

Abstract
Unwanted antibody responses significantly impact human health, and current options for treating deleterious antibody responses largely rely on broad immunosuppressants that can compromise overall immunity. A desirable alternative is to induce antigen-specific immune tolerance. We have shown that co-presentation of antigen and ligands of B cell Siglecs (sialic acid-binding immunoglobulin-like lectin) on a liposomal nanoparticle induces antigen-specific tolerance. Although Siglec-engaging tolerance-inducing antigenic liposomes (STALs) induce robust B cell tolerance in naïve mice, the full potential of STALs requires long-term tolerance induction and suppression of an ongoing immune response. We hypothesized that STALs encapsulated with rapamycin (RAPA), an immunomodulator, could improve the efficacy of STALs and potentially enable their use in the context of immunological memory. Here, we show that formulation of STALs with RAPA produces enhanced tolerance induction in naïve mice compared to STALs without RAPA, but has minimal impact on inducing tolerance in previously sensitized mice. These findings indicate that the addition of immunomodulator to STALs could be beneficial in tolerance induction and support future development of STALs for the treatment of allergy and autoimmune diseases.
Keywords: CD22, liposome, sialic acid, rapamycin, immune tolerance
Graphical abstract
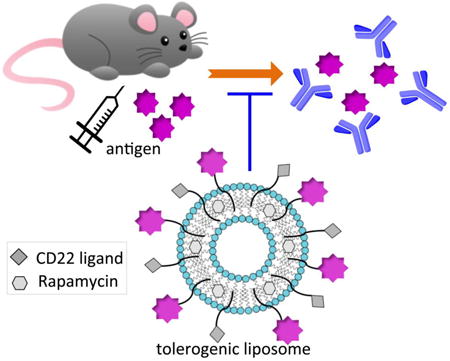
Enhanced tolerance: The formulation of Siglec-engaging tolerance inducing antigenic liposomes (STALs) encapsulated with immunosuppressant rapamycin (RAPA) was optimized and then investigated in vivo. As a result, STALs+RAPA has shown enhanced tolerance induction in naïve mice, which will support future development of STALs for the treatment of allergy and autoimmune diseases.
Introduction
Unwanted immune responses occur in numerous medical conditions, including autoimmune disease[1, 2], organ transplant rejection[3, 4], allergies[5, 6], and decreased efficacy of biotherapeutics[7]. Current therapeutic strategies largely rely on immunosuppressive drugs with chronic administration[8], which can compromise immunity.[9, 10] Preventing immune responses in a manner that is specific to a particular antigen, described as antigen-specific tolerance, is highly desirable from an efficacy and safety standpoint.[11]
Multiple approaches for inducing antigen-specific tolerance have been reported[11, 12], such as sustained antigen administration over a time course of months to years[13], expression or attachment of antigen to syngeneic cells[14, 15], loading particles with MHC complexes[16, 17], co-delivery of peptide or protein antigen with immunosuppressive drugs[18, 19], or co-administration of pharmacological agents in an enzyme-replacement therapy.[20] In the above approaches, tolerance induction is thought to stem from either by inhibition of the antigen-specific T cells, which can take place through T cell anergy or induction of regulatory T cells. As B cells are the precursors of the antibody-secreting plasma cells, directly targeting the antigen-reactive B cells offers an alternative and potentially more straightforward way for systematically inducing humoral immune tolerance to the desired antigens.[21]
B cells express a set of B cell receptor (BCR) inhibitory co-receptors[22], among them are CD22 and Siglec-G (Siglec-10 in humans), members of the Siglec (sialic acid-binding immunoglobulin-like lectin) family that recognize sialic acid-containing glycans of glycoproteins and glycolipids.[23, 24] Recent studies have demonstrated that co-presentation of antigens with ligands of CD22 or Siglec-G results in strong inhibition of B cell activation, inhibition of tonic BCR signaling through the PI3K/Akt survival pathway, apoptosis of antigen-reactive B cells and induction of B cell tolerance due to depletion of the antigen-specific B cells from the B cell repertoire.[25] Based on the above findings, Siglec-engaging tolerance-inducing antigenic liposomes (STALs) have been developed to force the co-localization of CD22 or Siglec-G with the BCR, thereby inducing antigen-specific B-cell tolerance.[26, 27] STALs were decorated with a high affinity and selective CD22 ligand (α2-6-linked sialic acid glycan 1, Figure 1A) and protein antigen (Figure 1B). Injecting mice with STALs bearing different antigens prevents B cell response to a subsequent challenge with the corresponding antigen, compared to mice injected with liposomes displaying antigen alone (immunogenic liposomes). Further studies in a mouse model of hemophilia A showed that STALs displaying FVIII and CD22 ligands induce tolerance in FVIII-deficient mice, which prevents the formation of high titers of FVIII-specific antibodies that can prevent efficacy of infused FVIII to prevent blood clotting. Likewise, STALs formulated with the major peanut allergen (Ara h 2) were shown to prophylactically protect mice from sensitization and subsequent allergic response to peanuts.[28] These results indicate that STALs have the ability to eliminate or prevent harmful B cell-mediated antibody responses in mice that are immunologically naïve to the antigen of interest. Nevertheless, many of the human conditions where unwanted antibody responses play a deleterious role involve a sensitized immune system that contains antigen-specific memory B and T cells.[11, 12] Since STALs were demonstrated to have a tolerogenic effect on both human naïve and memory B cells in vitro, yet excessive T cell help was revealed to diminish the tolerogenic properties of STALs in vivo, these two results suggest that the full realization of STALs for treating an immune system sensitized to the antigen of interest may require addressing both B and T cells.[26] Accordingly, we considered that STALs formulated with an immunosuppressive agent may enhance B cell tolerance, and therefore have the potential to allow for immune tolerance induction in an immune system previously sensitized to the antigen.
Figure 1.
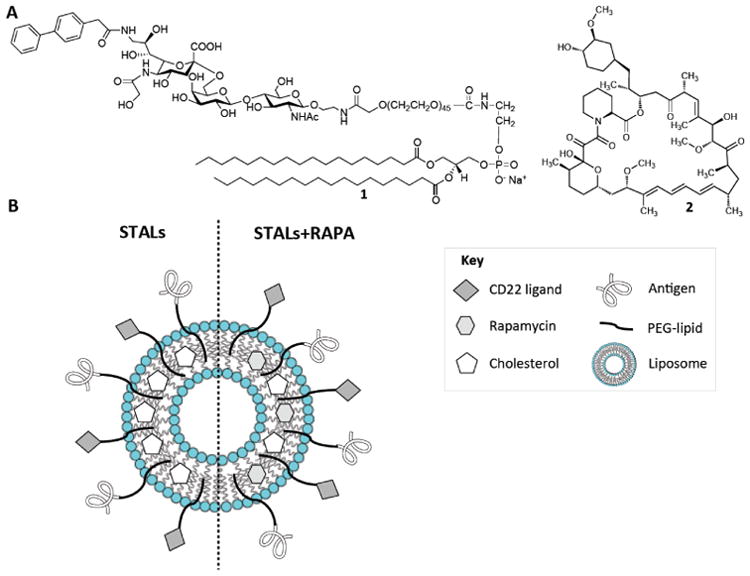
Antigen-specific liposome formulations with CD22 ligand and rapamycin. (A) Chemical structures of the murine CD22 ligand linked to PEGylated lipid (1) and rapamycin (2). (B) Schematic illustration of antigen-specific liposome formulations equipped with CD22 ligand (STALs) and the new platform (STALs+RAPA).
Rapamycin (RAPA, 2 in Figure 1A) is a potent immunosuppressive agent that is used clinically to prevent acute renal allograft rejection.[29] RAPA inhibits the mammalian target of rapamycin (mTOR) pathway, thus inducing cell-cycle arrest to a broad range of lymphocytes including B cells and T cells.[30-33] Recently, PLGA-nanoparticles encapsulated with both a protein or peptide antigen along with RAPA, were shown to induce antigen-specific immunological tolerance, an effect attributed to induction of T cell tolerance.[18, 19, 34] The proposed mechanism of action for these particles involves uptake of the particles by dendritic cells (DCs) and presentation to antigen-specific T cells in a tolerogenic manner which blunts T cells priming and induces regulatory T cells. Since STALs are actively taken up by B cells through recognition of CD22 ligands and the antigen, and are also expected to be non-specifically phagocytosed by macrophages and dendritic cells, we reasoned that addition of rapamycin to STALs might improve their ability to induce antigen specific tolerance.[30, 32, 33] Here, we report a new formulation of STALs containing rapamycin (Figure 1B). STALs with rapamycin exhibit enhanced tolerance induction in naïve mice compared to STALs without rapamycin. The same formulation exhibited minimal tolerance in sensitized mice, but not significantly better than STALs without rapamycin. These results demonstrate that co-delivery of an immuno-modulatory agent in STALs enhances their toleragenic capacity in naïve animals, suggesting they should be further explored for the treatment of conditions involving harmful antibodies.
Results and Discussion
Formulation of STALs encapsulated with rapamycin
Rapamycin is a macrolide with low oral bioavailability (< 15%) that is largely due to its poor water solubility (2.6 μg/mL) and high lipophilicity (logP 5.77).[35] Encapsulation of rapamycin into nanoparticles, such as liposomes[36-39], micelles[40], and poly(lactic-co-glycolic) acid (PLGA)-nanoparticles[18, 19], can improve its pharmacokinetic properties and, accordingly, its bioavailability. Previously described methods for encapsulating rapamycin have required elevated hydration temperature[36, 39], long sonication time[37, 38], or organic solvents[39], which are not ideal conditions for formulating STALs due to potential of denaturing the protein antigen. In order to develop a compatible encapsulation method with protein antigen and rapamycin, the liposome composition, such as type of lipid, lipid/cholesterol ratio and drug/lipid ratio, were varied to determine the conditions that encapsulate the greatest amount of RAPA while maintaining liposomal stability. Lipids in the composition are expressed as a molar percentage (mol %) of the total lipids in the formulation. Liposomes were routinely formulated at room temperature, with minimum sonication time, extruded through 100-nm pores to unify their size, purified on a CL-4B column to remove non-encapsulated RAPA, and tested for RAPA content by analytical HPLC.
Diverse phospholipids with different transition temperatures were tested, including 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), but none of these provide a major advantage on rapamycin encapsulation as determined by HPLC (Figure S1-3, supporting information), therefore, all liposomal preparations used DSPC since it is the standard phospholipid used in ‘stealth’ liposomes. On the other hand, varying the phospholipid:cholesterol and drug:lipid ratio did substantially influence the amount of encapsulated RAPA. In particular, we found that decreasing the cholesterol:phospholipid ratio was crucial for an optimal encapsulation of RAPA, which is consistent with the RAPA embedding into the lipid bilayer of the liposome due to lipid lipophilic nature. Specifically, using a fixed rapamycin to total lipid ratio of 3 mol% we found that drug encapsulation efficiency (EE%) could be improved from 7% to 60% by decreasing the amount of cholesterol in the formulation form 19 mol% to 9 mol% (Figure 2A). Nevertheless, further reducing cholesterol content was deleterious, since 5% cholesterol substantially decreased encapsulation of RAPA, which may have been due to instability of the lipid bilayer at low cholesterol concentration. Thus, we further optimized STALs using 9% cholesterol, STALs were prepared that contained 1 mol% of the high affinity CD22 ligand, 0.1 mol% ovalbumin (OVA) as the protein antigen. The CD22 ligand (1) and ovalbumin (OVA) were conjugated to DSPE through a 2K PEG linker according to previously reported procedures.[26] The total molar fraction of pegylated (PEG) lipids was always kept at 5 mol% for the formulation. STALs encapsulated with RAPA showed similar encapsulation efficiencies and were, therefore, characterized by transmission electron microscopy (TEM) in order to measure the particle size and to examine the membrane integrity over time. We found that the morphology of STALs+RAPA particles were stable, with only a slight increase in the size after 5 days (Figure 2B&C, S4). In order to demonstrate that the encapsulation of RAPA does not affect the ability of STALs to inhibit B cell activation, liposomes were formulated with duck egg lysozyme (DEL), instead of OVA, to exam their ability to stimulate hen egg lysozyme (HEL)-reactive B cells, from Hy10 mice[41], using a flow cytometry-based calcium flux assay (Figure 2D).[26] Indeed, co-presentation of CD22 ligand (mCD22L) and protein antigen (DEL) on STALs prevented activation of the antigen-specific B cells compared to liposomes that display antigen alone. Importantly, RAPA encapsulation showed no abrogation of this inhibitory effect. Therefore, STALs formulated with RAPA retain their ability to engage CD22 on the surface of antigen-specific B cells in order to prevent B cell activation, which is a key first step in inducing B cell tolerance.
Figure 2.
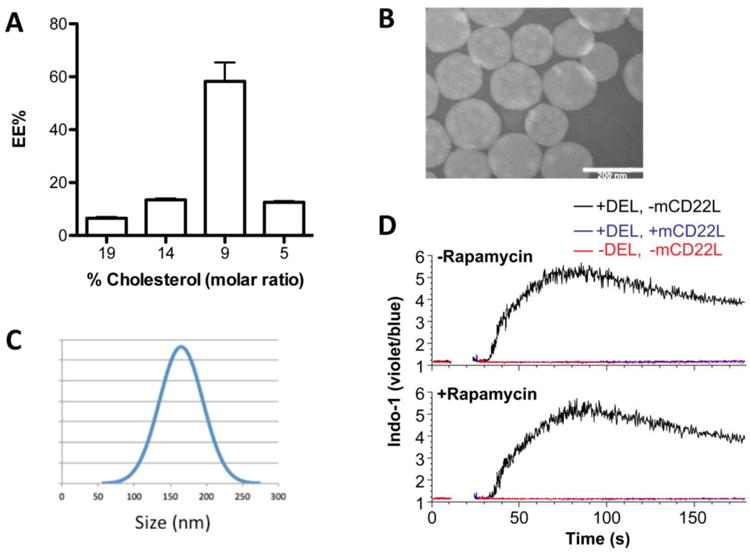
Characteristics of rapamycin encapsulated STALs. (A) RAPA encapsulation efficiency (EE%) related to the cholesterol/total lipid ratio. (B) Representative transmission electron microscopic image on day 5 after formulation and (C) the size distribution on day 5 (160±30nm). (D) Calcium flux in IgMHEL B cells stimulated with the indicated liposomes, with or without rapamycin encapsulation. DEL, duck egg lysozyme.
Antigen density plays a key role in tolerance induction
Previously, we showed that the antigen density on STALs greatly impacts the degree of tolerance induction.[26] Specifically, we had shown that 0.033% (molar ratio to total lipid) OVA was optimal. Therefore, we chose this density as a starting point and varied the antigen density 3-fold up and down to determine the impact of RAPA on STALs and immunogenic liposomes. To examine tolerance induction, mice were administered liposomes on day 0 and challenged with immunogenic OVA liposomes (without CD22 ligand and RAPA) two weeks later. Antibody production was followed on days 0, 7, 14, 21, and 28 by ELISA, which facilitated a direct comparison between formulations bearing different OVA density (Figure 3A&B). To ensure all the mice received the same initial amount of OVA (0.03 nmol OVA per mouse) in the immunization, the injection concentration of liposomes was calculated accordingly. Among three groups with different OVA density, 0.01% OVA STALs showed the least inhibition of antibody production before the challenge on day 14, while strong suppression of anti-OVA antibodies with 0.1% OVA STALs was observed (Figure 3A). The same trend was also observed with STALs+RAPA, but it is notable that among them 0.1% OVA STALs+RAPA showed the highest inhibition potency of any of the groups (Figure 3B). After the challenge with immunogenic liposomes, most groups of STALs showed a blunted antibody response compared to the PBS group, demonstrating that immunological tolerance had indeed been induced. The strongest tolerance was observed with the STALs containing 0.1% OVA, and tolerance induction was significantly stronger in OVA+RAPA group, as seen mostly clearly by comparing the 0.1% OVA groups on day 28 (Figure 3C). These data indicate that addition of RAPA to STALs improves immunological tolerance induction in naïve mice.
Figure 3.
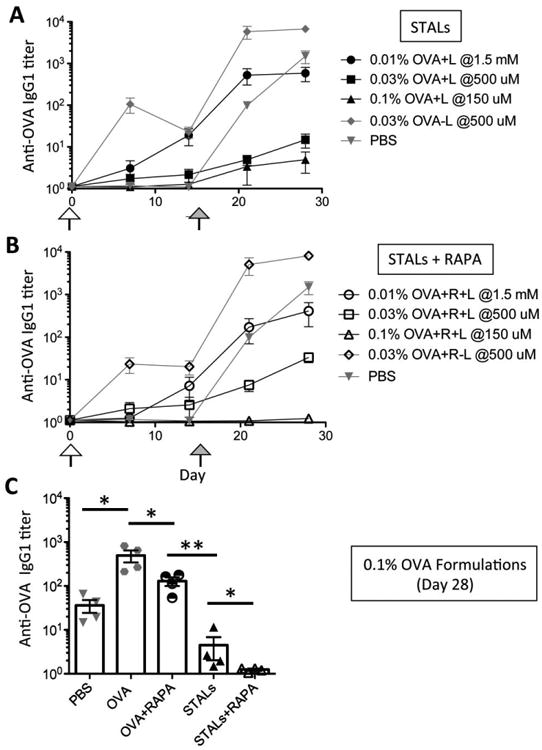
Tolerance induction in mice by STALs is improved with RAPA. (A-B) Mice were immunized on day 0 (blank arrow), and then challenged with OVA liposomes on day 14 (solid arrow). For the initial immunization, STALs were injected at different concentrations as indicated to be sure each mouse received the same amount of OVA antigen. (A) Anti-OVA titers (IgG1) of the test groups immunized with STALs have shown that antigen density on the STALs surface could influence the immunogenicity of the STALs. (B) 0.1% OVA STALs displaying both ligand and rapamycin showed the best tolerance among the groups. (C) Anti-OVA titers (IgG1) on day 28 were plotted for all groups containing 0.1% OVA. ** P ≤ 0.01, * P ≤ 0.05.
One versus two STALs+RAPA injections for tolerance induction
In all studies described to date with tolerance induction by STALs, only a single dose of STALs have been administered. However, since liposomes have a finite circulation time and antigen-specific B cells can be repopulated in the mouse following B cell tolerance induction by STALs[26], we considered that multiple injections could be beneficial in producing more robust tolerance. Accordingly, we investigated the effect of one versus two injections of STALs+RAPA. Both one and two injections showed significant reduction of antibody production two weeks after the challenge (day 42) compared to the groups given PBS on day 0 (Figure 4). In four out of five mice that received two injections of STALs+RAPA, anti-OVA antibody levels were at baseline with the other mouse only 3-fold higher than background. Alternatively, only 2 out of 5 mice that received only a single injection of STALs had baseline titers, while 2 out of 5 mice had significantly higher titers. Although the average titers between the two injection strategies were not significantly different, the low average antibody production of the two-injection strategy suggests that multiple injections of STALs+RAPA is well tolerated and may be therapeutically beneficial for generating robust and long-lasting immune tolerance (Figure 4B).
Figure 4.
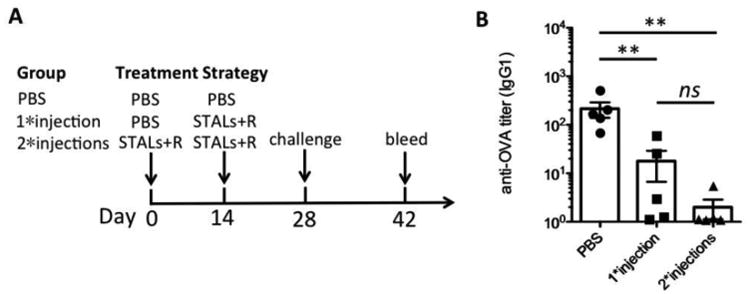
Mice (n=5) were immunized 0, 1, or 2 times before challenging with OVA antigen. Mice were challenged with OVA liposomes on day 28 and bled on day 42. (A) Treatment strategies for 0, 1, or 2 injections. (B) Anti-OVA IgG1 titers plotted on day 42. Results are representative of two experiments. ** P ≤ 0.01.
Evaluation of tolerance induction in sensitized mice
Memory B cells, generated in response to T-dependent antigen, are important in generating long-lasting protective immunity when exposed to the same antigens.[42] Clinically, memory B cells are critical for maintaining sensitivity of individuals to allergens[43], transplant antigens[4], autoimmune antigens[44], and biological drugs[45]. For this reason, a method to eliminate the antigen-specific memory B cells could be extraordinarily beneficial as a treatment strategy for such conditions. We had previously demonstrated that STALS were tolerogenic on human peripheral blood and tonsillar memory B cells in vitro, as measured by strong inhibition of B cell activation and induction of apoptosis.[26] However, to date the effect of STALs on memory B cells in vivo, where memory T cells will also be present, has not been reported.
To investigate the use of STALs in tolerizing memory B cells in a mouse sensitized to antigen, we developed sensitization strategy that uses soluble OVA as the antigen, rather than OVA emulsified in adjuvant, to prevent the formation of antigen depots that can sustain antibody levels for a long time. Using standard grade OVA, which is known to contain trace amounts of endotoxins that increases its immunogenicity[46], 200 μg OVA was delivered intraperitoneally to C57BJ/6J mice for three straight weeks, which induced substantial anti-OVA titers to titers of approximately 1×104 (Figure S5, supporting information). Antibody levels in a large cohort of mice immunized in this way were monitored over time post-immunization with the goal of waiting a period of time that allowed for antibodies to decrease approximately 100-fold, since we felt that high antibody levels may mask the antigen on STALs and not allow them to interact with the B cells of interest. After 8 months, the antibody levels had decreased 100-fold to an average titer of 1×102. Within the large cohort of mice, some variation existed in the titers; therefore, the mice were evenly distributed in the treatment groups such that the average titer was equal among the groups. Five groups of mice (n=6-9/group) were used to initiate treatment with PBS or OVA liposomes with and without the mCD22 Ligand and with and without RAPA. All the mice were challenged 26 days after initiation of treatment with 100 ug of soluble OVA. Mice were bled on days 0, 21, and 47 to evaluate the anti-OVA titers (Figure 5A).
Figure 5.
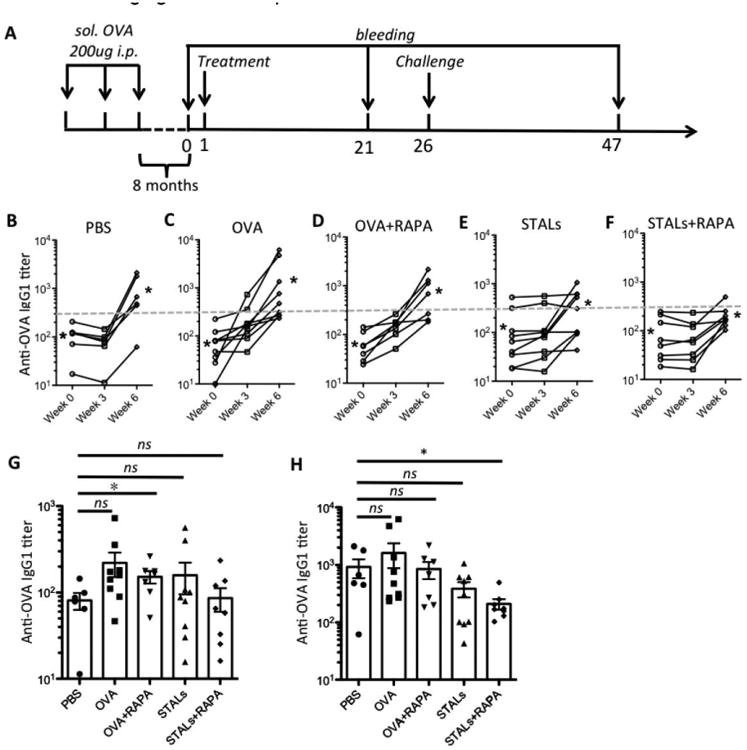
Tolerance induction in sensitized mice (n=6-9). (A) Mice were immunized three times, one week apart, then let settled for 8 months before treatment with OVA-liposomes (OVA) ± RAPA or STALs ± RAPA on day 1. The mice were challenged with 100 μg of soluble OVA (i.p.) on day 26. Mice were bled on days 0, 21, and 47, and anti-OVA IgG1 titers were plotted. (B-F) Levels of antibody production to OVA in mice (n=6-9) with different treatment strategies. Changes in the antibody production as measured by ELISA through weeks. Each line represents an individual mouse; asterisks represent the geometric means for each group on week 0 (left) and week 6 (right). The grey dashed line represents the antibody titer level at 300 of the ELISA assay. (G-H) Anti-OVA IgG1 titer of each test group was plotted on day 21 (G) and day 47 (H). * P ≤ 0.05.
Antibody levels of individual mouse in each group reveal several interesting features (Figure 5B-F). First, in the PBS group (Figure 5B), anti-OVA levels dropped in the first three weeks to approximately 2-fold, which is well inline with the half-life reported for IgG in mice.[47] For mice that received OVA liposomes without the mCD22 Ligand (Figure 5C&D), a robust increase in anti-OVA titers was observed. Alternatively, mice that received STALs had a minimal change in anti-OVA titers (Figure 5E&F). Plotting the titers of all 5 groups on day 21 indicates that the antibody production showed little difference between the treated groups and the PBS group (Figure 5G). Following the challenge on day 26, increases in anti-OVA antibodies were observed on day 47 in all test groups (week 3-6, Figure 5B-F). The STALs+RAPA group, showing the lowest average level of antibody production and with less variation than the other groups, gave statistically lower antibody production than the PBS control (Figure 5H). However, it should be noted that titers in the STALs+RAPA group did increase between day 21 and 47 and there was no statistical difference between STALs and STALs+RAPA (Figure 5F). Nonetheless, while the current formulation tolerance induction in sensitized mice is minimal, the results are encouraging for further optimization.
Conclusions
We find that formulation of STALs[26] with the immunosuppressive drug rapamycin enhances induction of antigen specific tolerance. Encapsulation of RAPA into STALs was maximized through fine-tuning the cholesterol/lipid ratio and this new formulation is compatible with both protein antigens and Siglec ligands presented on the surface of the liposome. Specifically, the new STALs+RAPA formulation induced more robust tolerance than regular STALs when administered to naïve mice. Although antigen specific tolerance was not formally tested here, we assume that tolerance induced was antigen specific since STALs alone induce antigen specific tolerance,[26] and as shown by others rapamycin loaded nanoparticles administered with antigen also induces antigen specific tolerance[18, 19].
Here we have also evaluated the ability of STALs to tolerize mice previously sensitized with an antigen. While neither STALs nor STALs+RAPA completely prevented a memory response induced by soluble antigen, there was a statistically significant reduction of the STALs+RAPA group over the untreated mice (PBS). In this regard, it is notable that mice injected with STALs+RAPA received approximately 0.5-1 μg of rapamycin. In contrast, nearly 100-fold higher doses of rapamycin were administered to induce antigen specific tolerance in mice using the PLGA-rapamycin nanoparticle platform that targets the T cell compartment.[18, 48] Although we have incorporated the maximum amount of rapamycin possible in the current STAL platform, it may be fruitful to consider alternative formulations that combine STALs with immuno-modulatory agents, that more effectively induce tolerance in human conditions with ongoing immune responses.
Experimental Section
Synthesis
The murine CD22 ligand (9-biphenylacetyl-N-glycolylneuraminic acid-α-2-6-galactose-β-1-4-N-acetylglucosamine-β-ethylamine (6′-BPANeuGc)) was reacted with NHS-PEG2000-DSPE (NOF America) to give sugar-lipid conjugate (1) as described previously.[49]
Protein-lipid conjugation
Ovalbumin (OVA) was purchased from Sigma-Aldrich, and DEL was purified from duck egg yolk. Proteins (OVA or DEL) were conjugated to pegylated distearoylphosethanolamine (PEG2000-DSPE) via maleimide chemistry.[50] For example, ovalbumin (4.8 mg, 0.12 μmol) was dissolved in PBS (480 μL) and then treated with N-succinimidyl 3-(2-pyridyldithio)-propionate (Pierce) (40 μL, 2 mg/mL in DMSO). The reaction mixture was gently shaken at RT for 1h and then desalted on Sephadex G-50 column. The obtained protein solution was then treated with 25 mM 1,4-Dithiothreitol at RT for 10 min. The amount of released thiol 2-pyridyl was quantified by absorbance at 343 nm to monitor the progress of protein modification. When the reaction was complete, desalted the thiol-derivatized protein with Sephadex G-50 column. Following desalting, the thiol-derivatized protein (1.5 mL, 35 μM) was immediately treated with maleimide-PEG2000-DSPE (NOF America) (26 μL, 20 mM in DMSO) under nitrogen and incubated at RT overnight. Lipid-modified OVA was purified from unmodified protein on Sephadex G-100 column and stored at 4 °C before use. Duck egg lysozyme was purified according to a published procedure[51] and was conjugated to PEG2000-DSPE according to similar procedure as ovalbumin. The final concentration of protein-lipid conjugates was calculated based on the absorbance at 280 nm.
Liposome formulation
All liposomes used for in vitro and in vivo studies were prepared following the thin film hydration method as reported previously. The appropriate amounts of lipid mixtures in the presence or absence of RAPA were firstly dissolved in chloroform and then dried by a stream of nitrogen gas. For the optimized formulations, distearoyl phospatidylcholine (DSPC) (Avanti Polar Lipids), cholesterol (Sigma-Aldrich) and pegylated lipid were in a molar ratio of 88:7:5. The total molar fraction of pegylated lipids was always kept at 5%; made up of the appropriate combination of PEG2000-DSPE (Avanti Polar Lipids), BPANeuGc-PEG2000-DSPE or protein-PEG2000-DSPE. To the dried lipid thin film, a DMSO stock of BPANeuGc-PEG2000-DSPE was added, and this mixture was lyophilized overnight. The dried lipids mixture were hydrated in PBS to give 5 mM lipid concentration and sonicated vigorously for a minimum of 5×30 seconds with 15 minutes intervals. Protein-PEG2000-DSPE was added at the time of hydration. The molar fraction of the protein on the liposomes varied during studies from 0.01 to 0.1%. To unify the size, liposomes were passed a minimum of 20 times through 800-nm, 200-nm and 100-nm polycarbonate membranes (Avanti Polar Lipids) using a hand-held mini-extrusion device (Avanti Polar Lipids). Extrusion was carried at 37-40 °C to give the best performance. The liposomes were then purified by CL-4B column and detected by Nanodrop2000 (Thermo Scientific) at 280 nm. The prepared liposomes were stored at 2.5 mM concentration in PBS buffer at 4 °C. The size of liposomes was routinely measured by dynamic light scattering (DLS) within one week targeting 100-200 nm (Figure S4). The image of STALs+RAPA was taken by transmission electron microscopic (Philips CM100 TEM) on day 5, showing in the range of 160±30 nm (Figure 2B).
HPLC analysis and encapsulation efficiency (EE)
An Agilent Technologies 1100 series with a quaternary pump, an auto-sampler and a UV detector at 278 nm was used for HPLC experiments. Column: Alltech C18, 5 μm, 4.6 × 150 mm. Mobile phases: A: H2O + 0.1% TFA; B: 95% MeCN/H2O + 0.1% TFA. Gradient: 0% B → 5% B (2 min); 5% B → 100% B (10 min); 100% B (6 min); 100% B → 5% B (5 min); 5% B → 0% B (2 min); flow rate: 1 mL/min. Calibration curves were constructed using standard solutions of know RAPA concentration at 2-60 μg/mL. RAPA shows a retention time at 13 min. To prepare the HPLC samples, 50 μL of liposome solutions (2.5 mM in PBS) was mixed with 150 μL of MeCN and then sonicated for 30 min. The mixture was filtered by 0.45 μm PVDF syringe filter for HPLC analysis. Chemstation software (Agilent) was used to calculate the peak area of each sample automatically. The amount of RAPA encapsulation efficiency (EE) was calculated by using the following equation:
Calcium flux assay
Splenocyte from Hy10 mice were re-suspended at 15 × 106 cells/mL in RPMI medium containing 1% FCS, 10 mM HEPES, 1 mM MgCl2, 1 mM EGTA, and 1 μM Indo-1 (Invitrogen). Cells were incubated in a 37°C water incubator for 30 minutes, followed by addition of a 5-fold volume of the same buffer (without Indo-1). The cells were centrifuged (270 g, 7 minutes), stained with antibodies to B220 (PE-Cy7) and CD5 (PE) for monitoring calcium flux on B cells, and then washed and stored on ice. An aliquot of 0.5 mL containing 1 × 106 cells was warmed (37°C, 5 minutes) prior to initiating calcium flux measurements. The cells stimulated with DEL-liposomes (ranging from 5–50 μM; total lipid) and Indo-1 fluorescence (violet versus blue) were monitored by flow cytometry (500–1000 events/s) for 3 minutes at 37 °C. Stimulation always took place 10 seconds after starting acquisition so that background could be established. Data were analyzed using FlowJo with the kinetics functions.
Immunization and blood collection
The Institutional Animal Care and Use Committee of The Scripps Research Institute (La Jolla, CA) approved all experimental procedures involving mice in this project. WT C57BL/6J mice were obtained from The Scripps Research Institute (TSRI) rodent breeding colony. Liposomes were delivered via the lateral tail vein in a volume of 200 μL. For sensitized study involving soluble OVA antigen, mice were injected with 100-200 μg of OVA dissolved in PBS and delivered intraperitoneally. Whole blood (40 μL) was collected from mice via a retro-orbital bleed to obtain the serum after centrifugation (17,000 g, 1 min). Serum was aliquoted and stored at -20 °C.
ELISAs
ELISA microplates were coated with ovalbumin (50 μL/well, 100 μg/ml in PBS) at 4 °C overnight. After being washed twice with TBS-T (0.1% Tween 20), plates were blocked with 1% BSA at RT for 1-2 hrs. Serum was initially diluted between 10- to 400-fold and then diluted in 2-fold serial dilution 7 times on the ELISA plate. Plates were incubated with serum (50 μL/well) at RT for 1-2 hrs, washed 4 times, and then incubated with anti-IgG1 mouse-horseradish peroxidase (HRP) conjugate (1:2000 dilution; Santa Cruz Biotechnology Inc.) at RT for 1 h. Following 5 times wash, plates were developed in 75 μL/well HRP at RT for 15 min, and quenched with 75 μL/well 2N H2SO4. Absorbance was measured at 450 nm with Synergy H1 microplate reader (BioTek). Anti-IgG1 titers were calculated with Prism (GraphPad Software) by applying a standard four-parameter IC50 function.
Statistics
Statistical significance was determined using an unpaired 2-tailed Student's t test. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Prof. Dr. Jason Cyster (UCSF) for providing us with Hy10 mice, Dr. Malcolm Robert Wood (TSRI) for microscopy, Anna Tran-Crie for help with the preparation of the manuscript, and Sarah Knight for technical help. Lijuan Pang was supported by a postdoctoral fellowship from the Swiss National Science Foundation (SNSF). This work is funded in part by NIH grant R01 AI099141.
References
- 1.Naparstek Y, Plotz PH. Annu Rev Immunol. 1993;11:79–104. doi: 10.1146/annurev.iy.11.040193.000455. [DOI] [PubMed] [Google Scholar]
- 2.Lleo A, Invernizzi P, Gao B, Podda M, Gershwin ME. Autoimmun Rev. 2010;9:A259–266. doi: 10.1016/j.autrev.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Kwun J, Bulut P, Kim E, Dar W, Oh B, Ruhil R, Iwakoshi N, Knechtle SJ. Semin Immunol. 2012;24:96–108. doi: 10.1016/j.smim.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clatworthy MR. Am J Transplant. 2011;11:1359–1367. doi: 10.1111/j.1600-6143.2011.03554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gould HJ, Sutton BJ. Nat Rev Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 6.Gauchat JF, Henchoz S, Mazzei G, Aubry JP, Brunner T, Blasey H, Life P, Talabot D, Flores-Romo L, Thompson J, et al. Nature. 1993;365:340–343. doi: 10.1038/365340a0. [DOI] [PubMed] [Google Scholar]
- 7.Singh SK. J Pharm Sci. 2011;100:354–387. doi: 10.1002/jps.22276. [DOI] [PubMed] [Google Scholar]
- 8.Allison AC. Immunopharmacology. 2000;47:63–83. doi: 10.1016/s0162-3109(00)00186-7. [DOI] [PubMed] [Google Scholar]
- 9.Carbone J, del Pozo N, Gallego A, Sarmiento E. Expert Rev Anti Infect Ther. 2011;9:405–413. doi: 10.1586/eri.10.178. [DOI] [PubMed] [Google Scholar]
- 10.Riminton DS, Hartung HP, Reddel SW. Curr Opin Neurol. 2011;24:217–223. doi: 10.1097/WCO.0b013e328346d47d. [DOI] [PubMed] [Google Scholar]
- 11.Miller SD, Turley DM, Podojil JR. Nat Rev Immunol. 2007;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 12.Sabatos-Peyton CA, Verhagen J, Wraith DC. Curr Opin Immunol. 2010;22:609–615. doi: 10.1016/j.coi.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, Förster R, Pabst O. J Exp Med. 2006;203:519–527. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skupsky J, Su Y, Lei TC, Scott DW. Curr Gene Ther. 2007;7:369–680. doi: 10.2174/156652307782151443. [DOI] [PubMed] [Google Scholar]
- 15.Smarr CB, Hsu CL, Byrne AJ, Miller SD, Bryce PJ. J Immunol. 2011;187:5090–5098. doi: 10.4049/jimmunol.1100608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irvine DJ, Swartz MA, Szeto GL. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Immunity. 2010;32:568–580. doi: 10.1016/j.immuni.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 18.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O'Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Proc Natl Acad Sci U S A. 2015;112:E156–165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishimoto TK, Ferrari JD, LaMothe RA, Kolte PN, Griset AP, O'Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, Maldonado RA. Nat Nanotechnol. 2016;11:890–899. doi: 10.1038/nnano.2016.135. [DOI] [PubMed] [Google Scholar]
- 20.Joseph A, Munroe K, Housman M, Garman R, Richards S. Clin Exp Immunol. 2008;152:138–146. doi: 10.1111/j.1365-2249.2008.03602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manjarrez-Orduño N, Quách TD, Sanz I. J Invest Dermatol. 2009;129:278–288. doi: 10.1038/jid.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsubata T. Infect Disord Drug Targets. 2012;12:181–190. doi: 10.2174/187152612800564455. [DOI] [PubMed] [Google Scholar]
- 23.Crocker PR, Paulson JC, Varki A. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 24.Macauley MS, Crocker PR, Paulson JC. Nat Rev Immunol. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson JC, Nemazee D. J Exp Med. 2010;207:173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. J Clin Invest. 2013;123:3074–3083. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfrengle F, Macauley MS, Kawasaki N, Paulson JC. J Immunol. 2013;191:1724–1731. doi: 10.4049/jimmunol.1300921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orgel KA, Duan S, Wright BL, Maleki SJ, Wolf JC, Vickery BP, Burks AW, Paulson JC, Kulis MD, Macauley MS. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touzot M, Soulillou JP, Dantal J. Curr Opin Organ Transplant. 2012;17:626–633. doi: 10.1097/MOT.0b013e32835a4be2. [DOI] [PubMed] [Google Scholar]
- 30.Brown VI, Fang J, Alcorn K, Barr R, Kim JM, Wasserman R, Grupp SA. Proc Natl Acad Sci U S A. 2003;100:15113–15118. doi: 10.1073/pnas.2436348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomson AW, Turnquist HR, Raimondi G. Nat Rev Immunol. 2009;9:324–327. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Limon JJ, So L, Jellbauer S, Chiu H, Corado J, Sykes SM, Raffatellu M, Fruman DA. Proc Natl Acad Sci U S A. 2014;111:E5076–5085. doi: 10.1073/pnas.1407104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Traitanon O, Mathew JM, La Monica G, Xu L, Mas V, Gallon L. PLoS One. 2015;10:e0129658. doi: 10.1371/journal.pone.0129658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang AH, Rossi RJ, Yoon J, Wang H, Scott DW. Cell Immunol. 2016;301:74–81. doi: 10.1016/j.cellimm.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Simamora P, Alvarez JM, Yalkowsky SH. Int J Pharm. 2001;213:25–29. doi: 10.1016/s0378-5173(00)00617-7. [DOI] [PubMed] [Google Scholar]
- 36.Onyesom I, Lamprou DA, Sygellou L, Owusu-Ware SK, Antonijevic M, Chowdhry BZ, Douroumis D. Mol Pharm. 2013;10:4281–4293. doi: 10.1021/mp400362v. [DOI] [PubMed] [Google Scholar]
- 37.Ghanbarzadeh S, Valizadeh H, Zakeri-Milani P. Bioimpacts. 2013;3:75–81. doi: 10.5681/bi.2013.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haeri A, Sadeghian S, Rabbani S, Anvari MS, Boroumand MA, Dadashzadeh S. Int J Pharm. 2011;414:16–27. doi: 10.1016/j.ijpharm.2011.04.055. [DOI] [PubMed] [Google Scholar]
- 39.Miao ZL, Deng YJ, Du HY, Suo XB, Wang XY, Wang X, Wang L, Cui LJ, Duan N. Exp Ther Med. 2015;9:941–946. doi: 10.3892/etm.2015.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nadig SN, Dixit SK, Levey N, Esckilsen S, Miller K, Dennis W, Atkinson C, Broome AM. RSC Adv. 2015;5:43552–43562. doi: 10.1039/C5RA04057D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen CD, Okada T, Tang HL, Cyster JG. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 42.Kurosaki T, Kometani K, Ise W. Nat Rev Immunol. 2015;15:149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- 43.Eriksson NE, Formgren H, Svenonius E. Allergy. 1982;37:437–443. doi: 10.1111/j.1398-9995.1982.tb02323.x. [DOI] [PubMed] [Google Scholar]
- 44.Browning JL. Nat Rev Drug Discov. 2006;5:564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 45.Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR, van Echo DA, von Hoff DD, Leyland-Jones B. J Clin Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 46.Mac Sharry J, Shalaby KH, Marchica C, Farahnak S, Chieh-Li T, Lapthorne S, Qureshi ST, Shanahan F, Martin JG. PLoS One. 2014;9:e98648. doi: 10.1371/journal.pone.0098648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vieira P, Rajewsky K. Eur J Immunol. 1988;18:313–316. doi: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 48.Guba M, Koehl GE, Neppl E, Doenecke A, Steinbauer M, Schlitt HJ, Jauch KW, Geissler EK. Transpl Int. 2005;18:89–94. doi: 10.1111/j.1432-2277.2004.00026.x. [DOI] [PubMed] [Google Scholar]
- 49.Chen WC, Completo GC, Sigal DS, Crocker PR, Saven A, Paulson JC. Blood. 2010;115:4778–4786. doi: 10.1182/blood-2009-12-257386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loughrey HC, Choi LS, Cullis PR, Bally MB. J Immunol Methods. 1990;132:25–35. doi: 10.1016/0022-1759(90)90394-b. [DOI] [PubMed] [Google Scholar]
- 51.Naknukool S, Hayakawa S, Uno T, Ogawa M. In: Global Issues in Food Science and Technology. Barbosa-Canovas G, Mortimer A, Lineback D, Spiess W, Buckle K, Colonna P, editors. Academic Press Inc; New York: 2009. pp. 293–307. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.