

Abstract
The inter- or intramolecular oxidative carboamination of alkynes catalyzed by [py2TiCl2NPh]2 is reported. These multicomponent reactions couple alkenes, alkynes and diazenes to form either α,β-unsaturated imines or α-(iminomethyl)cyclopropanes via a TiII/TiIV redox cycle. Each of these products is formed from a common azatitanacyclohexene intermediate that undergoes either β-H elimination or α,γ-coupling, wherein the selectivity is under substrate control.
Simple intermolecular alkyne carboamination reactions can potentially provide convenient access points to a range of important functional groups and reactive intermediates such as α,β-unsaturated imines, α-functionalized imines, or α-functionalized cyclopropanes.1 Although analogous alkyne hydro-functionalization reactions2 have been heavily studied, the current methods for alkyne carboamination are limited to coupling of diaryl aldimines and alkynes using early transition metals,3 through intramolecular reactions catalyzed by late transition metals,4 or through multistep processes catalyzed by Cu and Rh.5 Similarly, alkene carboamination6 has seen considerable advances recently, but these methods are still mainly limited to intramolecular cyclization reactions. Accessing practical, intermolecular multicomponent carboamination catalysis remains a significant challenge.
Recently, we reported a multicomponent, py3TiCl2(NPh)-catalyzed formal [2+2+1] reaction of alkynes and diazenes for the oxidative synthesis of penta- and trisubstituted pyrroles (Figure 1).7 In our preliminary studies of the mechanism, we found that an alkyne initially undergoes [2+2] cycloaddition with a Ti imido to generate an azatitanacyclobutene intermediate, I, which then undergoes insertion of a second alkyne to generate an azatitanacyclohexadiene, II. This species then reductively eliminates pyrrole, and the resulting TiII fragment is reoxidized to a TiIV imido by azobenzene. We anticipate that this new mode of TiII/TiIV redox reactivity has the potential to open up vast new classes of Ti-catalyzed reactions.
Figure 1.
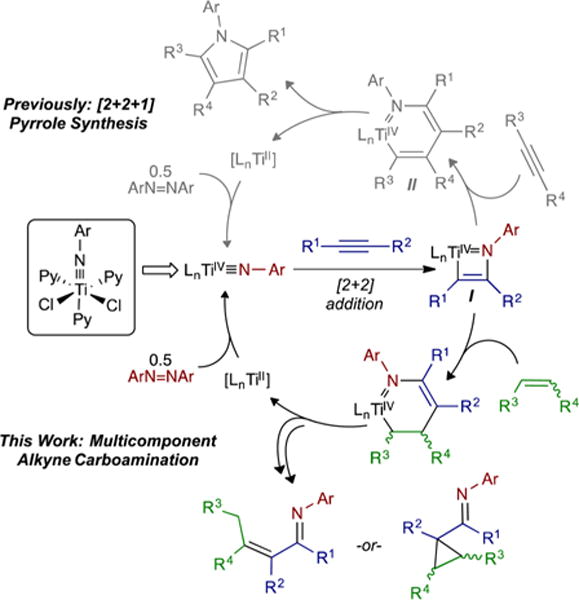
Overview of Ti-catalyzed nitrene transfer reactions.
Given that the mechanisms of each alkyne coupling step in the [2+2+1] pyrrole synthesis are different, we postulated that it should be possible to decouple the reacting partners and design multicomponent coupling reactions of different unsaturated substrates. Encouragingly, Odom,8 Livinghouse9 and Mindiola3c–e have recently demonstrated that isocyanides, nitriles and imines can intercept analogous [2+2] imide+alkyne azatitanacyclobutene intermediates in hydroamination-like reactions.
Our initial target of this strategy was the multicomponent coupling of an alkyne and an alkene with azobenzene. Alkenes were chosen as the third reacting partner because they readily undergo 1,2- and 2,1-insertion reactions, but typically do not undergo intermolecular [2+2] reactions with Ti imidos,10 thus limiting the potential for unwanted alkene homocoupling. Herein, we report our initial results on the intra- and intermolecular oxidative multicomponent coupling of alkynes, alkenes and diazenes, which yields formal alkyne carboamination products: either α,β-unsaturated imines or α-functionalized cyclopropanes.
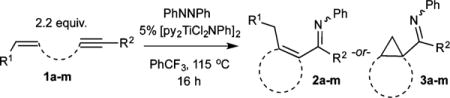
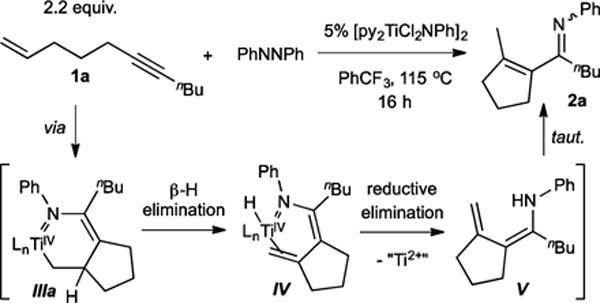
We initially focused on the [py2TiCl2(NPh)]2-catalyzed reaction of tethered enynes with azobenzene, envisioning that the intramolecular reactions would be less likely to suffer from competitive pyrrole formation or alkyne trimerization (Table 1). Reaction of 2.2 equiv undec-1-en-6-yne (1a) with 5 mol % [py2TiCl2(NPh)]2 in the presence of 1 equiv azobenzene at 115 °C gave the α,β-unsaturated imine 1-(2-methylcyclopent-1-en-1-yl)-N-phenylpentan-1-imine (2a) in 50% isolated yield (Figure 2).7
Table 1.
Scope of Multicomponent Carboamination of Tethered Enynes with PhNNPha
| |||
|---|---|---|---|
|
| |||
| Substrate | % Isolated Yieldb (2:3) | 1H NMR % Yield (2:3) | |
| 1a |
|
50 (>99:1) |
92 (85:15) |
| 1b |
|
86 (44:56) |
92 (53:47) |
| 1c |
|
37 (>99:1) |
–d |
| 1d |
|
13 (1: >99)e |
29 (1: >99) |
| 1e |
|
60 (>99:1) |
91 (>99:1) |
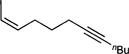
| 1f |
|
69 (>99:1) |
86 (>99:1) |
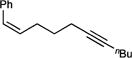
| 1g |
|
50 (>99:1) |
–d |
| 1h |
|
54 (49:51) |
–d |
| 1i |
|
57 (5:95)f |
–d |
| 1j |
|
36 (1:>99)g |
–d |
| 1k |
|
37 (1:>99)h |
–d |
| 11 |
|
0 | n.d. |
| 1m |
|
0 | n.d. |
Loading of [py2TiCl2NPh]2 and reaction yields with respect to PhNNPh.
Isolated as the ketone product after hydrolysis. See SI for details.
Determined by 1H NMR analysis of the crude reaction mixture.
Could not be determined due to peak overlap in the 1H NMR spectrum.
Isolated as the retro-ene product 3d′ (Figure 3).
As a mixture of 2i, 3i and 4i (Figure 5).
As 4j.
As a mixture of 3k and 4k.
Figure 2.
Tethered enynes yield α,β-unsaturated imines upon catalysis with PhNNPh.
This product likely forms through the expected azatitanacyclohexene intermediate III, but instead of C–N reductive coupling to form a dihydropyrrole, the metallacycle collapses via β-H elimination to give IV, followed by subsequent N–H reductive elimination to V and dienamine isomerization (Figure 2). Alternately, direct β-H abstraction by the amide from intermediate III could also form V.11 Unlike in the previously reported pyrrole synthesis, it is likely that the sp3-hybridized α-C is less prone to C–N reductive elimination due to poor orbital overlap,12 which allows for the β-H elimination pathway to kinetically outcompete direct C–N elimination.
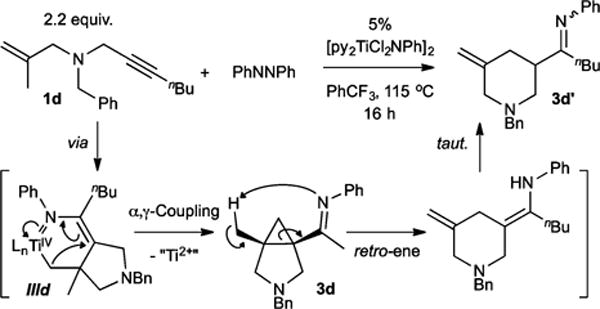
In an attempt to shut down β-H elimination, we next examined substrates that upon metalation would lack a β-H to eliminate. Treatment of N-benzyl-N-(2-methylallyl)hept-2-yn-1-amine (1d) under catalytic conditions gave 1-(1-benzyl-5-methylenepiperidin-3-yl)pentan-1-one (3d′) in low yield upon acidic workup. This product arises from isomerization via retro-ene ring opening of a cis-cyclopropane (3d), which is generated via catalysis (Figure 3).
Figure 3.
Internally substituted alkenes yield α-(iminomethyl)-cyclopropanes upon catalysis with PhNNPh as β-H elimination/abstraction is shut down.
Remarkably, by shutting down the β-H elimination process, the azatitanacyclohexene IIId collapses via attack of the α-C on the γ-C,13 resulting in reductive elimination of an α-imino functionalized cyclopropane. In fact, this cyclopropanation can also be observed when using the deuterated analogue 1b: because β-D elimination (which must occur in 1b) is typically slower than β-H elimination (in 1a), there should be a larger krel for forming the cyclopropane 3 versus the α,β-unsaturated imine 2 in the reaction of 1b. This is reflected in the 1H NMR product ratios, where 1a forms an 85:15 ratio of 2a:3a, whereas at similar overall conversion 1b forms a larger percentage of 3b, 50:50 2b:3b.
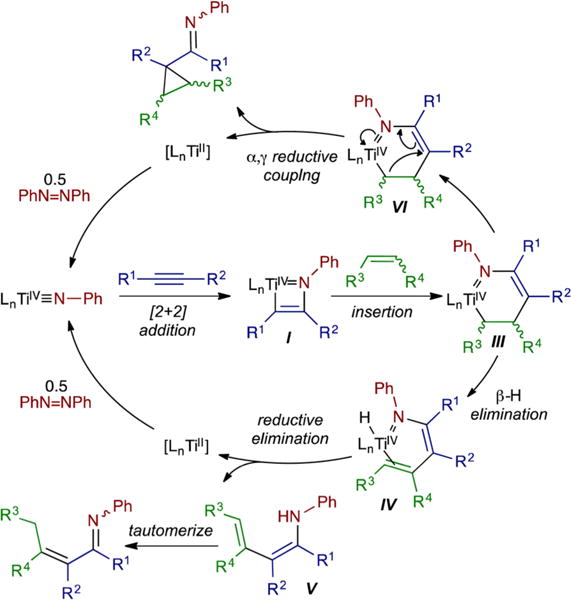
The overall preliminary mechanistic manifold of these carboaminations is presented in Figure 4. Azatitanacyclohexenes (III) are prone to metallacycle collapse via competitive α,γ-coupling (VI) or β-H elimination/abstraction (IV). These pathways are kinetically accessible because the α- and β-carbons are sp3-hybridized, making direct C–N reductive coupling more challenging while opening up alternative reductive cleavage pathways through the increased flexibility of the metallacycle. This is characteristic of all of the multicomponent reactions reported herein.
Figure 4.
Proposed mechanism for Ti-catalyzed alkyne carboaminations.
To probe the scope of carboamination and selectivity for β-H elimination vs α,γ-coupling, we examined catalysis with several more tethered enynes (Table 1). In most cases, isolated yields of the reactions were moderate due to the difficulty in separating the product isomers, but 1H NMR analysis of the crude mixtures generally indicated that the reactions proceeded to total conversion. Terminal and internal alkenes were competent for catalysis, and there was little difference in utilizing E or Z alkenes 1e and 1f. Only internal alkynes are currently compatible because their more-reactive terminal counterparts undergo [2+2+1] pyrrole synthesis and alkyne trimerization too rapidly.7
Interestingly, simply changing from a propyl linker (1a) to a butyl linker (1h) erodes selectivity for the α,β-unsaturated imine 2h from 85:15 to 50:50, indicating that there is a subtle steric balance between β-H elimination and α,γ-coupling. Shorter tethers (1l), as expected, do not undergo reaction and bulky substituents on the alkyne (1m), which enforce the wrong [2+2] regiochemistry necessary for alkene insertion, also do not react productively.
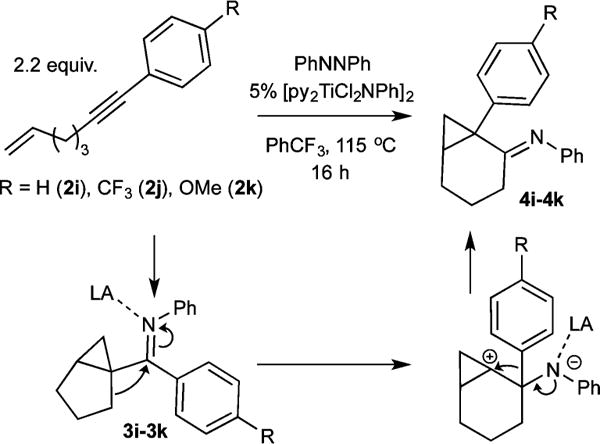
Aryl-substituted alkynes heavily favor α,γ-coupling due to increased electrophilicity of the γ-C caused by the aryl substituent (1i–1k). Furthermore, the resulting electrophilic bicyclo[3.1.0]hexane arylimines (3i–3k) undergo further reactivity in situ: titanium Lewis acid-catalyzed carbocation rearrangement yields the fused 1-arylbicyclo[4.1.0]heptan-2-imines 4i–4k (Figure 5).
Figure 5.
Phenyl-substituted alkynes yield bicyclo[3.1.0]hexane imines that can undergo Lewis acid-catalyzed carbocation rearrangement.
Next, intermolecular heterocouplings between internal alkynes and terminal unactivated alkenes were attempted (Table 2). Terminal alkenes compete effectively with alkynes for the second insertion into the azametallacyclobutene intermediate. Even at a 1:1 ratio of 1-octene:3-hexyne, moderate yields of the α,γ-unsaturated imine product were obtained, with the remaining mass balance undergoing competitive [2+2+1] pyrrole formation. The yield of the α,γ-unsaturated product 6b could be increased from 31% to 61% by doubling the concentration of 1-octene. In all cases, terminal alkenes react via 2,1-insertion, indicating that this step is likely under steric control where the alkene substituent orients preferentially toward an uncrowded Ti center rather than a 2° carbon substituent.
Table 2.
Intermolecular Multicomponent Carboamination of Alkynes and Alkenes with PhNNPha
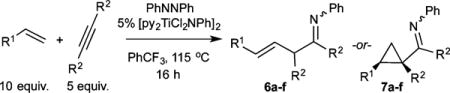
| ||||
|---|---|---|---|---|
|
| ||||
| Alkene | R2 | % Isolated Yield (6:7)b | 1H NMR % Yield (6:7)c | |
| 5a |
|
Me | 54 (40:60) | n.d. |
| 5b |
|
Et | 61 (>99:1) | n.d. |
| 5c |
|
Me | 42 (9:91) | 63 (15:85)d |
| 5d |
|
Et | 51 (>99:1) | 70 (71:29)e |
| 5ef |
|
Et | 40 (>99:1) | n.d. |
| 5f |
|
Et | 0 | n.d. |
Loading of [py2TiCl2NPh]2 and reaction yields with respect to PhNNPh.
Isolated as the ketone product after hydrolysis. See SI for details.
Determined by 1H NMR analysis of the crude reaction mixture.
96:4 ratio of cis:trans cyclopropane product.
74:26 ratio of cis:trans cyclopropane product.
Reaction run in neat alkene.
As was the case in the intramolecular multicomponent couplings, subtle structural changes in intermolecular hetero-couplings also lead to dramatic shifts in selectivity between β-H elimination/abstraction and α,γ-coupling products. This selectivity shift is apparent in the reaction of 4-allylanisole with internal alkynes: reaction with 3-hexyne gives a 71:29 ratio of 6d:7d, whereas reaction with 2-butyne inverts the selectivity and yields a 15:85 ratio of 6c:7c by 1H NMR analysis. The cis:trans selectivity of the cyclopropanes also varies heavily between the 3-hexyne product 7d (74:26) and 2-butyne product 7c (96:4), which has similarly been observed in Kulinkovich-type cyclopropanation reactions.14
In addition to unsubstituted linear terminal alkenes, terminal alkenes bearing 2° groups are also competent for catalysis. 4-Vinylcyclohex-1-ene undergoes reaction to give low yields of the product with exclusive reactivity at the terminal alkene. Bulkier alkenes, such as 3,3-dimethylbut-1-ene, fail to react.
In conclusion, we have demonstrated the first examples of a three-component oxidative alkyne carboamination, generating either α,β-unsaturated imines or α-functionalized cyclopropanes. Preliminary mechanistic studies indicate that these Ti-catalyzed reactions proceed through a common azametallacy-clohexene intermediate. Somewhat remarkably, both intra- and intermolecular reactions proceed in moderate to good yields and selectivities despite the large potential for the occurrence of undesired competitive processes such as alkyne homocoupling. We are currently examining new catalyst classes to further understand and increase control over the rate and selectivity of these unique transformations, as well as further pursuing new Ti redox catalytic reactions promoted by diazene oxidants.
Supplementary Material
Acknowledgments
Financial support was provided by the University of Minnesota (start-up funds) and the National Institutes of Health (1R35GM119457). Equipment for the Chemistry Department NMR facility were supported through a grant from the National Institutes of Health (S10OD011952) with matching funds from the University of Minnesota.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09939.
Full experimental procedures, characterization data and spectra (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.(a) Sandhu JS, Sain B. Heterocycles. 1987;26:777. [Google Scholar]; (b) Amslinger S. ChemMedChem. 2010;5:351–356. doi: 10.1002/cmdc.200900499. [DOI] [PubMed] [Google Scholar]; (c) Carson CA, Kerr MA. Chem Soc Rev. 2009;38:3051–3060. doi: 10.1039/b901245c. [DOI] [PubMed] [Google Scholar]; (d) Tang P, Qin Y. Synthesis. 2012;44:2969–2984. [Google Scholar]; (e) Okajima T. Nucl Acids Symp Series. 2007;51:215–216. doi: 10.1093/nass/nrm108. [DOI] [PubMed] [Google Scholar]; (f) Kumar AK. Int J Pharm Pharm Sci. 2013;5:467–472. [Google Scholar]
- 2.For a review of hydroamination, see:; Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ruck RT, Zuckerman RL, Krska SW, Bergman RG. Angew Chem, Int Ed. 2004;43:5372–5374. doi: 10.1002/anie.200461063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ruck RT, Bergman RG. Organometallics. 2004;23:2231–2233. doi: 10.1021/om0497994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Basuli F, Aneetha H, Huffman JC, Mindiola DJ. J Am Chem Soc. 2005;127:17992–17993. doi: 10.1021/ja0566026. [DOI] [PubMed] [Google Scholar]; (d) Aneetha H, Basuli F, Bollinger J, Huffman JC, Mindiola DJ. Organometallics. 2006;25:2402–2404. [Google Scholar]; (e) Basuli F, Wicker B, Huffman JC, Mindiola DJ. J Organomet Chem. 2011;696:235–243. [Google Scholar]; For a review on titanium carboamination, see:; (f) Mindiola DJ. Comments Inorg Chem. 2008;29:73–92. [Google Scholar]
- 4.(a) Kajita Y, Matsubara S, Kurahashi T. J Am Chem Soc. 2008;130:6058–6059. doi: 10.1021/ja7114426. [DOI] [PubMed] [Google Scholar]; (b) Yoshino Y, Kurahashi T, Matsubara S. J Am Chem Soc. 2009;131:7494–7495. doi: 10.1021/ja900805y. [DOI] [PubMed] [Google Scholar]; (c) Maizuru N, Inami T, Kurahashi T, Matsubara S. Org Lett. 2011;13:1206–1209. doi: 10.1021/ol200090g. [DOI] [PubMed] [Google Scholar]; (d) Zavesky BP, Babij NR, Wolfe JP. Org Lett. 2014;16:4952–4955. doi: 10.1021/ol502471x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Patil NT, Kavthe RD, Yamamoto Y. Adv Heterocycl Chem. 2010;101:75–95. [Google Scholar]; (f) Chemler SR, Fuller PH. Chem Soc Rev. 2007;36:1153–1160. doi: 10.1039/b607819m. [DOI] [PubMed] [Google Scholar]
- 5.Rh-catalyzed alkyne carboaminations are two-step reactions involving Cu-catalyzed alkyne/azide cycloaddition followed by Rh-catalyzed iminovinylidene formation:; Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J Am Chem Soc. 2008;130:14972–14974. doi: 10.1021/ja805079v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piou T, Rovis T. Nature. 2015;527:86–90. doi: 10.1038/nature15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert ZW, Hue RJ, Tonks IA. Nat Chem. 2016;8:63–68. doi: 10.1038/nchem.2386. [DOI] [PubMed] [Google Scholar]
- 8.Odom AL, McDaniel TJ. Acc Chem Res. 2015;48:2822–2833. doi: 10.1021/acs.accounts.5b00280. [DOI] [PubMed] [Google Scholar]
- 9.McGrane LP, Jensen M, Livinghouse T. J Am Chem Soc. 1992;114:5459–5460. [Google Scholar]
- 10.Only ethylene has been observed:; Polse JL, Andersen RA, Bergman RG. J Am Chem Soc. 1998;120:13405–13414. [Google Scholar]
- 11.Agapie T, Labinger JA, Bercaw JE. J Am Chem Soc. 2007;129:14281–14295. doi: 10.1021/ja073493h. [DOI] [PubMed] [Google Scholar]
- 12.(a) Mann G, Baranano D, Hartwig JF, Rheingold AL, Guzei IA. J Am Chem Soc. 1998;120:9205–9219. [Google Scholar]; (b) Low JJ, Goddard WA. Organometallics. 1986;5:609–622. [Google Scholar]
- 13.Suzuki K, Urabe H, Sato F. J Am Chem Soc. 1996;118:8729–8730. [Google Scholar]
- 14.Kulinkovich OG, de Meijere A. Chem Rev. 2000;100:2789–2834. doi: 10.1021/cr980046z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.