

SUMMARY
The first epithelial to mesenchymal transition (EMT) occurs in trophoblast stem (TS) cells during implantation. Inactivation of the serine/threonine kinase MAP3K4 in TS cells (TSKI4 cells) induces an intermediate state of EMT, where cells retain stemness, lose epithelial markers, and gain mesenchymal characteristics. Investigation of relationships between MAP3K4 activity, stemness, and EMT in TS cells may reveal key regulators of EMT. Here, we show that MAP3K4 activity controls EMT through the ubiquitination and degradation of HDAC6. Loss of MAP3K4 activity in TSKI4 cells results in elevated HDAC6 expression and the deacetylation of cytoplasmic and nuclear targets. In the nucleus, HDAC6 deacetylates the promoters of tight junction genes, promoting the dissolution of tight junctions. Importantly, HDAC6 knockdown in TSKI4 cells restores epithelial features including cell-cell adhesion and barrier formation. These data define an epigenetic role for HDAC6 in regulating gene expression during transitions between epithelial and mesenchymal phenotypes.
Graphical abstract
Mobley et al. find that HDAC6 is regulated by MAP3K4 during trophoblast stem cell differentiation and EMT. MAP3K4 promotes HDAC6 ubiquitination and degradation, maintaining the epithelial state. During EMT, HDAC6 directly deacetylates histones on epithelial gene promoters such as claudin6 and occludin, promoting the dissolution of tight junctions.
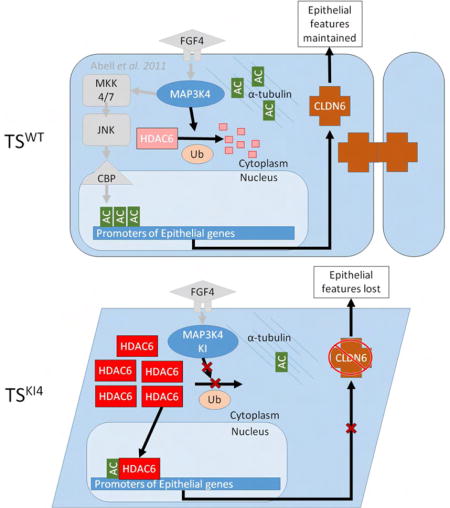
INTRODUCTION
Epithelial to mesenchymal transition (EMT) is a morphogenetic program controlling cellular phenotype that is essential for the development and maintenance of multicellular organisms (Thiery et al., 2009). During EMT, epithelial cells lose apical-basal polarity and cell-cell adhesion while acquiring a front-back, mesenchymal morphology. This transition converts nonmotile epithelial cells to invasive mesenchymal cells. In EMT, cells lose the expression of epithelial markers such as the cell-cell adhesion protein E-cadherin, and gain the expression of mesenchymal markers like N-cadherin and vimentin. In addition, several transcription factors promote EMT, including Lef1, Snai1, and Twist (Lamouille et al., 2014). EMT is responsible for the creation of many cell types during embryogenesis. Further, EMT is activated during tissue regeneration after injury and in disease states such as organ fibrosis and cancer metastasis.
One of the first developmental EMT events occurs during implantation (Thiery et al., 2009). A subset of epithelial, trophoblast stem (TS) cells in the trophectoderm convert to invasive trophoblast giant cells that invade the mother and establish placentation. TS cells can be isolated from pre-implantation blastocysts and cultured indefinitely. The stemness properties (self-renewal and potency) of TS cells are maintained by the presence of FGF4 and conditioned media from mitotically inactivated mouse embryonic fibroblasts (MEF-CM) (Tanaka et al., 1998). Removal of FGF4 and MEF-CM induces TS cell EMT. Cells lose the expression of E-cadherin, and convert into invasive trophoblast giant cells with mesenchymal properties, including expression of N-cadherin, vimentin, fibronectin, Snai1 and Lef1 (Abell et al., 2011). Recently, we reported the isolation of TS cells from conceptuses of mice with a targeted mutation of MAP3K4 that inactivates its kinase activity (Abell et al., 2009; Abell et al., 2011). Mice expressing kinase-inactive MAP3K4 (KI4 mice) display developmental phenotypes due to perturbed EMT including neural tube, skeletal, and implantation defects (Abell et al., 2009; Abell et al., 2005). TS cells isolated from KI4 conceptuses (TSKI4) exhibit characteristics of EMT while maintaining their stemness properties (Abell et al., 2011). Specifically, undifferentiated TSKI4 cells have a more mesenchymal morphology with reduced E-cadherin, and increased N-cadherin and vimentin relative to wild-type (TSWT) cells. Also, EMT-inducing transcription factors Snai1 and Twist are elevated in TSKI4 cells and cells display increased invasiveness compared to TSWT cells.
MAP3K4 activates CBP, a histone acetyltransferase (HAT), to promote the acetylation of histones H2A and H2B (H2A/H2BAc) on the promoters of genes critical for maintaining the epithelial phenotype (Abell et al., 2011). Loss of MAP3K4 activity results in reduced CBP-mediated acetylation and the conversion of epithelial stem cells to a more mesenchymal morphology. Because CBP acetylates numerous targets in addition to histones, we predicted that MAP3K4 regulates the acetylation of additional proteins. Herein, we show MAP3K4 activity represses HDAC6 expression and activity required for deacetylation of cytoplasmic and nuclear proteins important for EMT. Loss of MAP3K4 activity in TSKI4 cells increases HDAC6 expression and activity, and knockdown of HDAC6 in TSKI4 cells restores epithelial features. We define a key nuclear role for HDAC6 in the deacetylation of promoters of genes encoding tight junction proteins, resulting in diminished cell-cell adhesion characteristic of the mesenchymal phenotype. In addition, we identify MAP3K4/HDAC6 regulated genes with previously undescribed roles in EMT. Our findings demonstrate MAP3K4 coordinates HDAC and HAT activities for the transition of stem cells between epithelial and mesenchymal states.
RESULTS
Reduced Acetylation of Proteins in TSKI4 Cells Is Due to Increased Expression and Activity of HDAC6
MAP3K4 activates CBP-mediated H2A/H2BAc to promote the expression of genes essential for maintaining the epithelial phenotype (Abell et al., 2011). Inactivation of MAP3K4 kinase activity in TSKI4 cells results in the loss of H2A/H2BAc and the gain of mesenchymal features. To identify MAP3K4 dependent changes in protein acetylation, we treated TSWT and TSKI4 cells with the deacetylase inhibitor, Trichostatin A (TSA), and compared lysine acetylation by Western blotting. Acetylation of several proteins was reduced in TSKI4 cells compared to TSWT cells, including the acetylation of a 50 kDa protein identified as acetylated α-tubulin (Ac-tubulin)(Figure 1A). Ac-tubulin was decreased in untreated TSKI4 cells relative to TSWT cells, indicating a significant deregulation of tubulin acetylation with loss of MAP3K4 kinase activity (Figure 1B).
Figure 1. TSKI4 cells exhibit reduced protein acetylation due to increased expression of HDAC6.
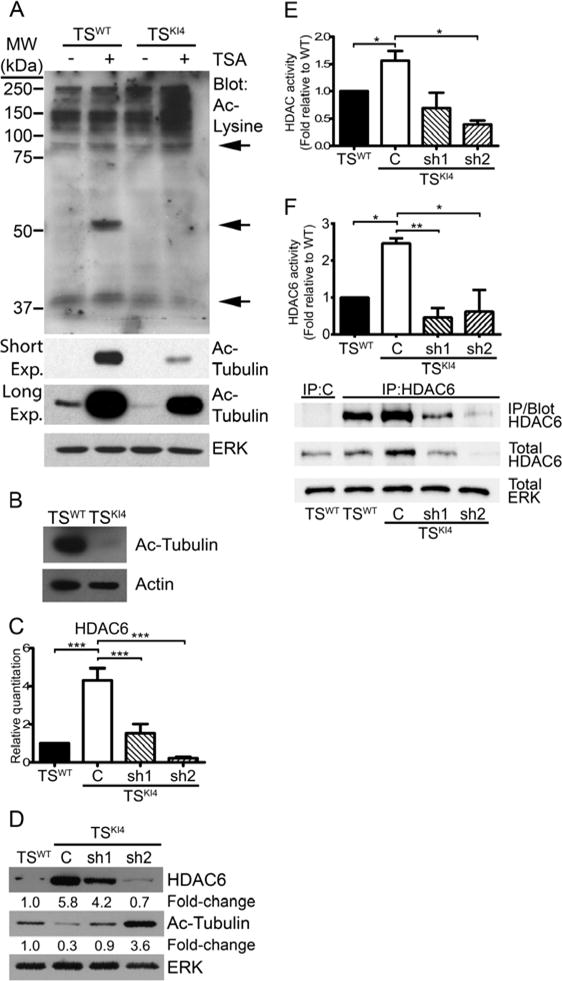
Ablation of MAP3K4 kinase activity reduces the acetylation of several proteins.
(A) Western blots of 40 μg of whole cell lysate from TSWT and TSKI4 cells treated with DMSO (−) or 50 nM pan-deacetylase inhibitor, TSA (+) are shown. Blots are representative of four experiments. Arrows denote proteins with altered acetylation. (Exp, exposure)
(B) Western blots of 40 μg untreated TSWT and TSKI4 whole cell lysates are representative of two independent experiments.
(C) HDAC6 transcript increases upon loss of MAP3K4 activity. qRT-PCR data are from TSWT and TSKI4 cells expressing a control shRNA (C) or TSKI4 cells expressing two independent HDAC6 shRNAs (sh1 and sh2). Data are expressed as a fold change relative to TSWT cells and are the mean ± SEM of between three and five independent experiments.
(D) HDAC6 protein increases with loss of MAP3K4 activity. Western blots of 10 μg whole cell lysates are representative of four independent experiments.
(E) Total cytoplasmic HDAC activity increases with loss of MAP3K4 activity. HDAC activity data show the mean ± SEM of four experiments.
(F) Cytoplasmic HDAC6 activity is increased in HDAC6 immunoprecipitates (IP) from TSKI4 relative to TSWT cytoplasmic lysate. Data show the mean ± SEM of three independent experiments. Blots show IP efficiency of the HDAC6 antibody relative to IgG control and are representative of three experiments.
Student’s t-test, *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001.
See also Figure S1.
α-tubulin can be deacetylated by both HDAC6 and SIRT2 (Hubbert et al., 2002; North et al., 2003). Real time quantitative PCR (qRT-PCR) showed that HDAC6 transcripts were increased four-fold in TSKI4 cells relative to TSWT cells, whereas SIRT2 transcripts were only modestly changed (Figures 1C and S1A). HDAC6 protein expression was also increased 5.8-fold in TSKI4 cells, whereas SIRT2 protein was not significantly changed (Figures 1D and S1B). To determine if decreased Ac-tubulin in TSKI4 cells was due to increased expression of HDAC6, we reduced HDAC6 expression in TSKI4 cells using two different HDAC6 lentiviral shRNAs. HDAC6 message and protein levels were partially reduced in TSKI4 cells with HDAC6 sh1, but HDAC6 levels remained above TSWT levels (Figures 1C and 1D). HDAC6 knockdown in TSKI4 cells with HDAC6 sh2 reduced HDAC6 message and protein levels below that of TSWT cells (Figures 1C and 1D). Knockdown of HDAC6 in TSKI4 cells restored Ac-tubulin to either near wild-type levels with sh1 or above wild-type levels with sh2 (Figure 1D). Examination of total cytoplasmic deacetylase activity revealed a 50% increase in TSKI4 cells relative to TSWT cells that was reduced by knockdown of HDAC6 in TSKI4 cells (Figure 1E). Further, HDAC6 activity was increased 2.6-fold in HDAC6 immunoprecipitates from TSKI4 cells relative to TSWT cells (Figure 1F). Together, these findings show MAP3K4 regulates the expression and activity of HDAC6.
Increased HDAC6 Expression and Activity Induce Specific Features of EMT in TSKI4 Cells
TSWT cells have an epithelial morphology, as compared to TSKI4 cells that have an intermediate phenotype with both epithelial and mesenchymal characteristics (Figures 2A and 2B). HDAC6 knockdown in TSKI4 cells reduced mesenchymal features and partially restored an epithelial morphology, suggesting that increased HDAC6 expression and activity in TSKI4 cells promote EMT (Figures 2C and 2D). Importantly, TSWT cells retained an epithelial morphology after HDAC6 knockdown in TSWT cells (Figure S2A–C).
Figure 2. HDAC6 represses epithelial features in TS cells.
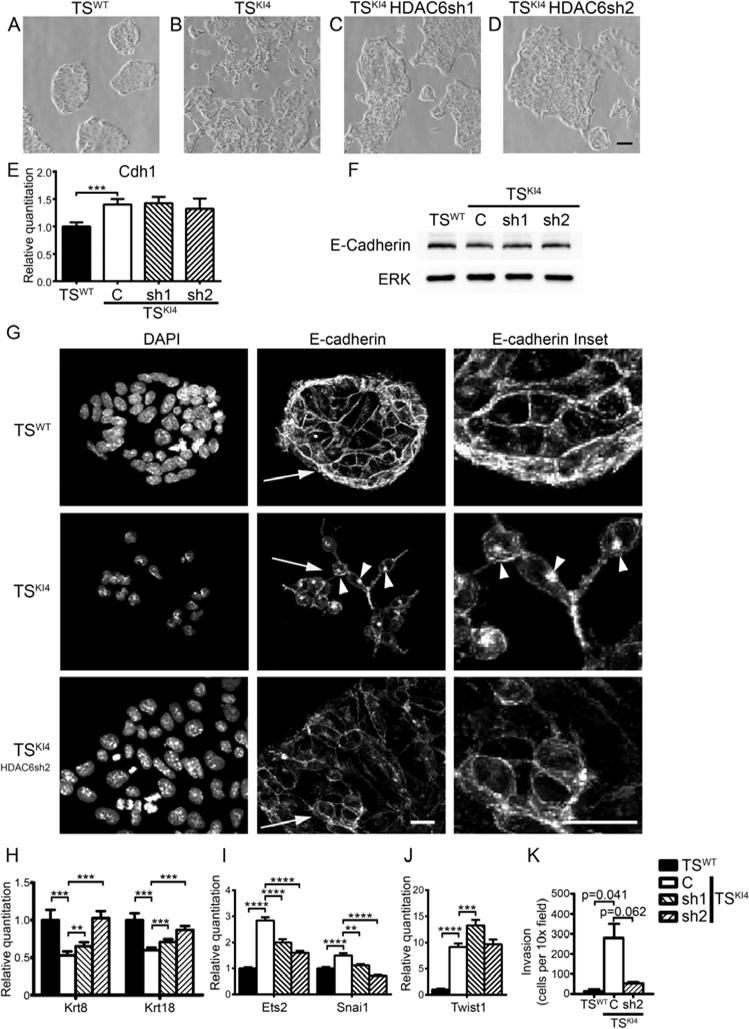
Knockdown of HDAC6 in TSKI4 cells partially restores the epithelial morphology
(A–D) Phase microscopy images of TSWT (A) and TSKI4 (B) cells expressing a control shRNA or TSKI4 cells expressing two independent HDAC6 shRNAs (sh1 and sh2) (C,D) are shown. Representative images from four independent experiments are shown. Black bar represents 200 μm.
(E) E-cadherin transcript levels are not HDAC6 dependent. qRT-PCR data show the mean ± SEM of three independent experiments.
(F) Western blots of whole cell lysates are representative of three independent experiments.
(G) E-cadherin localization is partially restored upon HDAC6 knockdown in TSKI4 cells. Confocal fluorescence images of E-cadherin staining are representative of three independent experiments. White bar represents 10 μm. Arrowheads indicate punctate intracellular localization of E-cadherin. Arrows indicate inset area.
(H) Krt8 and Krt18 transcript levels measured by qRT-PCR are dependent on HDAC6 expression.
(I,J) Selective regulation of EMT inducing transcription factor expression by HDAC6 as measured by qRT-PCR.
(E,H–J) Data are expressed as a fold-change relative to TSWT cells. Data are the mean ± SEM of between three and four independent experiments.
(K) Invasion through growth factor reduced Matrigel by undifferentiated cells is shown. Data show the mean ± range of two independent experiments performed in triplicate. Student’s t-test, **p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001
See also Figure S2.
As a measure of the epithelial state, we examined the expression and localization of E-cadherin, a key marker of epithelial cells. E-cadherin levels were similar between TSWT cells and TSKI4 cells and were not altered by HDAC6 knockdown (Figures 2E and 2F). However, E-cadherin localization was altered. Unlike TSWT cells with E-cadherin immunostaining at the plasma membrane, E-cadherin was mislocalized to cytoplasmic puncta in TSKI4 cells (Figure 2G). Knockdown of HDAC6 in TSKI4 cells partially restored E-cadherin localization to the plasma membrane (Figure 2G). Expression of epithelial keratins (Krt8 and Krt18) was reduced in TSKI4 cells and restored by HDAC6 knockdown, further showing that HDAC6 represses epithelial characteristics (Figure 2H). Krt8, Krt18 and Cdh1 levels were increased by knockdown of HDAC6 in TSWT cells (Figure S2D).
To further address the role of HDAC6 in TSKI4 cell EMT, we measured the expression of EMT inducing transcription factors and invasiveness. Snai1, Ets2 and Twist1 transcripts were elevated in TSKI4 cells, and HDAC6 knockdown partially reduced Snai1 and Ets2 expression, suggesting a modest role for HDAC6 in transcriptional regulation of EMT-inducing transcription factors (Figures 2I and 2J). TSKI4 cells were twenty times more invasive than TSWT cells and knockdown of HDAC6 in TSKI4 cells decreased invasiveness (Figure 2K). These data indicated that MAP3K4 regulates specific aspects of TS cell EMT through HDAC6.
HDAC6 Expression and Activity Are Induced by TS Cell Differentiation and EMT
Removal of FGF4, heparin, and MEF-CM induces TS cell differentiation and EMT (Abell et al., 2011). We hypothesized that HDAC6 expression and/or activity were regulated during differentiation. As shown in Figure 3A and 3B, HDAC6 transcript and protein were induced by TSWT cell differentiation for four days. This induction of HDAC6 expression was associated with increased cytoplasmic HDAC activity showing that normal stem cell differentiation and EMT were associated with increased HDAC6 expression and activity (Figure 3C). Coexpression of wild-type MAP3K4 with HDAC6 in 293 cells inhibited HDAC6 activity (Figure 3D). Conversely, HDAC6 activity was higher in the presence of kinase-inactive MAP3K4 as compared to wild-type MAP3K4, suggesting that kinase-inactive MAP3K4 (KI) failed to inhibit HDAC6 activity (Figure 3E).
Figure 3. MAP3K4 regulates HDAC6 expression and activity during normal TS cell differentiation and EMT through direct protein-protein interaction.

(A–C) HDAC6 expression and activity are increased upon differentiation of TS cells. Cells were either undifferentiated (0) or differentiated for four days (4) in medium lacking FGF4, heparin, and MEF-CM.
(A) qRT-PCR data show the mean ± SEM of three independent experiments.
(B) Western blots of whole cell lysates are representative of three independent experiments.
(C) Cytoplasmic HDAC activity is expressed as the mean ± SEM of four independent experiments.
(D) Co-expression of HA-MAP3K4 and FL-HDAC6 reduces cytoplasmic FL-HDAC6 activity. FL-HDAC6 was immunoprecipitated from transfected 293 cells and HDAC6 activity and expression were measured. Activity data are the mean ± range and blots are representative of two independent experiments.
(E) FL-HDAC6 was co-expressed with either wild-type or kinase-inactive HA-MAP3K4. FL-HDAC6 was immunoprecipitated and activity measured as described in (D). Activity data show the mean ± SEM of three independent experiments, relative to empty vector. Blots are representative of three independent experiments.
(F–H) The kinase domain of MAP3K4 binds HDAC6 independent of MAP3K4 kinase activity. 293 cells were transfected with the indicated constructs and blots were probed with the indicated antibodies.
(I,J) Endogenous MAP3K4 and HDAC6 co-immunoprecipitate in murine TS cells (I) and human 293 cells (J).
(F–J) Blots are representative of three independent experiments. (FL, Flag; KD, Kinase Domain; KI, Kinase Inactive; WT, wild-type; C, control; sh2, HDAC6 knockdown shRNA2).
Student’s t-test, *p-value < 0.05;***p-value < 0.001.
Interaction of MAP3K4 and HDAC6
Based on our findings that wild-type MAP3K4 inhibited HDAC6 activity, we determined if MAP3K4 and HDAC6 were bound in a protein complex. HA-tagged MAP3K4 was coexpressed with either Flag-tagged HDAC6 or HDAC7 and Flag-tagged proteins were immunoprecipitated with anti-Flag antibody. HA-MAP3K4 coimmunoprecipitated with Flag-HDAC6, but not Flag-HDAC7, demonstrating the specific binding of MAP3K4 to HDAC6 (Figure 3F). To define the domain of MAP3K4 interacting with HDAC6, we deleted the N-terminus of MAP3K4 leaving only the kinase domain (HA-MAP3K4 KD). The kinase domain alone bound HDAC6 similarly to full-length MAP3K4 demonstrating that the kinase domain of MAP3K4 was sufficient for HDAC6 binding (Figure 3G). TSKI4 cells have a targeted mutation of the catalytic lysine 1361 to arginine in the kinase domain of MAP3K4. Because binding of HDAC6 to MAP3K4 occurs through the kinase domain of MAP3K4, we determined if this mutation altered HDAC6 binding. As shown in Figure 3H, kinase-inactive HA-MAP3K4KI bound HDAC6 at levels similar to wild-type HA-MAP3K4, showing this kinase domain mutation does not impair binding. This interaction was confirmed by coimmunoprecipitation of endogenous MAP3K4 and HDAC6 in both murine TS cells and human 293 cells (Figures 3I and 3J). Endogenous KI MAP3K4 coimmunoprecipitated with HDAC6 in TSKI4 cells, further demonstrating that kinase-inactive MAP3K4 bound HDAC6 (Figure 3I).
MAP3K4 Promotes the Ubiquitination and Degradation of HDAC6
Although MAP3K4KI retained HDAC6 binding, it failed to inhibit HDAC6 activity. Further, the lack of MAP3K4 kinase activity resulted in increased HDAC6 protein levels. Elevated HDAC6 in TSKI4 cells could result from cells being in an intermediate state of EMT, and/or a direct effect of MAP3K4 on HDAC6 protein stability. It has previously been shown that HDAC6 is itself an E3 ligase capable of autoubiquitination (Zhang et al., 2014). In addition, HDAC6 is ubiquitinated by CHIP, an E3 ligase, and subsequently degraded (Cook et al., 2012). To determine the effect of MAP3K4 on HDAC6 ubiquitination, HA-MAP3K4 and Flag-HDAC6 were coexpressed with Myc-tagged ubiquitin. When expressed alone, both Flag-HDAC6 and HA-MAP3K4 were ubiquitinated (Figure 4A). Coexpression of HA-MAP3K4 with Flag-HDAC6 dramatically increased the ubiquitination of Flag-HDAC6 (Figure 4A, upper panel). In contrast, Flag-HDAC6 did not significantly alter the ubiquitination of HA-MAP3K4 (Figure 4A, lower panel). Based on the increased endogenous HDAC6 protein levels in TSKI4 cells, we predicted that kinase-inactive MAP3K4 might be deficient in promoting HDAC6 ubiquitination. When coexpressed, HA-MAP3K4KI failed to promote the ubiquitination of Flag-HDAC6 (Figure 4B). Further, the ubiquitination of HA-MAP3K4KI was decreased compared to HA-MAP3K4WT (Figure 4C). These changes were not HDAC6 dependent, suggesting the kinase activity of MAP3K4 regulates its ubiquitination independent of HDAC6 (Figure 4C). To examine the impact of MAP3K4 on endogenous HDAC6, TSWT cells and TSKI4 cells were treated with cycloheximide (CHX) to inhibit protein synthesis. At four and eight hours of CHX treatment, HDAC6 levels were significantly decreased in TSWT cells, whereas HDAC6 levels in TSKI4 cells remained elevated (Figure 4D). Together, these data suggested that MAP3K4 kinase activity decreases the stability of HDAC6 protein. Loss of HDAC6 degradation in TSKI4 cells resulted in elevated levels of HDAC6 protein.
Figure 4. MAP3K4 promotes the ubiquitination and degradation of HDAC6.

(A–C) 293 cells were transfected with the indicated constructs. Western blots were probed with the indicated antibodies. (FL, Flag; KI, Kinase Inactive; WT, wild-type)
(A) Co-expression of wild-type HA-MAP3K4 and FL-HDAC6 increases the ubiquitination of HDAC6 but not MAP3K4. Samples were immunoprecipitated with the indicated antibody and probed for Myc-ubiquitin. Blots shown are representative of two independent experiments.
(B) Kinase inactive MAP3K4 does not promote ubiquitination of HDAC6. Samples were immunoprecipitated using anti-FL antibody and probed for Myc-ubiquitin. Data shown are representative of four independent experiments.
(C) Ubiquitination of wild-type and kinase inactive MAP3K4 is not influenced by HDAC6. Samples were immunoprecipitated with anti-HA antibody and probed for Myc-ubiquitin. Blots shown are representative of three independent experiments.
(D) The degradation rate of endogenous HDAC6 is reduced in the absence of MAP3K4 kinase activity. TSWT or TSKI4 cells were treated for the indicated number of hours with cycloheximide (CHX), an inhibitor of translation. Representative blots from three independent experiments are shown. Fold change relative to wild-type 0 hours was calculated by normalizing HDAC6 densitometry to total ERK.
Loss of MAP3K4 Kinase Activity Increases HDAC6 Nuclear Localization and Function
Cytoplasmic functions of HDAC6 are well studied, while the roles of HDAC6 in the nucleus are not well defined (Valenzuela-Fernandez et al., 2008). TSKI4 cells have increased nuclear localization of HDAC6 as compared to TSWT cells (Figures 5A and 5B and see Figure S3A). Increased HDAC6 expression correlated with increased nuclear HDAC activity in TSKI4 cells that was reduced by HDAC6 knockdown (Figure 5C). TSKI4 cells display a selective loss of H2A/H2BAc, whereas histones H3 and H4 are not altered (Abell et al., 2011). To determine if increased HDAC6 contributed to loss of H2A/H2BAc, we examined histone acetylation after HDAC6 knockdown in TSKI4 cells. H2AK5 and H2BK5 acetylation was restored by HDAC6 knockdown in TSKI4 cells, demonstrating HDAC6 regulation of specific histone acetylation (Figure 5D). Similar to TSKI4 cell EMT, normal differentiation for four days also increased nuclear localization and activity of HDAC6 in TSWT cells (Figure 5E and 5F). Differentiation of TS cells reduced acetylation of histones H2A, H2B, H3 and H4 (Figure 5G). As HDAC6 expression and activity increase in the nucleus with differentiation, we determined if HDAC6 was involved in the deacetylation of histones during normal TS cell differentiation. As shown in Figure 5H, HDAC6 knockdown in TSKI4 cells resulted in the selective maintenance of H2A/H2BAc during differentiation, whereas deacetylation of H3 and H4 was not affected, suggesting that HDAC6 selectively deacetylated histones H2A and H2B (Figure 5H). Together, these data demonstrate a key role for HDAC6 during differentiation and EMT by promoting the selective deacetylation of H2A/H2B.
Figure 5. TSKI4 cells have increased nuclear HDAC6 localization and function.
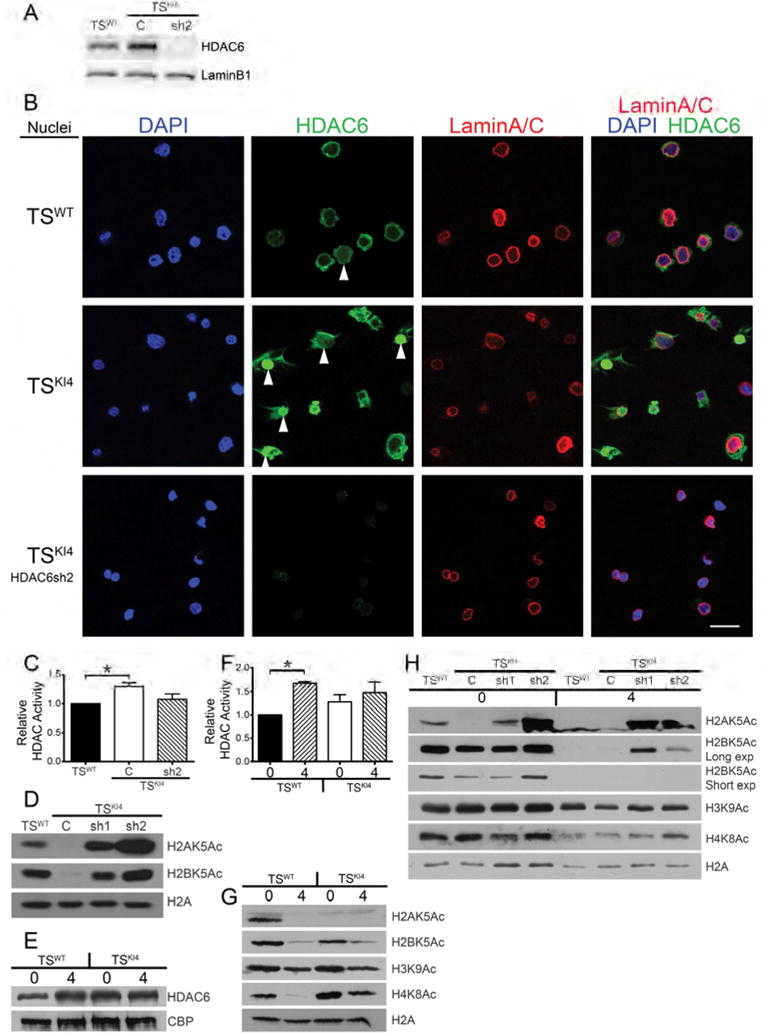
(A) Nuclear HDAC6 protein levels are elevated in TSKI4 cells. Western blots of nuclear lysates are from TSWT or TSKI4 cells expressing a control shRNA (C) or TSKI4 cells expressing HDAC6 shRNA 2 (sh2).
(B) Immunofluorescence microscopy images of isolated nuclei show increased nuclear HDAC6 (green) in TSKI4 cells relative to TSWT cells. Nuclei were stained with DAPI (blue) and LaminA/C (red). Arrowheads indicate nuclear HDAC6. White bar represents 50 μm.
(A,B) Images shown are representative of three independent experiments.
(C) Increased total nuclear HDAC activity in TSKI4 cells expressed as the mean ± range of two independent experiments.
(D) Histone acetylation is restored by knockdown of HDAC6 in TSKI4 cells. Western blots are representative of three independent experiments.
(E–F) Differentiation and EMT induce nuclear HDAC6 localization and activity.
(E) Western blots of nuclear lysates from undifferentiated TSWT or TSKI4 cells (0) or cells differentiated four days (4). Blots are representative of two independent experiments.
(F) Nuclear HDAC activity assays were performed using nuclear lysates from undifferentiated cells or cells differentiated as in (E). Data are the mean ± range of two independent experiments.
(G–H) Western blots from cells undifferentiated (0) or differentiated for four days (4) as in (E). Blots are representative of three independent experiments.
(G) Histone acetylation is reduced with differentiation and EMT in TS cells.
(H) HDAC6 shRNA knockdown prevents differentiation-induced deacetylation of H2A/H2B.
Student’s t-test, *p-value < 0.05.
See also Figure S3.
HDAC6 Inhibits the Expression of Tight Junction Proteins by Direct Promoter Binding and Deacetylation of H2BK5
Based on the presence of HDAC6 in the nucleus of TSKI4 cells and HDAC6 dependent changes in H2BK5Ac, we hypothesized that HDAC6 directly deacetylates H2BK5 on specific gene promoters in TSKI4 cells. To identify MAP3K4/HDAC6/H2BK5Ac-dependent genes in TSKI4 cells, we performed anti-H2BK5Ac chromatin immunoprecipitation coupled to deep sequencing (ChIP-seq) of TSWT, TSKI4 and TSKI4 HDAC6sh2 cells. Analysis of H2BK5Ac-ChIP-seq data identified 1817 genes with decreased H2BK5Ac in TSKI4 cells relative to TSWT cells that increased in TSKI4 HDAC6sh2 cells (Figures 6A and 6B). RNA-seq showed decreased transcripts of 2624 genes in TSKI4 cells relative to TSWT cells, whose expression increased in TSKI4 HDAC6sh2 cells, suggesting their HDAC6 dependence (Figure 6C). Comparison of genes with MAP3K4/HDAC6 dependent H2BK5Ac and genes with MAP3K4/HDAC6 dependent expression identified 514 MAP3K4/HDAC6/H2BK5Ac dependent genes (Figures 6C and 6D and Table S1). These genes showed HDAC6 dependent gene expression and promoter H2BK5Ac, implicating regulation by HDAC6 (Table S1). ToppFunn gene ontology (GO) analyses of Biological Processes of these 514 genes showed significant enrichment in several processes including the organization of cell-cell junctions and the actin cytoskeleton, tight junction assembly, and epithelium development (Figure S4A). Further, ToppFunn GO analyses of Cellular Components revealed enrichment in genes important to cell-cell, apical, and tight junctions (Figure 6E). Fold changes relative to TSWT cells in genes from the Cell-Cell Junction category showed decreased expression in TSKI4 cells that was restored by HDAC6 knockdown (Figure S4B). With the exception of the membrane raft category, all enriched Cellular Component categories included the gene Cldn6, a key tight junction protein mediating cell-cell adhesion in epithelial stem cells (Figure 6E). Read density plots showed reduced H2BK5Ac near the transcription start site (TSS) of several tight junction genes including Clnd6 and Tjp1 in TSKI4 cells relative to TSWT cells that was restored by HDAC6 knockdown (Figures 6F and S5A–D). Significantly, H2BK5Ac at the TSS of the stemness marker Cdx2 was not dependent on MAP3K4 or HDAC6 (Figure S5E). Reduced Cldn6 promoter H2BK5Ac correlated with decreased Cldn6 transcript and protein in TSKI4 cells (Figures 6G and 6H). Further, knockdown of HDAC6 to levels below wild-type (sh2) restored Cldn6 expression (Figures 6G and 6H). Immunofluorescence confocal microscopy revealed the co-localization of Cldn6 and ZO-1 at cell-cell junctions in TSWT cells (Figure 6I). In contrast, Cldn6 was undetectable in TSKI4 cells and ZO-1 (Tjp1) was mislocalized (Figures 6I). Expression and localization of Cldn6 and ZO-1 were partially restored in TSKI4 HDAC6sh2 cells, suggesting a key role for HDAC6 in the dissolution of tight junctions (Figure 6I). Three dimensional confocal microscopy images showed loss of tight junctions in TSKI4 cells that are restored by HDAC6 knockdown (Figures S5F and S5G). Using fluorescent dye exclusion assays to measure barrier function, we observed increased dye flux through confluent monolayers of TSKI4 cells relative to TSWT cells, suggesting that loss of MAP3K4 compromises barrier function (Figure 6J). Knockdown of HDAC6 in TSKI4 cells inhibited dye flux to levels below TSWT cells, suggesting that HDAC6 impairs barrier function (Figure 6J).
Figure 6. HDAC6 directly binds and deacetylates the promoters of genes encoding tight junction proteins critical for maintaining the epithelial phenotype.
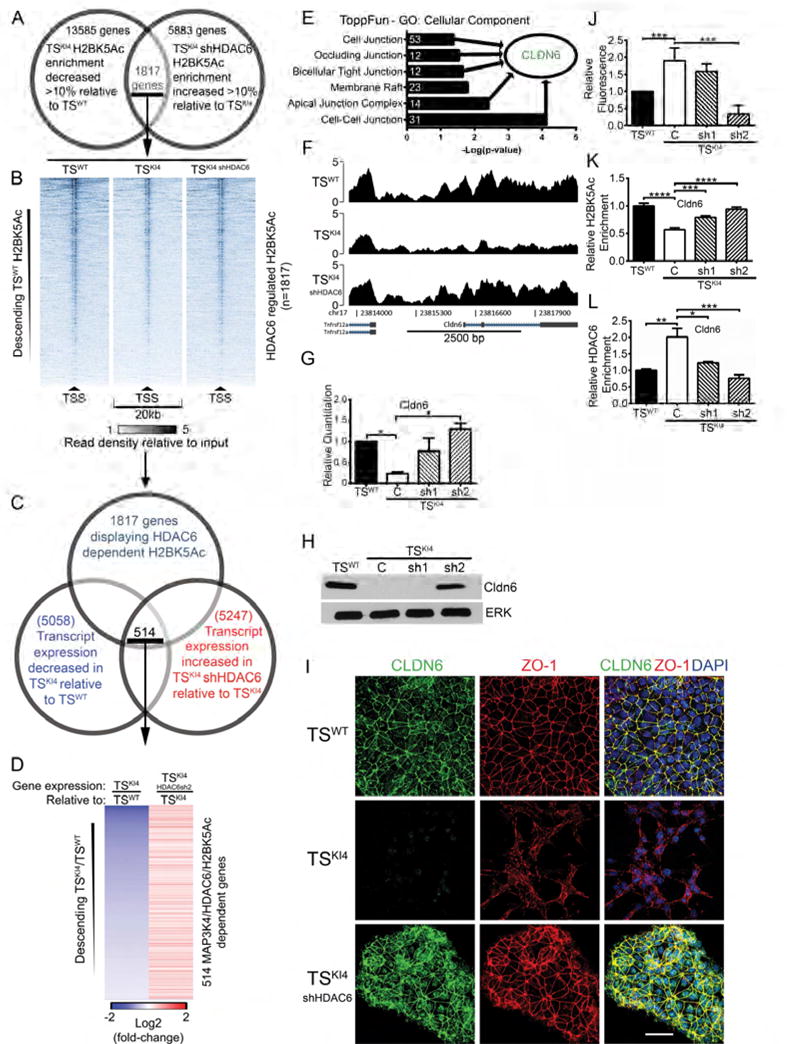
(A) Bioinformatic analysis of anti-H2BK5Ac ChIP-seq data from TSWT or TSKI4 cells expressing a control shRNA or TSKI4 cells expressing HDAC6 sh2. 1817 genes showed MAP3K4/HDAC6 dependent changes in enrichment of H2BK5Ac at their promoters.
(B) ChIP-seq read density plots showing H2BK5Ac at the TSS ± 10 kb of 1817 MAP3K4/HDAC6 dependent genes. Data are expressed as the intensity of the normalized read count of the IP divided by the normalized read count of the TSWT cell input.
(C) ChIP-seq and RNA-seq data identify 514 candidate genes showing MAP3K4/HDAC6 dependent changes in both H2BK5Ac and gene expression.
(D) Relative gene expression heat-map of 514 MAP3K4/HDAC6/H2BK5Ac dependent genes from RNA-seq data. Data columns show TSKI4 cell gene expression relative to TSWT cells and TSKI4 HDAC6sh2 cell gene expression relative to TSKI4 cells.
(E) ToppFun analysis of 514 MAP3K4/HDAC6/H2BK5Ac dependent genes. White numbers indicate the number of genes in each category.
(F) H2BK5Ac ChIP-seq read density plots at the Cldn6 TSS ± 2.5 kb. Data are expressed as in (B).
(G) Restored expression of Cldn6 upon knockdown of HDAC6 in TSKI4 cells. qRT-PCR data show fold changes relative to TSWT cells and are expressed as the mean ± SEM of three independent experiments.
(H) Knockdown of HDAC6 in TSKI4 cells to below wild-type levels restores Cldn6 protein.
(I) Confocal immunofluorescence microscopy showing restoration of tight-junction proteins Cldn6 (green) and ZO-1 (red) expression and localization in TSKI4 HDAC6sh2 cells. White bar represents 50 μm. Nuclei were stained with DAPI (blue). (H,I) Images shown are representative of four independent experiments.
(J) Reduced tight-junction barrier formation in TSKI4 cells is restored by HDAC6 knockdown. Diffusion of fluorescent dye across a confluent monolayer of cells is shown as a fold-change relative to TSWT cells.
(K) Acetylation of H2BK5 on the Cldn6 promoter is restored upon knockdown of HDAC6 in TSKI4 cells as measured by H2BK5Ac ChIP-PCR. Data shown are the mean ± SEM of between three and seven independent experiments.
(L) Enrichment of HDAC6 on the Cldn6 promoter in TSKI4 cells as measured by HDAC6 ChIP-PCR. Data shown are the mean ± SEM of between three and five independent experiments.
Student’s t-test, *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001.
The findings thus far implicated HDAC6 in the transcriptional regulation of Cldn6 and other tight junction proteins. To determine if HDAC6 represses Cldn6 expression through direct binding and deacetylation of the Cldn6 promoter, we compared H2BK5Ac and HDAC6 ChIP coupled to qRT-PCR (ChIP-PCR). Consistent with H2BK5Ac ChIP-seq, H2BK5Ac was decreased on the Cldn6 promoter in TSKI4 cells by H2BK5Ac ChIP-PCR (Figure 6K). Knockdown of HDAC6 in TSKI4 cells restored H2BK5Ac on the Cldn6 promoter, suggesting that HDAC6 might be positioned on the Cldn6 promoter in TSKI4 cells (Figure 6K). As measured by anti-HDAC6 ChIP-PCR, HDAC6 was enriched on the Cldn6 promoter in TSKI4 cells compared to TSWT cells (Figure 6L). Similar results were observed for the tight junction protein Ocln whose transcript levels were restored in TSKI4 HDAC6sh2 cells compared to TSKI4 cells (Figure S5H). ChIP-PCR showed reduced Ocln promoter H2BK5Ac and increased HDAC6 enrichment in TSKI4 cells (Figures S5I and S5J). Together, these data demonstrate MAP3K4 dependent regulation of tight junction gene expression by direct HDAC6 binding and H2BK5Ac promoter deacetylation.
Identification of HDAC6 Regulated Genes with Previously Undefined Roles in Maintaining the Epithelial Phenotype
We previously identified 31 genes downregulated in both TSKI4 and claudin-low breast cancer (CL) cells, also having reduced H2BK5 promoter acetylation in TSKI4 cells (Abell et al., 2011). Of these 31 genes, eight were also present in the 514 MAP3K4/HDAC6/H2BK5Ac gene list (Table S1). We predicted that a subset of these genes is directly downregulated by HDAC6 deacetylation of H2BK5 in TSKI4 cells, conferring a mesenchymal phenotype. To identify genes most likely to be directly downregulated by HDAC6 during EMT, we sorted the eight CL/MAP3K4/HDAC6/H2BK5Ac genes based on the restoration of promoter H2BK5Ac after HDAC6 knockdown. Ahnak, Pik3r3 and Suox showed the greatest restoration of H2BK5Ac after HDAC6 knockdown (Figure S6A–C). Unlike Cldn6 and Ocln, these genes have not been previously associated with EMT. Using qRT-PCR, we validated the reduced expression of Ahnak, Pik3r3, and Suox in TSKI4 cells that was restored by knockdown of HDAC6 (Figures 7A–C). Treatment of TSKI4 cells with tubastatin, an HDAC6 selective inhibitor, partially restored the expression of Ahnak, Pik3r3, and Suox (Figures 7D–F). Interestingly, tubastatin treatment also reduced HDAC6 transcript levels, suggesting that HDAC6 can regulate its own expression (Figure S6D). Individual shRNA knockdown of Ahnak, Pik3r3, and Suox in TSWT cells produced morphological changes characterized by a loss of epithelial morphology and a gain of mesenchymal morphology (Figure 7G–K). In addition, knockdown selectively altered the transcript levels of epithelial and mesenchymal markers, suggesting a role of these genes in maintaining the epithelial phenotype (Figures 7L and 7M).
Figure 7. HDAC6 directly deacetylates the promoters of genes important for EMT.
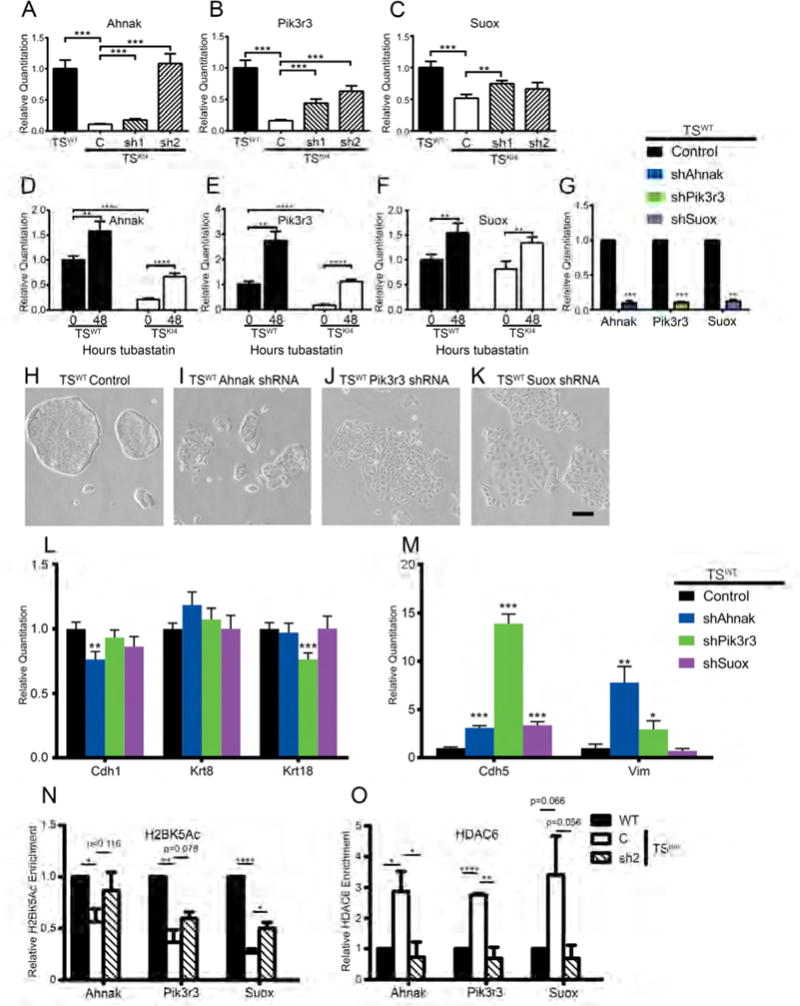
(A–C) Expression of genes predicted to maintain the epithelial phenotype is restored by knockdown of HDAC6 in TSKI4 cells. qRT-PCR data expressed as a fold change relative to TSWT cells are the mean ± SEM of three independent experiments.
(D–F) Expression of the indicated genes was measured by qRT-PCR. The indicated cell lines were treated for 48 hours with vehicle DMSO (0) or 10 nM Tubastatin A (48). Data show the mean ± SEM of between two (F) and three (D,E) independent experiments.
(G) Transcripts were measured by qRT-PCR in TSWT cells infected with a control vector or shRNAs for the indicated genes. Data show the mean ± SEM of three independent experiments.
(H–K) Shown are phase microscopy images of control infected TSWT cells (H) or TSWT cells expressing shRNAs for Ahnak (I), Pik3r3 (J), or Suox (K). Representative images from four independent experiments are shown. Black bar represents 200 μm.
(L–M) qRT-PCR data showing decreased epithelial markers (L) and increased endothelial and mesenchymal markers (M) for TSWT cells expressing the indicated shRNAs. Data shown are relative to control TSWT cells and are the mean ± SEM of three independent experiments.
(N–O) ChIP-PCR shows reduced H2BK5 acetylation (N) and enrichment of HDAC6 (O) on the promoters of specific genes in TSKI4 cells. Data shown are the mean ± SEM of three independent experiments.
Student’s t-test, *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001; ****p-value < 0.0001.
See also Figure S6.
HDAC6 Is Bound to the Promoters of Previously Unidentified Genes Important for Maintaining the Epithelial Phenotype
Consistent with H2BK5Ac ChIP-seq, H2BK5Ac ChIP-PCR demonstrated the loss of H2BK5Ac on the promoters of Ahnak, Pik3r3, and Suox in TSKI4 cells that increased in TSKI4 HDAC6sh2 cells (Figure 7N). HDAC6 ChIP-PCR revealed binding of HDAC6 to the promoters of Ahnak, Pik3r3, and Suox, demonstrating direct regulation by HDAC6 (Figure 7O). In TSKI4 cells, HDAC6 was enriched at these promoters, consistent with the reduction of H2BK5Ac on these genes. These findings suggest that HDAC6 directly represses the expression of Ahnak, Pik3r3, and Suox by deacetylation of H2BK5, contributing to EMT in TSKI4 cells.
DISCUSSION
Herein, we establish MAP3K4 as a key coordinator of chromatin modifiers during transitions between epithelial and mesenchymal states. Our previous work showed MAP3K4 activates CBP to promote H2A/H2BAc on epithelial genes (Abell et al., 2011). Here, our findings show MAP3K4 promotes the ubiquitination and degradation of HDAC6. Loss of MAP3K4 activity in TSKI4 cells results in increased cytoplasmic and nuclear expression and activity of HDAC6. In addition to showing that MAP3K4 regulates HDAC6 deacetylation of α-tubulin, we demonstrate that HDAC6 promotes differentiation and EMT in TS cells by deacetylating H2A/H2BAc on genes maintaining the epithelial phenotype. Specifically, HDAC6 is bound to promoters of several tight junction genes including Cldn6, repressing expression by directly deacetylating H2BK5. Importantly, we have used this innovative MAP3K4/HDAC6 regulated system to identify genes like Ahnak, Pik3r3, and Suox that regulate EMT. Pik3r3 encoding a PI3K regulatory subunit and Suox encoding a mitochondrial sulfite oxidase lack previously defined roles in EMT. Ahnak is a large scaffolding protein that colocalizes with ZO-1 in endothelial cell tight junctions, consistent with a role for Ahnak in EMT (Gentil et al., 2005). Together, these findings demonstrate that MAP3K4 maintains the epithelial state by preventing histone deacetylation by HDAC6, further implicating MAP3K4 as a key coordinator of cellular phenotype.
We have identified an interaction between HDAC6 and MAP3K4 mediated through the kinase domain of MAP3K4 in epithelial stem cells. There is only one other demonstration of MAP3K/HDAC complex formation. HDAC4 binds and deacetylates MAP3K2, promoting activation of MAP3K2 essential for the regulation of muscle atrophy (Choi et al., 2012). We predict that MAP3K/HDAC protein complex formation may represent conserved mechanisms controlling cellular function.
HDAC6 transcript and protein are increased in both TSKI4 cells and differentiating TS cells in EMT. TSKI4 cells are in an intermediate state of EMT, suggesting an explanation for elevated HDAC6 transcript and protein (Abell et al., 2011). Alternatively, MAP3K4 may repress HDAC6 transcripts through the regulation of MAPK-dependent transcription factors or AP1 controlled microRNAs (Bae et al., 2015). In addition, HDAC6 inhibition with tubastatin decreased HDAC6 transcript levels, suggesting that HDAC6 may regulate its own expression. Importantly, we show MAP3K4 promotes the ubiquitination and degradation of HDAC6 that is lost in TSKI4 cells, identifying a mechanism regulating HDAC6 protein expression.
The HDAC family of proteins has been implicated in EMT, primarily through the study of broad-spectrum HDAC inhibitors that inhibit and/or reverse fibrosis of the heart, lens, liver, lung, and kidney (Pang and Zhuang, 2010). For example, pan-HDAC inhibition suppresses TGFβ-induced EMT in the kidney, but the precise mechanisms are unknown. Two studies have implicated cytoplasmic HDAC6 in the regulation of EMT (Mak et al., 2012; Shan et al., 2008). Mak et al found that HDAC6 promotes the mesenchymal state in ovarian and colon cancer by preventing CD133 trafficking to and degradation by the endosome-lysosome. Importantly, HDAC6 knockdown in these cancer cells restored E-cadherin expression. In A549 lung adenocarcinoma cells, TGFβ-induced EMT increased HDAC6 deacetylation of α-tubulin, but not HDAC6 expression (Shan et al., 2008). In contrast, we show MAP3K4-dependent changes in the expression and activity of HDAC6. While previous studies defined cytoplasmic functions, we show a direct nuclear role of HDAC6 in TS cells, where HDAC6 is physically bound to and deacetylating promoters of tight junction genes, promoting EMT. Similar to CBP, HDAC6 selectively regulates H2A/H2BAc. Loss of H2A/H2BAc is associated with the initiation of EMT and the metastable state (Abell et al., 2011). In contrast, H3 and H4 acetylation is p300 dependent in epithelial TS cells, but the deacetylases for H3 and H4 in TS cells are undefined (Abell et al., 2011). Loss of acetylation of all four histones results in differentiation induced EMT (Abell et al., 2011). We predict that distinct acetyltransferases and deacetylases may control EMT initiation versus terminal differentiation EMT in TS cells.
KI4 mice lacking MAP3K4 activity and CBP deficient mice share several overlapping developmental phenotypes associated with EMT including neural tube, craniofacial and skeletal defects (Abell et al., 2005; Tanaka et al., 1997). In addition, KI4 mice display placental defects (Abell et al., 2009). In contrast, HDAC6 knockout mice develop normally (Zhang et al., 2008). Lack of placental phenotypes in CBP and HDAC6 knockouts may be due to compensation by other HATs and HDACs. Alternatively, the KI4 placental phenotype may reflect both loss of acetylation by CBP and gain of deacetylation by HDAC6, promoting EMT and trophoblast hyperinvasion.
We demonstrate an epigenetic role for HDAC6 in the nucleus of TS cells. Although HDAC6 has been found in the nucleus of MCF7 breast cancer cells and T cells, HDAC6 is located primarily in the cytoplasm of most cells (Riolo et al., 2012; Wang et al., 2009). Overexpression studies show that HDAC6 is retained in the cytoplasm by p300-mediated acetylation, which prevents interaction with importin-α (Liu et al., 2012). However, p300 activity is not altered in TSKI4 cells (Abell et al., 2011). As p300 and CBP have overlapping functions in some cell types, HDAC6 may be retained in the cytoplasm in TSWT cells by CBP dependent acetylation. In TSKI4 cells, HDAC6 may be hypoacetylated either due to lack of CBP activity, or an overall increase in cytoplasmic HDAC activity.
Our work defines a direct role of HDAC6 in regulating the dissolution of tight junctions, a key step in EMT. Claudins are key components of tight junctions in epithelial cells, promoting cell-cell adhesion and barrier function (Kwon, 2013). Claudins are lost in the highly invasive and metastatic claudin-low breast cancer subtype (Prat and Perou, 2011). Further, re-expression of Cldn6 in MCF7 breast cancer cells reduces invasiveness and restores drug sensitivity (Kwon, 2013). Cldn6 is expressed at higher levels in stem cells as compared to somatic cells, but transcriptional regulators of claudins are poorly defined (Furuse and Moriwaki, 2009; Kwon, 2013). In addition to Cldn6, MAP3K4 and HDAC6 control the expression of several other tight junction proteins including Cldn1, Cldn9, Ocln, Tjp1, Tjp2 and Tjp3, suggesting a key role for HDAC6 in disrupting tight junctions. We also demonstrate HDAC6 dependent changes in localization of the adherens junction protein E-cadherin, indicating additional non-transcriptional mechanisms controlled by HDAC6 that regulate EMT. These data suggest that MAP3K4 inhibition of HDAC6 is essential for maintaining cell-cell adhesion in TS cells.
In summary, we have defined a mechanism by which MAP3K4 directs epigenetic modifiers to control TS cell phenotype. Our data clearly demonstrate that MAP3K4 inhibits the activity of HDAC6, controlling H2A/H2BAc on genes important for the epithelial state. A key question remaining is whether CBP and HDAC6 coregulate the same genes. Further, how is the specificity and selectivity of CBP and HDAC6 binding to gene promoters mediated? Future studies will examine the coregulation of genes by MAP3K4, CBP and HDAC6, defining an epigenetic mechanism for controlling normal stem cell EMT during development. As MAP3K4, CBP and HDAC6 are therapeutically tractable targets, this work may provide insight into the study of pathological EMT states in which epithelial cells may adopt a metastable EMT phenotype.
EXPERIMENTAL PROCEDURES
Cell Lines, Culture Conditions, and Transfections
TSWT and TSKI4 cells were isolated as previously described from crosses of mice heterozygous for a targeted mutation of MAP3K4 (K1361R) (Abell et al., 2011). TS cells were cultured without feeders in 30% TS media (RPMI 1640, 20% fetal bovine serum, 1% penicillin and streptomycin, 1% L-glutamine, 1% sodium pyruvate, and 100 μM β-mercaptoethanol) and 70% MEF-CM. For maintenance of stemness, TS medium was supplemented with FGF4 (37.5 ng/ml) and Heparin (1 mg/ml). Invasion assays, differentiation, 293 culture, transfection, and plasmids are specified in Supplemental Experimental Procedures.
Lentivirus Production, Infections, and shRNA Knockdown of HDAC6
Replication incompetent lentivirus was produced as previously described (Abell et al., 2011). Briefly, 293 cells were transfected with control or shRNA constructs in pLKO.1 (Open Biosystems, TRCN0000008414 and TRCN0000008415), pMD2.G and psPAX2 (Addgene) using Lipofectamine plus (Invitrogen). Infections were performed as previously described (Abell et al., 2011).
Drug Treatment
Cells were treated for 24 hours with DMSO or 50 nM trichostatin A (TSA) (Sigma). Cells were treated every 24 hours for a total of 48 hours with DMSO or 10 nM Tubastatin A (Cayman Chemical). For the inhibition of protein synthesis, cells were treated with DMSO or 0.1 mg/ml cycloheximide (Fisher).
Cell Lysates, Immunoprecipitation, Western Blotting, and HDAC activity
Whole cell, cytoplasmic, and nuclear lysates, and acid histone extracts were isolated as previously described (Abell et al., 2009; Abell et al., 2011). Immunoprecipitation, Antibodies and Sources, and HDAC Activity are specified in Supplemental Experimental Procedures.
qRT-PCR
RNA isolation, cDNA preparation, qRT-PCR and primers are specified in Supplemental Experimental Procedures.
Immunofluorescence
Whole cell immunostaining was performed as previously described (Abell et al., 2009). For nuclear staining, cells were trypsinized for 4 minutes, pelleted at 6000 rpm, and suspended in homogenization buffer (10 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA). Cells were disrupted by passage five times through a 25-gauge needle and nuclei pelleted. Nuclei were fixed for 10 minutes in 3% paraformaldehyde, suspended in PBS, and dried onto coverslips. Immunostaining, Antibodies, and Confocal Imaging are specified in Supplemental Experimental Procedures.
RNA-seq and ChIP-seq
ChIP was modified from Abell et al 2011. Immunoprecipitation, antibodies, sequencing, analysis, visualization, and gene list enrichment are specified in Supplemental Experimental Procedures.
Barrier Function Measured by Transwell Solute Flux Assay
Cells were cultured at confluence for three days on Corning 3413 0.4 μm transwell permeable supports. 70 kDa rhodamine B isothiocynate-Dextran dye (1 mg/ml) was added to the top of each transwell and incubated for 2.5 hours. Fluorescence in the lower chamber was measured using a Synergy H1MD plate reader (BioTek).
Supplementary Material
Highlights.
MAP3K4 promotes HDAC6 ubiquitination and degradation to maintain the epithelial state
Trophoblast stem cell differentiation and EMT induce HDAC6 expression and activity
Nuclear HDAC6 promotes EMT by direct deacetylation of tight junction gene promoters
MAP3K4 inhibits HDAC6 to control tight junctions during EMT
Acknowledgments
A.N.A is supported by the Memphis Research Consortium and by National Institutes of Health grant GM116903. G.L.J is supported by National Institutes of Health grants GM101141, GM68820, and NCI Breast SPORE CA58223, the Komen Foundation, the Lustgarten Foundation, and the University Cancer Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
R.J.M., D.R., L.D.D., K.A., J.S.Z. and A.N.A. performed experiments and wrote the manuscript. J.S.Z., K.L., S.M.G., S.R. and R.H. analyzed sequencing data. R.J.M., G.L.J. and A.N.A. designed experiments and wrote the manuscript.
ACCESSION NUMBERS
The accession number for all sequencing data is GSE92426.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, one table and Supplemental Experimental Procedures.
References
- Abell AN, Granger DA, Johnson NL, Vincent-Jordan N, Dibble CF, Johnson GL. Trophoblast stem cell maintenance by fibroblast growth factor 4 requires MEKK4 activation of Jun N-terminal kinase. Molecular and cellular biology. 2009;29:2748–2761. doi: 10.1128/MCB.01391-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abell AN, Jordan NV, Huang W, Prat A, Midland AA, Johnson NL, Granger DA, Mieczkowski PA, Perou CM, Gomez SM, et al. MAP3K4/CBP-regulated H2B acetylation controls epithelial-mesenchymal transition in trophoblast stem cells. Cell stem cell. 2011;8:525–537. doi: 10.1016/j.stem.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abell AN, Rivera-Perez JA, Cuevas BD, Uhlik MT, Sather S, Johnson NL, Minton SK, Lauder JM, Winter-Vann AM, Nakamura K, et al. Ablation of MEKK4 kinase activity causes neurulation and skeletal patterning defects in the mouse embryo. Molecular and cellular biology. 2005;25:8948–8959. doi: 10.1128/MCB.25.20.8948-8959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae HJ, Jung KH, Eun JW, Shen Q, Kim HS, Park SJ, Shin WC, Yang HD, Park WS, Lee JY, et al. MicroRNA-221 governs tumor suppressor HDAC6 to potentiate malignant progression of liver cancer. J Hepatol. 2015;63:408–419. doi: 10.1016/j.jhep.2015.03.019. [DOI] [PubMed] [Google Scholar]
- Choi MC, Cohen TJ, Barrientos T, Wang B, Li M, Simmons BJ, Yang JS, Cox GA, Zhao Y, Yao TP. A direct HDAC4-MAP kinase crosstalk activates muscle atrophy program. Molecular cell. 2012;47:122–132. doi: 10.1016/j.molcel.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Gendron TF, Scheffel K, Carlomagno Y, Dunmore J, DeTure M, Petrucelli L. Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Human molecular genetics. 2012;21:2936–2945. doi: 10.1093/hmg/dds125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Moriwaki K. The role of claudin-based tight junctions in morphogenesis. Annals of the New York Academy of Sciences. 2009;1165:58–61. doi: 10.1111/j.1749-6632.2009.04441.x. [DOI] [PubMed] [Google Scholar]
- Gentil BJ, Benaud C, Delphin C, Remy C, Berezowski V, Cecchelli R, Feraud O, Vittet D, Baudier J. Specific AHNAK expression in brain endothelial cells with barrier properties. J Cell Physiol. 2005;203:362–371. doi: 10.1002/jcp.20232. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Kwon MJ. Emerging roles of claudins in human cancer. International journal of molecular sciences. 2013;14:18148–18180. doi: 10.3390/ijms140918148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. The Journal of biological chemistry. 2012;287:29168–29174. doi: 10.1074/jbc.M112.371120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, Gingras AC, Mazitschek R, Neel BG, Stagljar I, et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell reports. 2012;2:951–963. doi: 10.1016/j.celrep.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Molecular cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Pang M, Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. The Journal of pharmacology and experimental therapeutics. 2010;335:266–272. doi: 10.1124/jpet.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Molecular oncology. 2011;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riolo MT, Cooper ZA, Holloway MP, Cheng Y, Bianchi C, Yakirevich E, Ma L, Chin YE, Altura RA. Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. The Journal of biological chemistry. 2012;287:10885–10893. doi: 10.1074/jbc.M111.308791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Yao TP, Nguyen HT, Zhuo Y, Levy DR, Klingsberg RC, Tao H, Palmer ML, Holder KN, Lasky JA. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. The Journal of biological chemistry. 2008;283:21065–21073. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282:2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein-Taybi syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10215–10220. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends in cell biology. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Molecular cell. 2014;55:31–46. doi: 10.1016/j.molcel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Molecular and cellular biology. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.