractive option (Ngambenjawong et al., 2017). Also here, IVM could be used to investigate the dynamics of CTL responses when altering the microenvironment. Future IVM studies will provide further insights into the dynamics of lymphatic dissemination and metastasis in different models of cancer, opening the door to the development of novel strategies intended to control the progression of the disease.
Acknowledgements
The Fuxe laboratory is supported by the Swedish Research Council (2014‐3114), the Swedish Cancer Society (CAN 2015‐773), and the Karolinska Institutet. The Karlsson laboratory is supported by the Swedish Research Council, the Swedish Cancer Society, the Magnus Bergvall Foundation, and the Karolinska Institutet. The Gonzalez laboratory is supported by Swiss National Foundation grants R‐equipt (145038) and Ambizione (148183) and a European Commission Marie Curie Reintegration Grant (612742).
References
- Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW and Terstappen L (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S et al (2008) Induction of emt by twist proteins as a collateral effect of tumor‐promoting inactivation of premature senescence. Cancer Cell 14, 79–89. [DOI] [PubMed] [Google Scholar]
- Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E et al (2007) Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 204, 2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Hashizume H and McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15, 102–111. [DOI] [PubMed] [Google Scholar]
- Bertran E, Caja L, Navarro E, Sancho P, Mainez J, Murillo MM, Vinyals A, Fabra A and Fabregat I (2009) Role of cxcr4/sdf‐1 alpha in the migratory phenotype of hepatoma cells that have undergone epithelial‐mesenchymal transition in response to the transforming growth factor‐beta. Cell Signal 21, 1595–1606. [DOI] [PubMed] [Google Scholar]
- Biswas S, Sengupta S, Roy Chowdhury S, Jana S, Mandal G, Mandal PK, Saha N, Malhotra V, Gupta A, Kuprash DV et al (2014) Cxcl13‐cxcr5 co‐expression regulates epithelial to mesenchymal transition of breast cancer cells during lymph node metastasis. Breast Cancer Res Treat 143, 265–276. [DOI] [PubMed] [Google Scholar]
- Boissonnas A, Fetler L, Zeelenberg IS, Hugues S and Amigorena S (2007) In vivo imaging of cytotoxic t cell infiltration and elimination of a solid tumor. J Exp Med 204, 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers RM, Weber RS, Andrews T, McGill D, Kare R and Wolf P (1997) Frequency and therapeutic implications of ‘skip metastases’ in the neck from squamous carcinoma of the oral tongue. Head Neck 19, 14–19. [DOI] [PubMed] [Google Scholar]
- Calhoun KH, Fulmer P, Weiss R and Hokanson JA (1994) Distant metastases from head and neck squamous‐cell carcinomas. Laryngoscope 104, 1199–1205. [DOI] [PubMed] [Google Scholar]
- Carter CL, Allen C and Henson DE (1989) Relation of tumor size, lymph‐node status, and survival in 24,740 breast‐cancer cases. Cancer 63, 181–187. [DOI] [PubMed] [Google Scholar]
- Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B et al (2011) Visualization of lymphatic vessels by prox1‐promoter directed gfp reporter in a bacterial artificial chromosome‐based transgenic mouse. Blood 117, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J and Weissleder R (2010) In vivo imaging in cancer. Cold Spring Harbor Persp Biol 2, a003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham HD, Shannon LA, Calloway PA, Fassold BC, Dunwiddie I, Vielhauer G, Zhang M and Vines CM (2010) Expression of the c‐c chemokine receptor 7 mediates metastasis of breast cancer to the lymph nodes in mice. Translat Oncol 3, 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Sarrou E, Podgrabinska S, Cassella M, Mungamuri SK, Feirt N, Gordon R, Nagi CS, Wang YR, Entenberg D et al (2013) Tumor cell entry into the lymph node is controlled by ccl1 chemokine expressed by lymph node lymphatic sinuses. J Exp Med 210, 1509–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene B and Berx G (2013) Regulatory networks defining emt during cancer initiation and progression. Nat Rev Cancer 13, 97–110. [DOI] [PubMed] [Google Scholar]
- Deguine J, Breart B, Lemaitre F, Di Santo JP and Bousso P (2010) Intravital imaging reveals distinct dynamics for natural killer and cd8(+) t cells during tumor regression. Immunity 33, 632–644. [DOI] [PubMed] [Google Scholar]
- Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R and Davidson B (2005) Snail, slug, and smad‐interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 103, 1631–1643. [DOI] [PubMed] [Google Scholar]
- Elston CW and Ellis IO (1991) Pathological prognostic factors in breast‐cancer.1. The value of histological grade in breast‐cancer ‐ experience from a large study with long‐term follow‐up. Histopathology 19, 403–410. [DOI] [PubMed] [Google Scholar]
- Fidler IJ and Balch CM (1987) The biology of cancer metastasis and implications for therapy. Curr Probl Surg 24, 137–209. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D and Birchmeier W (1991) E‐cadherin‐mediated cell cell‐adhesion prevents invasiveness of human carcinoma‐cells. J Cell Biol 113, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxe J and Karlsson MC (2012) Tgf‐beta‐induced epithelial‐mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol 22, 455–461. [DOI] [PubMed] [Google Scholar]
- Fuxe J, Vincent T and Garcia de Herreros A (2010) Transcriptional crosstalk between tgf‐beta and stem cell pathways in tumor cell invasion: Role of emt promoting smad complexes. Cell Cycle 9, 2363–2374. [DOI] [PubMed] [Google Scholar]
- Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H and Moustakas A (2008) Sustained tgf beta exposure suppresses smad and non‐smad signalling in mammary epithelial cells, leading to emt and inhibition of growth arrest and apoptosis. Oncogene 27, 1218–1230. [DOI] [PubMed] [Google Scholar]
- Gerli R, Solito R, Weber E and Agliano M (2000) Specific adhesion molecules bind anchoring filaments and endothelial cells in human skin initial lymphatics. Lymphology 33, 148–157. [PubMed] [Google Scholar]
- Gonzalez SF, Degn SE, Pitcher LA, Woodruff M, Heesters BA and Carroll MC (2011) Trafficking of b cell antigen in lymph nodes. Annu Rev Immunol 29, 215–233. [DOI] [PubMed] [Google Scholar]
- Gujam FJ, Going JJ, Edwards J, Mohammed ZM and McMillan DC (2014) The role of lymphatic and blood vessel invasion in predicting survival and methods of detection in patients with primary operable breast cancer. Crit Rev Oncol Hematol 89, 231–241. [DOI] [PubMed] [Google Scholar]
- Heesters BA and Carroll MC (2016) The role of dendritic cells in s. Pneumoniae transport to follicular dendritic cells. Cell Rep 16, 3130–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa S, Detmar M, Kerjaschki D, Nagamatsu S, Matsuo K, Tanemura A, Kamata N, Higashikawa K, Okazaki H, Kameda K et al (2009) Nodal lymphangiogenesis and metastasis role of tumor‐induced lymphatic vessel activation in extramammary Paget's disease. Am J Pathol 175, 2235–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, RiethmUller G et al (2008) Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68. [DOI] [PubMed] [Google Scholar]
- Ji RC (2012) Macrophages are important mediators of either tumor‐ or inflammation‐induced lymphangiogenesis. Cell Mol Life Sci 69, 897–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson J, Tabor V, Wikell A, Jalkanen S and Fuxe J (2015) Tgf‐β1‐induced epithelial‐ mesenchymal transition promotes monocyte/macrophage properties in breast cancer cells. Front Oncol 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong RJB, Hermans J, Molenaar J, Briaire JJ and le Cessie S (2001) Prediction of survival in patients with head and neck cancer. Head Neck 23, 718–724. [DOI] [PubMed] [Google Scholar]
- Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, Parker A, Nguyen A, Sun S, Kashemirov B et al (2015) Real‐time intravital imaging establishes tumor‐associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov 5, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A and Kikkawa F (2007) Chemoresistance to paclitaxel induces epithelial‐mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 31, 277–283. [PubMed] [Google Scholar]
- Kalluri R and Weinberg RA (2009) The basics of epithelial‐mesenchymal transition. J Clin Investig 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataru RP, Lee YG and Koh GY (2014) Interactions of immune cells and lymphatic vessels. Adv Anat Embryol Cell Biol 214, 107–118. [DOI] [PubMed] [Google Scholar]
- Khalil DN, Smith EL, Brentjens RJ and Wolchok JD (2016) The future of cancer treatment: Immunomodulation, cars and combination immunotherapy. Nat Rev Clin Oncol 13, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T and Miyazono K (2008) Snail is required for tgfbeta‐induced endothelial‐mesenchymal transition of embryonic stem cell‐derived endothelial cells. J Cell Sci 121, 3317–3324. [DOI] [PubMed] [Google Scholar]
- Kriehuber E, Breiteneder‐Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D and Maurer D (2001) Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med 194, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J and Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDran H, Gataker T, Chelsden W, Hitch C and Dosley R (1752) Traite des operations de chirurgie (English translation Gataker), London. [Google Scholar]
- Leemans CR, Tiwari R, Nauta JJP, Vanderwaal I and Snow GB (1993) Regional lymph‐node involvement and its significance in the development of distant metastases in head and neck‐carcinoma. Cancer 71, 452–456. [DOI] [PubMed] [Google Scholar]
- Leon X, Quer M, Orus C, Venegas MD and Lopez M (2000) Distant metastases in head and neck cancer patients who achieved loco‐regional control. Head Neck 22, 680–686. [DOI] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF and Groom AC (1998) Multistep nature of metastatic inefficiency ‐ dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TA, Goyal A, Watkins G and Jiang WG (2005) Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol 12, 488–496. [DOI] [PubMed] [Google Scholar]
- Massague J (2008) Tgf beta in cancer. Cell 134, 215–230.18662538 [Google Scholar]
- Mempel TR, Henrickson SE and Von Andrian UH (2004) T‐cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159. [DOI] [PubMed] [Google Scholar]
- Mihira H, Suzuki HI, Akatsu Y, Yoshimatsu Y, Igarashi T, Miyazono K and Watabe T (2012) Tgf‐beta‐induced mesenchymal transition of ms‐1 endothelial cells requires smad‐dependent cooperative activation of rho signals and mrtf‐a. J Biochem 151, 145–156. [DOI] [PubMed] [Google Scholar]
- Moalli F, Proulx ST, Schwendener R, Detmar M, Schlapbach C and Stein JV (2015) Intravital and whole‐organ imaging reveals capture of melanoma‐derived antigen by lymph node subcapsular macrophages leading to widespread deposition on follicular dendritic cells. Front Immunol 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed RA, Martin SG, Gill MS, Green AR, Paish EC and Ellis IO (2007) Improved methods of detection of lymphovascular invasion demonstrate that it is the predominant method of vascular invasion in breast cancer and has important clinical consequences. Am J Surg Pathol 31, 1825–1833. [DOI] [PubMed] [Google Scholar]
- Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD and Chodosh LA (2005) The transcriptional repressor snail promotes mammary tumor recurrence. Cancer Cell 8, 197–209. [DOI] [PubMed] [Google Scholar]
- Moustakas A and Heldin CH (2016) Mechanisms of tgfbeta‐induced epithelial‐mesenchymal transition. J Clin Med 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngambenjawong C, Gustafson HH and Pun SH (2017) Progress in tumor‐associated macrophage (tam)‐targeted therapeutics. Adv Drug Deliv Rev https://doi.org/10.1016/j.addr.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesner RA and Hauser AE (2011) Recent advances in dynamic intravital multi‐photon microscopy. Cytometry A 79, 789–798. [DOI] [PubMed] [Google Scholar]
- Nieto MA and Cano A (2012) The epithelial‐mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol 22, 361–368. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA and Thiery JP (2016) Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Oliver G and Detmar M (2002) The rediscovery of the lymphatic system: Old and new insights into the development and biological function of the lymphatic vasculature. Genes Dev 16, 773–783. [DOI] [PubMed] [Google Scholar]
- Paez‐Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D and Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang MF, Georgoudaki AM, Lambut L, Johansson J, Tabor V, Hagikura K, Jin Y, Jansson M, Alexander JS, Nelson CM et al (2016) Tgf‐beta1‐induced emt promotes targeted migration of breast cancer cells through the lymphatic system by the activation of ccr7/ccl21‐mediated chemotaxis. Oncogene 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piera‐Velazquez S, Mendoza FA and Jimenez SA (2016) Endothelial to mesenchymal transition (endomt) in the pathogenesis of human fibrotic diseases. J Clin Med 5, E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AM and Randolph GJ (2013) Dendritic cell migration through the lymphatic vasculature to lymph nodes. Adv Immunol 120, 51–68. [DOI] [PubMed] [Google Scholar]
- Podgrabinska S, Braun P, Velasco P, Kloos B, Pepper MS, Jackson DG and Skobe M (2002) Molecular characterization of lymphatic endothelial cells. Proc Natl Acad Sci U S A 99, 16069–16074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Angeli V and Swartz MA (2005) Dendritic‐cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol 5, 617–628. [DOI] [PubMed] [Google Scholar]
- Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R and Hauser AE (2014) Tracking plasma cell differentiation and survival. Cytometry A 85, 15–24. [DOI] [PubMed] [Google Scholar]
- Sharma P and Allison JP (2015) The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Shibue T and Weinberg RA (2017) Emt, cscs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol. https://doi.org/10.1038/nrclinonc.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields JD (2011) Lymphatics: at the interface of immunity, tolerance, and tumor metastasis. Microcirculation 18, 517–531. [DOI] [PubMed] [Google Scholar]
- Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ and Swartz MA (2007) Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine ccr7 signaling. Cancer Cell 11, 526–538. [DOI] [PubMed] [Google Scholar]
- Singh A and Settleman J (2010) Emt, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt M, Kanazawa N, Seig M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB and Sorokin L (2005) The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the t cell area of the lymph node. Immunity 22, 19–29. [DOI] [PubMed] [Google Scholar]
- Sleeman JP (2000) The lymph node as a bridgehead in the metastatic dissemination of tumors. Lymph Metastas Sent Lymphonodect 157, 55–81. [DOI] [PubMed] [Google Scholar]
- Sommers CL, Heckford SE, Skerker JM, Worland P, Torri JA, Thompson EW, Byers SW and Gelmann EP (1992) Loss of epithelial markers and acquisition of vimentin expression in adriamycin‐resistant and vinblastine‐resistant human breast‐cancer cell‐lines. Can Res 52, 5190–5197. [PubMed] [Google Scholar]
- Stein JV and Gonzalez S (2017) Dynamic intravital imaging of cell‐cell interactions in the lymph node. J Allergy Clin Immunol 139, 12–20. [DOI] [PubMed] [Google Scholar]
- Steinman RM and Nussenzweig MC (2002) Avoiding horror autotoxicus: the importance of dendritic cells in peripheral t cell tolerance. Proc Natl Acad Sci U S A 99, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki M, Higashikawa K, Yoneda S, Ono S, Shigeishi H, Nagayama M and Kamata N (2008) Up‐regulation of stromal cell‐derived factor‐1alpha and its receptor cxcr4 expression accompanied with epithelial‐mesenchymal transition in human oral squamous cell carcinoma. Oncol Rep 19, 993–998. [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RYJ and Nieto MA (2009) Epithelial‐mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Vargas P, Maiuri P, Bretou M, Saez PJ, Pierobon P, Maurin M, Chabaud M, Lankar D, Obino D, Terriac E et al (2016) Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol 18, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudev NS and Reynolds AR (2014) Anti‐angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis 17, 471–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R and Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL et al (2009) A snail1‐smad3/4 transcriptional repressor complex promotes tgf‐beta mediated epithelial‐mesenchymal transition. Nat Cell Biol 11, 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WS, Wagner W and Ye T (2003) The prospects for high resolution optical brain imaging: the magnetic resonance perspective. Magn Reson Imaging 21, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino‐Catania GV, Chikina M and Shlomchik MJ (2016) A temporal switch in the germinal center determines differential output of memory b and plasma cells. Immunity 44, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worbs T, Hammerschmidt SI and Forster R (2017) Dendritic cell migration in health and disease. Nat Rev Immunol 17, 30–48. [DOI] [PubMed] [Google Scholar]
- Yang AD, Fan F, Camp ER, van Buren G, Liu WB, Somcio R, Gray MJ, Cheng HY, Hoff PM and Ellis LM (2006) Chronic oxaliplatin resistance induces epithelial‐to‐mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 12, 4147–4153. [DOI] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumsteg A and Christofori G (2012) Myeloid cells and lymphangiogenesis. Cold Spring Harb Perspect Med 2, a006494. [DOI] [PMC free article] [PubMed] [Google Scholar]