

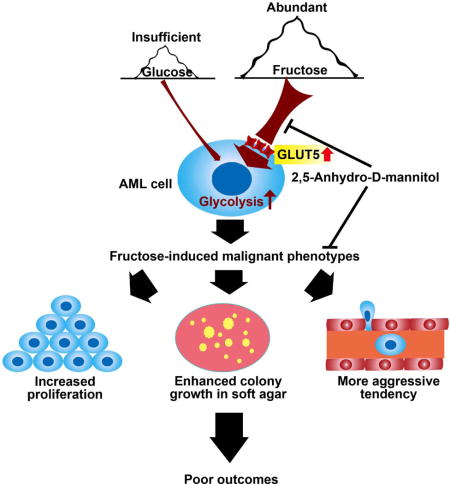
SUMMARY
Rapidly proliferating leukemic progenitor cells consume substantial glucose that may lead to glucose insufficiency in bone marrow. We show that acute myeloid leukemia (AML) cells are prone to fructose utilization with an upregulated fructose transporter GLUT5, compensating for glucose deficiency. Notably, AML patients with upregulated transcription of GLUT5-encoding gene SLC2A5 or increased fructose utilization have poor outcomes. Pharmacological blockage of fructose uptake ameliorates leukemic phenotypes and potentiates the cytotoxicity of antileukemic agent, Ara-C. In conclusion, this study highlights enhanced fructose utilization as a metabolic feature of AML and a potential therapeutic target.
eTOC Blurb
Chen et al. show that AML cells exhibit enhanced fructose utilization under low-glucose conditions via upregulating the fructose transporter GLUT5, exacerbating leukemic phenotypes. Pharmacologic blockade of fructose utilization selectively eliminates AML cells and enhances the efficacy of Ara-C.
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous disorder of hematopoietic stem cells characterized by uncontrolled proliferation of aberrant clones of myeloid progenitor cells with impaired differentiation and by suppressed production of healthy hematopoietic cells. The pathogenesis of AML involves two types of gene mutations, class I mutations which induce cellular proliferation, and class II mutations which compromise normal differentiation (Ferrara and Schiffer, 2013). In addition, a third class of genes encoding epigenetic modifiers, such as DNMT3A, IDH1, IDH2, and TET2, are found to play a major role in leukemogenesis (Ferrara and Schiffer, 2013). Recent studies showed that aberrant metabolism was also critically involved in AML pathogenesis and progression (Chen et al., 2014; Losman et al., 2013).
Conventional drug therapy for AML usually includes a cell cycle inhibitor, arabinofuranosyl cytidine (Ara-C), which targets cells in S or DNA synthesis phase, and a topoisomerase II inhibitor, such as an anthracycline derivative (usually daunorubicin). The anthracyclines also target dividing cells preferentially by inhibiting DNA transcription/replication but are classified as cell cycle independent drugs (Martincic and Hande, 2005). Due to the diverse genetic and epigenetic abnormalities of individual patients, treatment efficacy and prognosis may vary significantly and there is no single treatment to cure AML patients of different molecular subtypes (Ferrara and Schiffer, 2013; Patel et al., 2012). On the other hand, the downstream metabolic alterations involve a more limited number of core pathways that show relatively low diversity (Cairns et al., 2011; Jang et al., 2013). For example, increased glycolysis (Warburg effect) is a common metabolic feature in various cancer cells, which has been under extensive investigation as a therapeutic target for cancer.
A good strategy to inhibit accelerated glycolysis is to block excessive metabolic fuel uptake. Inhibition of glycolysis using a glucose analog, 2-deoxyglucose (2-DG) has already been shown to re-sensitize acute lymphoblastic leukemia (ALL) cells to prednisolone therapy (Hulleman et al., 2009). The combination of 2-DG and an MCL1 anti-apoptotic protein inhibitor is proved to be synergistic in overcoming prednisolone resistance in ALL cells (Aries et al., 2013). Our recent study of AML cell lines revealed enhanced expression of genes relating to both glycolysis and the TCA cycle (Chen et al., 2014). The same study also identified a panel of 6 elevated serum metabolites related to glycolysis and the TCA cycle in a cohort of 223 de novo cytogenetically normal AML patients that predicted poor survival outcomes independent of cytogenetic risk. Accordingly, reducing glycolysis which is upstream of the TCA cycle may prove beneficial in AML.
Normal, non-proliferating cells switch to glycolysis only under hypoxic conditions (Lum et al., 2007). Hypoxia promotes the switch to glycolytic metabolism via the hypoxia-inducible factor-1 (HIF1) pathway, and progression of AML has been linked to the expansion of hypoxia in the subendosteal bone marrow niche relative to normal bone marrow (Tabe and Konopleva, 2014). It has been reported that AML cells display a highly active glycolytic metabolism (Herst et al., 2011). Such an increase in glycolytic flux in AML cells would accelerate glucose consumption and lead to glucose insufficiency in the bone marrow. In fact, significantly lower glucose levels are found in leukemic bone marrow relative to peripheral blood (Tiziani et al., 2013). Thus, a flexible metabolic program is required for AML cells to accommodate a low-glucose environment. Fructose is the second most abundant blood sugar in human beings with a physiologically normal range of 0.5–1.0 mM (Barone et al., 2009; Liu et al., 2010). Based on these previous observations, we hypothesized that AML cells will show increased expression of the fructose transporter, GLUT5 and exhibit enhanced fructose utilization relative to normal cells especially under low-glucose conditions.
RESULTS
AML Cells Exhibited Increased Fructose Utilization under Low-glucose Conditions
For the first set of experiments we hypothesized that AML cells would switch to fructose utilization especially in the absence or low level of glucose. In order to assess fructose utilization in AML cells, 2 parameters were selected, fructose uptake and fructose-induced cell proliferation. Four AML cell lines with distinct genetic backgrounds along with 4 different batches of normal monocytes as controls were employed for both assays (Table S1). All of the cells were able to import 13C-fructose (Figure 1A). However, AML cells displayed a higher fructose transport rate than normal monocytes under conditions of glucose deficiency (0 mM or 0.75 mM). Fructose uptake was significantly increased in the leukemic cells under condition of glucose deficiency (0 mM or 0.75 mM) compared to glucose sufficiency (6 mM) (p < 0.01).
Figure 1. Fructose utilization by AML cells.
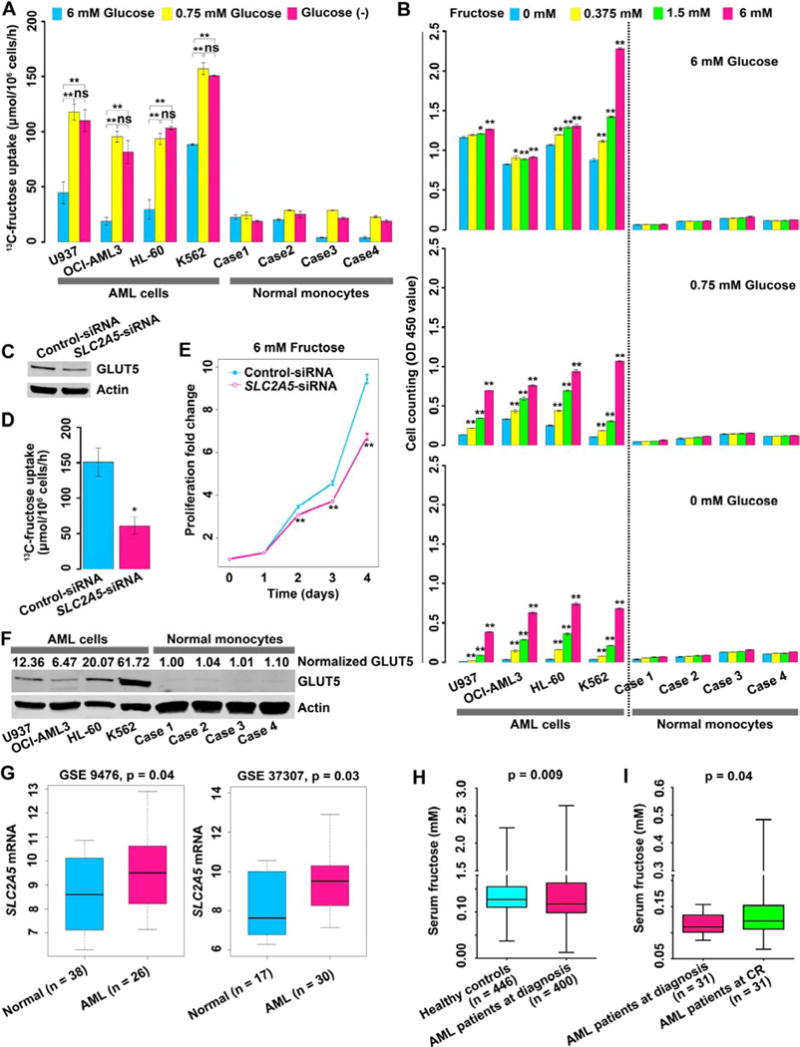
(A) 13C-fructose uptake by AML cells and normal monocytes.
(B) Fructose-induced proliferation of AML cells and normal monocytes under the conditions of different glucose levels. Cells were grown in the media for 72 hours. For each glucose condition, p values were obtained by comparison with the cell proliferation in 0 mM fructose.
(C) Western blot showing GLUT5 knockdown in K562 cells by siRNA targeting SLC2A5.
(D) Uptake of 13C-fructose by K562 cells with or without SLC2A5 inhibition.
(E) Fructose-induced proliferation of K562 cells with or without SLC2A5 inhibition.
(F) GLUT5 expression between AML cells and normal monocytes. Relative GLUT5 expression was computed by dividing the value of GLUT5 signal intensity by the value of actin signal intensity. Normalized GLUT5 was obtained by normalizing each relative GLUT5 expression to the mean of relative GLUT5 expression of 4 cases of normal monocytes.
(G) The expression of GLTU5-encoding gene SLC2A5 between normal hematopoietic cells of healthy controls and AML blast cells of patients. The data were obtained from a public microarray database.
(H) Serum fructose concentration comparison between healthy controls and AML patients.
(I) Serum fructose concentration of paired samples from 31 AML patients at diagnosis and at CR.
Error bars represent mean ± SEM. * p < 0.05, ** p < 0.01 (Student’s t test).
See also Figure S1 and Tables S1 and S2.
The next objective was to determine if fructose uptake led to enhanced proliferation of AML cells under glucose deficient conditions. The results showed that AML cells exhibited a significant increase (p < 0.01) in proliferation induced by fructose under conditions of low glucose or glucose deprivation, indicating that fructose was an important fuel source for AML cells when glucose was limited (Figure 1B). In contrast, in the presence of fructose, normal monocytes displayed little or no increases in proliferation under any of the tested glucose conditions, indicating that normal monocytes showed significantly lower fructose utilization compared to AML cells (Figure 1B). We concluded from these studies that AML cells not only readily utilize fructose as a metabolic fuel for survival but also that their ability to proliferate is actually enhanced by increased fructose under glucose-limiting conditions, whereas, normal monocytes hardly rely on fructose for growth.
GLUT5 mediated Fructose Uptake and Had Increased Expression in AML Cells
Having established that AML cells readily utilize fructose, the next step was to identify the sugar transporter that was being used for fructose uptake. GLUT5, encoded by the SLC2A5 gene, has been reported to be the main transporter for fructose with high selectivity in various cells (Burant et al., 1992; Zhao and Keating, 2007). Thus we hypothesized that the sugar transporter involved in AML cell fructose utilization was in fact, GLUT5. We tested this hypothesis by knocking down the SLC2A5 gene in K562 cells using an RNAi technique (Figure 1C). The results showed that both 13C-fructose uptake and fructose-induced proliferation were suppressed (Figures 1D and 1E). SLC2A5 gene silencing did not affect glucose uptake and glucose-induced proliferation in K562 cells (Figures S1A and S1B) indicating the selectivity of this transporter for fructose. These data demonstrated that GLUT5 mediated the fructose uptake in AML cells.
Increased GLUT5 expression was observed in all 4 of the AML cell lines compared to normal monocytes (Figure 1F). The next question was whether GLUT5 or its encoding gene SLC2A5 was also upregulated in primary AML blast cells from de novo AML patients. We analyzed gene expression patterns of the major sugar transporter genes in primary AML blast cells using published microarray data sets (Stirewalt et al., 2008; Stirewalt et al., 2012) and determined that SLC2A5 gene expression was significantly increased in blast cells compared to normal hematopoietic cells, indicating increased fructose uptake capability of patient-derived AML cells (Figure 1G). In contrast, for the well-known glucose transporter genes, SLC2A1, SLC2A2 and SLC2A4, there were no significant differences between primary AML cells and normal hematopoietic cells while SLC2A3 showed significant reduction in primary AML cells (Figure S1C). Subsequently, we explored the possible mechanism for upregulating SLC2A5 in primary AML blast cells. We analyzed the data sets including GSE1159, GSE425 and TCGA data (Bullinger et al., 2004; Valk et al., 2004) and found that SLC2A5 expression was positively correlated to AML1-ETO, NPM1 mutations and RUNX1 mutations, whereas negatively linked to PML-RARA and CEBPA biallelic mutations, indicating that overexpression of SLC2A5 may be secondary to these gene events (Table S2).
Based on above observations, we hypothesized that increased expression of SLC2A5 in primary AML blast cells would accelerate fructose utilization thus resulting in reduced circulating fructose in peripheral blood. To test this hypothesis, serum samples from healthy controls (n = 446) and AML patients (n = 400) were analyzed using GC-TOFMS. To avoid the well-known confounding factors influencing circulating fructose level, including hepatic function, dietary and therapy, we executed the following strategies. First, we reviewed the key parameters of hepatic function including serum alanine aminotransferase and aspartate aminotransferase in all enrolled subjects and confirmed that there was no significant difference for hepatic function between AML patients and healthy controls (Chen et al., 2014). Secondly, all serum samples were collected from 12-hour fasting peripheral blood to minimize dietary impact. Thirdly, all AML serum samples were obtained from patients at diagnosis without any therapeutic interventions. Data from GC-TOFMS showed reduced serum fructose concentrations in AML patients relative to controls (Figure 1H), suggesting elevated fructose utilization of leukemic blast cells in these patients. Hypothesizing that AML blast cells were responsible for the reduction of serum fructose, when these neoplastic cells were eliminated by chemotherapy, the serum fructose level would be expected to increase. To test this hypothesis, we analyzed paired serum samples from AML patients (n = 31) at diagnosis and at complete remission (CR) and found that serum fructose was significantly raised in the cases at CR compared to those at diagnosis (p = 0.04, Figure 1I), supporting our hypothesis that AML blast cells contributed to the decrease of serum fructose.
In summary, these results provided convincing evidence that an increase in the sugar transporter GLUT5 encoded by SLC2A5, was responsible for the increased uptake and utilization of fructose in our test set of AML cell lines and also in the primary leukemic cells of AML patients.
High Expression of SLC2A5 and Enhanced Fructose Utilization Were Associated with Poor Outcomes of AML Patients
Based on the above observations of increased fructose utilization and upregulated fructose transporter GLUT5 and its encoding gene SLC2A5 in AML cells, we asked if there was an association between the modified SLC2A5 expression/fructose utilization and the therapeutic outcomes of AML patients. We analyzed the relevance of SLC2A5 expression to the survival of AML patients in 5 previously published gene-expression data sets (Bullinger et al., 2004; Metzeler et al., 2008; Valk et al., 2004). Among those cases, patients with intermediate-risk AML were selected for analysis due to the relatively low heterogeneity in this subgroup. The patients of each set were divided into two groups: those with above-median SLC2A5 expression, and those with below-median SLC2A5 expression. Above-median SLC2A5 expression was associated with inferior overall survival, and a formal meta-analysis of all 5 data sets indicated an overall hazard ratio of 1.49 [95% confidence interval (CI): 1.41–1.57] in a univariate Cox model and 1.48 (95% CI: 1.34–1.62) in a multivariate Cox model after adjustment for clinical confounding factors including age, white blood cell counting and FLT3-ITD mutations (Figure 2A). To avoid the potential statistical bias, we used the mean instead of median value of SLC2A5 expression as the cut-off point in each data set. Above-mean SLC2A5 expression was also closely linked to poor overall survival, and a formal meta-analysis of all 5 data sets demonstrated an overall hazard ratio of 1.87 (95% confidence interval, 1.24–2.50) in the Cox model (Figure S2A).
Figure 2. Prognostic value of SLC2A5 expression levels and fructose utilization rates in AML patients.
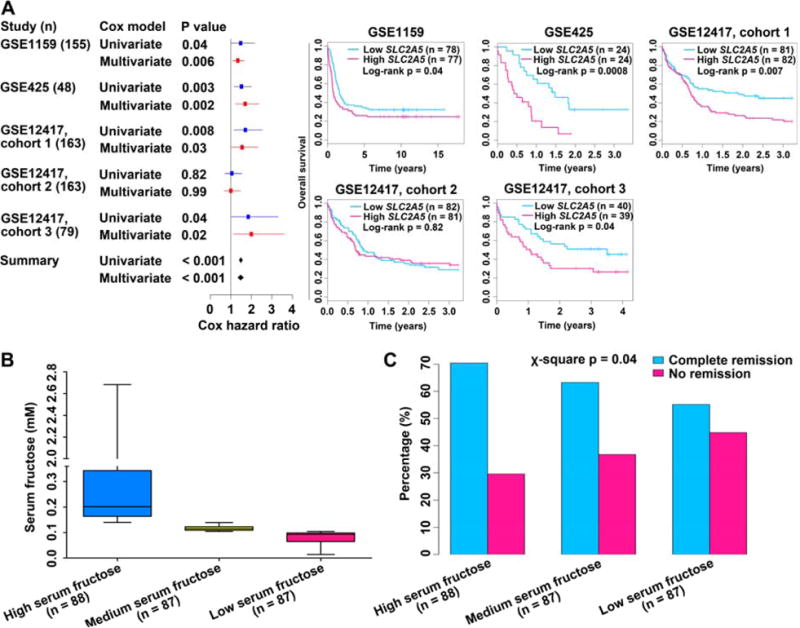
(A) (Left) Meta-analysis of Cox hazard ratio of SLC2A5 expression levels in 5 data sets containing patients with intermediate-risk AML. Solid horizontal lines denoted 95% confidence intervals; boxes denoted the relative influence of each set over the results; diamond indicates the summary 95% confidence interval. The continuous SLC2A5 value was used in the analysis. (Right) Kaplan-Meier survival curves of 5 cohorts of patients in the left meta-analysis. Patients were separated into above-median (deep pink line) and below-median (deep sky blue line) of SLC2A5 expression.
(B) AML patients were evenly divided into 3 groups according to the serum levels of fructose.
(C) The complete remission and no remission rates of 3 AML groups in (B).
See also Figure S2.
Subsequently, we analyzed the potential correlation between fructose utilization and therapeutic response in 262 de novo AML patients from data collected in a previous study (Chen et al., 2014). These patients were evenly divided into 3 groups based on their serum fructose concentrations: high, medium and low fructose (Figure 2B), representing low, medium, and high fructose utilization, respectively. We found that the rate of treatment failure was increased and the CR rate was reduced when fructose utilization was elevated (Figure 2C), demonstrating that enhanced fructose utilization was associated with poor therapeutic response in AML patients. Of note, there was no significant difference for the overall survival and event-free survival among these 3 groups of AML patients (Figures S2B and S2C).
Enhanced Fructose Utilization Mediated by SLC2A5 Exacerbated Leukemic Phenotypes
High SLC2A5 expression and enhanced fructose utilization were closely associated with the poor outcomes of AML patients. Additionally, downregulation of SLC2A5 in K562 cells significantly suppressed fructose-induced colony formation (Figure S3A). Thus, we hypothesized that enhanced fructose utilization mediated by SLC2A5 would result in a more malignant phenotype. We ectopically expressed SLC2A5 in U937, K562 and OCI-AML3 cells (Figures 3A, S3B and S3C) as they were typical representatives of AML with distinct genetic aberrations and risk status (Table S1). To determine what fructose concentration to be used for the in vitro study, we first investigated the level of this sugar in AML patient bone marrow which hosted living primary AML blast cells. There were only 3 AML patients who had paired bone marrow and peripheral blood samples in our previous metabolomic study, and the data showed that fructose concentration was elevated in bone marrow with a range of 0.71–4.74 mM compared to the serum levels ranging from 0.02 to 0.41 mM (Figure S3D). Additionally, we assayed unpaired bone marrow (n = 7) and peripheral blood (n = 10) samples from patients with hematological malignancies and also found elevated fructose levels in bone marrow (p = 0.007, Table S3). Hence, we used 6 mM fructose, which was pathophysiologically relevant to AML, to perform the in vitro assays. As compared with the control cells (U937-MigR1, K562-MigR1 and OCI-AML3-MigR1), gene modified cells (U937-SLC2A5, K562-SLC2A5 and OCI-AML3-SLC2A5) displayed increased 13C-fructose uptake (Figures 3B, S3E and S3F). Enforced SLC2A5 expression also increased fructose-induced proliferation under distinct glucose conditions (Figures 3C–3E, and S3G–S3L). These results indicated enhanced fructose utilization mediated by SLC2A5 conferred a proliferation advantage on AML cells even in the presence of glucose.
Figure 3. Enhanced fructose utilization mediated by SLC2A5 exacerbates the leukemic phenotypes of AML cells.
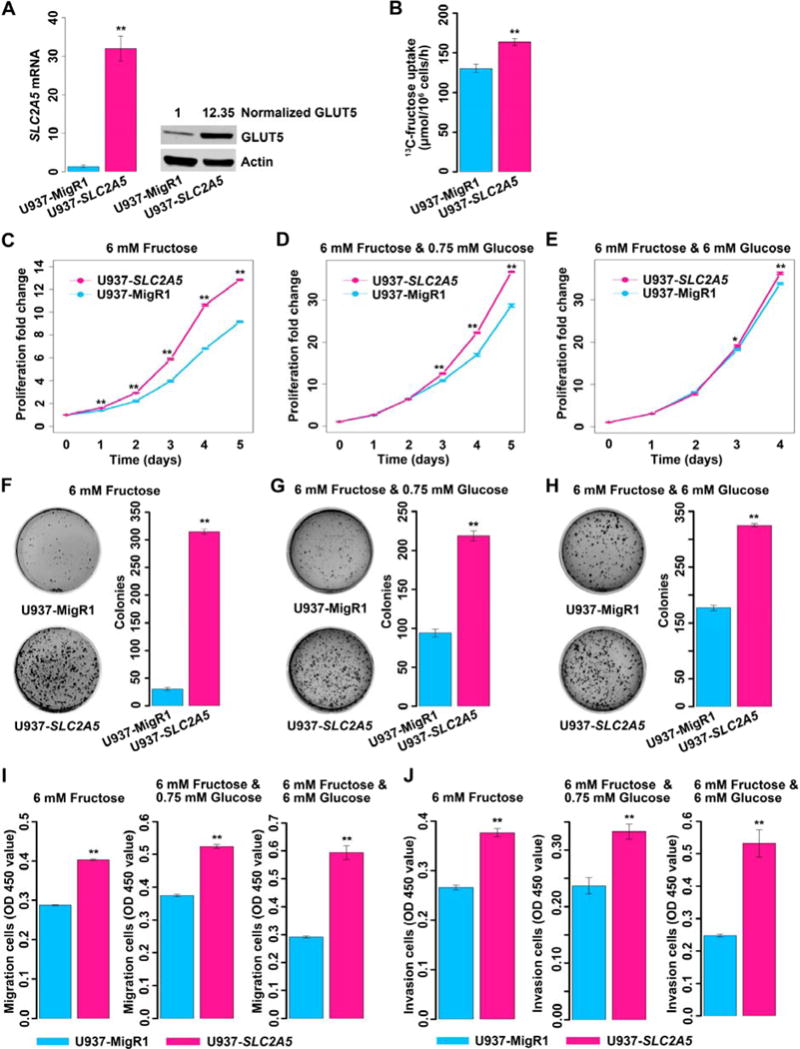
(A) Measurement of the expression of SLC2A5/GLUT5 in U937 cells transfected with the control MigR1 retrovirus (U937-MigR1) or MigR1-SLC2A5 retrovirus (U937-SLC2A5) by Q-PCR and western blot.
(B) Uptake of 13C-labeled fructose by U937-MigR1 and U937-SLC2A5 cells.
(C–E) Proliferation of U937-MigR1 and U937-SLC2A5 cells in complete media containing 6 mM fructose without glucose or with distinct levels of glucose.
(F–H) Colony formation of U937-MigR1 and U937-SLC2A5 cells in soft agar fed with complete media containing 6 mM fructose without glucose or with distinct levels of glucose. Cells were seeded at a density of 3,000/well (F) or 2,000/well (G) or 1,000/well (H). Colonies were assayed at day 23 (F) or day 16 (G and H).
(I) Migration of U937-MigR1 and U937-SLC2A5 cells fed with complete medium containing 6 mM fructose without glucose or with different levels of glucose.
(J) Invasion of U937-MigR1 and U937-SLC2A5 cells fed with complete medium containing 6 mM fructose without glucose or with different levels of glucose.
Error bars represent mean ± SEM. * p < 0.05, ** p < 0.01 (Student’s t test).
See also Figures S3 and S4 and Table S3.
We then investigated the proliferation of transfected AML cells under low fructose conditions (0.375 mM) in the absence of glucose and found that ectopic expression of SLC2A5 accelerated the proliferation of K562 and OCI-AML3, whereas transfected U937 cells were unable to grow under such a fructose-limiting condition (Figures S3M–S3O).
The next question was whether enhanced fructose utilization influenced the malignancy of AML cells. To determine this, we assayed the influence of enhanced fructose utilization ability on colony formation in soft agar, a parameter positively associated with increased cancer cell malignancy (Zebisch et al., 2012). As depicted by the data of Figures 3F–3H and S4A–S4F, AML cells with ectopic SLC2A5 fed with fructose exhibited increased colony growth relative to control cells under distinct glucose conditions. This indicated enhanced fructose utilization mediated by SLC2A5 exacerbated the malignant phenotype of AML cells even in the presence of glucose.
Finally, we investigated if enhanced fructose utilization was able to promote the capability of migration and invasion. As shown in Figures 3I, 3J and S4G–S4J, enhanced fructose utilization mediated by SLC2A5 significantly increased the migratory and invasive tendency of AML cells under reduced or normal glucose conditions, implicating enhanced fructose utilization causes a more aggressive phenotype regardless of glucose concentration.
Enhanced Fructose Utilization Mediated by SLC2A5 Strongly Activated Glycolytic Flux
Logically, one would suppose that enhanced proliferation and enhanced fuel uptake in the form of fructose would cause increased glycolytic flux and result in increased amounts of metabolites from glycolysis. We performed a metabolic flux assay to establish whether an increased glycolytic flux was a possible explanation for the more aggressive tendency exhibited by AML cells. We compared the metabolic flux of 13C-labeled fructose between nontransfected AML control cells and AML cells with ectopic SLC2A5. All the cells were cultured in 6 mM 13C-fructose for 96 hours and then the spent media were collected for analysis. AML cells with enhanced fructose utilization mediated by ectopic SLC2A5 produced more 13C-pyruvate, 13C-lactate and 13C-alanine than the control cells (Figures 4A–4C). This indicated that enhanced fructose utilization in AML cells strongly activated glycolytic flux (Figure 4D).
Figure 4. Enhanced fructose utilization mediated by SLC2A5 activates glycolytic flux.
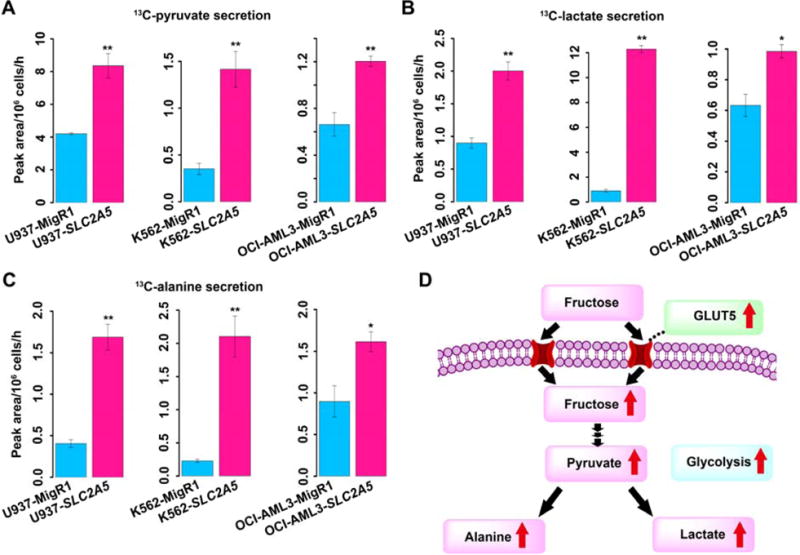
(A) Secretion of 13C-labeled pyruvate derived from 13C-fructose tracer by AML control cells and AML cells with ectopic SLC2A5.
(B) Secretion of 13C-labeled lactate derived from 13C-fructose tracer by AML control cells and AML cells with ectopic SLC2A5.
(C) Secretion of 13C-labeled alanine derived from 13C-fructose tracer by AML control cells and AML cells with ectopic SLC2A5.
(D) Proposed metabolic scheme depicting increased glycolytic flux activated by enhanced fructose utilization mediated by SLC2A5.
Error bars represent mean ± SEM. * p < 0.05, ** p < 0.01 (Student’s t test).
Pharmacological Blockage of Fructose Utilization Alleviated Leukemic Phenotypes and Potentiated the Cytotoxicity of an Antileukemic Drug, Ara-C in vitro
Due to the causal relation between enhanced fructose utilization and exacerbation of leukemic phenotypes, we asked if inhibiting fructose utilization with a pharmacological agent would be able to alleviate the leukemic phenotypes. We used 2,5-anhydro-D-mannitol (2,5-AM), a fructose analogue with high affinity for GLUT5 (Yang et al., 2002), to treat AML cells with enhanced fructose utilization mediated by ectopic SLC2A5 expression. Data showed that 2,5-AM treatment significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or in the presence of low level of glucose (Figures 5A–5D, and Figures S5A–S5H). The combination of the results supported the hypothesis that inhibition of fructose utilization decreases the malignant leukemic phenotypes of AML cells with enhanced fructose utilization.
Figure 5. Response of AML cells to pharmacological blockage of fructose utilization.
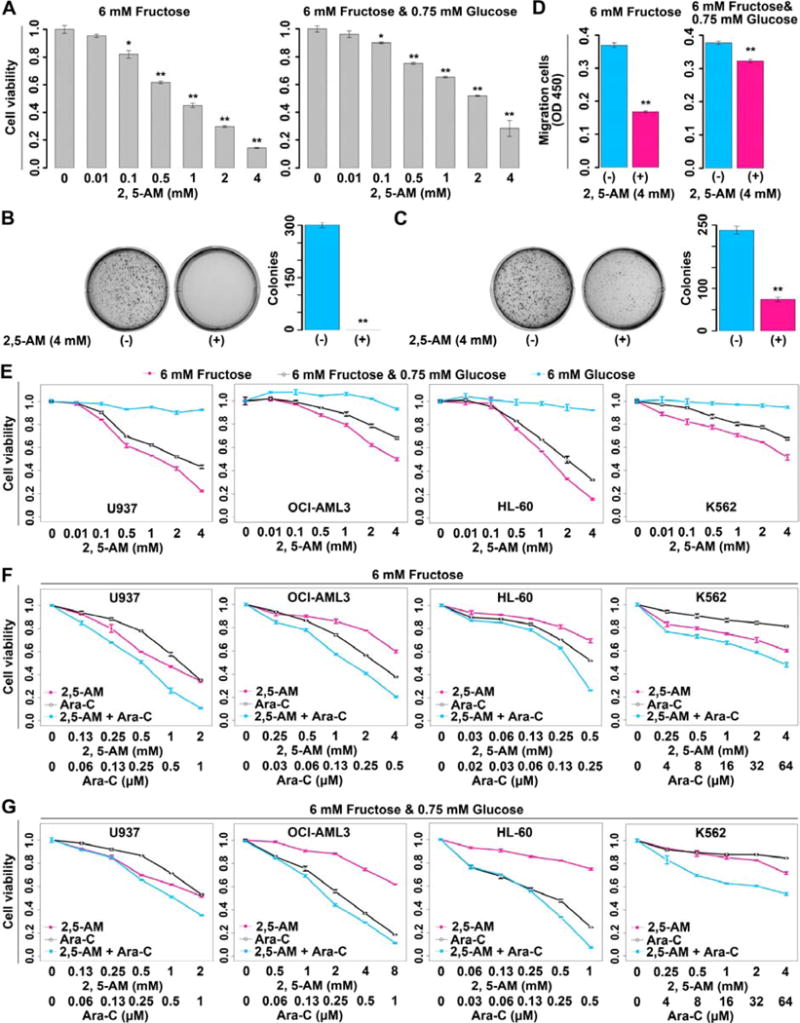
(A) Suppressed proliferation of U937-SLC2A5 cells treated with the fructose analogue 2,5-AM. Two carbon source conditions, 6 mM fructose and 6 mM fructose plus 0.75 mM glucose, were tested. P values were obtained by comparison with the proliferation of U937-SLC2A5 cells in 0 mM 2,5-AM.
(B–C) The colony growth of U937-SLC2A5 cells in soft agar under the condition of 6 mM fructose (B) or 6 mM fructose plus 0.75 mM glucose (C) with or without 2,5-AM treatment.
(D) Migration assay of U937-SLC2A5 cells with or without 2,5-AM treatment in medium containing 6 mM fructose or 6 mM fructose plus 0.75 mM glucose.
(E) Proliferation of AML cells treated with 2,5-AM. Three carbon resource conditions were tested.
(F–G) The synergistic effect between 2,5-AM and Ara-C in AML cells cultured in complete medium containing 6 mM fructose (F) or 6 mM fructose plus 0.75 mM glucose (G).
Error bars represent mean ± SEM. * p < 0.05, ** p < 0.01 (Student’s t test).
See also Figure S5 and Table S4.
Next, we investigated the efficacy of 2,5-AM on all of the 4 AML cell lines enrolled in this study. This drug suppressed fructose-induced cell proliferation in a dose dependent manner in all AML cell lines under glucose-limiting conditions, whereas it had little influence on the glucose-induced cell proliferation (Figure 5E). Normal monocytes were also tested using 2,5-AM and there was negligible effect on glucose-induced cell growth (Figure S5I). Together, these results demonstrated a high specificity for the GLUT5 inhibitor, 2,5-AM on inhibition of fructose utilization.
AML is usually treated with a combination of a cell-cycle independent drug such as daunorubicin and a cell-cycle specific inhibitor such as Ara-C (Martincic and Hande, 2005; Momparler, 2013). Because cancer therapy rarely involves administration of a single drug, we chose to replace daunorubicin with 2,5-AM and co-administer it with Ara-C to our 4 cell lines. Of note, a synergistic effect between 2,5-AM and Ara-C was observed in all AML cell lines grown in fructose without glucose or with low level of glucose, demonstrating that 2,5-AM potentiated the cytotoxicity of the currently used AML drug, Ara-C (Figures 5F and 5G, Table S4). To summarize these results, 2,5-AM, a GLUT5 inhibitor was cytotoxic to AML cells but not to normal monocytes and also decreased the malignant phenotypes of AML cells with enhanced fructose utilization observed in these studies. The combination of 2,5-AM and Ara-C acts in a synergistic way to eradicate AML cells.
Fructose Utilization Was Enhanced in AML Mice and Pharmacological Blockage of This Metabolic Pathway Showed Therapeutic Potential
We performed a study using an AML mouse model driven by the fusion gene AML1-ETO and mutated C-KIT (Wang et al., 2011) to investigate the activity of fructose utilization and the therapeutic potential of pharmacological blockage of this metabolic pathway in vivo. It was reasonable to choose this mouse model since that SLC2A5 expression was positively associated with AML1-ETO in AML patients (Table S2) and the patients with AML1-ETO and C-KIT mutations exhibited increased fructose utilization (Figure S6). Five mouse groups were enrolled, including a normal control group, AML mice treated with vehicle (vehicle group), AML mice treated with 2,5-AM (2,5-AM group), AML mice treated with Ara-C (Ara-C group) and AML mice treated with 2,5-AM and Ara-C (2,5-AM & Ara-C group). We assayed the parameters of these 5 groups of mice 17 days after leukemic cell transplantation (Table S5). Compared to the normal controls, AML mice showed increased SLC2A5 expression in bone marrow cells (Figure 6A). AML mice also exhibited reduced serum fructose levels (Figure 6B). These data indicated that fructose utilization was enhanced in AML mice. Administration of 2,5-AM significantly upregulated serum fructose concentrations in AML mice of vehicle group (p < 0.01) and 2,5-AM & Ara-C group (p < 0.001) (Figure 6B), indicating that fructose utilization in vivo was suppressed by this reagent. Inhibition of fructose utilization by 2,5-AM in AML mice significantly suppressed the growth of bone marrow and peripheral AML blast cells, and ameliorated splenomegaly (Figures 6C–6E). Additionally, 2,5-AM administration improved the impaired peripheral blood cell counts as shown by the decreased white blood cells (p < 0.001) and increased red blood cells (p = 0.01), hemoglobin (p = 0.01) and platelets (p = 0.04) (Figures 6F–6I). Furthermore, the use of 2,5-AM prolonged the overall survival of AML mice (p = 0.01, Figure 6J). Of note, the combination of 2,5-AM and Ara-C showed an increased efficacy on alleviating leukemic phenotypes as compared to 2,5-AM or Ara-C alone (Figures 6C, 6D and 6F–6I). Moreover, the effect of 2,5-AM or Ara-C alone on overall survival of the AML mice was enhanced by the concurrent use of 2,5-AM and Ara-C (p < 0.001 and p = 0.08, respectively) (Figure 6J), demonstrating a synergistic antitumor effect between these two agents against AML.
Figure 6. Enhanced fructose utilization and the therapeutic potential of pharmacological blockage of this metabolic pathway in AML mice.
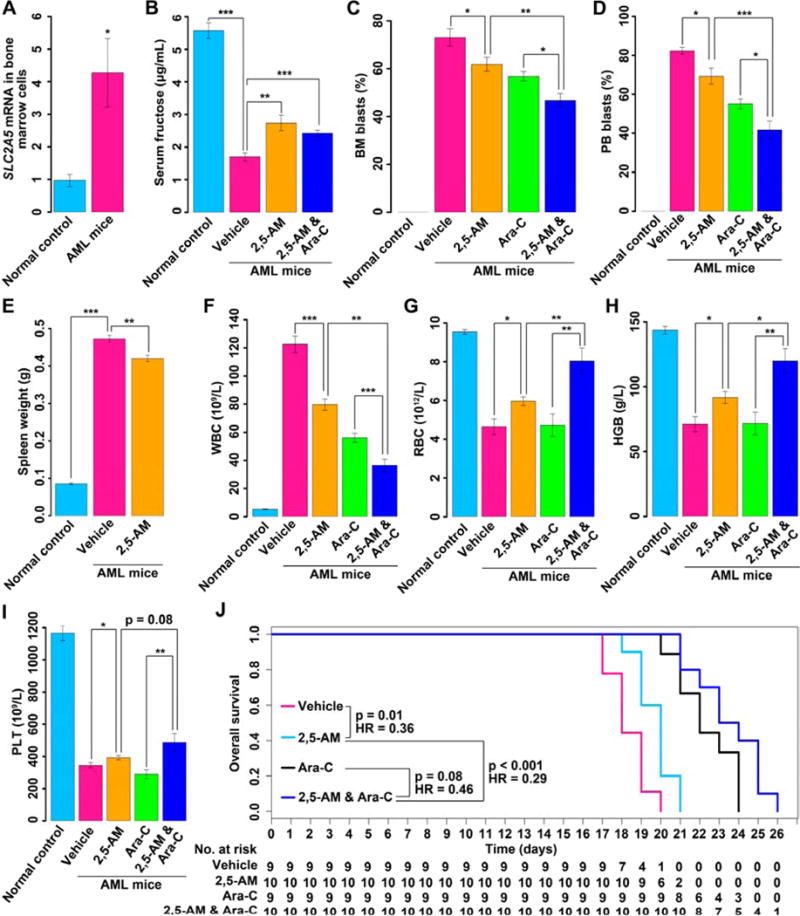
(A) SLC2A5 expression in bone marrow cells between normal controls (n = 6) and AML mice (n = 6).
(B) Serum fructose concentration measurements for normal controls (n = 6), AML mice treated with vehicle (n = 6), AML mice treated with 2,5-AM (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(C) The percentage of BM blast cells in normal controls (n = 6), AML mice treated with vehicle (n = 6), AML mice treated with 2,5-AM (n = 6), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(D) The percentage of PB blast cells in normal controls (n = 6), AML mice treated with vehicle (n = 6), AML mice treated with 2,5-AM (n = 6), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(E) Spleen weight measurements for normal controls (n = 6), AML mice treated with vehicle (n = 6) and AML mice treated with 2,5-AM (n = 6).
(F) WBC counts in PB for normal controls (n = 6), AML mice treated with vehicle (n = 8), AML mice treated with 2,5-AM (n = 8), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(G) RBC counts in PB for normal controls (n = 6), AML mice treated with vehicle (n = 8), AML mice treated with 2,5-AM (n = 8), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(H) HGB measurements in PB for normal controls (n = 6), AML mice treated with vehicle (n = 8), AML mice treated with 2,5-AM (n = 8), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(I) PLT counts in PB for normal controls (n = 6), AML mice treated with vehicle (n = 8), AML mice treated with 2,5-AM (n = 8), AML mice treated with Ara-C (n = 6) and AML mice treated with 2,5-AM and Ara-C (n = 6).
(J) Overall survival curves of AML mice treated with vehicle (n = 9), AML mice treated with 2,5-AM (n = 10), AML mice treated with Ara-C (n = 9) and AML mice treated with 2,5-AM and Ara-C (n = 10).
Abbreviation: BM, bone marrow; PB, peripheral blood; WBC, white blood cell; RBC, red blood cell; HGB, hemoglobin; PLT, platelet; HR, hazard ratio.
Error bars represent mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 (Student’s t test).
See also Figure S6 and Table S5.
DISCUSSION
Considerable attention has been paid to glycolysis due to its high activity in cancer cells and its close association with therapeutic resistance and clinical outcome (Zhao et al., 2013). Glycolysis has been found to be enhanced in AML and is linked to poor survival in patients partially via contribution to Ara-C resistance (Chen et al., 2014). Thus, drugs, such as 2-DG, aiming at interrupting glucose utilization in cancer cells may have potential for therapeutic outcome improvement (Chen et al., 2014). Unfortunately, since normal cells also use glucose as their main metabolic fuel, inhibiting glucose utilization with 2-DG causes substantial adverse side effects, including hypoglycemia-like symptoms, gastrointestinal bleeding, hypotension, decreased respiratory frequency as well as, hematologic and biochemical toxicity (Raez et al., 2013; Vijayaraghavan et al., 2006). It is therefore important to fully understand the distinct metabolic features of fuel utilization for both AML and normal cells.
In the current study in vitro AML cell line models and an in vivo AML mouse model were chosen to investigate whether fructose, an alternative substrate for glycolysis, would exhibit enhanced utilization by AML cells and normal monocyte controls in a glucose-limiting environment. The AML cell lines were chosen to represent 4 different French-American-British subtypes, and to highlight a variety of mutations and individual different pathways leading towards leukemogenesis. We found that these 4 AML cell lines highly expressed GLUT5 and switched to fructose utilization under glucose-limiting conditions, whereas normal monocytes showed a low dependence on fructose for cell survival and growth. For AML mice, their bone marrow blast cells exhibited increased SLC2A5 expression and elevated fructose utilization as measured by mRNA and serum levels, respectively.
Fructose has previously been reported to alter the glycan structures on the cell surface of tumor cells and increase their proliferative and invasive properties when compared to glucose in vitro (Monzavi-Karbassi et al., 2010). Our data demonstrated that altered fructose utilization plays a key role in AML progression as evidenced by an exacerbated leukemic phenotype regardless of the concentration of glucose present. In the fructose-containing media with or without glucose, AML cells with high SLC2A5 expression showed elevated fructose uptake, increased proliferation and colony growth, and enhanced abilities of migration and invasion. As colony formation in soft agar is linked to cancer stem cell properties (Matsubara et al., 2013), the high SLC2A5 expression may contribute to leukemic stem cell compartment expansion. Metabolic flux analysis demonstrated that high fructose utilization strongly activated glycolytic flux with increased pyruvate and lactate production in AML cells. The increased glycolytic activity has been implicated in promoting aggressiveness of cancer cells (Diers et al., 2012; Doherty and Cleveland, 2013). Consequently, enhanced fructose utilization or high SLC2A5 expression in AML cells may be linked to poor outcomes in AML patients.
Evidence of this is provided in this study through analysis of five gene expression data sets previously published which showed that above median/mean SLC2A5 expression was associated with inferior overall survival. Analysis of serum fructose concentrations in 262 de novo AML patients revealed an association of serum fructose levels with the efficacy of inducing remission using standard AML 3+7 treatment. Those patients who had higher serum fructose concentrations had higher rates of complete remission compared to those with low or medium serum fructose levels. These results link enhanced AML cell fructose utilization to a worse patient clinical outcome. Of note, the investigation of the association between SLC2A5 expression/fructose utilization and AML patient prognosis is a retrospective analysis. A blinded prospective study is needed to validate these findings.
In conclusion, the unique fructose utilization feature of AML cells provides a promising cancer target, and fructose inhibition may thus provide a viable, cell-cycle independent therapy for AML. A fructose analogue, 2,5-AM, was effective in alleviating the neoplastic phenotypes of AML cells in vitro and in vivo. We also observed a significant synergistic effect between 2,5-AM and Ara-C on the elimination of AML cells. It was noticeable that fructose utilization was heterogeneous in AML patients, as shown by the highly diverse expression of SLC2A5 and serum fructose in these patients. Our findings suggested only those AML patients whose blast cells highly expressed SLC2A5 and exhibited enhanced fructose utilization would be able to obtain therapeutic benefit using a fructose utilization inhibitor in combination with a chemotherapeutic agent.
EXPERIMENTAL PROCEDURES
AML Cell Lines, Normal Monocytes, Reagents and Antibodies
Four representative AML cell lines, U937, OCI-AML3, HL-60 and K562, were chosen for this study. U937 and OCI-AML3 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), while HL-60 and K562 were obtained from National Cancer Institute (NCI)-Frederick. All cell lines were maintained in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies) and antibiotics. A total of 4 different control samples of normal monocytes were enrolled in this study. One control sample was purchased from Stemcell technologies. The remaining controls were provided by 3 healthy volunteers. All the normal monocytes were used to perform assays at once after arrival. Reagents used in the study included D-fructose (Sigma-Aldrich), D-glucose (Sigma-Aldrich), GLUT5 antibody (Santa Cruz), Actin antibody (Li-cor), fructose analogue 2,5-anhydro-D-mannitol (2,5-AM, Santa Cruz), and antileukemic agent arabinofuranosyl cytidine (Ara-C, Sigma-Aldrich).
Knockdown of SLC2A5 Expression by RNAi in AML cells
The fructose transporter gene SLC2A5 was knocked down in K562 AML cells using RNAi technology. K562 cells were transfected with a siRNA targeting SLC2A5 (Qiagen) or a non-targeting control siRNA (Qiagen). Transfections were conducted using a Lipofectamine 3000 Transfection Reagent kit (Life Technologies) following the manufacturer’s protocol. After 96 hour incubation, cells were harvested and protein extracts were prepared for western blot analysis of GLUT5 expression. Meanwhile, the proliferation of the transfected cells fed with fructose or glucose was measured at different time points using a Cell Counting Kit-8 (CCK-8, Dojindo Laboratories) following the manufacturer’s protocol.
Overexpression of SLC2A5 in AML Cells
Enforced expression of SLC2A5 in AML cells was achieved using a retrovirus system consisting of MigR1 vector (a gift from Warren Pear) (Pear et al., 1998) and VSVG and gag/pol packaging vectors. A more detailed description of the procedure is provided in the Supplemental Information procedures.
Measurement of Serum Fructose
Serum fructose concentrations of AML patients and healthy volunteers were examined as part of the metabolomic study involving a large series of AML cases previously described (Chen et al., 2014; Wang et al., 2013). A total of 400 de novo AML patients and 446 age- and gender-matched healthy controls were enrolled in this study from the hematology centers of Shanghai, Hangzhou, Suzhou, Shenyang, Nanjing, Dalian and Beijing. The clinical data of these subjects were accessed in our previously published study (Chen et al., 2014). For therapeutic response analysis, 262 cases who received standard DA regimen (daunorubicin 40 mg/m2/day for 3 days and Ara-C 100 mg/m2/day for 7 days) in remission-induction phase were analyzed. All participants provided informed written consent in accordance with the regulation of the Institutional Review Boards of the related Universities/Hospitals in agreement with the Declaration of Helsinki. Mouse peripheral blood serum samples were collected as mentioned below.
Serum samples of human subjects and mice were analyzed using gas chromatograph-time-of-flight mass spectrometry (GC-TOFMS) as previously described (Chen et al., 2014; Wang et al., 2013). Fructose was identified by library searching and then confirmed by the standard (Sigma-Aldrich). Calibration of fructose concentration data was performed as previously reported (Chen et al., 2014). The absolute concentration of serum fructose was determined from the calibration curve.
Analysis of 13C-labeled Fructose/Glucose Uptake and 13C-labeled Fructose-derived Metabolites by GC-TOFMS
To assay the fructose/glucose uptake and fructose-derived metabolites, cells were cultured in glucose-free RPMI 1640 medium supplemented with 10% dialyzed FBS (dFBS) and 6 mM [U-13C6] fructose (Cambridge Isotope Labs) or 6 mM [U-13C6] glucose (Cambridge Isotope Labs). After incubation for 96 hours, cell culture media were collected. Then, 20 μL of cell medium was used for extraction of metabolites of 13C-fructose and 13C-glucose. Metabolite derivatization and GC-TOFMS assays were performed as previously described (Chen et al., 2014; Wang et al., 2013). 13C-fructose and 13C-glucose were monitored using ions at m/z 220 and 323 respectively. 13C-fructose-derived metabolites, including 13C-pyruvate, 13C-lactate and 13C-alanine were monitored at m/z 177, 222 and 118, respectively. Metabolite consumption and release were calculated based on the algorithm previously reported (Jain et al., 2012).
AML Mouse Study
We enrolled 5 groups of mice in this study, including a normal control group, AML mice treated with vehicle (vehicle group), AML mice treated with the fructose utilization inhibitor 2,5-AM (2,5-AM group), AML mice treated with Ara-C (Ara-C group) and AML mice treated with 2,5-AM and Ara-C (2,5-AM & Ara-C group). 8-week-old female BALB/c mice were selected for this study. Mice were maintained under specific pathogen-free conditions, kept on a 12-h light-dark cycle, and fed normal diet. AML mice were generated using a previously reported procedure (Wang et al., 2011) with minor modifications. Briefly, 1 × 105 GFP-positive murine leukemic cells (splenic cells) with AML1-ETO and mutated C-KIT were injected into the tail vein of each sublethally irradiated (3.5 Gy) mouse. For the 2,5-AM treatment group, the reagent was dissolved in physiological saline and intraperitoneally injected into AML mice at a dose of 150 mg/kg/d at day 5 after leukemic cell transplantation until the mice were sacrificed or died. For the Ara-C treatment group, Ara-C was dissolved in physiological saline and intraperitoneally injected into AML mice at a dose of 25 mg/kg/d at day 3 after leukemic cell transplantation for 5 days. For the 2,5-AM & Ara-C treatment group, 2,5-AM and Ara-C were given to the mice according to the methods described in the 2,5-AM group and Ara-C group. The same volume of physiological saline was given to the vehicle group. Seventeen days after leukemic cell transplantation, 5 groups of mice, including 6 normal controls, 8 AML mice treated with vehicle, 8 AML mice treated with 2,5-AM, 6 AML mice treated with Ara-C and 6 mice treated with 2,5-AM and Ara-C, were sacrificed to obtain peripheral blood, serum, bone marrow and spleen for analysis. Additionally, 9 AML mice treated with vehicle, 10 AML mice treated with 2,5-AM, 9 AML mice treated with Ara-C, and 10 AML mice treated with a combination of 2,5-AM and Ara-C were maintained for investigating overall survival. The animal experiments were approved by the Department of Animal Experimentation at Shanghai Jiao Tong University School of Medicine.
Statistical Analysis
Significant differences between groups were determined using the Student’s t test. The difference for survival time was assessed using log-rank test. Meta-analysis of survival data was performed by use of survcomp package in R software (version 2.15.0, www.r-project.org). The difference for therapeutic response among AML patient groups with distinct serum fructose levels was assessed using χ-square test. The significance level was set up at p < 0.05. The evaluation of synergistic effect between fructose analogue 2,5-AM and Ara-C was executed in CompuSyn software (ComboSyn, Inc., Paramus, NJ).
Supplementary Material
Significance.
Glucose, a well-known carbon source, has been extensively studied in various cancers. However, little is known about the role of fructose, the second most abundant blood sugar, in cancers including AML. This study shows that AML cells predominantly use fructose as an important glucose alternative to promote cell proliferation. Enhanced fructose utilization aggravates the leukemic phenotypes of AML cells thus causing poor outcomes in patients. This unique metabolic feature may create an important biological target since most normal cells including normal monocytes do not use fructose as the main metabolic fuel. Small molecule chemical agents or antibodies that can inhibit or block fructose uptake will selectively inhibit AML cell growth and synergize with antileukemic drugs.
Highlights.
AML cells are prone to fructose utilization to offset glucose insufficiency
Increased SLC2A5 transcription and fructose utilization predict poor patient outcomes
Enhanced fructose utilization exacerbates leukemic cell phenotypes
Inhibition of fructose uptake shows therapeutic potential for AML
Acknowledgments
This work was partially supported by NIH/NCI grant P30 CA071789 and the National Natural Science Foundation of China for Excellent Young Scholars Grant 81222004. We thank Dr. Peter J.M. Valk (Erasmus University Medical Center, Rotterdam, Netherlands) and Dr. Jonathan R. Pollack (Stanford University, Stanford, US) for sharing the clinical data of microarray data sets GSE1159 and GSE425 respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental information
Supplemental Information includes extended experimental procedures, figures and tables.
Author Contribution
W.J. and S.-J.C. were the principal investigator of this study, and conceived the research together with W.-L.C.; W.-L.C. performed the molecular and cellular biology experiments and analyzed the data; W.-L.C., A.Z., J.L. and Q.C. did serum fructose measurement; G.X., M.S., and L.Z. carried out 13C-labeled sugar uptake and glycolytic flux assays; Z. Chen and S.-J.C. provided the clinical data that involved 400 AML patients for this study; W.-L.C, Y.-Y.W. and L.X. performed mouse study; W.-L.C, R.W. and Y.N. performed statistical analysis; W.-L.C. and W.J. wrote the manuscript; C.R. and Z. Cheng contributed to manuscript revision.
References
- Aries IM, Hansen BR, Koch T, van den Dungen R, Evans WE, Pieters R, den Boer ML. The synergism of MCL1 and glycolysis on pediatric acute lymphoblastic leukemia cell survival and prednisolone resistance. Haematologica. 2013;98:1905–1911. doi: 10.3324/haematol.2013.093823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, Wu X, Yu Y, Amlal H, Seidler U, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284:5056–5066. doi: 10.1074/jbc.M808128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- Burant CF, Takeda J, Brot-Laroche E, Bell GI, Davidson NO. Fructose transporter in human spermatozoa and small intestine is GLUT5. J Biol Chem. 1992;267:14523–14526. [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Chen WL, Wang JH, Zhao AH, Xu X, Wang YH, Chen TL, Li JM, Mi JQ, Zhu YM, Liu YF, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124:1645–1654. doi: 10.1182/blood-2014-02-554204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers AR, Broniowska KA, Chang CF, Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. Biochem J. 2012;444:561–571. doi: 10.1042/BJ20120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–495. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- Herst PM, Howman RA, Neeson PJ, Berridge MV, Ritchie DS. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. Journal of leukocyte biology. 2011;89:51–55. doi: 10.1189/jlb.0710417. [DOI] [PubMed] [Google Scholar]
- Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R, Den Boer ML. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009;113:2014–2021. doi: 10.1182/blood-2008-05-157842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Experimental & molecular medicine. 2013;45:e45. doi: 10.1038/emm.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010;70:6368–6376. doi: 10.1158/0008-5472.CAN-09-4615. [DOI] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincic D, Hande KR. Topoisomerase II inhibitors. Cancer chemotherapy and biological response modifiers. 2005;22:101–121. doi: 10.1016/s0921-4410(04)22005-1. [DOI] [PubMed] [Google Scholar]
- Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Scientific reports. 2013;3:3230. doi: 10.1038/srep03230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momparler RL. Optimization of cytarabine (ARA-C) therapy for acute myeloid leukemia. Exp Hematol Oncol. 2013;2:20. doi: 10.1186/2162-3619-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzavi-Karbassi B, Hine RJ, Stanley JS, Ramani VP, Carcel-Trullols J, Whitehead TL, Kelly T, Siegel ER, Artaud C, Shaaf S, et al. Fructose as a carbon source induces an aggressive phenotype in MDA-MB-468 breast tumor cells. International journal of oncology. 2010;37:615–622. doi: 10.3892/ijo_00000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer chemotherapy and pharmacology. 2013;71:523–530. doi: 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary AR, Hockenbery D, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47:8–20. doi: 10.1002/gcc.20500. [DOI] [PubMed] [Google Scholar]
- Stirewalt DL, Pogosova-Agadjanyan EL, S O. 2012 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37307.
- Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. British journal of haematology. 2014;164:767–778. doi: 10.1111/bjh.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiziani S, Kang Y, Harjanto R, Axelrod J, Piermarocchi C, Roberts W, Paternostro G. Metabolomics of the tumor microenvironment in pediatric acute lymphoblastic leukemia. PLoS One. 2013;8:e82859. doi: 10.1371/journal.pone.0082859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan R, Kumar D, Dube SN, Singh R, Pandey KS, Bag BC, Kaushik MP, Sekhar K, Dwarakanath BS, Ravindranath T. Acute toxicity and cardio-respiratory effects of 2-deoxy-D-glucose: a promising radio sensitiser. Biomedical and environmental sciences: BES. 2006;19:96–103. [PubMed] [Google Scholar]
- Wang JH, Chen WL, Li JM, Wu SF, Chen TL, Zhu YM, Zhang WN, Li Y, Qiu YP, Zhao AH, et al. Prognostic significance of 2-hydroxyglutarate levels in acute myeloid leukemia in China. Proc Natl Acad Sci U S A. 2013;110:17017–17022. doi: 10.1073/pnas.1315558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Zhao LJ, Wu CF, Liu P, Shi L, Liang Y, Xiong SM, Mi JQ, Chen Z, Ren R, Chen SJ. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A. 2011;108:2450–2455. doi: 10.1073/pnas.1019625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Dowden J, Tatibouet A, Hatanaka Y, Holman GD. Development of high-affinity ligands and photoaffinity labels for the D-fructose transporter GLUT5. Biochem J. 2002;367:533–539. doi: 10.1042/BJ20020843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebisch A, Wolfler A, Fried I, Wolf O, Lind K, Bodner C, Haller M, Drasche A, Pirkebner D, Matallanas D, et al. Frequent loss of RAF kinase inhibitor protein expression in acute myeloid leukemia. Leukemia. 2012;26:1842–1849. doi: 10.1038/leu.2012.61. [DOI] [PubMed] [Google Scholar]
- Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007;8:113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.