

Abstract
Epithelial‐to‐mesenchymal transition (EMT) and its reverse mesenchymal‐to‐epithelial transition (MET) have been suggested to play crucial roles in metastatic dissemination of carcinomas. These phenotypic transitions between states are not binary. Instead, carcinoma cells often exhibit a spectrum of epithelial/mesenchymal phenotype(s). While epithelial/mesenchymal plasticity has been observed preclinically and clinically, whether any of these phenotypic transitions are indispensable for metastatic outgrowth remains an unanswered question. Here, we focus on epithelial/mesenchymal plasticity in metastatic dissemination and propose alternative mechanisms for successful dissemination and metastases beyond the traditional EMT/MET view. We highlight multiple hypotheses that can help reconcile conflicting observations, and outline the next set of key questions that can offer valuable insights into mechanisms of metastasis in multiple tumor models.
Keywords: epithelial‐to‐mesenchymal transition, hybrid epithelial/mesenchymal, mesenchymal‐to‐epithelial transition, metastasis, phenotypic plasticity
Abbreviations
- AT3
anaplastic tumor 3
- CTC
circulating tumor cell
- E
epithelial
- ECM
extracellular matrix
- EMT
epithelial‐to‐mesenchymal transition
- EMT‐TF
EMT‐inducing transcription factor
- Fsp1
fibroblast‐specific protein 1
- M
mesenchymal
- MET
mesenchymal‐to‐epithelial transition
- NEPC
neuroendocrine prostate cancer
- PDAC
pancreatic ductal adenocarcinoma
1. Introduction
Epithelial‐to‐mesenchymal transition (EMT) is a cellular process loosely defined as a loss of the epithelial traits of tight cell–cell adhesion and apico‐basal polarization and a gain of mesenchymal traits of motility and invasion (Savagner, 2015). The concept of EMT evolved from initial observations that embryonic and adult epithelial cells converted to migratory and invasive fibroblast‐like cells when embedded in 3D collagen gels (Greenburg and Hay, 1982). Defined then as a ‘transformation’, EMT has since been well studied in gastrulation, neural crest migration, heart development, branching morphogenesis, wound healing, fibrosis, and cancer metastasis. ‘Transformation’ has given way to ‘transition’ and more recently ‘plasticity’ to accurately represent its reversibility as well as its nonbinary nature (Jolly et al., 2015a; Nieto et al., 2016). In the context of cancer, the hypothesis that EMT and mesenchymal‐to‐epithelial transition (MET) drive the invasion–metastasis cascade (Thiery, 2002) has been pursued enthusiastically for over a decade (Hartwell et al., 2006; Jung et al., 2008; Mani et al., 2007; Ocaña et al., 2012; Onder et al., 2008; Spaderna et al., 2008; Stankic et al., 2013; Tsai et al., 2012; Yang et al., 2004), but recent studies have questioned the indispensability of these transitions in establishing metastasis (Fischer et al., 2015; Shamir et al., 2014; Somarelli et al., 2016a; Zheng et al., 2015). These results have stimulated provocative discussions on what steps are necessary and sufficient to establish macrometastases in vivo. Here, we attempt to reconcile some apparent contradictions, and highlight key unanswered questions that need to be addressed for a better understanding of the contribution of EMT and MET in metastasis in multiple tumor types.
2. EMT and MET are not binary processes
A tacit assumption in the proposed role of EMT and MET during the metastasis–invasion cascade was that, similar to the distinct developmental lineages – epithelium and mesenchyme – carcinoma cells can attain either a fully epithelial or a fully mesenchymal state (Thiery, 2002). This assumption was supported by the labeling of phenotypes co‐expressing canonical epithelial and mesenchymal markers as ‘metastable’, strongly suggesting that these observations were a snapshot en route to full EMT/MET and thus could not reflect a stable state or an end point of a transition in itself (Lee et al., 2006). Only recently has the concept of a hybrid epithelial/mesenchymal (E/M) state been revisited in cancer (Bronsert et al., 2014; Chao et al., 2012; Grosse‐Wilde et al., 2015; Huang et al., 2013; Lecharpentier et al., 2011; McCart Reed et al., 2016; Naber et al., 2013; Sampson et al., 2014; Schliekelman et al., 2015; Strauss et al., 2011), and shown to be stable over multiple passages in vitro (Jolly et al., 2016). This revised understanding of cancer cell plasticity has been at least in part driven by computational modeling efforts of EMT/MET regulatory networks (Jia et al., 2015; Li et al., 2016; Lu et al., 2013; Zadran et al., 2014) that have triggered investigations of single‐cell phenotypes in terms of their EMT status (Andriani et al., 2016; Grosse‐Wilde et al., 2015).
In the context of wound healing and embryonic development, the intermediate state(s) of EMT has (have) been well studied (Arnoux et al., 2008; Futterman et al., 2011; Johnen et al., 2012; Kuriyama et al., 2014; Leroy and Mostov, 2007; Micalizzi et al., 2010; Revenu and Gilmour, 2009; Shaw and Martin, 2016; Somarelli et al., 2013). The idea that EMT need not be an ‘all‐or‐none’ process (Nieto, 2013) has motivated a detailed dissection of different axes that cumulatively define EMT – basement membrane remodeling, motility, cell–cell adhesion, apical constriction, and loss of apico‐basal polarity – in sea urchin embryo. Each of these axes is regulated by a distinct set of transcription factors, and the subcircuits corresponding to each axes are interconnected and overlapping. Intriguingly, no single EMT‐inducing transcription factor (EMT‐TF) is involved in all of these subcircuits, highlighting the complexity of cellular plasticity even in relatively simpler organisms such as sea urchin (Saunders and McClay, 2014). These axes are likely to influence one another, but for the sake of a better comprehension, even if we imagine these axes to be independent, EMT is at least a process happening in a five‐dimensional space (Fig. 1A). Induction of different EMT‐TFs may affect these five subcircuits or axes differently, and thus, there may be varying degrees of overlap in the gene expression profiles obtained after overexpression of EMT‐TFs. For example, as shown in Fig. 1B, EMT‐TF1 and EMT‐TF2 have both overlapping and distinct influences in gene expression landscape, but EMT‐TF3 has no overlap with gene expression changes driven by EMT‐TF1 and EMT‐TF2.
Figure 1.
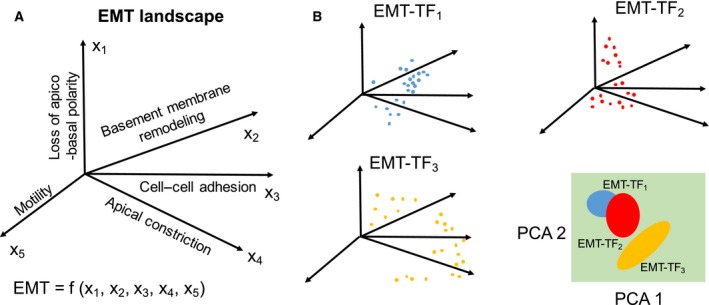
Representing EMT as a multidimensional nonlinear process. (A) EMT phenotypic landscape may contain multiple axes (x1–x5). (B) Induction of EMT by different EMT‐TFs may drive epithelial cells into different regions on this multidimensional landscape (shown by different colored dots). There may be some overlap in the effect of more than one EMT‐TFs in regulation of one or more of these axes contributing to EMT, as can be realized by projecting this multidimensional space into two principal component axes (PCA).
Thus, EMT progression is not a unidimensional linear process, but a navigation through a rugged highly nonlinear landscape (Fig. 1A,B). Also, although assumed here as independent axes, these five aspects of EMT may affect one another too, thus compounding the nonlinearity of the process. Another aspect of complexity underlying EMT may well result from heterogeneity in cancer subtypes; for instance, the landscape underlying this five‐dimensional space may be different for luminal vs. basal connections. Thus, it may be extremely tricky to define EMT, but for practical purposes, we will consider here single‐cell migration and/or invasion with the loss of cell–cell adhesion as EMT, as was postulated initially (reviewed in Cheung and Ewald, 2014). This loss of cell–cell adhesion typically co‐occurs with a decrease in other epithelial traits such as loss of apico‐basal polarity, and a concomitant increase in genes often expressed specifically in mesenchymal cells and tissues (Kalluri and Weinberg, 2009).
Moreover, the epigenetic reprogramming accompanying many of these key developmental events may rewire EMT regulatory networks differently in different tissue types, further amplifying the heterogeneity and context dependence of EMT states. For instance, in breast cancer, basal cells exhibit bivalent chromatin states, with both activating and repressive marks for a key EMT‐TF, ZEB1, but luminal cells only have repressive marks for ZEB1 (Chaffer et al., 2013). Thus, basal cells are already poised to display stronger EMT traits upon exposure to EMT‐inducing cytokines such as TGF‐β, as compared to luminal cells. Similarly, GRHL2 – a transcription factor that can inhibit EMT (Varma et al., 2012; Walentin et al., 2015) – can be methylated in sarcomas as compared to carcinoma (Somarelli et al., 2016b), thus rewiring the circuit regulating EMT in sarcomas.
Therefore, with such ubiquitous tissue‐ or even subtype‐specific complexity and heterogeneity being revealed, binning carcinoma phenotypes into either fully epithelial or fully mesenchymal, and dismissing all hybrid phenotypes as ‘metastable’, can only hamper a better understanding of both the nuances of EMT and MET, and how these processes may impinge on metastasis.
3. Role of EMT‐TFs in metastasis: necessary or permissive?
In the context of cancer, Snail (SNAI1) was identified as the first EMT‐TF that directly repressed transcription of the epithelial cell–cell adhesion molecule, E‐cadherin. Overexpression of SNAI1 in MDCK and many carcinoma cell lines led to the loss of cell–cell adhesion mediated by E‐cadherin, transformed the morphology of cells from epithelial to spindle‐like mesenchymal, and enhanced their migratory and invasive traits in vitro (Batlle et al., 2000; Cano et al., 2000). Further work revealed a similar, but less potent role of another EMT‐TF Slug (SNAI2, a member of the Snail family) both in vitro and in vivo (Bolós et al., 2003; Hajra et al., 2002). SNAI1 was also shown to induce the expression of mesenchymal markers fibronectin and Zeb1 (Guaita et al., 2002), the latter of which is an EMT‐TF that can promote tumor invasiveness in vitro and is correlated with tumor cell differentiation in vivo (Aigner et al., 2007; Spaderna et al., 2008). Later, Twist was identified as yet another EMT‐TF that inhibited E‐cadherin as well as regulated other components of EMT in MDCK cells, mammary epithelial cells (Yang et al., 2004), and breast cancer cell lines (Vesuna et al., 2008). Silencing Twist in 4T1 cells suppressed the number of lung metastases significantly, however, did not completely inhibit them (Yang et al., 2004), still leaving open the possibility that Twist, and potentially other EMT‐TFs, may act more as catalysts rather than drivers of metastasis (Fig. 2A). In other words, just as a catalyst can lower the activation energy barrier for a chemical reaction, these EMT‐TFs may make a cell more poised or prone to undergo EMT. These abovementioned studies confirmed that the EMT‐TFs that governed developmental EMT also contributed to one or more aspects of EMT in carcinomas in vitro, a claim that was substantiated by in vivo negative correlation between these EMT‐TFs and E‐cadherin expression. Thus, these studies led to a conceptual framework suggesting that aberrant activation of one or more EMT‐TFs (resulting from many potential microenvironmental factors such as hypoxia, secreted EMT‐inducing cytokines from the stroma, for example, TGF‐β, or altered degradation rate of EMT‐TFs) was a necessary and sufficient condition for metastasis.
Figure 2.
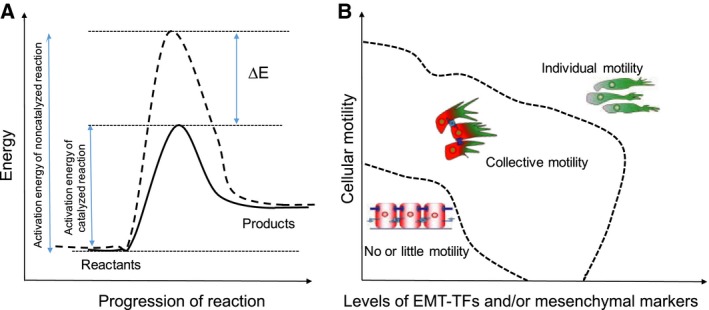
Role of EMT‐TFs. (A) EMT‐TFs can act as catalysts of cellular plasticity. A catalyst reduces the activation energy (by an amount of ∆E) required for the progression of a reaction. (B) Phase diagram showing different types of motility that can be possible at varying levels of EMT‐TFs and/or mesenchymal markers, and cellular motility. Dotted lines represent phase separations.
The conceptual framework that EMT promotes metastasis and invasion was recently challenged by two lineage‐tracing studies in mouse models of pancreatic and breast cancer. Zheng et al. (2015) genetically knocked out either Twist or Snail in a spontaneous pancreatic ductal adenocarcinoma (PDAC) model – KPC model, but the incidence of metastasis was not altered significantly. Multiple alternative interpretations have been proposed for this observation – (a) knockdown of one EMT‐TF need not be sufficient to ablate EMT completely, and compensatory EMT‐inducing pathways may be present (Li and Kang, 2016), (b) the marker used for lineage tracing of cells undergoing EMT in this study – α‐smooth muscle actin – is rarely induced spontaneously upon activation of EMT in this particular mouse model (Pattabiraman and Weinberg, 2017). The other study (Fischer et al., 2015) focused on spontaneous breast‐to‐lung metastasis mouse models and used fibroblast‐specific protein 1 (Fsp1) as a lineage‐tracing marker of cells undergoing an EMT. The authors found many Fsp1‐negative cells metastasizing to lung, suggesting that not even a transient activation of EMT was essential for metastasis. Although the specificity and sensitivity of Fsp1 to mark cells undergoing EMT and/or fibroblasts may be called into question (De Chiara and Crean, 2016; Pattabiraman and Weinberg, 2017), this study also demonstrated that overexpression of miR‐200 suppressed multiple levels of many EMT‐TFs, including ZEB1, yet did not affect lung metastasis (Zheng et al., 2015), thus providing a stronger argument for alternative mechanisms of dissemination.
In contrast, knockdown of Zeb1 in HCT116 and SW480 cells has been shown to inhibit lung metastases after intrasplenic or intravenous injection in nude mice (Spaderna et al., 2008). Similarly, deletion of TWIST1 drastically inhibited lung metastasis of 4T1 cells implanted in the mammary gland of recipient mice (Yang et al., 2004), emphasizing a causal role of EMT‐TFs in metastasis. Technically speaking, these studies were conducted with different approaches compared to spontaneous metastasis genetically engineered mouse models discussed above (Fischer et al., 2015; Zheng et al., 2015). Another recent study performed in the same KPC mouse model illustrates that depletion of ZEB1, in sharp contrast to that of Snail or Twist, suppresses stemness, colonization, invasion, and metastasis (Krebs et al., 2017). However, ZEB1 depletion fails to suppress metastasis completely, thereby falling somewhat short of confirming ZEB1 as necessary and sufficient for establishing metastasis, and leaving open the possibility that other modes of migration may also be important for metastasis, at least in this mouse model.
Together, these data suggest that metastasis for all carcinoma cells need not require an overt upregulation of various EMT markers to gain migratory and invasive traits. For instance, in tumor organoids, breast cancer cells can invade the extracellular matrix (ECM) by three modes – collective invasion, mesenchymal invasion, and amoeboid invasion. In this model system, only cells undergoing mesenchymal invasion utilize an EMT‐like program (Nguyen‐Ngoc et al., 2012). Conversely, collectively invading cells do not typically express vimentin or Twist1 and maintain E‐cadherin‐mediated contacts with follower cells. Rather than undergoing an EMT, the cells undergoing collective invasion appear to undergo a transition toward a more basal‐like phenotype, expressing K14 and p63 (Cheung et al., 2013). Put together, it still remains a possibility that the traits needed for successful metastasis can be gained by altering cellular adhesion and invasion through pathways that do not necessitate supraphysiological or aberrant overexpression of one or more EMT‐TFs identified so far (Fig. 2B). In other words, morphological changes associated with EMT can occur without an overt upregulation of any mesenchymal markers (Cheung and Ewald, 2014). Further, an overt or a complete EMT may not be as efficient for metastasis as the scenario when some molecular and/or morphological epithelial traits are retained (Biddle et al., 2011; Jolly et al., 2015a; Shamir et al., 2014).
4. Has a full EMT ever been seen in vivo?
Recent progress in considering EMT as more of a spectrum of phenotypes instead of a binary process has driven an emerging notion that unlike during development, in which terminally differentiated epithelial and mesenchymal states exist, carcinoma cells might undergo more partial transitions to an incomplete mesenchymal phenotype (Lambert et al., 2017; Nieto et al., 2016). This notion is supported by observations that induction of a fully mesenchymal state through overexpression of an EMT‐TF may lead to a loss of tumor‐initiating potential and thus the ability to colonize (Celià‐Terrassa et al., 2012; Ocaña et al., 2012; Ruscetti et al., 2015; Tsai et al., 2012). Earlier studies based on similar overexpression of EMT‐TFs proposed an increase in tumor‐initiating potential (Mani et al., 2008). Reconciling these contradictions, recent studies that categorized cells into E (epithelial), M (mesenchymal), and hybrid E/M, instead of just E and M, have proposed that tumor‐initiating potential might be maximum when cells are in a hybrid E/M state (Grosse‐Wilde et al., 2015; Jolly et al., 2014; Ombrato and Malanchi, 2014; Ruscetti et al., 2015). Such hybrid E/M cells co‐expressing various epithelial and mesenchymal markers have been observed in breast, ovarian, lung, and renal cell carcinoma cell lines (Andriani et al., 2016; Grosse‐Wilde et al., 2015; Huang et al., 2013; Sampson et al., 2014; Schliekelman et al., 2015), in mouse models of prostate cancer and PDAC (Rhim et al., 2013; Ruscetti et al., 2015), primary breast and ovarian cancer tissue (Strauss et al., 2011; Yu et al., 2013), in the bloodstream of breast, lung, and prostate cancer patients (Armstrong et al., 2011; Lecharpentier et al., 2011; Yu et al., 2013), and in metastatic brain tumors (Jeevan et al., 2016). More importantly, triple‐negative breast cancer patients had a significantly higher number of such hybrid E/M cells as compared to other subtypes, suggesting a correlation between a hybrid E/M phenotype and tumor aggressiveness (Yu et al., 2013).
Although it is likely that many carcinomas undergo only a partial transition, some cancers reflect a more complete phenotypic transition based on typical morphological and molecular readouts. For example, Beerling et al. (2016) identified a rare population of E‐cadlo cells that underwent spontaneous full EMT without any exogenous induction of EMT‐TFs, and converted to an epithelial state upon reaching the metastatic site. Another model system that tends to exhibit a more complete EMT is the Dunning model of prostate cancer that was derived in 1961 from a spontaneous prostate adenocarcinoma in a Copenhagen rat (Dunning, 1963; Issacs et al., 1978). The DT cell line established from this model expresses numerous epithelial biomarkers, including E‐cadherin, claudin 4, and pan‐cytokeratin (Oltean et al., 2008), possesses a cobblestone‐like appearance (Oltean et al., 2006; Somarelli et al., 2013, 2016a), and, when implanted back into syngeneic rats, produces an extremely slow‐growing, indolent tumor (Presnell et al., 1998). Serial passage in castrated rats of this tumor led to a diverse family tree of increasingly aggressive tumors and derivative cell lines (Issacs et al., 1982; Smolev et al., 1977; Tennant et al., 2000). One of these cell lines, derived from an anaplastic, highly aggressive variant, led to the development of the anaplastic tumor 3 (AT3) cell line. Compared to pre‐EMT DT cells, AT3 cells exhibit a post‐EMT phenotype (Oltean et al., 2006, 2008; Somarelli et al., 2013), with spindle‐like morphology, low cell–cell attachment, enhanced invasion (Schaeffer et al., 2014), and metastatic capacity (Oltean et al., 2006). Consistent with these observations, microarray analysis of DT and AT3 cells revealed distinct epithelial and mesenchymal biomarker expression, with robust expression of multiple epithelial markers in the DTs and mesenchymal markers in the AT3s (Oltean et al., 2008). These analyses suggest that in vivo serial passage under androgen‐deprived conditions induces a phenotypic transition consistent with EMT in the AT3 line. Thus, AT3 cells tend to reflect ‘epigenetically fixed’ EMT, reminiscent of the ‘epigenetically fixed’ mesenchymal state observed for human non‐small‐cell lung cancer H1703 or Calu6 cells upon prolonged exposure to TGF‐β (Thomson et al., 2011). Yet, unlike the findings discussed above in which a complete EMT reduces the metastatic capacity of the cells, AT3 cells are highly metastatic and remain in a ‘fixed’ mesenchymal state during metastatic colonization (Somarelli et al., 2016a).
Clinically, EMT has been suggested to play a role in promoting the admixed phenotypes observed in the case of carcinosarcomas – rare cancers comprised both carcinomatous and sarcomatous elements. Interestingly, cells expressing markers and/or morphological features of an intermediate or hybrid epithelial/mesenchymal state have been observed (Bittermann et al., 1990; DeLong et al., 1993; Haraguchi et al., 1999; Paniz Mondolfi et al., 2013), suggesting that the mesenchymal component is derived via EMT from the carcinomatous component. Furthermore, genetic analyses support a clonal origin of both epithelial and stromal elements within these tumors (Somarelli et al., 2015). While it remains to be conclusively tested whether carcinosarcomas represent tumors in which a portion of the cells underwent EMT, the majority of data suggest that, in most cases, the mesenchymal element is likely derived from a carcinoma (Somarelli et al., 2015).
Similar to carcinosarcomas, in which tumors exhibit admixture of two phenotypes, prostate tumors with areas of adenocarcinoma and neuroendocrine prostate cancer (NEPC) have also been observed. Both the adenocarcinomatous and NEPC phenotypes share common mutations, suggesting a common cell of origin (Beltran et al., 2011; Hansel et al., 2011; Tan et al., 2014). Likewise, a longitudinal analysis of patients with adenocarcinoma that progresses to NEPC indicated that NEPC results from clonal evolution of an original adenocarcinoma through phenotypic plasticity (Beltran et al., 2016). Further lineage‐tracing studies support this finding, with combined genetic loss of Pten/Rb1/Trp53 inducing an NEPC‐like transition by upregulating stemness factor Sox2 and epigenetic remodeling protein Ezh2 (Ku et al., 2017; Mu et al., 2017). While not a classic example of EMT, NEPC‐like tumors represent similar phenotypic plasticity, and some players implicated in EMT such as Snail have also been reported in the context of NEPC‐like tumors and neuroendocrine differentiation (McKeithen et al., 2011).
Taken together, although induction of at least a partial EMT at the invasive edges in primary xenografts has been observed in vivo (Bonnomet et al., 2012; Klymkowsky and Savagner, 2009), a careful investigation of partial vs. full EMT needs to be conducted in vivo to dissect the contributions of these phenotypic transitions to invasion, dissemination, and metastasis. It is also likely that each tumor's requirements for EMT/MET are slightly different depending on the original cell of origin (e.g., basal vs. luminal), its unique mutation profile (e.g., p53 loss), and its epigenetics (e.g., bivalent vs. monovalent chromatin). A more sophisticated understanding of the hybrid E/M phenotype and its molecular underpinnings will surely help to further elucidate the context‐dependent requirements for plasticity during various stages of the metastatic cascade.
5. Cohesive cell migration and EMT: mutually exclusive migration modes?
Many recent reports have suggested alternative mechanisms for the escape of carcinoma cells, besides the single‐cell dissemination enabled by EMT. Specifically, collectively invading cells have been shown to migrate through the ECM with intact cell–cell junctions (Clark and Vignjevic, 2015; Friedl et al., 2012). Collective invasion need not always exhibit significant changes in canonical epithelial and mesenchymal markers (Cheung et al., 2013; Shamir et al., 2014), but cells at the leading edge of these cohorts may express certain EMT traits (Westcott et al., 2015). A three‐dimensional reconstruction of serial section samples of many tumors has suggested that cell clusters are the predominant agents of invasion and that single‐cell dissemination is extremely rare (Bronsert et al., 2014). Some of these collectively invading cohorts – referred as ‘tumor buds’ – displayed loss of cell polarity, reduced total levels and membrane localization of E‐cadherin, and increased nuclear ZEB1. However, because these cells were not spindle‐shaped and maintained E‐cadherin levels at least partially, they were labeled as a hybrid E/M phenotype, instead of a full EMT (Bronsert et al., 2014; Grigore et al., 2016). It is expected that collectively invading strands and tumor buds are precursors of clusters of circulating tumor cells (CTCs) also called as tumor emboli, as observed in patients with invasive melanoma, lung cancer, inflammatory breast cancer, and clear cell renal cancer (Hou et al., 2012; Jolly et al., 2015a; Kats‐Ugurlu et al., 2009; Ye et al., 2010), thereby suggesting that the clusters of tumor cells retaining some of their epithelial traits can complete the metastasis–invasion cascade and give rise to polyclonal metastatic colonies (Cheung et al., 2016). However, whether the clusters need upregulation of any mesenchymal markers still remains to be investigated extensively.
These clusters of CTCs, although much less prevalent than individually migrating CTCs, can act as primary ‘villains’ of metastasis by forming 50 times more tumors as compared to individual CTCs (Aceto et al., 2014). In addition, clusters may be more efficient in resisting cell death during circulation and associate with significantly worse outcome in patients (Cheung and Ewald, 2016). Inhibiting players that mediate cell–cell adhesion directly or indirectly in these clusters such as plakoglobin or keratin 14 (K14) compromised their metastatic potential (Aceto et al., 2014; Cheung et al., 2016). These results are reminiscent of the essential role of E‐cadherin in forming tumor emboli and distant metastasis in inflammatory breast cancer (Tomlinson et al., 2001) – a highly aggressive cancer that predominantly metastasizes via clusters (Kleer et al., 2001). Thus, retention of cell–cell adhesion as an epithelial trait may actually be crucial to successful metastasis in many aggressive cancers.
Activation of an EMT program – either fully or partially – at the invasive edge can alter the ability of primary tumor cells to intravasate and disseminate as individual CTCs (Bonnomet et al., 2012; Roth et al., 2016), and CTCs can display a dynamic spectrum of EMT phenotypes (Yu et al., 2013). But, any causal role of EMT‐TFs, and by extension, of a partial or full EMT in mediating CTC cluster formation still remains to be thoroughly investigated. This issue is convoluted by observations that CTC clusters may contain platelets that are known to secrete TGF‐β (Aceto et al., 2014), a potent mediator of EMT. Recently developed technologies to isolate CTC clusters, such as Cluster‐Chip, may be critical in this endeavor (Sarioglu et al., 2015).
6. Is MET required for metastasis?
While many studies have focused on the importance of EMT during metastasis (Tsai and Yang, 2013), it has also been hypothesized that cells transition back to an epithelial state through MET to form macrometastases (Thiery, 2002). This hypothesis is based upon the observation that many metastases express epithelial markers (Christiansen and Rajasekaran, 2006).
For example, Chao et al. (2010) examined E‐cadherin expression in primary breast tumors and matched metastases and found that 62% of cases had increased E‐cadherin at the metastatic site compared to the primary tumor. Although metastatic tumors commonly display an epithelial phenotype, it has also long been known that undifferentiated/mesenchymal metastases also occur in patients with cancer. Even in a single patient, there is heterogeneity in the phenotypic status of multiple metastases (Spremulli and Dexter, 1983). These observations lead us to inquire about the requirement of MET for metastasis. Do some disseminated tumor cells not require MET to colonize secondary sites? Or do colonized tumor cells retain a high level of phenotypic plasticity, thereby priming them for multiple rounds of MET and EMT subsequent to metastatic seeding?
Thomas Brabletz postulated two types of metastatic progression – one based on phenotypic plasticity and the other plasticity independent. Metastatic progression that is based on phenotypic plasticity would require MET in order to colonize secondary sites. On the other hand, tumor cells can acquire genetic alterations that confer upon the cell all the necessary traits for dissemination and metastatic seeding in one go and do not require MET (Brabletz, 2012). In vivo experimental evidence for these two models of metastatic progression was demonstrated using lethal reporters of MET that kill all the cells undergoing MET. These reporters revealed the existence of both MET‐dependent and MET‐independent paths to metastatic progression – an MET‐dependent path in carcinosarcomas, whereas an MET‐independent path in prostate cancer (Somarelli et al., 2016a). It is likely that EMT‐TFs and microRNA families that maintain an epithelial phenotype (Bracken et al., 2008; Burk et al., 2008; Lu et al., 2013) regulate MET‐dependent metastatic mechanisms. Indeed, it was recently shown in a spontaneous squamous cell carcinoma model that Twist1 activation promoted EMT and CTCs. However, turning off Twist1 at distant sites allowed MET and was essential for disseminated tumor cells to proliferate and form macrometastases (Tsai et al., 2012), reminiscent of observations that EMT typically arrests the cell cycle (Vega et al., 2004).
Mechanisms underlying MET‐independent metastasis still remain elusive. One hypothesis is based on recent observations that cells that fail to undergo cell cycle arrest upon induction of EMT accumulate genomic instability (Comaills et al., 2016). Therefore, the cells metastasizing independent of MET may be genomically unstable. This instability may serve to enrich for the rare subset of cells that are likely to lead to dedifferentiated and highly metastatic tumors that are cross‐resistant to next‐line therapies (Creighton et al., 2009; Sun et al., 2012). Therefore, therapies used to treat cancer cells may also select for genetic alterations that allow for both the maintenance of an EMT and sustained uncontrolled proliferation, thus potentially obviating the need for MET.
An alternative explanation of the results presented above is that cells might undergo a partial MET, which reporters could miss capturing, just as many reporter systems may be less sensitive in capturing a partial EMT (Li and Kang, 2016; Pattabiraman and Weinberg, 2017). In partial MET, cells are likely to retain their mesenchymal traits and gain their proliferative ability without the acquisition of any genetic alterations. In a study comparing primary and metastatic tissue from breast and prostate cancer, E‐cadherin was found at the cellular membranes more often in metastases than in primary tumors. However, metastases also retained mesenchymal markers vimentin and Fsp1 (Chao et al., 2012). This study suggests that some metastases may maintain a high amount of phenotypic plasticity and are primed to switch between states as selection occurs during growth or by treatment. Thus, it is not necessarily the phenotype that favors metastasis, but the acquisition of the suite of traits needed to metastasize.
A central question that remains unanswered is whether partial EMT is the same as partial MET in its phenotypic consequence. Most phenotypic studies have been performed in carcinomas, which are derived from epithelial cells. As discussed above, these cells likely retain intrinsic epithelial phenotype and acquire migratory and invasive traits, leading to a partial EMT that can promote tumor dissemination (Jolly et al., 2015a). Yet, as these cells are still epithelial in origin, they are probably often less likely to undergo a complete epigenetic reprogramming to acquire a phenotype similar to that of normal mesenchymal tissues. Thus, it is not surprising that many carcinoma cells revert to an epithelial‐like state when arriving to an epithelial environment to form metastases. It is crucial that these cells are able to reactivate the cell cycle to proliferate and colonize; if the cells become fixed in a mesenchymal‐like phenotype and break the connection between the epithelial phenotype and cell cycle activation, either by mutation or by epigenetic reprogramming, their metastatic potential might be severely compromised (Fig. 3).
Figure 3.
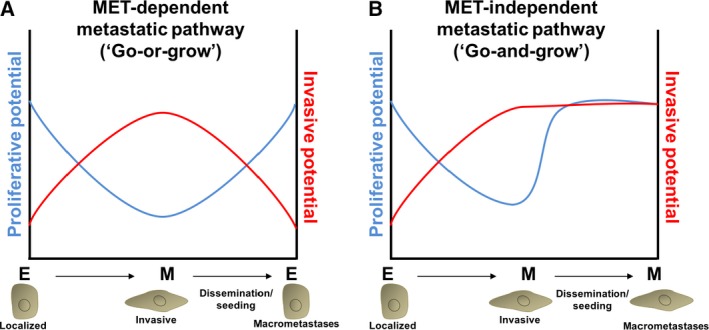
Plasticity‐dependent and plasticity‐independent pathways to metastasis. (A) In MET‐dependent metastasis, post‐EMT‐like cancer cells upregulate invasive programs that facilitate dissemination and seeding (red curve). The invasive program comes at a cost; EMT induction leads to downregulation of proliferative potential (blue curve). Re‐establishment of an epithelial‐like phenotype via MET at the metastatic site awakens the proliferative potential necessary for the formation of macrometastases. (B) In MET‐independent metastasis, therapy, epigenetic reprogramming, acquisition of novel mutations, or other mechanisms induce a post‐EMT state that becomes fixed in a proliferationhigh/invasionhigh phenotype. Cells metastasizing via an MET‐independent pathway may be more aggressive, stem‐like, chemorefractory, and more likely to seed and re‐seed further metastases.
Interestingly, sarcomas provide a unique perspective on the need for MET during metastasis. Sarcomas are cancers of a mesenchymal lineage. These cancers are highly aggressive and metastatic, and upregulation of mesenchymal biomarkers is observed in metastases compared to primary tumors (Shen et al., 2011; Wiles et al., 2013), suggesting that these tumors metastasize via an MET‐independent route. It is possible that sarcoma cells are primed for enhanced metastatic capacity because of their mesenchymal lineage and that the acquisition of growth advantages during cancer initiation enables these cancers to metastasize readily via an MET‐independent route. Clinically, sarcomas occur in younger patients and have a shorter overall survival compared to carcinomas (Siegel et al., 2017), suggesting that the rate‐limiting step for metastasis of these cancers may indeed be tumor initiation. Conversely, in carcinomas, sustained cell growth is commonly coupled to MET during the formation of macrometastases. In this scenario, induction of a MET might be the rate‐limiting step in metastasis.
7. Role of the microenvironment
Phenotypic plasticity can be influenced by the tumor microenvironment; for instance, upregulation of hypoxia (Sun et al., 2009) and soluble factors released by macrophages and other infiltrating immune cells (Huang and Du, 2008; Toh et al., 2011) leads to upregulation of EMT‐TFs and EMT induction. The importance of the microenvironment in driving a metastatic phenotype is underscored by the presence of ‘tumor microenvironment of metastasis’ (TMEM), in which the surrounding microenvironmental niche promotes metastatic dissemination and colonization. For example, factors such as hypoxia (Ju et al., 2017), tumor‐infiltrating neutrophils (Gordon‐Weeks et al., 2017), and radiation treatment (Bouchard et al., 2017; Ruegg et al., 2011) have been demonstrated to generate a metastasis‐promoting microenvironment. Not only do these microenvironmental factors play important roles in creating a prometastasis environment, but also the spatial relationships among these factors are critical. Along these lines, spatial proximity of an endothelial cell, a perivascular macrophage, and an invasive cancer cell overexpressing Mena (a key actin polymerization regulatory protein) – as identified by intravital imaging – was highly correlated with metastasis (Robinson et al., 2009). Based on these insights, it has been suggested that normalizing the tumor microenvironment could be a potential therapeutic strategy to improve patient outcomes (Jain, 2013).
Dynamics of the microenvironment can enable a passive shedding of cancer cells into circulation. This mode would be instead of a postulated active crawling or migration of cancer cells into the circulation or toward any nutrient or chemokine gradient and cleavage of ECM by secreting proteases (Bockhorn et al., 2007) For instance, blood vessels have been proposed to engulf clusters of cancer cells, thus obviating the need for EMT (Fang et al., 2015). These clusters may avoid cell death in circulation by cell–cell contact‐mediated survival signals (Shen and Kramer, 2004) and may already be enriched for players such as Jag1 (Cheung et al., 2016) that can help them evade multiple therapies (Boareto et al., 2016; Li et al., 2014; Shen et al., 2015; Simões et al., 2015) and colonize successfully (Sethi et al., 2011). Not surprisingly, Jag1 is enriched in aggressive cancers such as basal‐like breast cancer (BLBC) (Reedijk et al., 2008) and can contribute to the abnormal vasculature typically observed in cancers (Benedito et al., 2009; Boareto et al., 2015a). Moreover, Fringe, a glycosyltransferase that inhibits the binding of Notch to Jag1 (Boareto et al., 2015b; Jolly et al., 2015b), is lost in BLBC (Zhang et al., 2014).
Therefore, active crawling or migration of cells driven by a partial or full EMT, often activated by overexpression of EMT‐TFs, is not certainly the only route to metastasis. It is not inconceivable that tumor cell dissemination – particularly cluster‐based dissemination – is a passive process where cells that can navigate the fitness bottlenecks from an evolutionary standpoint eventually form metastases (Amend et al., 2016). Both genetic and nongenetic heterogeneity may be crucial or even synergistic in conferring a rare subpopulation of cells with high adaptability or plasticity that lets them transit the entire invasion–metastasis cascade. Such plasticity may coincide with co‐expression of many epithelial and mesenchymal markers, owing to phenotypic alterations that accumulate over multiple steps of the entire metastatic cascade.
8. Conclusion
Single‐cell dissemination as enabled by EMT followed by a MET has been considered to be a hallmark of metastasis. However, alternative modes of dissemination, such as collective or cluster‐based migration and invasion, can exist where cells need not shed cell–cell adhesion completely, and may not even exhibit an overt upregulation of mesenchymal markers, while having gained the traits of migration and invasion. Furthermore, disseminated cancer cells may undergo metastatic colonization via an MET‐independent pathway. Together, the wealth of data acquired thus far support a more nuanced view of the role of EMT/MET in cancer metastasis. While in some cases, EMT/MET are critically important, in other scenarios EMT and MET may not be playing a necessary role, but more of permissive and potentially catalytic roles by regulating phenotypes that speed the processes necessary to escape and colonize.
Acknowledgements
This work was supported by the National Science Foundation (NSF) Center for Theoretical Biological Physics (NSF PHY‐1427654), NSF PHY‐1605817, and NSF DMS‐1361411. HL was also supported as a CPRIT (Cancer Prevention and Research Institute of Texas) Scholar in Cancer Research of the State of Texas at Rice University. JAS acknowledges support from the Duke Cancer Institute, the Duke Genitourinary Oncology Laboratory, the Department of Orthopaedics, and the Triangle Center for Evolutionary Medicine (TriCEM). MKJ was supported by a training fellowship from the Gulf Coast Consortia on the Computational Cancer Biology Training Program (CPRIT Grant No. RP170593). MKJ would like to thank Anne Grosse‐Wilde and Adrian Biddle for valuable discussions.
References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H et al (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P et al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26, 6979–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amend SR, Roy S, Brown JS and Pienta KJ (2016) Ecological paradigms to understand the dynamics of metastasis. Cancer Lett 380, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani F, Bertolini G, Facchinetti F, Baldoli E, Moro M, Casalini P, Caserini R, Milione M, Leone G, Pelosi G et al (2016) Conversion to stem‐cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol 10, 253–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ and Garcia‐Blanco MA (2011) Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res 9, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoux V, Nassour M, L'Helgoualc'h, Hipskind RA and Savagner P (2008) Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell 19, 4738–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J and De Herreros AG (2000) The transcription factor snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2, 84–89. [DOI] [PubMed] [Google Scholar]
- Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, Maynard C, Schäfer R, van Diest P, Voest E, van Oudenaarden A et al (2016) Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis‐enhancing stem cell capacity. Cell Rep 14, 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S et al (2016) Divergent clonal evolution of castration‐resistant neuroendocrine prostate cancer. Nat Med 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST et al (2011) Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M and Adams RH (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135. [DOI] [PubMed] [Google Scholar]
- Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, Costea DE and Mackenzie IC (2011) Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res 71, 5317–5326. [DOI] [PubMed] [Google Scholar]
- Bittermann P, Chun B and Kurman R (1990) The significance of epithelial differentiation in mixed mesodermal tumors of the uterus. A clinicopathologic and immunohistochemical study. Am J Surg Pathol 14, 317–328. [DOI] [PubMed] [Google Scholar]
- Boareto M, Jolly MK, Ben‐Jacob E and Onuchic JN (2015a) Jagged mediates differences in normal and tumor angiogenesis by affecting tip‐stalk fate decision. Proc Natl Acad Sci U S A 112, E3836–E3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boareto M, Jolly MK, Goldman A, Pietila M, Mani SA, Sengupta S, Ben‐Jacob E, Levine H and Onuchic JN (2016) Notch‐Jagged signaling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J R Soc Interface 13, 20151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boareto M, Jolly MK, Lu M, Onuchic JN, Clementi C and Ben‐Jacob E (2015b) Jagged‐Delta asymmetry in Notch signaling can give rise to a Sender/Receiver hybrid phenotype. Proc Natl Acad Sci U S A 112, E402–E409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockhorn M, Jain RK and Munn LL (2007) Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol 8, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolós V, Peinado H, Pérez‐Moreno MA, Fraga MF, Esteller M, Cano A (2003) The transcription factor Slug represses E‐cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116, 499–511. [DOI] [PubMed] [Google Scholar]
- Bonnomet A, Syne L, Brysse A, Feyereisen E, Thompson EW, Noël A, Foidart J‐M, Birembaut P, Polette M and Gilles C (2012) A dynamic in vivo model of epithelial‐to‐mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 31, 3741–3753. [DOI] [PubMed] [Google Scholar]
- Bouchard G, Therriault H, Bujold R, Saucier C and Paquettte B (2017) Induction of interleukin‐1β by mouse mammary tumor irradiation promotes triple negative breast cancer cells invasion and metastasis development. Int J Radiat Biol 93, 507–516. [DOI] [PubMed] [Google Scholar]
- Brabletz T (2012) To differentiate or not — routes towards metastasis. Nat Rev Cancer 12, 425–436. [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF and Goodall GJ (2008) A double‐negative feedback loop between ZEB1‐SIP1 and the microRNA‐200 family regulates epithelial‐mesenchymal transition. Cancer Res 68, 7846–7854. [DOI] [PubMed] [Google Scholar]
- Bronsert P, Enderle‐Ammour K, Bader M, Timme S, Kuehs M, Csanadi A, Kayser G, Kohler I, Bausch D, Hoeppner J et al (2014) Cancer cell invasion and EMT marker expression: a three‐dimensional study of the human cancer‐host interface. J Pathol 234, 410–422. [DOI] [PubMed] [Google Scholar]
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S and Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR‐200 family promotes EMT and invasion in cancer cells. EMBO Rep 9, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Pérez‐Moreno Ma, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial‐mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol 2, 76–83. [DOI] [PubMed] [Google Scholar]
- Celià‐Terrassa T, Meca‐Cortés Ó, Mateo F, De Paz AM, Rubio N, Arnal‐Estapé A, Ell BJ, Bermudo R, Díaz A, Guerra‐Rebollo M et al (2012) Epithelial‐mesenchymal transition can suppress major attributes of human epithelial tumor‐initiating cells. J Clin Invest 122, 1849–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA and Weinberg RA (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao YL, Shepard CR and Wells A (2010) Breast carcinoma cells re‐express E‐cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 9, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y, Wu Q, Acquafondata M, Dhir R and Wells A (2012) Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron 5, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ and Ewald AJ (2014) Illuminating breast cancer invasion: diverse roles for cell‐cell interactions. Curr Opin Cell Biol 30, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ and Ewald AJ (2016) A collective route to metastasis: seeding by tumor cell clusters. Science 352, 167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Gabrielson E, Werb Z and Ewald AJ (2013) Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155, 1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS et al (2016) Polyclonal breast cancer metastases arise from collective dissemination of keratin 14‐expressing tumor cell clusters. Proc Natl Acad Sci U S A 113, E854–E863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen JJ and Rajasekaran AK (2006) Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 66, 8319–8326. [DOI] [PubMed] [Google Scholar]
- Clark AG and Vignjevic DM (2015) Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol 36, 13–22. [DOI] [PubMed] [Google Scholar]
- Comaills V, Kabeche L, Morris R, Zou L, Daniel A, Yu M, Madden MW, Licausi JA, Boukhali M, Tajima K et al (2016) Genomic instability is induced by persistent proliferation of cells undergoing epithelial‐to‐mesenchymal transition. Cell Rep 17, 2632–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI et al (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor‐initiating features. Proc Natl Acad Sci U S A 106, 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara L and Crean J (2016) Emerging transcriptional mechanisms in the regulation of epithelial to mesenchymal transition and cellular plasticity in the kidney. J Clin Med 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong W, Grignon D, Eberwein P, Shum D and Wyatt J (1993) Sarcomatoid renal cell carcinoma. An immunohistochemical study of 18 cases. Arch Pathol Lab Med 117, 636–640. [PubMed] [Google Scholar]
- Dunning W (1963) Prostate cancer in the rat. Natl Cancer Inst Monogr 12, 351–369. [PubMed] [Google Scholar]
- Fang JH, Zhou HC, Zhang C, Shang LR, Zhang L, Xu J, Zheng L, Yuan Y, Guo RP, Jia WH et al (2015) A novel vascular pattern promotes metastasis of hepatocellular carcinoma in an epithelial‐mesenchymal transition‐independent manner. Hepatology 62, 452–465. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Locker J, Sahai E and Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14, 777–783. [DOI] [PubMed] [Google Scholar]
- Futterman MA, García AJ and Zamir EA (2011) Evidence for partial epithelial‐to‐mesenchymal transition (pEMT) and recruitment of motile blastoderm edge cells during avian epiboly. Dev Dyn 240, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon‐Weeks AN, Lim SY, Yuzhalin AE, Jones K, Markelc B, Kim KJ, Buzzelli JN, Fokas E, Cao Y, Smart S et al (2017) Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2‐dependent angiogenesis in mice. Hepatology 65, 1920–1935. [DOI] [PubMed] [Google Scholar]
- Greenburg G and Hay ED (1982) Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol 95, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigore A, Jolly MK, Jia D, Farach‐Carson M and Levine H (2016) Tumor budding: the name is EMT. Partial EMT. J Clin Med 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse‐Wilde A, Fouquier d’ Herouei A, McIntosh E, Ertaylan G, Skupin A, Kuestner RE, del Sol A, Walters K‐A, Huang S (2015) Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS One 10, e0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AGG and Baulida J (2002) Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem 277, 39209–39216. [DOI] [PubMed] [Google Scholar]
- Hajra KM, David YSC and Fearon ER (2002) The SLUG zinc‐finger protein represses E‐cadherin in breast cancer. Cancer Res 62, 1613–1618. [PubMed] [Google Scholar]
- Hansel DE, Nakayama M, Luo J, Abukhdeir AM, Park BH, Bieberich CJ, Hicks JL, Nelson WG, Mostwin JL, Phil D et al (2011) Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 69, 603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi S, Fukuda Y, Sguisaki Y and Yamanaka N (1999) Pulmonary carcinosarcoma: immunohistochemical and ultrastructural studies. Pathol Int 49, 903–908. [DOI] [PubMed] [Google Scholar]
- Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC and Weinberg RA (2006) The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A 103, 18969–18974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJC, Greystoke A, Zhou C, Morris K et al (2012) Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small‐cell lung cancer. J Clin Oncol 30, 525–532. [DOI] [PubMed] [Google Scholar]
- Huang D and Du X (2008) Crosstalk between tumor cells and microenvironment via Wnt pathway in colorectal cancer dissemination. World J Gastroenterol 14, 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY‐J, Wong MK, Tan TZ, Kuay KT, Ng AHC, Chung VY, Chu Y‐S, Matsumura N, Lai H‐C, Lee YF et al (2013) An EMT spectrum defines an anoikis‐resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e‐cadherin restoration by a src‐kinase inhibitor, saracatinib (AZD0530). Cell Death Dis 4, e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issacs J, Heston W, Weissman R and Coffey DS (1978) Animal models of the hormone‐sensitive and ‐insensitive prostatic adenocarcinomas, Dunning R‐3327‐H, R‐3327‐HI, and R‐3327‐AT. Cancer Res 38, 4353–4359. [PubMed] [Google Scholar]
- Issacs J, Wake N, Coffey DS and Sandberg A (1982) Genetic instability coupled to clonal selection as a mechanism for tumor progression in the Dunning R‐3327 rat prostatic adenocarcinoma system. Cancer Res 42, 2353–2371. [PubMed] [Google Scholar]
- Jain RK (2013) Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol 31, 2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevan DS, Cooper JB, Braun A, Murali RAJ and Jhanwar‐Uniyal M (2016) Molecular pathways mediating metastases to the brain via epithelial‐to‐mesenchymal transition: genes, proteins, and functional analysis. Anticancer Res 36, 523–532. [PubMed] [Google Scholar]
- Jia D, Jolly MK, Boareto M, Parsana P, Mooney SM, Pienta KJ, Levine H and Ben‐Jacob E (2015) OVOL guides the epithelial‐hybrid‐mesenchymal transition. Oncotarget 6, 15436–15448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnen N, Francart M‐E, Thelen N, Cloes M and Thiry M (2012) Evidence for a partial epithelial‐mesenchymal transition in postnatal stages of rat auditory organ morphogenesis. Histochem Cell Biol 138, 477–488. [DOI] [PubMed] [Google Scholar]
- Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben‐Jacob E, Onuchic JN and Levine H (2015a) Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol 5, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly MK, Boareto M, Lu M, Onuchic JN, Clementi C and Ben‐Jacob E (2015b) Operating principles of Notch‐Delta‐Jagged module of cell‐cell communication. New J Phys 17, 55021. [Google Scholar]
- Jolly MK, Huang B, Lu M, Mani SA, Levine H and Ben‐Jacob E (2014) Towards elucidating the connection between epithelial − mesenchymal transitions and stemness. J R Soc Interface 11, 20140962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben‐Jacob E and Levine H (2016) Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 7, 27067–27084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JA, Godet I, Ye IC, Byun J, Jayatilaka H, Lee SJ, Xiang L, Samanta D, Lee MH, Wu P‐H et al (2017) Hypoxia selectively enhances integrin α5β1 receptor expression in breast cancer to promote metastasis. Mol Cancer Res 15, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee KP, Park SJ, Park JH, Jang YS, Choi SY, Jung JG, Jo K, Park DY, Yoon JH et al (2008) TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial‐mesenchymal transition. Oncogene 27, 2635–2647. [DOI] [PubMed] [Google Scholar]
- Kalluri R and Weinberg RA (2009) The basics of epithelial‐mesenchymal transition. J Clin Invest 119, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kats‐Ugurlu G, Roodnik I, de Weijert M, Tiemessen D, Maass C, Verrjip K, van der Laak J, de Waal R, Mulders P, Oosterwijk E et al (2009) Circulating tumour tissue fragments in patients with pulmonary metastasis of clear cell renal cell carcinoma. J Pathol 219, 287–293. [DOI] [PubMed] [Google Scholar]
- Kleer CG, van Golen KL, Braun T and Merajver SD (2001) Persistent E‐cadherin expression in inflammatory breast cancer. Mod Pathol 14, 458–464. [DOI] [PubMed] [Google Scholar]
- Klymkowsky MW and Savagner P (2009) Epithelial‐mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol 174, 1588–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AM, Mitschke J, Losada ML, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P et al (2017) The EMT‐activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 19, 518–529. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J et al (2017) Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama S, Theveneau E, Benedetto A, Parsons M, Tanaka M, Charras G, Kabla A and Mayor R (2014) In vivo collective cell migration requires an LPAR2‐dependent increase in tissue fluidity. J Cell Biol 206, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR and Weinberg RA (2017) Emerging biological principles of metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecharpentier A, Vielh P, Perez‐Moreno P, Planchard D, Soria JC and Farace F (2011) Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non‐small cell lung cancer. Br J Cancer 105, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Dedhar S, Kalluri R and Thompson EW (2006) The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy P and Mostov KE (2007) Slug is required for cell survival during partial epithelial‐mesenchymal transition of HGF‐induced tubulogenesis. J Cell Sci 18, 1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Hong T and Nie Q (2016) Quantifying the landscape and kinetic paths for epithelial‐mesenchymal transition from a core circuit. Phys Chem Chem Phys 18, 17949–17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W and Kang Y (2016) Probing the fifty shades of EMT in metastasis. Trends Cancer 2, 65–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Masiero M, Banham AH and Harris AL (2014) The notch ligand JAGGED1 as a target for anti‐tumor therapy. Front Oncol 4, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Jolly MK, Levine H, Onuchic JN and Ben‐Jacob E (2013) MicroRNA‐based regulation of epithelial‐hybrid‐mesenchymal fate determination. Proc Natl Acad Sci U S A 110, 18144–18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao M‐J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL and Weinberg RA (2007) Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal‐like breast cancers. Proc Natl Acad Sci U S A 104, 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCart Reed AE, Kutasovic JR, Vargas AC, Jayanthan J, Al‐Murrani A, Reid LE, Chambers R, Da Silva L, Melville L, Evans E et al (2016) An epithelial to mesenchymal transition programme does not usually drive the phenotype of invasive lobular carcinomas. J Pathol 238, 489–494. [DOI] [PubMed] [Google Scholar]
- McKeithen D, Graham T, Chung LWK and Odero‐Marah V (2011) Snail transcriptional factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate 70, 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Farabaugh SM and Ford HL (2010) Epithelial‐mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 15, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C‐C, Wongvipat J, Ku S‐Y, Gao D, Cao Z et al (2017) SOX2 promotes lineage plasticity and antiandrogen resistance in TP53‐ and RB1‐deficient prostate cancer. Science 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naber HPH, Drabsch Y, Snaar‐Jagalska BE, ten Dijke P and van Laar T (2013) Snail and Slug, key regulators of TGF‐b‐induced EMT, are sufficient for the induction of single‐cell invasion. Biochem Biophys Res Commun 435, 58–63. [DOI] [PubMed] [Google Scholar]
- Nguyen‐Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, Werb Z and Ewald AJ (2012) ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A 109, E2595–E2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA (2013) Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342, 1234850. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA and Thiery JP (2016) EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Ocaña OH, Córcoles R, Fabra A, Moreno‐Bueno G, Acloque H, Vega S, Barrallo‐Gimeno A, Cano A and Nieto MA (2012) Metastatic colonization requires the repression of the epithelial‐mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724. [DOI] [PubMed] [Google Scholar]
- Oltean S, Febbo PG and Garcia‐Blanco MA (2008) Dunning rat prostate adenocarcinomas and alternative splicing reporters: powerful tools to study epithelial plasticity in prostate tumors in vivo. Clin Exp Metastasis 25, 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean S, Sorg BS, Albrecht T, Bonano VI, Brazas RM, Dewhirst MW and Garcia‐Blanco MA (2006) Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial mesenchymal plasticity. Proc Natl Acad Sci U S A 103, 14116–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ombrato L, Malanchi I (2014) The EMT universe: space between cancer cell dissemination and metastasis initiation. Crit Rev Oncog 19, 349–361. [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES and Weinberg RA (2008) Loss of E‐cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68, 3645–3654. [DOI] [PubMed] [Google Scholar]
- Paniz Mondolfi AE, Jour G, Johnson M, Reidy J, Cason RC, Barkoh BA, Benaim G, Singh R and Luthra R (2013) Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next‐generation sequencing. Hum Pathol 44, 2853–2860. [DOI] [PubMed] [Google Scholar]
- Pattabiraman DR and Weinberg RA (2017) Targeting the epithelial‐to‐mesenchymal transition: the case for differentiation‐based therapy. Cold Spring Harb Symp Quant Biol LXXXI, 30957 https://doi.org/10.1101/sqb.2016.81.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnell S, Borchkert K, Glover W, Gregory C, Mohler J and Smith G (1998) Isolation and characterization of propagable cell lines (HUNC) from the androgen‐sensitive Dunning R3327H rat prostatic adenocarcinoma. Carcinogenesis 19, 585–590. [DOI] [PubMed] [Google Scholar]
- Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, O'Malley FP, Egan SE and Andrulis IL (2008) JAG1 expression is associated with a basal phenotype and recurrence in lymph node‐negative breast cancer. Breast Cancer Res Treat 111, 439–448. [DOI] [PubMed] [Google Scholar]
- Revenu C and Gilmour D (2009) EMT 2.0: shaping epithelia through collective migration. Curr Opin Genet Dev 19, 338–342. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Jennifer M, Mccallister F, Reichert M, Beatty GL, Anil K, Vonderheide RH et al (2013) EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS and Jones JG (2009) Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res 15, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B, Jayaratna I, Sundi D, Cheng T, Melquist J, Choi W, Porten S, Nitti G, Navai N, Wszolek M et al (2016) Employing an orthotopic model to study the role of epithelial‐mesenchymal transition in bladder cancer metastasis. Oncotarget 8, 34205–34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg C, Monnier Y, Kuonen F and Imaizumi N (2011) Radiation‐induced modifications of the tumor microenvironment promote metastasis. Bull Cancer 98, 47–57. [DOI] [PubMed] [Google Scholar]
- Ruscetti M, Quach B, Dadashian EL, Mulholland DJ and Hong W (2015) Tracking and functional characterization of epithelial‐mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res 75, 2749–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson VB, David JM, Puig I, Patil PU, de Herreros AG, Thomas GV and Rajasekaran AK (2014) Wilms’ tumor protein induces an epithelial‐mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS One 9, e102041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT et al (2015) A microfluidic device for label‐free, physical capture of circulating tumor cell clusters. Nat Methods 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR and McClay DR (2014) Sub‐circuits of a gene regulatory network control a developmental epithelial‐mesenchymal transition. Development 141, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P (2015) Epithelial‐mesenchymal transitions: from cell plasticity to concept elasticity. Curr Top Dev Biol 112, 273–300. [DOI] [PubMed] [Google Scholar]
- Schaeffer D, Somarelli JA, Hanna G, Palmer GM and Garcia‐Blanco MA (2014) Cellular migration and invasion uncoupled: increased migration is not an inexorable consequence of epithelial‐to‐mesenchymal transition. Mol Cell Biol 34, 3486–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliekelman MJ, Taguchi A, Zhu J, Dai X, Rodriguez J, Celiktas M, Zhang Q, Chin A, Wong C, Wang H et al (2015) Molecular portraits of epithelial, mesenchymal and hybrid states in lung adenocarcinoma and their relevance to survival. Cancer Res 75, 1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG and Kang Y (2011) Tumor‐derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS and Ewald AJ (2014) Twist1‐induced dissemination preserves epithelial identity and requires E‐cadherin. J Cell Biol 204, 839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TJ and Martin P (2016) Wound repair: a showcase for cell plasticity and migration. Curr Opin Cell Biol 42, 29–37. [DOI] [PubMed] [Google Scholar]
- Shen Y, Chen H, Zhang J, Chen Y, Wang M, Ma J, Hong L, Liu N, Fan Q, Lu X et al (2015) Increased Notch signaling enhances radioresistance of malignant stromal cells induced by glioma stem/progenitor cells. PLoS One 10, e0142594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X and Kramer RH (2004) Adhesion‐mediated squamous cell carcinoma survival through ligand‐independent activation of epidermal growth factor receptor. Am J Pathol 165, 1315–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Zhang Y, Yang H, Xu R and Huang G (2011) Overexpression of ZEB1 relates to metastasis and invasion in osteosarcoma. J Surg Oncol 105, 830–834. [DOI] [PubMed] [Google Scholar]
- Siegel R, Miller K and Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67, 7–30. [DOI] [PubMed] [Google Scholar]
- Simões B, O'Brien C, Eyre R, Silva A and Yu L (2015) Anti‐estrogen resistance in human breast tumors is driven by JAG1‐NOTCH4‐dependent cancer stem cell activity. Cell Rep 29, 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolev J, Heston W, Scott W and Coffey D (1977) Characterization of the Dunning R3327H prostatic adenocarcinoma: an appropriate animal model for prostatic cancer. Cancer Treat Rep 61, 273–287. [PubMed] [Google Scholar]
- Somarelli JA, Boss M‐K, Epstein JI, Armstrong AJ and Garcia‐Blanco MA (2015) Carcinosarcomas: tumors in transition? Histol Histopathol 30, 673–687. [DOI] [PubMed] [Google Scholar]
- Somarelli JA, Schaeffer D, Bosma R, Bonano VI, Sohn JW, Kemeny G, Ettyreddy A and Garcia‐Blanco MA (2013) Fluorescence‐based alternative splicing reporters for the study of epithelial plasticity in vivo. RNA 19, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarelli JA, Schaeffer D, Marengo MS, Bepler T, Rouse D, Ware KE, Hish AJ, Zhao Y, Buckley AF, Epstein JI et al (2016a) Distinct routes to metastasis: plasticity‐dependent and plasticity‐independent pathways. Oncogene 35, 4302–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarelli JA, Shelter S, Jolly MK, Wang X, Bartholf Dewitt S, Hish AJ, Gilja S, Eward WC, Ware KE, Levine H et al (2016b) Mesenchymal‐epithelial transition in sarcomas is controlled by the combinatorial expression of miR‐200s and GRHL2. Mol Cell Biol 36, 2503–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A et al (2008) The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68, 537–544. [DOI] [PubMed] [Google Scholar]
- Spremulli E and Dexter D (1983) Human tumor cell heterogeneity and metastasis. J Clin Oncol 1, 496–509. [DOI] [PubMed] [Google Scholar]
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J and Benezra R (2013) TGF‐b‐Id1 signaling opposes twist1 and promotes metastatic colonization via a mesenchymal‐to‐epithelial transition. Cell Rep 5, 1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss R, Li Z‐Y, Liu Y, Beyer I, Persson J, Sova P, Möller T, Pesonen S, Hemminki A, Hamerlik P et al (2011) Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS One 6, e16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, True L and Nelson PS (2012) Treatment‐induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ning X, Zhang Y, Lu Y, Nie Y, Han S, Liu L, Du R, Xia L, He L et al (2009) Hypoxia‐inducible factor‐1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial‐to‐mesenchymal transition. Kidney Int 75, 1278–1287. [DOI] [PubMed] [Google Scholar]
- Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, Mosier S, Gocke CD, Epstein JI, Netto GJ et al (2014) Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res 20, 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant T, Kim H, Sokoloff M and Rinker‐Schaeffer C (2000) The Dunning model. Prostate 43, 295–302. [DOI] [PubMed] [Google Scholar]
- Thiery JP (2002) Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2, 442–454. [DOI] [PubMed] [Google Scholar]
- Thomson S, Petti F, Sujka‐Kwok I, Mercado P, Bean J, Monaghan M, Seymour SL, Argast GM, Epstein DM and Haley JD (2011) A systems view of epithelial‐mesenchymal transition signaling states. Clin Exp Metastasis 28, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, Kato M, Prevost‐Blondel A, Thiery JP and Abastado JP (2011) Mesenchymal transition and dissemination of cancer cells is driven by myeloid‐derived suppressor cells infiltrating the primary tumor. PLoS Biol 9, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson JS, Alpaugh ML and Barsky SH (2001) An intact overexpressed E‐cadherin/α, β‐catenin axis characterizes the lymphovascular emboli of inflammatory breast carcinoma. Cancer Res 61, 5231–5241. [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S and Yang J (2012) Spatiotemporal regulation of epithelial‐mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH and Yang J (2013) Epithelial‐mesenchymal plasticity in carcinoma metastasis. Genes Dev 27, 2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma S, Cao Y, Tagne JB, Lakshminarayanan M, Li J, Friedman TB, Morell RJ, Warburton D, Kotton DN and Ramirez MI (2012) The transcription factors grainyhead‐like 2 and NK2‐homeobox 1 form a regulatory loop that coordinates lung epithelial cell morphogenesis and differentiation. J Biol Chem 287, 37282–37295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I and Nieto MA (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesuna F, van Diest P, Chen JH and Raman V (2008) Twist is a transcriptional repressor of E‐cadherin gene expression in breast cancer. Biochem Biophys Res Commun 367, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentin K, Hinze C, Werth M, Haase N, Varma S, Morell R, Aue A, Pötschke E, Warburton D, Qiu A et al (2015) A Grhl2‐dependent gene network controls trophoblast branching morphogenesis. Development 142, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott JM, Prechtl AM, Maine EA, Dang TT, Esparza MA, Sun H, Zhou Y, Xie Y and Pearson GW (2015) An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J Clin Invest 125, 1927–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles ET, Bell R, Thomas D, Beckerle M and Lessnick SL (2013) ZEB2 represses the epithelial phenotype and facilitates metastasis in ewing sarcoma. Genes Cancer 4, 486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939. [DOI] [PubMed] [Google Scholar]
- Ye Y, Tellez JD, Durazo M, Belcher M, Yearsley K and Barsky SH (2010) E‐cadherin accumulation within the lymphovascular embolus of inflammatory breast cancer is due to altered trafficking. Anticancer Res 30, 3903–3910. [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM et al (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadran S, Arumugam R, Herschman H, Phelps ME and Levine RD (2014) Surprisal analysis characterizes the free energy time course of cancer cells undergoing epithelial‐to‐mesenchymal transition. Proc Natl Acad Sci U S A 111, 13235–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chung W, Wu G, Egan SE and Xu K (2014) Tumor‐suppressive activity of lunatic fringe in prostate through differential modulation of notch receptor activation. Neoplasia 16, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu C‐C, LeBleu VS and Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]