

Abstract
Novel immunotherapy approaches have provided durable remission in a significant number of cancer patients with cancers previously considered rapidly lethal. Nonetheless, the high degree of nonresponders, and in some cases the emergence of resistance in patients who do initially respond, represents a significant challenge in the field of cancer immunotherapy. These issues prompt much more extensive studies to better understand how cancer cells escape immune surveillance and resist immune attacks. Here, we review the current knowledge of how cellular heterogeneity and plasticity could be involved in shaping the tumor microenvironment (TME) and in controlling antitumor immunity. Indeed, recent findings have led to increased interest in the mechanisms by which cancer cells undergoing epithelial‐mesenchymal transition (EMT), or oscillating within the EMT spectrum, might contribute to immune escape through multiple routes. This includes shaping of the TME and decreased susceptibility to immune effector cells. Although much remains to be learned on the mechanisms at play, cancer cell clones with mesenchymal features emerging from the TME seem to be primed to face immune attacks by specialized killer cells of the immune system, the natural killer cells, and the cytotoxic T lymphocytes. Recent studies investigating patient tumors have suggested EMT as a candidate predictive marker to be explored for immunotherapy outcome. Promising data also exist on the potential utility of targeting these cancer cell populations to at least partly overcome such resistance. Research is now underway which may lead to considerable progress in optimization of treatments.
Keywords: antitumor immunity, EMT, immune escape, tumor microenvironment
Abbreviations
- APCs
antigen‐presenting cells
- CAFs
cancer‐associated fibroblasts
- CCL
C‐C motif chemokine ligand
- CCR
C‐C motif chemokine receptor
- COX2
cyclooxygenase‐2
- cSMAC
central supramolecular activation cluster
- CTC
circulating tumor cell
- CTLA‐4
cytotoxic T lymphocyte‐associated protein 4
- CTL
cytotoxic T lymphocyte
- CXCL
C‐X‐C motif chemokine ligand
- DC
dendritic cells
- EMT
epithelial‐mesenchymal transition
- EMT‐TF
epithelial‐mesenchymal transition transcription factor
- Epi
epithelial
- Erbb2
Erb‐b2 receptor tyrosine kinase 2
- ESRP1
epithelial splicing regulatory protein 1
- FFPE
formalin‐fixed paraffin‐embedded
- FOXC2
forkhead box C2
- FOXP3
forkhead box P3
- GRHL2
grainyhead‐like transcription factor 2
- GZM
granzyme
- HIF‐1α
hypoxia‐induced factor 1 alpha
- HNSCC
head and neck squamous cell carcinoma
- ICAM
intercellular adhesion molecule‐1
- ICOS
inducible T‐cell costimulator
- IDO
indoleamine‐2,3‐dioxygenase
- IFN
interferon
- IL
interleukin
- KLF8
kruppel‐like factor 8
- LAG3
lymphocyte activating 3
- LFA‐1
lymphocyte function‐associated antigen‐1
- MDSC
myeloid‐derived suppressor cells
- Mes
mesenchymal
- MET
mesenchymal‐epithelial transition
- MHC
major histocompatibility complex
- MIC
MHC class I chain‐related protein
- miRs
microRNA
- MSI
microsatellite instability
- NK
natural killer
- NSCLC
non‐small‐cell lung cancers
- PD‐1
programmed cell death protein‐1
- PD‐L1
programmed cell death 1 ligand 1
- PGE
prostaglandin E
- PI3K
phosphatidyl inositol‐3‐kinase
- Prf
perforin
- PRRX1
paired‐related homeobox 1
- pSMAC
peripheral supramolecular activation cluster
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- SNAIL1
snail family zinc finger 1
- TCR
T‐cell receptor
- TGF‐β
tumor growth factor beta
- TIL
tumor‐infiltrating lymphocyte
- TIM3
T‐cell immunoglobulin mucin 3
- TME
tumor microenvironment
- TNC
tenascin‐C
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- TSP
thrombospondin
- TWIST1
Twist family bHLH transcription factor 1
- VEGF
vascular endothelial growth factor
- ZEB
zinc finger E‐box binding homeobox
1. Introduction
Here, in a first part, we consider the role of the tumor microenvironment (TME) in the field of Immuno‐oncology, and then, introduce key notions regarding the epithelial‐mesenchymal transition (EMT), cell plasticity, resistance to chemotherapy and radiotherapy. In a second part, we review the current state of knowledge regarding the multifaceted manner by which EMT, and cancer cells with mesenchymal features, could influence the TME and modulate antitumor immunity. Finally, we discuss the potential impact and the need to integrate these discoveries to develop effective therapeutic strategies, as well as for the development of biomarkers of response for immunotherapies.
2. Tumor microenvironment and antitumor immunity
Although immune checkpoint blockers (anti‐PD‐1, anti‐PD‐L1, anti‐CTLA‐4) on T cells result in significantly improved survival in various metastatic cancer types [non‐small‐cell lung cancers (NSCLC), melanoma, renal carcinoma, bladder carcinoma, head and neck squamous cell carcinoma (HNSCC), and lymphoma] (Ansell, 2017; Borghaei et al., 2015; Brahmer et al., 2015; Burstein et al., 2017; Ferris et al., 2016; Garon et al., 2015; Hamid et al., 2013; Motzer et al., 2015; Reck et al., 2016; Rosenberg et al., 2016; Sharma et al., 2017b; Younes et al., 2016), a high fraction of patients with cancer fail to respond to these therapeutic interventions. This is manifested through different forms including intrinsic resistance, or through acquired resistance in patients initially responding (Restifo et al., 2016; Sharma et al., 2017a). Moreover, in the current setting, a number of cancer types respond poorly, such as prostate, breast, non‐microsatellite instability (non‐MSI) colon, and pancreatic cancers. While the exact reasons for this lack of response are largely unknown, it is interesting to note that even in good responders, the responses can significantly differ between cancer lesions in a given patient (O'Donnell et al., 2017). This underlies the importance of the TME and cancer niches establishing during cancer initiation and progression. Indeed, a developing tumor, to sustain its growth, must establish an immunosuppressive TME to neutralize the activation of immune responses (Gajewski et al., 2006; Joyce and Fearon, 2015; Kerkar and Restifo, 2012; Mantovani et al., 2008; Vasievich and Huang, 2011). All immune cell types [including dendritic cells (DC), natural killer (NK) cells, macrophages, neutrophils, B and T lymphocytes (CD4+ T helper 1 (TH1) and 2 (TH2), CD8+ cytotoxic T cells (CTL), memory cells, and regulatory T (Treg) cells)] may be present within or at the edge of a given tumor, or in a form of tertiary lymphoid structures located in the stromal compartment that gather multiple immune components similar to that found in secondary lymphoid organs (Fridman et al., 2012). The immune infiltrates may vary strikingly during stages of tumor development, from one patient to another, between different cancers, or cancer histotypes. Analysis of the ‘immune contexture’ in patient tumors revealed that the TME comprises a mosaic of immunosuppressive cells such as myeloid‐derived suppressor cells (MDSC) (Kumar et al., 2016; Marvel and Gabrilovich, 2015), cancer‐associated fibroblasts (CAFs) (Kraman et al., 2010; Salmon et al., 2012), tumor‐associated macrophages (Allavena and Mantovani, 2012; Ruffell et al., 2012), and Treg cells (Ghiringhelli et al., 2005; Whiteside, 2008). Immunoregulatory enzymes (arginase, COX2, INOS) and immunosuppressive substances produced by these cells such as IL‐10, tumor growth factor beta (TGF‐β), vascular endothelial growth factor, PGE2, or PD‐L1 can impede both the innate and adaptive immunities by inhibiting NK cells, CD4+ and CD8+ effector T cells, or by inhibiting DC maturation, through reducing major histocompatibility complex (MHC) molecules on the latter and costimulatory signals essential to activate naïve T cells. Together with this growing knowledge, a new hope has emerged that efficient targeting of the components of the TME could affect cancer in all of its stages.
Besides these different cell types, hypoxia is an essential metabolic element of the TME that shapes cellular plasticity and tumor heterogeneity (Keith and Simon, 2007; Pouyssegur et al., 2006). During tumor development, while cells residing close to blood vessels are relatively well oxygenated, those at more distant sites often face hypoxic stress, coinciding with O2 deprivation. Hypoxia conditioning leads to stabilization of HIF‐α proteins in these cells which mediate cellular adaptation to this stress by inducing expression of target genes, associated with changes in cell metabolism, behavior, and phenotype (Keith and Simon, 2007; Majmundar et al., 2010; Pouyssegur et al., 2006). The intensity and the duration of stress vary among the different cell types. Considerable evidence now suggests that hypoxia also impairs antitumor immunity through different mechanisms involving cancer cell plasticity or education of nonimmune cells toward an immunosuppressive phenotype (Corzo et al., 2010; Kumar and Gabrilovich, 2014; Noman et al., 2015).
The concept of immunoediting, originally embodied by the notion of tumor immunosurveillance, can recapitulate many of the salient features observed in the natural history of a cancer. It highlights the important interactions between the immune system and the tumor at various stages of development (Dunn et al., 2002; Mittal et al., 2014). In the first phase, ‘elimination’, a substantial number malignant cells are recognized by competent immune effector killer cells such as CTLs and NK cells through the activation of innate and adaptive immune routes. In a second phase, sporadic cancer cells managing to survive the immune attacks enter an ‘equilibrium’ phase where editing of the tumor occurs. The establishment of an immunosuppressive TME probably begins during this phase and will be further accentuated in final phase of the immunoediting process, called the ‘escape’ phase. This phase is characterized by malignant clones shaping the TME under a severe selection pressure and adaptation, and developing intrinsic mechanisms to escape immune detection and destruction (Zitvogel et al., 2006). This gives rise to clinically significant immunologically edited tumors. More research is required to decrypt the complex network of interactions between tumor, immune and nonimmune cells in the clinical setting (Galon et al., 2013). It is unlikely that immunoediting completely eradicates the cancer cell clones with highly immunogenic epitopes, as evidenced by notable responses observed in some patients under anti‐CTLA4 and anti‐PD1 therapies. Additionally, it is still unclear in this context why most patient tumors maintain high cancer cell heterogeneity and how this impacts on TME changes (and vice versa) as well as in mounting immune resistance (Holzel et al., 2013).
Over the past decade, EMTs, and transitional states between epithelial (Epi) and mesenchymal (Mes) states, have been suggested as critical mediators in metastatic progression, and therapy resistance, including chemo‐, radio‐, and targeted therapy resistances. In light of the current knowledge, cancer cells undergoing EMT, or harboring Mes characteristics, may also have profound influence on the cellular components present in the TME, including immune cells. Evidence is now accumulating that such cross‐talks might be key determinants in facilitating immune escape by tumors, with the potential to regulate immunotherapy efficacy.
3. EMT, cell plasticity, and cancer: basic principles
Early embryonic developmental stages are characterized by extensive plasticity in cellular organizations. Following cellularization or cleavages, cells expand their intercellular contact to progressively form a polarized layer called an epithelium. The cells within these sheets develop extensive apico‐basal cell surface polarity with actin microfilaments that are mostly concentrated at the cortex of the basolateral membranes, and with microtubules oriented baso‐apically that position cytoplasmic organelles, such as the Golgi, above the nucleus along the baso–apical axis. In polarized Epi cells, junctional complexes are localized on the lateral domain, and the basal domain interacts exclusively with the extracellular matrix forming a basal lamina through integrin receptors. These distinct junctional complexes, particularly E‐cadherin‐associated adherens junctions, contribute to maintaining apico‐basal polarity (Huang et al., 2012). Mes cells, on the contrary, are formed during gastrulation through EMT, a fundamental mechanism of development in most multicellular organisms (Lim and Thiery, 2012; Nieto et al., 2016). During gastrulation in higher vertebrates, Mes mesodermal and endodermal cells dissociate from an Epi cell layer called the epiblast and ingress through the primitive streak. Mes cells lose their apico‐basal polarity, redistribute their actin cytoskeleton throughout the cytoplasm, and remain connected to the basal surface through focal adhesions and intercellular punctate adhesions. These features contribute to the development of a front‐rear polarity during the acquisition of migratory behavior. Most of the ingressed cells then engage in the reverse process named mesenchymal‐epithelial transition (MET) to establish paraxial and lateral mesodermal structures. Organogenesis subsequently involves cycles of EMT and MET, such as seen in heart development. These successive rounds of EMT and MET in development are driven by multiple signaling pathways, including canonical Wnt, TGF‐β, and RTK pathways (Lim and Thiery, 2012). One of the most striking examples of EMT is the formation and subsequent extensive migrations of cells of the neural crest, a transient, embryonic structure in the dorsal neuroepithelium. These properties are proposed to have been hijacked by cancer cells to initiate invasion and metastasis (Thiery, 2002). Our current understanding of the mechanisms driving EMT in carcinoma has predominantly come from in vitro studies using a limited number of carcinoma cell lines. EMT is classically driven by transcriptional repressors commonly referred to as EMT transcription factors (EMT‐TF) including SNAIL1/2 and ZEB1/2, which directly repress E‐cadherin expression by binding to E‐boxes on its proximal promoter. TWIST and several other transcription factors (FOXC2, E47 (TFC3), KLF8, and PRRX1) also induce EMT. Although it is still unclear whether these factors directly regulate E‐cadherin expression (De Craene and Berx, 2013), they have multiple other target genes and may function downstream in canonical RTK, TGF‐β, and Wnt receptor signaling, among others (Lamouille et al., 2014). The miRs (microRNA) are also critically involved in maintaining the Epi state, with ZEB1 and miR‐200 family members forming negatively regulated feed‐back loops (Zhang and Ma, 2012). In addition to transcriptional and miR regulation, splicing mechanisms, post‐translational modifications, and epigenetic changes also significantly contribute to the EMT phenotype (De Craene and Berx, 2013). Epigenetics ensures a more stable position in the EMT spectrum as compared with the continuous expression of transcriptional repressors. For instance, DNA methylation and histone repressive marks are associated with a more Mes phenotype, whereas poised chromatin allows for a more plastic phenotype, residing in the intermediate EMT stages (Tam and Weinberg, 2013). Some transcription factors, such as GRHL2, originally shown to sustain E‐cadherin gene expression through its binding to its second intron (Cieply et al., 2012), may also participate in maintaining the stability of Epi state. GRHL2 thus protects carcinoma cells from transitioning from the intermediate Epi state to the Mes states. However, the forced expression of GRHL2 in Mes ovarian carcinoma cells cannot revert the cells to an Epi state (Chung et al., 2016). Evidence suggests that this irreversibility is primarily caused by heterochromatization of GRHL2 target genes. In recent years, studies have emphasized that Mes carcinoma cells have acquired stem cell properties (Chaffer et al., 2011; Mani et al., 2008) and a drug‐resistant phenotype (Gupta et al., 2009; Mitra et al., 2015; Singh and Settleman, 2010). However, there remain numerous questions regarding when and how carcinoma cells acquire a Mes phenotype, drug resistance, and stemness. For example, does it happen before or during dissemination through the lymph and blood vessels? Approximately 3% of luminal breast carcinoma cells express Mes markers and a significantly higher percentage is detected in triple‐negative breast tumors (Sarrio et al., 2008; Tan et al., 2014; Yu et al., 2013a). A longitudinal analysis of patients with metastatic breast cancer, who underwent several cycles of different targeted therapeutics to overcome refractoriness, finally became fully refractory once their tumors reached a Mes phenotype (Yu et al., 2013a). EMT in these carcinoma cells may be induced by the local microenvironment and hypoxia, priming certain cells for disseminating from the primary tumor. Stromal cells, including myofibroblast and inflammatory cells, could contribute as EMT inducers by secreting numerous growth factors and cytokines (Bai et al., 2015; Wyckoff et al., 2007). Overall, it appears that the intermediate Epi and Mes carcinoma phenotypes are more suitable for tumor progression and distant dissemination, thus making the EMT process a promising target for therapeutic intervention, to prevent or suppress these deleterious effects. A major issue is whether we can develop new therapeutic strategies based on the EMT concept (Antony et al., 2016; Chua et al., 2012). Moreover, the concept that EMT confers invasion, metastasis, stemness, and drug resistance need to be addressed in the clinical setting rather than with murine models. These studies will pave the way toward a deeper understanding of the most important properties conferred by EMT during dissemination.
4. EMT and resistance to chemotherapy and radiotherapy
EMT‐TFs have been found to mingle with stemness pathways and to induce resistance to chemotherapy and radiotherapy in various cancer models, in vitro and in vivo (Dave et al., 2012; Fischer et al., 2015; Sanchez‐Tillo et al., 2012; Zheng et al., 2015). The molecular pathways connecting chemotherapy/radiotherapy and EMT‐TFs strongly depend on the molecular type of the tumor (Deng et al., 2016; Tan et al., 2014). TGF‐β, NF‐kB, Wnt, FGF, and EGF/HER2 pathways, found to be activated in response to chemotherapy/radiotherapy, can stimulate EMT‐TF expression. Regulation by miRs is also to consider as certain miRs, found to be downregulated in resistant cells, directly suppress EMT‐TF expression, while increasing chemosensitivity in cells with a more Epi phenotype (Nantajit et al., 2015). A deregulation of the miR‐200 family/ZEBs axis appears to mediate docetaxel resistance of prostate cancer cells (Hanrahan et al., 2017; Puhr et al., 2012). miR‐21, a well‐known ‘oncomir’ (i.e., a miR with oncogenic properties), is associated with EMT, resistance to trastuzumab, as well as to chemotherapy (paclitaxel or doxorubicin) through the PTEN/AKT pathway in HER2‐positive breast cancer cells (De Mattos‐Arruda et al., 2015). A primary mechanism of chemoresistance involves drug elimination by increasing efflux of hydrophobic drugs regulated by ATP‐binding cassette (ABC) transporters, a family of energy‐dependent transporters including multidrug resistance protein 1 MDR1/P‐glycoprotein (ABCB1 gene) and multidrug resistance protein MRP (ABCC1 gene). EMT‐TFs including SNAIL and TWIST families bind to E‐boxes found in promoter regions of ABC genes (Saxena et al., 2011). Radiation and hypoxia also generate reactive oxygen species (ROS), and it was suggested that EMT programs can be activated by ROS elevation with potential consequences on drug or radiation resistance (Nantajit et al., 2015). The EMT‐TF SLUG has been found to antagonize p53‐mediated apoptosis by repressing PUMA, therefore promoting BCL2 function and cell survival (Kurrey et al., 2009). Radiotherapy and several chemotherapy agents affect genome integrity. Recent evidence shows that EMT‐TFs regulate genomic instability in various model systems by activating DNA damage response genes such as ERCC1, DNA ligase 1, and ATM (Deng et al., 2016; Hsu et al., 2010), or by regulating BRCA1 expression in breast carcinomas in the case of SLUG (Wu et al., 2012).
5. Cancer cell immune resistance
Aside from the creation of an immunosuppressive TME that one could consider as providing with tumor‐extrinsic mechanisms (van der Burg et al., 2016), immune escape can also be engaged by cancer cells themselves, as they face and trick the immune system. Identification and characterization of cancer cell‐intrinsic mechanisms have been the subject of intense research (van der Burg et al., 2016; Crespo et al., 2013; Dunn et al., 2002; Khong and Restifo, 2002; O'Donnell et al., 2017). For instance, cancer cells can escape the immune system of the host by hiding their tumor‐specific antigens. In particular, the immune cells are greedy for cancer cell expressing neoantigens, produced as a result of tumor‐specific ‘nonsynonymous’ mutations in gene‐coding regions, and cross‐presented by the antigen‐presenting cells (APCs) (Rooney et al., 2015). It was recently found that the tumor neoantigen spectrum strongly correlates with immunogenicity and with response to anti‐PD1 immunotherapy (McGranahan et al., 2016; Rizvi et al., 2015; Snyder et al., 2014). By losing some of their neoantigens, cancer cells thus manage to evade the immune system and immunotherapy treatments (Anagnostou et al., 2017). This can be achieved through different molecular events. Downregulation or loss of MHC class I proteins was observed a while ago and confirmed by high‐throughput studies (Garrido et al., 2010; Korkolopoulou et al., 1996; Rooney et al., 2015). MHC class I proteins associate with small peptide antigens (8–10 amino acids in length) to present them on the surface of cells and activate CD8+ T lymphocytes via the T‐cell receptor (TCR). Of note, TEIPP (T‐cell epitopes associated with impaired peptide processing) antigens may be presented by the residual MHC class I molecules of immune‐edited cancer cells, which could be exploited by vaccination approaches (van der Burg et al., 2016). Other defects in the antigen‐processing machinery involving beta‐2‐microglobulin (B2M), or transporter associated with antigen processing (TAP‐1 and TAP‐2), are also regarded as important mechanisms of immune escape (del Campo et al., 2014; Korkolopoulou et al., 1996). Likewise, alterations in the interferon‐gamma (IFN‐γ) signaling pathway are known to impact on antitumor responses (Gao et al., 2016). Cancer cells can also hinder the immune attacks by overexpressing antiapoptotic proteins such as BCL‐XL (Jazirehi et al., 2011).
6. The acquisition of a mesenchymal phenotype is associated with resistance to CTLs
The hypothesis that EMT might also contribute to immune escape of tumors was addressed by exploiting the human mammary carcinoma model MCF7 which underwent EMT, following stable expression of SNAIL or after prolonged exposure to tumor necrosis factor alpha (TNF‐α), exhibited reduced susceptibility to CTL‐mediated lysis (Akalay et al., 2013). MCF7 cells display pronounced Epi traits as assessed by their EMT scores when the EMTed MCF7 derivatives displayed various Mes states (Akalay et al., 2013). In this case, the protection from CTL‐mediated lysis appeared to be linked with the activation of an autophagic program, which may contribute to promote survival in these cells. A similar impairment of CTL‐mediated lysis was also noted in another model of MCF7 cells undergoing EMT (Akalay et al., 2015) wherein silencing of WISP2 (WNT1‐inducible signaling pathway protein 2) coincides with hyperactivity of TGF‐β signaling, EMT, and acquisition of stemness properties (Ferrand et al., 2014; Fritah et al., 2008). In this instance, the autophagy status did not appear to be involved in the resistant phenotype; however, a disruption of TGF‐β signaling in these cells by treatment with A83‐01, an inhibitor of TGF‐β‐related type I receptors ALK5, ALK4, and ALK7, effectively decreased resistance to CTLs. These findings suggest that key developmental pathways such as TGF‐β signaling in Mes cancer cells can mediate immune resistance to CTLs. This further suggests that different Mes cancer cell variants along the EMT spectrum could engage various mechanisms of resistance (Fig. 1). In this regard, we recently investigated the expression levels of PD‐L1 in the Mes MCF7 derivatives (Noman et al., 2017). While PD‐L1 was highly expressed in MCF7 cells silenced for WISP2, other cell lines had little or no expression of PD‐L1 similar to that found in the parental MCF7 cells. Additionally, it was demonstrated that PD‐L1 production by these MCF7‐shWISP2 cells participates in modulating immunoresistance toward CTLs. The EMT‐TF ZEB1 was identified as an important regulator of PD‐L1 expression in this system, supporting a rationale for EMT blockers as an alternative to control PD‐L1 expression and boost immunotherapeutic responses.
Figure 1.
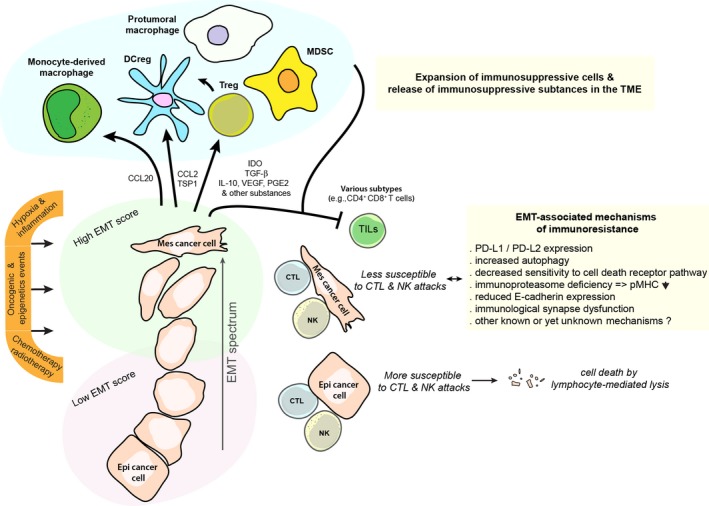
Cross‐talks between carcinoma cells, immunosuppressive cells, and immune effector cells within the tumor microenvironment in controlling antitumor immunity and immune escape.
Tumor hypoxia is an important parameter to consider as a driver of EMT, tumor heterogeneity, and tumor immune escape. Using a model of lung adenocarcinoma (IGR‐Heu) cells derived from a nonmetastatic patient, we noted hypoxic stress‐induced phenotypic diversification along the EMT spectrum (Terry et al., 2017). We observed that the shift toward a more Mes phenotype in hypoxia‐exposed cells is only observed in a fraction of these NSCLC cells. In other words, while some cells undergo an EMT, others do not, or not to the same extent, therefore promoting cancer cell heterogeneity. Further analysis of cancer subclones with pronounced Epi or Mes phenotypes emerging from such hypoxic stress revealed that Mes subclones exhibited an increased propensity to resist CTL cell‐mediated lysis. The observed resistance to the autologous CTL Heu171 clone attack can be partially explained by the absence of detectable E‐cadherin in the Mes IGR‐Heu cancer variants, as the cytolytic function of CTL Heu171 relies on integrin CD103 (αΕβ7 integrin), and its interactions with its preferred ligand, E‐cadherin (Franciszkiewicz et al., 2013; Le Floc'h et al., 2007). Of note, CD103 is preferentially expressed by tumor‐infiltrating lymphocytes (TILs), and its expression in tumor tissue in NSCLC and ovarian cancers was associated with a good outcome (Djenidi et al., 2015; Webb et al., 2014). We also suspect the contribution of resistance mechanisms independent of the E‐cadherin–CD103 interaction, as inhibition of TGF‐β signaling minimized resistance to CTL‐mediated killing without apparent changes in expression of E‐cadherin in Mes cancer clones (Terry et al., 2017).
It is known that CTLs mainly use the perforin/granzyme pathway to destroy target cells (Barry and Bleackley, 2002). However, especially when this pathway is affected, the death may also be triggered through the activation of caspase‐dependent or caspase‐independent death receptor pathways with engagement of TNF‐related apoptosis‐inducing ligand (TRAIL) or FAS at the surface of cancer cells (Barry and Bleackley, 2002). Using the pancreatic PANC1 model, it has been demonstrated that cells with forced expression of Brachyury, an EMT inducer, had decreased susceptibility to lymphocyte‐mediated killing compared to control cells (Hamilton et al., 2014). By using experimental conditions in which target cancer cells were cocultured with effector CTLs for a relatively long period (Palena et al., 2007), Hamilton and colleagues were able to demonstrate that the poor killing observed was due in great part to a defect in caspase‐dependent apoptotic death in the cells, despite immune antigenicity (Hamilton et al., 2014).
Interestingly, in other cellular contexts, such as NSCLC cells, defects in antigen‐presenting machinery associated with immunoproteasome deficiency were found to be a common event in cancers with a more Mes profile and could affect T cell‐mediated cytotoxicity (Tripathi et al., 2016).
7. Alterations of cell–cell interactions and immunological synapses
Immune killer cells, such as CTLs and NK cells, highly rely on their physical contacts with APCs or target cancer cells for their activity, maturation, production of IFN‐γ and TNF‐α, and lytic functions (Dustin and Long, 2010). These interactions may occur in a form of an ‘immunological synapse’ (IS) (Dustin, 2014; Dustin et al., 2010) (Fig. 2). Originally described for T‐cell activation and cytotoxicity, the IS is also critical for NK cell cytotoxicity (Dustin and Long, 2010; Mace et al., 2014; Orange, 2008). Formation of the IS in T cells required the coordinated interactions of MHC‐TCR, regulation of cytoskeletal elements such as actin, and the integration of integrin‐based signals and forces in part generated when integrin molecules such as lymphocyte function‐associated antigen‐1 on the T cell, interacted with intercellular adhesion molecule 1 (ICAM‐1) on the target cell. During peptide‐MHC/TCR ligation, integrins undergo conformational changes mediated by phosphorylation cascades, including phosphotyrosine kinase activation, linking integrins to the actin cytoskeleton. The actin cytoskeleton undergoes polymerization at the edge of these active synapses and this further coincides with immune cell flattening and a size increase in the synaptic diameter (Comrie and Burkhardt, 2016). This also leads to the emergence of TCR microclusters coalescing toward the center of the IS into the zone referred to as the central supramolecular activation cluster (Yu et al., 2013b). On the other hand, integrin microclusters generally stand in the periphery of the synapse to form a highly contractile zone termed as peripheral supramolecular activation cluster.
Figure 2.
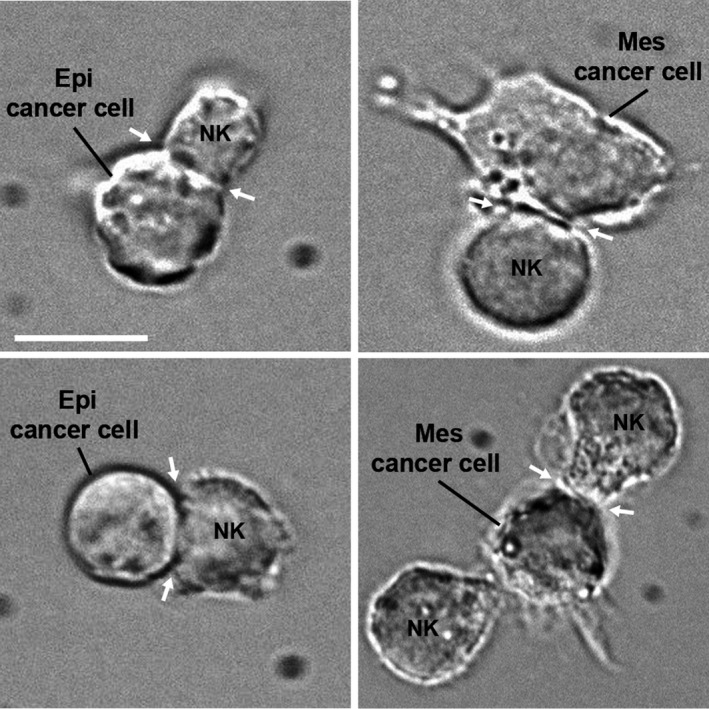
Phase‐contrast microscopy showing conjugates of Epi cancer cell, or Mes cancer cells, with NK lymphocytes (NK92) formed after coculturing the cells for 30 min. Immunological synapses (arrows) are indicated. Scale bar, 10 μm.
Mechanical forces generated at the synapse through intercellular adhesion are also important to regulate adhesion‐based signals and the rearrangement of the actin cytoskeleton. IS involving NK cells seems to obey similar rules except that these cells do not express TCR receptors. Instead, they express activating and inhibitory receptors (DNAM1, NKG2D, 2B4) that may likewise regulate signaling, activity, and dynamic changes in the integrin–actin network at the different stages of NK cell cytotoxicity. Thus again, actin dynamics are tightly controlled at the IS to achieve optimal effector functions. Numerous genetic aberrations have been identified and may alter various stages in NK and CTL cytotoxicity, recognition, F‐actin and microtubule networks, trafficking and docking of lytic granules (not discussed above) (Milner and Holland, 2013; Orange, 2008), thus leading to T cell or NK disorders and severe immunodeficiency. These examples highlight the critical requirement of an operational IS to generate an effective immune response.
Whether some cancer cells act by impeding the IS remains largely unappreciated, although some facts support this hypothesis. For instance, loss of MHC and subsequent MHC/TCR complexes will inevitably hinder much of the activity of the IS involving T cells, as well as its architecture. In line with this, we denoted an alteration of the IS signaling in CTL clones cocultured with Mes MCF7 derivatives, concurrent with reduced expression of MHC class I by the latter, when compared to Epi parental MCF7 cells (Akalay et al., 2013). In this line, Dongre et al. (2017) recently observed a drop in MHC class I expression in Mes breast carcinoma cells deriving from MMTV‐PyMT mice compared to their more Epi counterparts.
To note, Mes carcinoma cells often show loss of cell polarity, with striking differences in their cytoskeletal organization and content compared to more Epi carcinoma cells (Schliekelman et al., 2015). Although we possess only incomplete understanding of the mechanisms involved, we hypothesize that carcinoma cells with a more Mes differentiation could be refractory to establishing mature IS when interacting with CTL. As the establishment of the IS, and activation cascades, relies in part on heterophilic interactions between integrins from the immune effector and ICAM‐1 on target cells, it is easy to foresee how loss of ICAM1 will impede IS formation (Anikeeva et al., 2005; Hamai et al., 2008). Moreover, changes in the mechanical forces coupled to the actin network could contribute to affect the integrity of the IS and the lytic commitment. Thus, it is likely that perturbation of the actin network in certain cancer cells will make them either more susceptible or more resistant to lymphocyte‐mediated lysis. It was previously observed that strong morphological changes in NSCLC IGR‐Heu cells, associated with alterations of the actin cytoskeleton, promoted resistance to CTL killing (Abouzahr et al., 2006). This appeared to occur despite the formation of apparently normal conjugates. At the other end, the recent report by Jachetti and colleagues is relevant in showing how cancer cells can perturb the actin network of effector T cells. By investigating a model of mouse cancer stem‐like cells (CSCs), the investigators elegantly showed that abnormal production of extracellular matrix protein tenascin‐C by CSCs, through interactions with integrin α5β1 at the surface of T cells and interfering with reorganization of the actin cytoskeleton, contributes to inhibition of T‐cell activation (with consequences on their proliferation, and cytokine production). Such observations still need to be fully addressed in the context of Mes carcinoma cells. Additionally, not only are these mechanisms pertinent for T cells and CTL‐mediated killing, but in some circumstances, they may also be pertinent in altering significantly cell‐mediated lysis by NK or other killer cells.
8. Immune resistance to NK cells
Natural killer cells are effector lymphocytes of the innate immune system controlling tumor growth during cancer initiation and progression (Vivier et al., 2008). Although the contribution of NK cells during the different steps of tumor progression is still unclear, presumably varying between histological and cancer types (Gentles et al., 2015), their behavior with Mes carcinoma cells remains to be fully addressed. Relevant for this discussion is the observation that NK cells in cancer tissues are rarely found in direct contact with the tumor mass, but rather present in the stroma (Halama et al., 2011). Other studies showed that NK cells can infiltrate tumors, but in this case, NK cells may have impaired capabilities to kill cancer cells (Mamessier et al., 2011; Platonova et al., 2011). In this context, one issue to consider is that cancer cells undergoing EMT, in association with their increased capacity to invade the stroma, and propensity to become circulating tumor cells (CTCs), are hardly detectable in the stromal compartment or in the bloodstream, due to common features with fibroblastic cells and lack of optimized approaches to detect them.
To note, reduction of MHC class I by certain cancer cells can be perceived as a ‘missing self’ by the inhibitory receptors KIRs of NK cells that usually interact with MHC class I (Vivier et al., 2008). As a result, this could render cancer cells susceptible to NK cells. However, because NK cell activation and killing is regulated by a combination of activating and inhibiting receptors and their related ligands (Chester et al., 2015), reduction or loss of MHC class I is rarely sufficient to trigger NK cell‐mediated lysis. Cancer cells may again exploit various strategies to compensate this loss such as upregulation of the nonclassical MHC class I proteins HLA‐G (Carosella et al., 2015) and HLA‐E (Braud et al., 1998), able to restrain NK activity through KIRs and CD94/NKG2A, respectively. There is also evidence in some cancer cells for an increase in resistance to perforin/granzyme B‐mediated cell death by immune cells (Ben Safta et al., 2015), reduction in cell surface MHC class I‐related chain (MIC) proteins, MICA and MICB, and UL16 binding proteins, ligands for NK‐activating receptor‐activating NKG2D (Malladi et al., 2016). Furthermore, the production by certain cancer cells of soluble forms of MICs could act as a decoy for NK cells while promoting degradation of NKG2D (Groh et al., 2002; Raffaghello et al., 2004).
9. EMT‐mediated immune resistance to NK cells
In a recent report, we observed that NSCLC tumor subclones with Mes features exhibited an increased propensity to resist NK cell‐mediated lysis compared to more Epi subclones. This immunoresistant phenotype was at least partly due to defective immune synapse signaling (Terry et al., 2017). Further investigations are needed to unravel the detailed mechanisms as to why those carcinoma cells are more prone to inactivate the IS when interacting with NK cells. Aside from our observation, one significant breakthrough came from Hamilton et al. who demonstrated that high levels of the EMT‐related factor Brachyury reduced the susceptibility of carcinoma cells not only to CTLs, but also to NK cells, lymphokine‐activated killer, FAS, and TRAIL‐induced cell death. In a recent report investigating the lung cancer setting, the same group further found a potential role for estrogen receptor α in promoting both Mes characteristics and resistance to immune‐mediated cytotoxicity (Hamilton et al., 2016). By contrast, the study of López‐Soto et al. (2013) described how EMT in colon cancer cells, following SNAIL overexpression, could instead render cancer cells more sensitive to NK‐mediated lysis in a manner that appears dependent on NKG2D ligands upregulated in the cells undergoing EMT. These investigators obtained similar results in immortalized keratinocytes stimulated by TGF‐β.
10. EMT, immune checkpoints, and immunosuppression
Similar to MDSC or Treg cells present in the TME, cancer cells can express immunoregulatory enzymes, immunosuppressive cytokines, or immune checkpoint ligands to modulate efficacy of the immune response and its duration. Among those, TGF‐β has been one of the most studied so far (Massague, 2008). It is a known EMT driver whose expression is often upregulated in Mes cancer cells. Moreover, TGF‐β can impair maturation, differentiation, or activation of both innate and adaptive immune cells, including NK cells, DCs, macrophages, neutrophils, and CD4+ and CD8+ T cells reviewed in Tu et al. (2014). TGF‐β can inhibit the cytotoxic T‐cell functions by altering the expression of cytotoxic gene products including perforin, granzyme A, granzyme B, Fas ligand, as well as IFN‐γ (Joffroy et al., 2010; Thomas and Massague, 2005). In addition, it can inhibit their differentiation into central memory T cells (Takai et al., 2013). In prostate cancer cell lines, TGF‐β led to MHC class I downregulation (Chen et al., 2015). TGF‐β secreted by cancer cells was also documented to induce Treg cells from CD4+ T cells, based on induction of Foxp3 expression in cocultures (Joffroy et al., 2010). In NK cells, TGF‐β impaired the cytokine production (Wilson et al., 2011) and their cytolytic activity at least by downregulating expression of NKG2D (Lee et al., 2004).
Clinical success of novel immunotherapies targeting immune checkpoints also underlines the importance of immune checkpoint ligands in the control of the immune response (Burstein et al., 2017). Expression of checkpoint molecules such as PD‐L1 dampens the immune response and can hold T cells from killing target cancer cells. Indeed, PD‐L1 is a ligand for the cell surface receptor PD‐1 especially expressed by T lymphocytes which upon signaling (Crespo et al., 2013; Zou et al., 2016) will reduce T‐cell activity, and in the case of CTLs, inhibit T‐cell cytotoxic activity, eventually enforcing an exhaustion state (van der Burg et al., 2016; Zou et al., 2016). When antibodies block these proteins, T cells may at least partially restore their activity. PD‐L1 expression can be naturally induced by inflammatory cytokines including IFN‐γ and TNF‐α, which limits immune responses upon inflammatory conditions. In cancer, various signals and alterations can drive PD‐L1 expression as an immunosuppression mechanism (Chen et al., 2016a; O'Donnell et al., 2016). Interestingly, both TGFβ1 and CD274 (encoding for PD‐L1) genes can be induced under hypoxic conditions, either directly via hypoxia‐induced factors (HIFs) or indirectly through related factors (Barsoum et al., 2014; Hasmim et al., 2013; Noman et al., 2014).
Chen et al. (2014) have identified a molecular link between EMT and more abundant expression of PD‐L1 in human lung tumors. In vitro and in vivo experiments demonstrated that downregulation of miR‐200s and ZEB1 overexpression not only drive EMT but also may lead to upregulation of PD‐L1. Beyond showing the regulation of PD‐L1 by the ZEB1/miR‐200 axis, perhaps one of the most intriguing observations was the association of these events with exhaustion of intratumoral CD8+ T lymphocytes, which ultimately promoted the development of metastases in mice. Further work by this group also indicated a role for bone morphogenetic protein‐4 (BMP4) to regulate PD‐L1 expression (Chen et al., 2016b). As a follow‐up question, it would be interesting to see whether additional immune checkpoints, other than PD‐L1, likewise associated with a more Mes tumor phenotype (Mak et al., 2016), play a role in T‐lymphocyte exhaustion.
These studies are also important because it is expected that tumors with high PD‐L1 expression, or other inhibitory checkpoints ligands, will be the best protected against the endogenous immune response, while patients with such tumor characteristics are also believed to be good candidates for immunotherapy, with better efficacy of immune checkpoint blockade, and reinvigoration of the immune response. Consistent with this notion, high expression of PD‐L1 in metastatic NSCLC patients was able to predict, at least to some degrees, response to anti‐PD1 treatment (Garon et al., 2015). This led to the development of clinical studies using predefined PD‐L1 expression level into the inclusion criteria for patients. Recent approval of pembrolizumab in the first‐line metastatic NSCLC has been established under such conditions (Reck et al., 2016). Nevertheless, PD‐L1 alone might not be sufficient as a predictive marker of response to anti‐PDL1 monoclonal antibodies (Topalian et al., 2016). Promising markers of response are currently under evaluation including the mutational load (Hugo et al., 2016; McGranahan et al., 2016; Rizvi et al., 2015; Rosenberg et al., 2016; Snyder et al., 2014); the MSI (Le et al., 2015), somatic copy number alterations (Davoli et al., 2017), or immune‐related signatures (Prat et al., 2017). The presence of tumor‐resident CD103+ TILs could also represent a good candidate marker of response to immunotherapy as this TIL population seems to express PD‐1 (Djenidi et al., 2015; Komdeur et al., 2016).
We should consider that optimal response would likely depend on the global expression pattern of the immune checkpoint receptors and the combined expressions of their ligands. In addition to PD‐L1, ligands expressed at the surface of cancer cells and immunosuppressive cells include PD‐L2, galectin‐9, CEACAM1, CD70, CD137L, OX40L, ICOSL, and corresponding receptors: T‐cell immunoglobulin mucin receptor 3 (TIM3; HAVCR2), lymphocyte activation gene 3 (LAG3) regarding the coinhibitory receptors, and costimulatory receptors CD27, CD137, OX40, GITRL, or ICOS (Mahoney et al., 2015; Melero et al., 2015).
Current studies aimed at unraveling how EMT may promote immunosuppression and to expand the list of the TME cellular components involved (Fig. 1). Kudo‐Saito et al. (2009) showed that forced expression of Snail in mouse melanoma B16F10 cells enhanced the EMT program in these cells, in conjunction with increased thrombospondin 1 production, induced an impairment of DC maturation while allowing expansion of a population of suppressive Treg‐like CD4+ Foxp3+ cells in cell cultures. Similarly, in syngeneic mice, tumors formed by mock‐transfected B16F10 cells revealed infiltrating CD8+ T cells, whereas tumors derived from Snail‐transduced B16F10 showed less tumor‐infiltrating CD8+ T cells, more Treg, and more metastasis to the lungs. Subsequent work from this group also pointed out the critical role of the chemokine CCL2 in mediating the described immunosuppressive effects, with possible effects on recruitment of other immunosuppressive CCR2+ populations such as MDSC and macrophages (Kudo‐Saito et al., 2013). Others found in the MMTV‐PyMT mouse model of breast cancer that tumors arising from Mes carcinoma cell lines, or Snailhigh cell populations, exhibited more Tregs and protumoral macrophages compared to their more epithelial, or snailLow, counterparts (Dongre et al., 2017). Using various cancer models, Ricciardi and colleagues observed that exposure to inflammatory cytokines can endow cancer cells undergoing EMT with a number of immunomodulatory effects depending on the cancer cell origin. This included interference with proliferation, differentiation, and apoptosis of NK, T‐, and B‐cell populations (Ricciardi et al., 2015). They underlined a role of the indoleamine‐2,3‐dioxygenase (IDO) pathway to explain the deleterious effects observed on T cells after inflammatory‐induced EMT (Platten et al., 2012; Ricciardi et al., 2015). Yu et al. reported evidence suggesting that hypoxia‐induced EMT in hepatocellular carcinoma (HCC) induces an immunosuppressive TME to promote metastasis (Ye et al., 2016). On a mechanistic standpoint, they showed that hypoxia‐induced EMT in HCC cells promoted increased expression of CCL20 in a HIF‐1α dependent manner, which through its action on monocyte‐derived macrophages, and expression of IDO by these cells, led to decreased proliferation of CD4+ and CD8+ T cells, while promoting the expansion of immunosuppressive Treg cells.
Collectively, these studies provide important insights into the diversity of actors and routes exploited by Mes cancer cells to escape the immune system. Moreover, this highlights the need to gain further knowledge on the composition of TME niches in various tumor sites and how those establish.
11. Cancer EMT signatures and immune activation
EMT scoring based on cancer‐specific transcriptomic signatures was pioneered by various groups (Byers et al., 2013; Mak et al., 2016; Tan et al., 2014). This EMT score was developed based on expression profiling of several hundred genes (Tan et al., 2014), and this score has been used to assess (semiquantitatively) the position of cancer cells in the EMT spectrum (Fig. 3). Most tumor types and corresponding cell lines exhibit a wide range of EMT scores, with values ranging from −0.8 for most Epi to +0.6 for most Mes features. While the majority of colon carcinomas and corresponding cell lines appear more Epi on average, some tumors do exhibit Mes‐like phenotypes likely corresponding to the newly established Mes molecular subtype (Punt et al., 2017). Other tumor types such as breast, ovarian, pancreatic, lung, and prostate tumors and cell lines have an average value of 0 but show a large spectrum of values. Remarkably, renal carcinomas display strong Mes features, perhaps because they derive from an epithelium initially originating from the metanephric mesodermal mesenchyme. On the other hand, melanomas are expectedly quite Mes but not as mesenchymal as osteosarcoma. The EMT score reflects, in part, the EMT status of the cancer cells but also contributions from the stromal fibroblasts in the tumor sample. Indeed, transcriptomes of breast tumors depleted from their stroma by microdissection show that luminal and ErbB2 subtypes are Epi‐like, whereas basal‐like and claudin‐low tumors have pronounced EMT scores. Generally, the EMT spectrum of cell lines spans the same spectrum as their corresponding tumor types, suggesting that cell lines maintain the EMT features from their tumors of origin (Tan et al., 2014).
Figure 3.
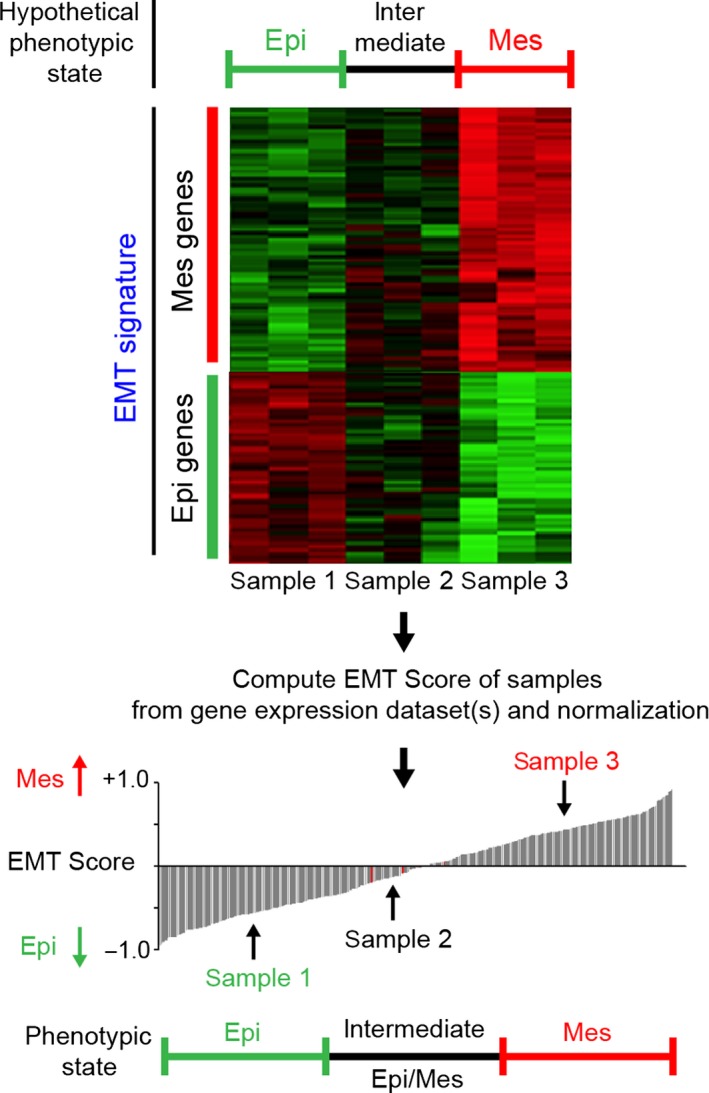
Schematic diagram illustrating the potential utility of EMT scoring. An EMT score is inferred after interrogating a gene expression dataset for previously published or in‐house‐generated EMT signatures. Cell line or tumor samples can be compared and sorted from most Epi to most Mes based on their EMT score. An arbitrary threshold may be used to define Epi, intermediate Epi/Mes, or more advanced Mes states.
Among molecular alterations associated with EMT, pathway analysis revealed association between high EMT score and high expression of several immune checkpoints including PD1, PD‐L1, PD‐L2, B7‐H3, OX40, OX40L, CD137, TIM3, LAG3, and CTLA4 (Mak et al., 2016). Lou et al. (2016) confirmed these observations in three independent datasets of lung cancer patients with early or advanced NSCLC adenocarcinomas. Additional reports suggested an association between PD‐L1 expression and EMT in NSCLC and HNSCC (Kim et al., 2016; Ock et al., 2016b). In their study, Lou et al. further found that tumors harboring high Mes content were associated with increased infiltration by TILs and Treg cells, and increased expression of costimulatory molecules (CD80, CD86). Furthermore, they showed elevated endogenous immune activation in these tumors, as revealed by a higher expression of IFN‐γ, IFN‐γ‐inducible CXCL10 and IDO, among many other upregulated genes (Lou et al., 2016). In the same study, the B7 family of immunomodulatory molecule, B7‐H3 (CD276), was correlated with the EMT score and found to have a prognostic value for disease‐free survival and overall survival. By contrast, neither PD‐L1, other immune checkpoints, nor EMT were found to have such prognostic value. No data were available on whether some of these patients received immunotherapy regimens during their treatment history. Nevertheless, these studies highlight the rationale to test gene expression‐based EMT signatures as a predictive marker to select patients for anticheckpoint therapies. Importantly, the investigators demonstrated no association between EMT and mutational burden, which raises the interesting possibility that evaluation of both mutational load and EMT status could help improve patient selection (Lou et al., 2016).
By investigating the Epi marker ESRP1 into The Cancer Genome Atlas dataset, Yao et al. (2016) identified a link between enhanced Mes features in ESRP1‐low melanoma and high infiltrating lymphocyte activity, as assessed by a two‐gene signature (PRF1 encoding for perforin and GZMA encoding for granzyme A). The ESRP1‐low/Mes high group further showed better overall survival compared to groups expressing full‐length or truncated forms of ESRP1. The authors suggested this subgroup of melanoma patients as well suited for immunotherapy intervention. In this regard, in the recent study of Hugo et al. (2016) analyzing biopsies of anti‐PD1‐treated advanced melanoma patients, EMT signatures and Mes‐related genes were found to be upregulated among nonresponding relative to responding patients, thus suggesting that the Mes features of the primary tumor may be associated with innate anti‐PD‐1 resistance. However, T cell‐related genes such as CD8A/B, PD‐L1, LAG3 (T‐cell checkpoint genes), and IFN‐γ did not appear differentially expressed in nonresponding compared to responsive tumors.
In bladder cancers, patients with tumors characterized by an Epi (luminal) phenotype had a better response rate while treated with anti‐PD‐L1 therapy compared to those harboring basal subtype (Rosenberg et al., 2016) expressing numerous Mes markers (Choi et al., 2014; Kamat et al., 2016). Reports also suggest that, similar to what has been found in the claudin‐low subtype of breast cancer, there exists a claudin‐low subtype of high‐grade muscle‐invasive bladder cancer, defined by highly apparent Mes features. Moreover, despite being infiltrated by immune cells, this subtype was actively immunosuppressed (Kardos et al., 2016).
In considering EMT signatures, it is important to remember that a rich stromal contingent present within the tumor specimen can contribute to disturbing the assessment of the cancer EMT score. In particular, it remains extremely complicated in this type of analysis to clearly distinguish Mes cancer cells from CAFs. EMT‐TFs are expressed by Mes cells in the normal and tumor stroma; moreover, they may contribute to the emergence of CAF populations, providing a specialized microenvironment also with a potential role in resistance to treatment and immune response. To our knowledge, the published data regarding association between EMT and immune signatures have not totally addressed the potential contribution of cancer vs stroma in Mes features. Interestingly, in colon cancer, where the stromal fraction should account for most of the Mes contents, Becht et al. (2016a), using transcriptomics, were able to identify an immune‐related stromal/Mes subtype, characterized by an immunosuppressive signature correlating with a high density of fibroblasts likely producing chemokines, cytokines, inflammatory, and angiogenic factors.
Altogether, these reports indicate that there is some need for caution when considering EMT scoring as a predictor of clinical response for immunotherapy. More research and prospective studies are urgently needed to test this hypothesis across numerous cancer types (and disease subtypes).
12. Targeting EMT to attenuate immunoresistance and improve susceptibility to cytotoxic antitumor immune response
In our previous work, we observed that inhibition of TGF‐β signaling can increase susceptibility of Mes carcinoma cells to CTL and NK cell‐mediated lysis (Akalay et al., 2015; Terry et al., 2017). In theory, such a strategy could be compatible with immunotherapy to facilitate response at multiple levels. Notably, Mes carcinoma cells often display elevated TGF‐β signaling contributing to maintain both cell plasticity and immune resistance of these cells. TGF‐β is likewise a well‐known immunosuppressive substance. Of therapeutic relevance, multiple molecules targeting TGF‐β signaling are currently tested in clinical studies (Chretien et al., 2014; de Gramont et al., 2017). We envision that Mes carcinoma may be avid targets for such compounds. Because phenotypic and functional EMT‐driven cancer cell plasticity allows malignant cells to adapt to selective pressures including the immune response, and leads to the development of resistance to targeted therapies, targeting EMT‐driven cancer cell plasticity may represent an innovative approach to cancer therapy. Given the dynamic nature of the EMT‐driven epithelial plasticity, targeting EMT may request to concurrently target several processes in order to be successful. Agents that can be used to target the EMT may include extracellular inducers of EMT, the transcription factors that orchestrate the EMT transcriptome, the downstream effectors of EMT, and the canonical pathways known to be involved in EMT, such as the signal transduction pathways driven by activation of TGF‐βR, EGFR, and AXL, as well as emerging pathways such as epigenetic therapies, glycosylation pathways, or EMT‐associated metabolic alterations (Davis et al., 2014; Malek et al., 2017). Recent studies from high‐throughput analyses have now selected a number of candidate compounds that could preferentially alter the viability of carcinoma cells with high Mes features (Byers et al., 2013; Chua et al., 2012; Ock et al., 2016a; Tan et al., 2014). T cells specifically educated or engineered to target Mes cancer cell populations, or vaccines designed to target those, could also represent interesting strategies to control the expansion of Mes cancer cell populations. Previous studies demonstrated some efficacy using vaccines directed to the EMT drivers Brachyury, TWIST1, or CRIPTO (Ardiani et al., 2014; Hamilton et al., 2013; Ligtenberg et al., 2016; Palena et al., 2007). More experimental studies are now needed to evaluate the clinical efficacy in monotherapy and in combination with approved immune checkpoint blockers.
Recent reports suggested that two widely used compounds, the EGFR inhibitor erlotinib, and fulvestrant, an antagonist of ER, may be efficient at decreasing Mes features of lung cancer cells as well as for improving their lysis by the immune effector cells (Dominguez et al., 2016; Hamilton et al., 2016). In addition, as suggested in our previous studies, it could be interesting to target autophagy when Mes carcinoma cells exhibit an autophagic state (Akalay et al., 2013).
Aside from the context of EMT, other compounds have been tested that should be considered to enhance susceptibility to immune killer cells (Bommarito et al., 2016; He et al., 2013). Agents inhibiting the expression of PD‐L1 through blockade of the AKT/PI3K/MTOR or the RAS/MEK signaling pathways, and more generally drivers of PD‐L1 expression, could help sensitize cancer cells to immunotherapy at the onset in various tumor types (Alsuliman et al., 2015; Lastwika et al., 2016; Loi et al., 2016; Peng et al., 2016). Of note, immune cells could be pharmacologically manipulated. Elegant work by Parameswaran et al. (2016) showed how pharmacological GSK3 inactivation in NK cells can restore NK cell cytotoxicity in patients with AML.
Although all these approaches could be efficient, heterogeneity of tumors, the diversity of these cancer cell populations, their high plasticity, and adaptability to stress will remain a major issue. Therefore, to achieve a durable response, it will be essential to develop strategies able to limit the tumor heterogeneity and cell plasticity. We presume that novel strategies targeting both tumor contingent and microenvironmental context might improve efficacy of cancer treatments. In this case, hypoxia could be an attractive target to limit tumor heterogeneity, cell plasticity, the expansion of immunosuppressive cells, and secretion of immunosuppressive substances, while rendering cancer cells more susceptible to immune attacks (Noman et al., 2015; Paolicchi et al., 2016; Wilson and Hay, 2011). One of the main caveats will be to design potent and specific inhibitors of HIFs. Alternatively, drugs affecting developmental pathways and plasticity of cells must be considered (Malladi et al., 2016; Pattabiraman and Weinberg, 2014). Similarly, epigenetic modifiers such as demethylating agents, or histone deacetylase inhibitors, could be exploited for blocking this plasticity or as priming agents to boost the efficacy of immunotherapy (Chretien et al., 2014; Saleh et al., 2016).
13. Surrogate biomarkers of EMT‐driven cancer cell plasticity and the immune microenvironment
The recent success of immunotherapies provides some evidence that additional approaches to genomic‐driven precision medicine may be rewarding. Indeed, although recent progress in the characterization of genetic abnormalities has resulted in the multiplication of clinical trials driven by genomic alterations (e.g., ProfiLER, SHIVA, MOSCATO, and MD Anderson genomic‐driven clinical trials), an important limitation of this approach is the relatively low average (30–40%) of targetable genomic alterations, leading to poor recruitment rate in early‐phase clinical trials, and potentially, a limited number of patients who may benefit from the targeted therapy (Barlesi et al., 2016; Massard et al., 2017). Concurrently, a platform that will allow screening real‐life biospecimens for surrogates of EMT‐driven cancer cell plasticity, to foster inclusions in clinical trials evaluating drugs targeting EMT and the immune microenvironment, is urgently needed. Of note, the immune microenvironment including the degree of activation of specific pathways and immune subpopulations is the subject of active bioinformatic research (Becht et al., 2016b; Galon et al., 2013; Gentles et al., 2015). However, this approach has some limitations: (i) the unknown relevance of the gene signature for the study of EMT in advanced stages of the disease; (ii) technological challenge to generate gene expression profiles from formalin‐fixed paraffin‐embedded (FFPE) samples; (iii) the inability to distinguish the contribution of stroma when no selection of the region of interest is performed; (iv) transcriptional EMT signatures identify phenotypic changes, but it is now recognized that overexpression of EMT‐TFs without phenotypic changes may be associated with functional plasticity; (v) the challenge to evaluate the dynamics of gene expression changes in serial tissue biopsies. In order to address these challenges, an effort to characterize metastatic disease, the development of technologies dedicated to FFPE samples (NanoString and HTG EdgeSeq technologies), and the identification of new surrogate biomarkers of EMT‐driven cancer cell plasticity in carcinoma cells that are not expressed by stroma cells are needed.
Finally, because EMT is reversible, it is critical to consider the dynamics of this process. While it is difficult to perform serial tumor biopsies, the use of liquid biopsies for a noninvasive monitoring of disease evolution is increasingly implemented (Haber and Velculescu, 2014; McGranahan and Swanton, 2017). CTCs consist of cancer cells with very different EMT phenotypes along the EMT spectrum including transitional/hybrid states between Epi and Mes (Kang and Pantel, 2013; Vona et al., 2000). Methods that rely on the display of cell surface Epi markers such as EpCAM by CTCs may well miss capturing a sizeable, clinically relevant portion of the CTCs (Farace et al., 2011). ISET is an antibody‐independent whole blood filtration‐based approach for CTC isolation that relies on the larger size of all types of circulating rare cells compared to most leukocytes (Laget et al., 2017). Feasibility of immunostaining for vimentin, pan‐cytokeratin, EpCAM, EGFR, Ki67, and c‐MET has been previously reported (Krebs et al., 2012; Laget et al., 2017; Lecharpentier et al., 2011). EMT can potentially occur in blood or lymph vessels; however, given that the half‐life of single CTC is very short (Meng et al., 2004), it is unlikely that these cells can be induced to undergo EMT. Nonetheless, they could do so beforehand, or once aggregated into microemboli which can reside for much longer in capillaries in close contact with platelets that release TGF‐β (Labelle et al., 2011). Much work remains to be conducted to adequately assess the role of EMT in CTCs and whether they can mark refractoriness to targeted therapeutics and immunotherapies (Alix‐Panabieres et al., 2017). Longitudinal studies evaluating changes over time of EMT‐TFs expression and other surrogates of EMT‐driven cancer cell plasticity may be informative, but are lacking information on the immune microenvironment. Immunomonitoring in blood may provide additional information compared to the in situ immune microenvironment, although this remains to be shown.
14. Conclusion
Recent advances in the field of cancer immunotherapy have revolutionized the management of patients with melanoma, NSCLC, renal cell carcinomas, bladder carcinomas, HNSCC, ovarian carcinomas, and lymphomas (Burstein et al., 2017). We are still at the beginning of an exciting period of discovery and improvement of these therapies. One of the biggest challenges toward such improvement is to better understand the mechanisms at play in the naturally acquired resistance seen in some patients, as well as in therapy‐induced resistance seen in subgroups of patients, on or after treatment, who do initially respond to immunotherapy. Additionally, it will be critical in this effort to identify potential targets responsible for this resistance and develop new strategies able to eliminate the cancer cell‐resistant clones or prevent their emergence. The link between EMT and immune recognition and killing of cancer cells is now well established. Numerous observations now provide relevant clues to how Mes carcinoma cells could contribute such resistance, while pointing those as promising targets to consider for improving immunotherapy regimens and develop predictive markers of response. In this perspective, we reason that epithelial‐mesenchymal plasticity, a critical program for carcinoma progression and metastasis, is a central driver of not only tumor malignancy but also immune regulation and antitumor response shaping. Clearly, EMT is a key process that is critical for immune resistance but also a potent driver for the activation of an immunosuppressive network within the TME. Therefore, targeting EMT may offer important perspectives for the current immunotherapy approaches for advanced tumors.
Acknowledgements
We are grateful to the Ligue Contre le Cancer (EL2015.LNCC/SaC), Institut National du Cancer (PLBIO15‐266), and the SIRIC‐SOCRATE program for their support.
Contributor Information
Stéphane Terry, Email: stephane.terry@gustaveroussy.fr.
Salem Chouaib, Email: salem.chouaib@gustaveroussy.fr.
References
- Abouzahr S, Bismuth G, Gaudin C, Caroll O, Van Endert P, Jalil A, Dausset J, Vergnon I, Richon C, Kauffmann A et al (2006) Identification of target actin content and polymerization status as a mechanism of tumor resistance after cytolytic T lymphocyte pressure. Proc Natl Acad Sci U S A 103, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AK et al (2013) Epithelial‐to‐mesenchymal transition and autophagy induction in breast carcinoma promote escape from T‐cell‐mediated lysis. Cancer Res 73, 2418–2427. [DOI] [PubMed] [Google Scholar]
- Akalay I, Tan TZ, Kumar P, Janji B, Mami‐Chouaib F, Charpy C, Vielh P, Larsen AK, Thiery JP, Sabbah M et al (2015) Targeting WNT1‐inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF‐4 and miR‐7 expression. Oncogene 34, 2261–2271. [DOI] [PubMed] [Google Scholar]
- Alix‐Panabieres C, Mader S and Pantel K (2017) Epithelial‐mesenchymal plasticity in circulating tumor cells. J Mol Med (Berl) 95, 133–142. [DOI] [PubMed] [Google Scholar]
- Allavena P and Mantovani A (2012) Immunology in the clinic review series; focus on cancer: tumour‐associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol 167, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsuliman A, Colak D, Al‐Harazi O, Fitwi H, Tulbah A, Al‐Tweigeri T, Al‐Alwan M and Ghebeh H (2015) Bidirectional crosstalk between PD‐L1 expression and epithelial to mesenchymal transition: significance in claudin‐low breast cancer cells. Mol Cancer 14, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N et al (2017) Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov 7, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikeeva N, Somersalo K, Sims TN, Thomas VK, Dustin ML and Sykulev Y (2005) Distinct role of lymphocyte function‐associated antigen‐1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 102, 6437–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell SM (2017) Nivolumab in the treatment of Hodgkin lymphoma. Clin Cancer Res 23, 1623–1626. [DOI] [PubMed] [Google Scholar]
- Antony J, Tan TZ, Kelly Z, Low J, Choolani M, Recchi C, Gabra H, Thiery JP and Huang RY (2016) The GAS6‐AXL signaling network is a mesenchymal (Mes) molecular subtype‐specific therapeutic target for ovarian cancer. Sci Signal 9, ra97. [DOI] [PubMed] [Google Scholar]
- Ardiani A, Gameiro SR, Palena C, Hamilton DH, Kwilas A, King TH, Schlom J and Hodge JW (2014) Vaccine‐mediated immunotherapy directed against a transcription factor driving the metastatic process. Cancer Res 74, 1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Adriani G, Dang TM, Tu TY, Penny HX, Wong SC, Kamm RD and Thiery JP (2015) Contact‐dependent carcinoma aggregate dispersion by M2a macrophages via ICAM‐1 and beta2 integrin interactions. Oncotarget 6, 25295–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, Ouafik L, Besse B, Rouquette I, Westeel V et al (2016) Routine molecular profiling of patients with advanced non‐small‐cell lung cancer: results of a 1‐year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 387, 1415–1426. [DOI] [PubMed] [Google Scholar]
- Barry M and Bleackley RC (2002) Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2, 401–409. [DOI] [PubMed] [Google Scholar]
- Barsoum IB, Smallwood CA, Siemens DR and Graham CH (2014) A mechanism of hypoxia‐mediated escape from adaptive immunity in cancer cells. Cancer Res 74, 665–674. [DOI] [PubMed] [Google Scholar]
- Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautes‐Fridman C, Laurent‐Puig P and Fridman WH (2016a) Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res 22, 4057–4066. [DOI] [PubMed] [Google Scholar]
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent‐Puig P, Sautes‐Fridman C, Fridman WH et al (2016b) Estimating the population abundance of tissue‐infiltrating immune and stromal cell populations using gene expression. Genome Biol 17, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Safta T, Ziani L, Favre L, Lamendour L, Gros G, Mami‐Chouaib F, Martinvalet D, Chouaib S and Thiery J (2015) Granzyme B‐activated p53 interacts with Bcl‐2 to promote cytotoxic lymphocyte‐mediated apoptosis. J Immunol 194, 418–428. [DOI] [PubMed] [Google Scholar]
- Bommarito D, Martin A, Forcade E, Nastke MD, Ritz J and Bellucci R (2016) Enhancement of tumor cell susceptibility to natural killer cell activity through inhibition of the PI3K signaling pathway. Cancer Immunol Immunother 65, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghaei H, Paz‐Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E et al (2015) Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med 373, 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E et al (2015) Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med 373, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH et al (1998) HLA‐E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391, 795–799. [DOI] [PubMed] [Google Scholar]
- van der Burg SH, Arens R, Ossendorp F, van Hall T and Melief CJ (2016) Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer 16, 219–233. [DOI] [PubMed] [Google Scholar]
- Burstein HJ, Krilov L, Aragon‐Ching JB, Baxter NN, Chiorean EG, Chow WA, De Groot JF, Devine SM, DuBois SG, El‐Deiry WS et al (2017) Clinical cancer advances 2017: Annual Report on Progress Against Cancer from the American Society of Clinical Oncology. J Clin Oncol, 35, 1341–1367. [DOI] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK et al (2013) An epithelial‐mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo AB, Kyte JA, Carretero J, Zinchencko S, Mendez R, Gonzalez‐Aseguinolaza G, Ruiz‐Cabello F, Aamdal S, Gaudernack G, Garrido F et al (2014) Immune escape of cancer cells with beta2‐microglobulin loss over the course of metastatic melanoma. Int J Cancer 134, 102–113. [DOI] [PubMed] [Google Scholar]
- Carosella ED, Rouas‐Freiss N, Tronik‐Le Roux D, Moreau P and LeMaoult J (2015) HLA‐G: an immune checkpoint molecule. Adv Immunol 127, 33–144. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K et al (2011) Normal and neoplastic nonstem cells can spontaneously convert to a stem‐like state. Proc Natl Acad Sci U S A 108, 7950–7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W et al (2014) Metastasis is regulated via microRNA‐200/ZEB1 axis control of tumour cell PD‐L1 expression and intratumoral immunosuppression. Nat Commun 5, 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Jiang CC, Jin L and Zhang XD (2016a) Regulation of PD‐L1: a novel role of pro‐survival signalling in cancer. Ann Oncol 27, 409–416. [DOI] [PubMed] [Google Scholar]
- Chen XH, Liu ZC, Zhang G, Wei W, Wang XX, Wang H, Ke HP, Zhang F, Wang HS, Cai SH et al (2015) TGF‐beta and EGF induced HLA‐I downregulation is associated with epithelial‐mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol Immunol 65, 34–42. [DOI] [PubMed] [Google Scholar]
- Chen L, Yi X, Goswami S, Ahn YH, Roybal JD, Yang Y, Diao L, Peng D, Peng D, Fradette JJ et al (2016b) Growth and metastasis of lung adenocarcinoma is potentiated by BMP4‐mediated immunosuppression. Oncoimmunology 5, e1234570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester C, Fritsch K and Kohrt HE (2015) Natural killer cell immunomodulation: targeting activating, inhibitory, and co‐stimulatory receptor signaling for cancer immunotherapy. Front Immunol 6, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman‐Censits J, Roth B, Cheng T, Tran M, Lee IL et al (2014) Identification of distinct basal and luminal subtypes of muscle‐invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chretien AS, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C and Olive D (2014) Cancer‐induced alterations of NK‐mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK‐mediated anti‐tumor activity. Front Immunol 5, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KN, Sim WJ, Racine V, Lee SY, Goh BC and Thiery JP (2012) A cell‐based small molecule screening method for identifying inhibitors of epithelial‐mesenchymal transition in carcinoma. PLoS One 7, e33183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT, Yang Z, Ye J, Muller J, Koh CM, Guccione E et al (2016) GRHL2‐miR‐200‐ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci Rep 6, 19943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieply B, Riley PT, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, Denvir J and Frisch SM (2012) Suppression of the epithelial‐mesenchymal transition by Grainyhead‐like‐2. Cancer Res 72, 2440–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comrie WA and Burkhardt JK (2016) Action and traction: cytoskeletal control of receptor triggering at the immunological synapse. Front Immunol 7, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T et al (2010) HIF‐1alpha regulates function and differentiation of myeloid‐derived suppressor cells in the tumor microenvironment. J Exp Med 207, 2439–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo J, Sun H, Welling TH, Tian Z and Zou W (2013) T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 25, 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave B, Mittal V, Tan NM and Chang JC (2012) Epithelial‐mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res 14, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Stewart TA, Thompson EW and Monteith GR (2014) Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci 35, 479–488. [DOI] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC and Elledge SJ (2017) Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355, eeaf8399, https://doi.org/10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene B and Berx G (2013) Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13, 97–110. [DOI] [PubMed] [Google Scholar]
- De Mattos‐Arruda L, Bottai G, Nuciforo PG, Di Tommaso L, Giovannetti E, Peg V, Losurdo A, Perez‐Garcia J, Masci G, Corsi F et al (2015) MicroRNA‐21 links epithelial‐to‐mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2‐positive breast cancer patients. Oncotarget 6, 37269–37280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Wang L, Chen H, Hao J, Ni J, Chang L, Duan W, Graham P and Li Y (2016) Targeting epithelial‐mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 7, 55771–55788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, Validire P, Besse B and Mami‐Chouaib F (2015) CD8+CD103+ tumor‐infiltrating lymphocytes are tumor‐specific tissue‐resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194, 3475–3486. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Tsang KY and Palena C (2016) Short‐term EGFR blockade enhances immune‐mediated cytotoxicity of EGFR mutant lung cancer cells: rationale for combination therapies. Cell Death Dis 7, e2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL and Weinberg RA (2017) Epithelial‐to‐mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res, Advance online publication. https://doi.org/10.1158/0008-5472.can-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ and Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3, 991–998. [DOI] [PubMed] [Google Scholar]
- Dustin ML (2014) What counts in the immunological synapse? Mol Cell 54, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Chakraborty AK and Shaw AS (2010) Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol 2, a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML and Long EO (2010) Cytotoxic immunological synapses. Immunol Rev 235, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farace F, Massard C, Vimond N, Drusch F, Jacques N, Billiot F, Laplanche A, Chauchereau A, Lacroix L, Planchard D et al (2011) A direct comparison of Cell Search and ISET for circulating tumour‐cell detection in patients with metastatic carcinomas. Br J Cancer 105, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrand N, Gnanapragasam A, Dorothee G, Redeuilh G, Larsen AK and Sabbah M (2014) Loss of WISP2/CCN5 in estrogen‐dependent MCF7 human breast cancer cells promotes a stem‐like cell phenotype. PLoS One 9, e87878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C et al (2016) Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med 375, 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciszkiewicz K, Le Floc'h A, Boutet M, Vergnon I, Schmitt A and Mami‐Chouaib F (2013) CD103 or LFA‐1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T‐cell effector functions. Cancer Res 73, 617–628. [DOI] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes‐Fridman C and Galon J (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306. [DOI] [PubMed] [Google Scholar]
- Fritah A, Saucier C, De Wever O, Bracke M, Bieche I, Lidereau R, Gespach C, Drouot S, Redeuilh G and Sabbah M (2008) Role of WISP‐2/CCN5 in the maintenance of a differentiated and noninvasive phenotype in human breast cancer cells. Mol Cell Biol 28, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J and Harlin H (2006) Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 213, 131–145. [DOI] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D and Marincola FM (2013) The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE et al (2016) Loss of IFN‐gamma pathway genes in tumor cells as a mechanism of resistance to anti‐CTLA‐4 therapy. Cell 167, 397–404 e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L et al (2015) Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Garrido F, Cabrera T and Aptsiauri N (2010) “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer 127, 249–256. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD et al (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E et al (2005) CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor‐beta‐dependent manner. J Exp Med 202, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gramont A, Faivre S and Raymond E (2017) Novel TGF‐beta inhibitors ready for prime time in onco‐immunology. Oncoimmunology 6, e1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V, Wu J, Yee C and Spies T (2002) Tumour‐derived soluble MIC ligands impair expression of NKG2D and T‐cell activation. Nature 419, 734–738. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA and Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high‐throughput screening. Cell 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber DA and Velculescu VE (2014) Blood‐based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov 4, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, Koch M, Weitz J, Kloor M, Zoernig I et al (2011) Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res 17, 678–689. [DOI] [PubMed] [Google Scholar]
- Hamai A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F, Lecluse Y, Vielh P, Avril MF, Robert C et al (2008) ICAM‐1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res 68, 9854–9864. [DOI] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS et al (2013) Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N Engl J Med 369, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DH, Griner LM, Keller JM, Hu X, Southall N, Marugan J, David JM, Ferrer M and Palena C (2016) Targeting estrogen receptor signaling with fulvestrant enhances immune and chemotherapy‐mediated cytotoxicity of human lung cancer. Clin Cancer Res 22, 6204–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DH, Huang B, Fernando RI, Tsang KY and Palena C (2014) WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial‐mesenchymal transition. Cancer Res 74, 2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DH, Litzinger MT, Jales A, Huang B, Fernando RI, Hodge JW, Ardiani A, Apelian D, Schlom J and Palena C (2013) Immunological targeting of tumor cells undergoing an epithelial‐mesenchymal transition via a recombinant brachyury‐yeast vaccine. Oncotarget 4, 1777–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan K, O'Neill A, Prencipe M, Bugler J, Murphy L, Fabre A, Puhr M, Culig Z, Murphy K and Watson RW (2017) The role of epithelial‐mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel‐resistant prostate cancer. Mol Oncol 11, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmim M, Noman MZ, Messai Y, Bordereaux D, Gros G, Baud V and Chouaib S (2013) Cutting edge: hypoxia‐induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF‐beta1. J Immunol 191, 5802–5806. [DOI] [PubMed] [Google Scholar]
- He S, Yin T, Li D, Gao X, Wan Y, Ma X, Ye T, Guo F, Sun J, Lin Z et al (2013) Enhanced interaction between natural killer cells and lung cancer cells: involvement in gefitinib‐mediated immunoregulation. J Transl Med 11, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzel M, Bovier A and Tuting T (2013) Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Hsu DS, Lan HY, Huang CH, Tai SK, Chang SY, Tsai TL, Chang CC, Tzeng CH, Wu KJ, Kao JY et al (2010) Regulation of excision repair cross‐complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin Cancer Res 16, 4561–4571. [DOI] [PubMed] [Google Scholar]
- Huang RY, Guilford P and Thiery JP (2012) Early events in cell adhesion and polarity during epithelial‐mesenchymal transition. J Cell Sci 125, 4417–4422. [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S, Berent‐Maoz B, Pang J, Chmielowski B, Cherry G et al (2016) Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazirehi AR, Baritaki S, Koya RC, Bonavida B and Economou JS (2011) Molecular mechanism of MART‐1+/A*0201+ human melanoma resistance to specific CTL‐killing despite functional tumor‐CTL interaction. Cancer Res 71, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Joffroy CM, Buck MB, Stope MB, Popp SL, Pfizenmaier K and Knabbe C (2010) Antiestrogens induce transforming growth factor beta‐mediated immunosuppression in breast cancer. Cancer Res 70, 1314–1322. [DOI] [PubMed] [Google Scholar]
- Joyce JA and Fearon DT (2015) T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80. [DOI] [PubMed] [Google Scholar]
- Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmstrom PU, Choi W, Guo CC, Lotan Y and Kassouf W (2016) Bladder cancer. Lancet 388, 2796–2810. [DOI] [PubMed] [Google Scholar]
- Kang Y and Pantel K (2013) Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, Iglesia MD, Milowsky MI, Parker JS, Kim WY et al (2016) Claudin‐low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 1, e85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B and Simon MC (2007) Hypoxia‐inducible factors, stem cells, and cancer. Cell 129, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkar SP and Restifo NP (2012) Cellular constituents of immune escape within the tumor microenvironment. Cancer Res 72, 3125–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong HT and Restifo NP (2002) Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 3, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Koh J, Kim MY, Kwon D, Go H, Kim YA, Jeon YK and Chung DH (2016) PD‐L1 expression is associated with epithelial‐to‐mesenchymal transition in adenocarcinoma of the lung. Hum Pathol 58, 7–14. [DOI] [PubMed] [Google Scholar]
- Komdeur FL, Wouters MC, Workel HH, Tijans AM, Terwindt AL, Brunekreeft KL, Plat A, Klip HG, Eggink FA, Leffers N et al (2016) CD103+ intraepithelial T cells in high‐grade serous ovarian cancer are phenotypically diverse TCRalphabeta+ CD8alphabeta+ T cells that can be targeted for cancer immunotherapy. Oncotarget 7, 75130–75144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkolopoulou P, Kaklamanis L, Pezzella F, Harris AL and Gatter KC (1996) Loss of antigen‐presenting molecules (MHC class I and TAP‐1) in lung cancer. Br J Cancer 73, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA and Fearon DT (2010) Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein‐alpha. Science 330, 827–830. [DOI] [PubMed] [Google Scholar]
- Krebs MG, Hou JM, Sloane R, Lancashire L, Priest L, Nonaka D, Ward TH, Backen A, Clack G, Hughes A et al (2012) Analysis of circulating tumor cells in patients with non‐small cell lung cancer using epithelial marker‐dependent and ‐independent approaches. J Thorac Oncol 7, 306–315. [DOI] [PubMed] [Google Scholar]
- Kudo‐Saito C, Shirako H, Ohike M, Tsukamoto N and Kawakami Y (2013) CCL2 is critical for immunosuppression to promote cancer metastasis. Clin Exp Metastasis 30, 393–405. [DOI] [PubMed] [Google Scholar]
- Kudo‐Saito C, Shirako H, Takeuchi T and Kawakami Y (2009) Cancer metastasis is accelerated through immunosuppression during Snail‐induced EMT of cancer cells. Cancer Cell 15, 195–206. [DOI] [PubMed] [Google Scholar]
- Kumar V and Gabrilovich DI (2014) Hypoxia‐inducible factors in regulation of immune responses in tumour microenvironment. Immunology 143, 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Patel S, Tcyganov E and Gabrilovich DI (2016) The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol 37, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY and Bapat SA (2009) Snail and slug mediate radioresistance and chemoresistance by antagonizing p53‐mediated apoptosis and acquiring a stem‐like phenotype in ovarian cancer cells. Stem Cells 27, 2059–2068. [DOI] [PubMed] [Google Scholar]
- Labelle M, Begum S and Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial‐mesenchymal‐like transition and promotes metastasis. Cancer Cell 20, 576–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laget S, Broncy L, Hormigos K, Dhingra DM, BenMohamed F, Capiod T, Osteras M, Farinelli L, Jackson S and Paterlini‐Brechot P (2017) Technical insights into highly sensitive isolation and molecular characterization of fixed and live circulating tumor cells for early detection of tumor invasion. PLoS One 12, e0169427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J and Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S et al (2016) Control of PD‐L1 expression by oncogenic activation of the AKT‐mTOR pathway in non‐small cell lung cancer. Cancer Res 76, 227–238. [DOI] [PubMed] [Google Scholar]
- Le Floc'h A, Jalil A, Vergnon I, Le Chansac Maux B, Lazar V, Bismuth G, Chouaib S and Mami‐Chouaib F (2007) Alpha E beta 7 integrin interaction with E‐cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 204, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D et al (2015) PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecharpentier A, Vielh P, Perez‐Moreno P, Planchard D, Soria JC and Farace F (2011) Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non‐small cell lung cancer. Br J Cancer 105, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Lee KM, Kim DW and Heo DS (2004) Elevated TGF‐beta1 secretion and down‐modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol 172, 7335–7340. [DOI] [PubMed] [Google Scholar]
- Ligtenberg MA, Witt K, Galvez‐Cancino F, Sette A, Lundqvist A, Lladser A and Kiessling R (2016) Cripto‐1 vaccination elicits protective immunity against metastatic melanoma. Oncoimmunology 5, e1128613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J and Thiery JP (2012) Epithelial‐mesenchymal transitions: insights from development. Development 139, 3471–3486. [DOI] [PubMed] [Google Scholar]
- Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV et al (2016) RAS/MAPK activation is associated with reduced tumor‐infiltrating lymphocytes in triple‐negative breast cancer: therapeutic cooperation between MEK and PD‐1/PD‐L1 immune checkpoint inhibitors. Clin Cancer Res 22, 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Soto A, Huergo‐Zapico L, Galvan JA, Rodrigo L, de Herreros AG, Astudillo A and Gonzalez S (2013) Epithelial‐mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J Immunol 190, 4408–4419. [DOI] [PubMed] [Google Scholar]
- Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C et al (2016) Epithelial‐mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res 22, 3630–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, Forbes LR, Watkin LB and Orange JS (2014) Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol 92, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD and Freeman GJ (2015) Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14, 561–584. [DOI] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ and Simon MC (2010) Hypoxia‐inducible factors and the response to hypoxic stress. Mol Cell 40, 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, Rodriguez‐Canales J, Wistuba II et al (2016) A patient‐derived, pan‐cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial‐to‐mesenchymal transition. Clin Cancer Res 22, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek R, Wang H, Taparra K and Tran PT (2017) Therapeutic targeting of epithelial plasticity programs: focus on the epithelial‐mesenchymal transition. Cells Tissues Organs 203, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E and Massague J (2016) Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, Goncalves A, Andre P, Romagne F, Thibault G et al (2011) Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest 121, 3609–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Romero P, Palucka AK and Marincola FM (2008) Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 371, 771–783. [DOI] [PubMed] [Google Scholar]
- Marvel D and Gabrilovich DI (2015) Myeloid‐derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 125, 3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2008) TGFbeta in Cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Michiels S, Ferte C, Le Deley MC, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S, Ammari S et al (2017) High‐throughput genomics and clinical outcome in hard‐to‐treat advanced cancers: results of the MOSCATO 01 Trial. Cancer Discov 7, 586–595, https://doi.org/10.1158/2159-8290.cd-16-1396. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal‐Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N and Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628. [DOI] [PubMed] [Google Scholar]
- Melero I, Berman DM, Aznar MA, Korman AJ, Perez Gracia JL and Haanen J (2015) Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 15, 457–472. [DOI] [PubMed] [Google Scholar]
- Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, Beitsch PD, Leitch M, Hoover S, Euhus D et al (2004) Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 10, 8152–8162. [DOI] [PubMed] [Google Scholar]
- Milner JD and Holland SM (2013) The cup runneth over: lessons from the ever‐expanding pool of primary immunodeficiency diseases. Nat Rev Immunol 13, 635–648. [DOI] [PubMed] [Google Scholar]
- Mitra A, Mishra L and Li S (2015) EMT, CTCs and CSCs in tumor relapse and drug‐resistance. Oncotarget 6, 10697–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal D, Gubin MM, Schreiber RD and Smyth MJ (2014) New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol 27, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF et al (2015) Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J Clin Oncol 33, 1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nantajit D, Lin D and Li JJ (2015) The network of epithelial‐mesenchymal transition: potential new targets for tumor resistance. J Cancer Res Clin Oncol 141, 1697–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA and Thiery JP (2016) EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V and Chouaib S (2014) PD‐L1 is a novel direct target of HIF‐1alpha, and its blockade under hypoxia enhanced MDSC‐mediated T cell activation. J Exp Med 211, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B and Chouaib S (2015) Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol 309, C569–C579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami‐Chouaib F, Thiery JP and Chouaib S (2017) The immune checkpoint ligand PD‐L1 is upregulated in EMT‐activated human breast cancer cells by a mechanism involving ZEB‐1 and miR‐200. OncoImmunology 6, e1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ock CY, Keam B, Kim S, Lee JS, Kim M, Kim TM, Jeon YK, Kim DW, Chung DH and Heo DS (2016a) Pan‐cancer immunogenomic perspective on the tumor microenvironment based on PD‐L1 and CD8 T‐cell infiltration. Clin Cancer Res 22, 2261–2270. [DOI] [PubMed] [Google Scholar]
- Ock CY, Kim S, Keam B, Kim M, Kim TM, Kim JH, Jeon YK, Lee JS, Kwon SK, Hah JH et al (2016b) PD‐L1 expression is associated with epithelial‐mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 7, 15901–15914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell JS, Long GV, Scolyer RA, Teng MW and Smyth MJ (2017) Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 52, 71–81. [DOI] [PubMed] [Google Scholar]
- O'Donnell JS, Smyth MJ and Teng MW (2016) Acquired resistance to anti‐PD1 therapy: checkmate to checkpoint blockade? Genome Med 8, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS (2008) Formation and function of the lytic NK‐cell immunological synapse. Nat Rev Immunol 8, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palena C, Polev DE, Tsang KY, Fernando RI, Litzinger M, Krukovskaya LL, Baranova AV, Kozlov AP and Schlom J (2007) The human T‐box mesodermal transcription factor Brachyury is a candidate target for T‐cell‐mediated cancer immunotherapy. Clin Cancer Res 13, 2471–2478. [DOI] [PubMed] [Google Scholar]
- Paolicchi E, Gemignani F, Krstic‐Demonacos M, Dedhar S, Mutti L and Landi S (2016) Targeting hypoxic response for cancer therapy. Oncotarget 7, 13464–13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran R, Ramakrishnan P, Moreton SA, Xia Z, Hou Y, Lee DA, Gupta K, deLima M, Beck RC and Wald DN (2016) Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat Commun 7, 11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR and Weinberg RA (2014) Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov 13, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X et al (2016) Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov 6, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platonova S, Cherfils‐Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, Andre P, Dieu‐Nosjean MC, Alifano M, Regnard JF et al (2011) Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 71, 5412–5422. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W and Van den Eynde BJ (2012) Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res 72, 5435–5440. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J, Dayan F and Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443. [DOI] [PubMed] [Google Scholar]
- Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, Martinez A, Nuciforo P, Comerma L, Alos L et al (2017) Immune‐related gene expression profiling after PD‐1 blockade in non‐small cell lung carcinoma, head and neck squamous cell carcinoma and melanoma. Cancer Res, Advance online publication. https://doi.org/10.1158/0008-5472.can-16-3556. [DOI] [PubMed] [Google Scholar]
- Puhr M, Hoefer J, Schafer G, Erb HH, Oh SJ, Klocker H, Heidegger I, Neuwirt H and Culig Z (2012) Epithelial‐to‐mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR‐200c and miR‐205. Am J Pathol 181, 2188–2201. [DOI] [PubMed] [Google Scholar]
- Punt CJ, Koopman M and Vermeulen L (2017) From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol 14, 235–246. [DOI] [PubMed] [Google Scholar]
- Raffaghello L, Prigione I, Airoldi I, Camoriano M, Levreri I, Gambini C, Pende D, Steinle A, Ferrone S and Pistoia V (2004) Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 6, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Rodriguez‐Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S et al (2016) Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med 375, 1823–1833. [DOI] [PubMed] [Google Scholar]
- Restifo NP, Smyth MJ and Snyder A (2016) Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 16, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, Bifari F and Krampera M (2015) Epithelial‐to‐mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell‐like immune‐modulatory properties in cancer cells. Br J Cancer 112, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G and Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg JE, Hoffman‐Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, Dawson N, O'Donnell PH, Balmanoukian A, Loriot Y et al (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet 387, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Affara NI and Coussens LM (2012) Differential macrophage programming in the tumor microenvironment. Trends Immunol 33, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MH, Wang L and Goldberg MS (2016) Improving cancer immunotherapy with DNA methyltransferase inhibitors. Cancer Immunol Immunother 65, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Franciszkiewicz K, Damotte D, Dieu‐Nosjean MC, Validire P, Trautmann A, Mami‐Chouaib F and Donnadieu E (2012) Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 122, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A and Postigo A (2012) EMT‐activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci 69, 3429–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrio D, Rodriguez‐Pinilla SM, Hardisson D, Cano A, Moreno‐Bueno G and Palacios J (2008) Epithelial‐mesenchymal transition in breast cancer relates to the basal‐like phenotype. Cancer Res 68, 989–997. [DOI] [PubMed] [Google Scholar]
- Saxena M, Stephens MA, Pathak H and Rangarajan A (2011) Transcription factors that mediate epithelial‐mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2, e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliekelman MJ, Taguchi A, Zhu J, Dai X, Rodriguez J, Celiktas M, Zhang Q, Chin A, Wong CH, Wang H et al (2015) Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res 75, 1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu‐Lieskovan S, Wargo JA and Ribas A (2017a) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Retz M, Siefker‐Radtke A, Baron A, Necchi A, Bedke J, Plimack ER, Vaena D, Grimm MO, Bracarda S et al (2017b) Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single‐arm, phase 2 trial. Lancet Oncol 18, 312–322. [DOI] [PubMed] [Google Scholar]
- Singh A and Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al (2014) Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S, Schlom J, Tucker J, Tsang KY and Greiner JW (2013) Inhibition of TGF‐beta1 signaling promotes central memory T cell differentiation. J Immunol 191, 2299–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam WL and Weinberg RA (2013) The epigenetics of epithelial‐mesenchymal plasticity in cancer. Nat Med 19, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY and Thiery JP (2014) Epithelial‐mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med 6, 1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB, Mami‐Chouaib F, Thiery JP and Chouaib S (2017) Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: consequences on susceptibility to cell‐mediated cytotoxicity. OncoImmunology 6, e1271858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP (2002) Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2, 442–454. [DOI] [PubMed] [Google Scholar]
- Thomas DA and Massague J (2005) TGF‐beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA and Pardoll DM (2016) Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 16, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, Jolly MK, Celiktas M, Rodriguez‐Canales J, Liu H et al (2016) Immunoproteasome deficiency is a feature of non‐small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc Natl Acad Sci U S A 113, E1555–E1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu E, Chia PZ and Chen W (2014) TGFbeta in T cell biology and tumor immunity: angel or devil? Cytokine Growth Factor Rev 25, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasievich EA and Huang L (2011) The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm 8, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Tomasello E, Baratin M, Walzer T and Ugolini S (2008) Functions of natural killer cells. Nat Immunol 9, 503–510. [DOI] [PubMed] [Google Scholar]
- Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, Capron F, Franco D, Pazzagli M, Vekemans M et al (2000) Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells. Am J Pathol 156, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JR, Milne K, Watson P, Deleeuw RJ and Nelson BH (2014) Tumor‐infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high‐grade serous ovarian cancer. Clin Cancer Res 20, 434–444. [DOI] [PubMed] [Google Scholar]
- Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB, El‐Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL and Cook GP (2011) Human tumour immune evasion via TGF‐beta blocks NK cell activation but not survival allowing therapeutic restoration of anti‐tumour activity. PLoS One 6, e22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WR and Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11, 393–410. [DOI] [PubMed] [Google Scholar]
- Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG and Weiss SJ (2012) Canonical Wnt signaling regulates Slug activity and links epithelial‐mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci U S A 109, 16654–16659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW and Condeelis J (2007) Direct visualization of macrophage‐assisted tumor cell intravasation in mammary tumors. Cancer Res 67, 2649–2656. [DOI] [PubMed] [Google Scholar]
- Yao J, Caballero OL, Huang Y, Lin C, Rimoldi D, Behren A, Cebon JS, Hung MC, Weinstein JN, Strausberg RL et al (2016) Altered expression and splicing of ESRP1 in malignant melanoma correlates with epithelial‐mesenchymal status and tumor‐associated immune cytolytic activity. Cancer Immunol Res 4, 552–561. [DOI] [PubMed] [Google Scholar]
- Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, Sun X, Li GG, Hu QD, Fu QH et al (2016) Hypoxia‐induced epithelial‐to‐mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res 76, 818–830. [DOI] [PubMed] [Google Scholar]
- Younes A, Santoro A, Shipp M, Zinzani PL, Timmerman JM, Ansell S, Armand P, Fanale M, Ratanatharathorn V, Kuruvilla J et al (2016) Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem‐cell transplantation and brentuximab vedotin: a multicentre, multicohort, single‐arm phase 2 trial. Lancet Oncol 17, 1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM et al (2013a) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Smoligovets AA and Groves JT (2013b) Modulation of T cell signaling by the actin cytoskeleton. J Cell Sci 126, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Zhang J and Ma L (2012) MicroRNA control of epithelial‐mesenchymal transition and metastasis. Cancer Metastasis Rev 31, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R (2015) Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Tesniere A and Kroemer G (2006) Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6, 715–727. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD and Chen L (2016) PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]