

Abstract
Many therapeutic agents that are used in patients with diabetes mitigate oxidative stress. These agents are of particular interest because oxidative stress is elevated in diabetes and is thought to contribute to vascular dysfunction. Agents that merely quench already formed reactive oxygen species have demonstrated limited success in improving cardiovascular outcomes. Thus, although vitamin E, C, and alpha lipoic acid appeared promising in animal models and initial human studies, subsequent larger trials have failed to demonstrate improvement in cardiovascular outcomes. Drugs that limit the production of oxidative stress are more successful in improving vascular outcomes in patients with diabetes. Thus, although statins, ACE inhibitors, ARBs and thiazolinediones are used for varied clinical purposes, their increased efficacy in improving cardiovascular outcomes is likely related to their success in reducing the production of reactive oxygen species at an earlier part of the cascade, thereby more effectively decreasing the oxidative stress burden. In particular, statins and ACE inhibitors/ARBs appear the most successful at reducing oxidative stress and vascular disease and have potential for synergistic effects.
Keywords: Diabetes, Oxidative stress, Endothelial function, Endothelium, Vitamin E, Vitamin C, Alpha-lipoic acid, Statin, ACE-inhibitor, Angiotensin II blocker, Thiazolinedione, Review
2. INTRODUCTION
The unifying theory that hyperglycemia induced elevations in superoxide production underlie the activation of many pathways involved in the pathogenesis of diabetic vascular disease naturally raised an interest in the role of antioxidant treatment. However, it appears that not all antioxidants improve vascular function. In fact, antioxidants that simply neutralize the excess oxidative burden present in diabetes appear less effective than antioxidants that block production of reactive oxygen species and limit the cascading increase in oxidative stress. In the following review, we briefly discuss the importance of oxidative stress in mediating vascular dysfunction in diabetes, common surrogate measures of vascular disease and finally, we review the vascular benefits of several therapeutic agents that have known antioxidant properties.
3. DIABETES IS AN OXIDATIVE STRESS DISORDER
3.1. Increased superoxide production
The diabetic state is associated with excess superoxide production. The failure of insulin to stimulate glucose uptake by fat and muscle tissues, results in hyperglycemia; this causes an increase in intracellular glucose concentrations in insulin-independent cell types, such as endothelium. Increased intracellular glucose concentrations result in an increased rate of glycolysis, which in turn increases the flux of pyruvate (the product of glycolysis) through the tricarboxylic acid (TCA) cycle. It is the increased flux of pyruvate through the TCA cycle that appears responsible for over-production of superoxide. The mitochondrial electron transport chain prematurely transfers electrons to oxygen thereby generating excess superoxide (1). It should be noted that hyperglycemia is not the only mechanism by which diabetes causes increased superoxide production. Diabetes is also associated with increased levels of free fatty acids, which contribute to increased superoxide production (2).
3.2. Oxidative stress and NO
Increased superoxide and reactive oxygen species negatively affect vascular health by downregulating endothelial dervived nitric oxide (NO). NO plays a key role in vasodilation as well as in maintaining a healthy vessel wall by inhibiting inflammation, cellular proliferation and thrombosis. Decreased NO bioavailability not only increases vascular tone, but also promotes structural and biological changes that lead to atherosclerosis. Decreased bioavailability is a result of both NO quenching by peroxynitrite and decreased NO production (1–3)
3.3. Quenching of NO to form peroxynitrite
The superoxide anion reacts with NO to form peroxynitrite, thus reducing the quantity of NO available to the vasculature “see Figure 1”. Peroxynitrite post-translationally modifies macromolecules, resulting in impaired protein and lipid function which promotes vascular dysfunction and atherosclerosis (4). The degradation of tyrosine nitrated proteins produces free nitrotyrosine. This marker of nitrosative stress has been found in tissues, atherosclerotic lesions and blood (5, 6, 7). In addition to modification of biomolecules, peroxynitrite may also modulate important signaling pathways and trigger mitochondrial dysfunction and cell death in endothelial cells and cardiomyocytes as described further below (8).
Figure 1.
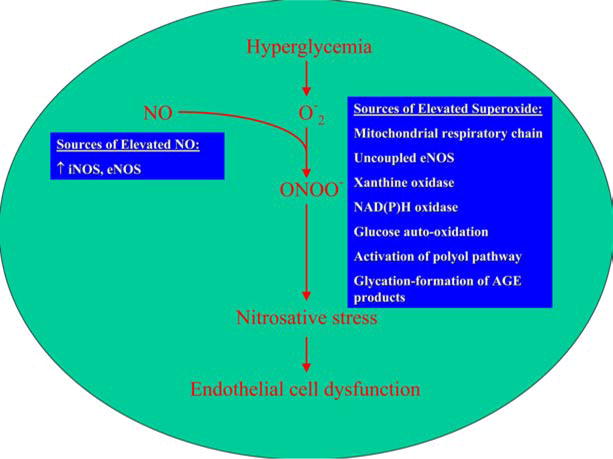
Hyperglycemia induced endothelial dysfunction. Superoxide produced secondary to hyperglycemia combines with NO to form peroxynitrite. This reduces the bioavailability of NO and induces nitrosative stress by multiple mechanisms including modifications of macromolecules and PARP induction.
3.4. Decreased NO production
Peroxynitrite also inactivates (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4), a cofactor involved in the production of NO. BH4 oxidation uncouples endothelial NO synthase (eNOS), the enzyme responsible for NO production. Normally, production of NO requires dimerization of eNOS, the presence of L-arginine, and the cofactor BH4. When these conditions are present, eNOS oxidizes its substrate L-arginine to produce L-citrulline and NO. BH4 deficiency uncouples the eNOS complex and promotes production of superoxide by eNOS, thus producing more oxidative stress “see Figure 2”.
Figure 2.
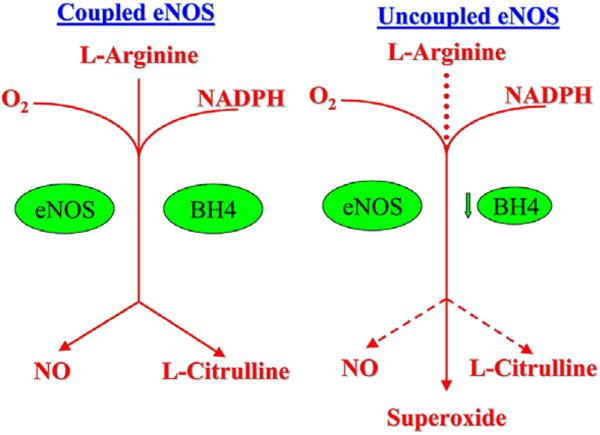
Coupled and uncoupled eNOS. (a) Coupled eNOS utilizes O2, L-Arginine and NADPH to produce NO and L-Citrulline (b) eNOS can be uncoupled by BH4 deficiency to produce superoxide rather than NO, which may further reduce available NO by combining with it to form peroxynitrite.
3.5. Oxidative stress or nitrosative stress and PARP activation
Oxidative stress or nitrosative stress, in the form of peroxynitrite also causes DNA single strand breaks and is one source of poly(ADP-ribose) polymerase (PARP) activation (4). The activation of PARP is an important mediator of vascular dysfunction in diabetes (8–10). PARP activation initiates a series of cell cycle events “see Figure 3” that deplete intracellular nicotinamide adenine dinucleotide (NAD) and adenosine 5′-triphosphate (ATP) pools, thus limiting glycolysis and mitochondrial respiration, leading to vascular cell dysfunction and death (3). Protein kinase C (PKC) activity, advanced glycation end-product/receptor for advanced glycation end-product (AGE/RAGE) interactions, and the hexosamine pathway can also be activated by PARP activation as a result of Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) dysfunction (11, 12). PARP activation is elevated in subjects with diabetes and is associated with an impairment in vascular reactivity (13). Recent evidence suggests that increased PARP acitivity is present in subjects with diabetes even prior to the onset of microvascular disease (14).
Figure 3.
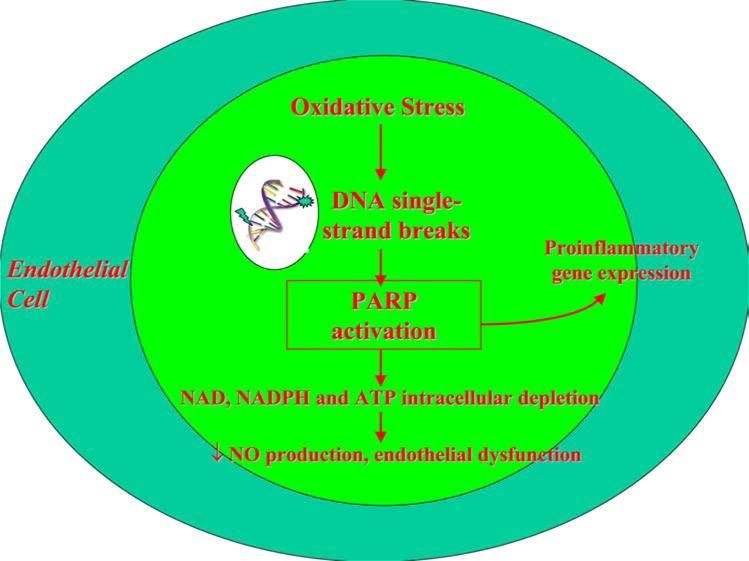
The role of peroxynitrite and PARP. Peroxynitrite induced PARP activation depletes intracellular NAD, NADPH and ATP pools leading to endothelial dysfunction.
3.6. Further pathways activated by increased superoxide production
In addition to elevated levels of peroxynitrite, decreased NO and PARP activation, at least four cellular processes have been previously noted to contribute to diabetic microvascular complications. These include: 1) increased flux through the polyol/aldose pathway, 2) activation of PKC, 3) increased production of AGEs and 4) increased flux through the hexosamine pathway. All of these pathways are activated by hyperglycemia induced superoxide production (15). “see Figure 4”
Figure 4.
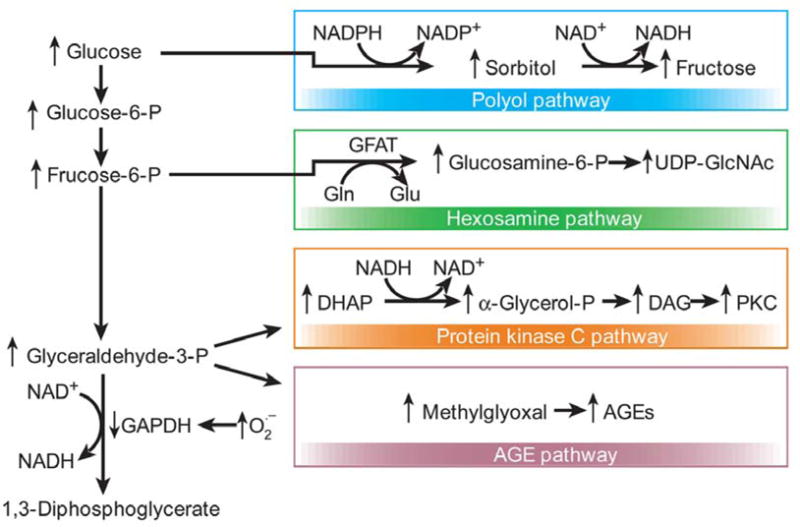
Hyperglycemia induced cellular pathways. Hyperglycemia-induced mitochondrial superoxide overproduction activates the polyol, hexosamine, protein kinase C and AGE pathways. Additionally, excess superoxide production inhibits GAPDH, thus diverting upstream metabolites from glycolysis to the above mentioned pathways. Figure adapted with permission from the publisher (15).
3.7. PKC activation
Hyperglycemia induced elevations in superoxide anion activate PKC, and activation of PKC further contributes to superoxide generation (1). PKC may also be activated by chronically elevated diacylglycerol (DAG) levels from increased de novo synthesis of DAG from glycolytic intermediates, increased activity of the polyol pathway, and via ligation of RAGE (16). The DAG-PKC pathway is activated to maximal levels in three to five days after the initiation of hyperglycemia and remains elevated for many years (17, 18). The activation of PKC increases the activity of membrane associated nicotinamide adenine dinucleotide phosphate (NADPH) oxidases which generate superoxide anion (19). Thus, PKC activation by oxidative stress generates more oxidative stress, creating a vicious circle of positive feedback.
Increased PKC activity is associated with abnormal vascular function and although blocking PKC activity appears to improve microvascular function in animal models, it has little benefit in humans. Activation of PKC results in abnormal vasodilation, increased vascular permeability, increased microvascular protein accumulation, increased plasminogen activator inhibitor-1 (PAI-1) expression, and activation of nuclear factor-kappa B (NF-kB) in endothelial cells and vascular smooth muscle cells. Inhibition of PKC with ruboxistaurin (or LY333531) greatly improves microvascular flow to the retina, kidney, endoneural blood supply and mesenteric bed in animal models (15, 20, 21). Despite these promising findings, ruboxistaurin has had less robust results in humans (22).
3.8. Advanced glycation end products and receptor for advanced glycation end products
AGEs are formed intra- and extracellularly non-enzymatically when reducing sugars combine with free amino groups of proteins, lipids, and guanyl nucleotides. These reactions are irreversible for the most part and accumulate with time. AGEs can alter the structure and function of intra- and extracellular proteins by forming covalent crosslinks. In addition, AGEs help make lipids more atherogenic by glycation and subsequent oxidation. AGEs also cause production of reactive oxygen species and block endothelial NO activity (23).
In addition to their direct effects on macromolecules, AGEs also bind and activate RAGE. Activation of RAGE by AGEs results in sustained activation of NF-kB and its target genes (24). AGE-bound RAGE also increases endothelial cell permeability to macromolecules. Elevated levels of AGEs have been noted in the serum of diabetic patients and correlate with progression of diabetic complications such as nephropathy (25, 26). Treatment of animals with inhibitors of AGE formation, such as aminoguanide, can prevent diabetic microvascular complications (27).
3.9. Polyol pathway
Increased intracellular glucose generates increased flux through the polyol pathway, by engaging the key enzyme, aldose reductase, which usually has a low affinity for glucose. Aldose reductase reduces glucose to sorbital, which is further oxidized to fructose, which consumes cellular NADPH, increasing cellular oxidative stress. Increased flux through the polyol pathway has been implicated in activation of PKC. Inhibition of aldose reductase has been shown to prevent diabetic nephropathy, retinopathy, and neuropathy in animal models (15). Larger clinical trials in humans, however, have had mixed results, thus raising questions regarding the importance of this mechanism (28, 29).
3.10. Hexosamine pathway
Hyperglycemia also shunts glucose through the hexosamine pathway. A glycolytic intermediate, fructose-6-phosphate (Fruc-6P) is converted with glucosamine-6-phosphate, and ultimately to N-acetylglucosamine. Hyperglycemia is associated with an increase in O-linked N-acetylglucosamine modification and decreases O-linked phosphorylation of the transcription factor Sp1, resulting in increased gene expression of transforming growth factor beta (TGF-beta) and PAI-1.(15) Elevated glucose levels also result in inhibition of eNOS, which is accompanied by a twofold increase in O-linked N-acetylglucosamine modification of eNOS and a reciprocal decrease in O-linked serine phosphorylation (30).
4. VASCULAR DISEASE IN DIABETES
Endothelial dysfunction in both the micro- and macro-circulation is the final result of oxidative stress initiated, self perpetuating cascade of events (31). Progressive capillary changes including neovasculariztion in retinopathy, and narrowing and/or microthrombosis in peripheral neuropathy are the result of hyperglycemia induced increases in endothelial cell permeability, vascular inflammation, and other structural changes. A reduction in hyperglycemia by intensive glycemic control protocol has been shown in two separate landmark trials to decrease progression and occurrence of microvascular complications (retinopathy, neuropathy, and nephropathy) in both type 1 and 2 diabetes (32, 33).
In contrast, glycemic control has been demonstrated to conclusively improve macrovascular outcomes in only type 1 diabetes. Despite this, macrovascular disease such as myocardial infarction (MI), cerebrovascular accidents, and peripheral arterial disease continues to account for a substantial portion of the mortality and morbidity in both type 1 and 2 diabetes. Improved glycemic control in type 1 diabetes has been associated with dramatically lower rates of macrovascular disease (42% decrease) (34, 35). However, despite reductions in all cause mortality associated with tighter glycemic control macrovascular event rates in type 2 diabetes are not improved with tighter glycemic control unless metformin was part of the regimen (33, 36). Patients with metformin included as part of their regimen are better able to maintain glycemic control over 3 years compared to other regimens and have greater improvements in all cause mortality and decrease in stroke rates (37). Thus, treatment of usual cardiovascular risk factors such as hyperlipidemia and hypertension in type 2 diabetes plays a larger role in lowering the risk of macrovascular events, suggesting that oxidative stress induced by these traditional cardiovascular risk factors appears more important than that induced by hyperglycemia in such patients.
5. METHODS OF ASSESSING ENDOTHELIAL FUNCTION
Prior to the development of macrovascular and microvascular clinical disease early changes in endothelial function can be measured. These changes reflect alterations in the regulation of vascular tone or reactivity which is influenced by endothelial NO production (endothelial-dependent vasoreactivity) as well as vascular smooth muscle relaxation in response to NO (endothelial-independent vasoreactivity). In endothelial dependent vasodilation, acetylcholine, shear stress or hypoxia can activate endothelial cells to release NO. The stimuli of shear stress and hypoxia are utilized in the flow mediated dilation (FMD) technique to produce endothelium-dependent vasodilation. In contrast, endothelium-independent vasodilation occurs as a result of smooth muscle cell relaxation in direct response to exogenous NO (from NO donors such as nitroglycerin or nitroprusside). Vasoreactivity, which refers to both endothelial dependent and independent vasodilation in response to a stimulus, is a means to quantify endothelial cell and vascular smooth muscle function.
5.1. Macrocirculatory measurements
Macrovascular disease is most commonly assessed by ultrasound measurements of brachial artery diameter and the common carotid intima-media thickness (IMT). Changes in brachial artery diameter after stimuli measure early functional changes associated with atherosclerosis. Endothelium-dependent vasodilation of the brachial artery can be assessed by intra-arterial infusion of substances that act on the endothelium to release NO, such as acetylcholine, or by FMD. FMD is induced by occluding the brachial artery with a pneumatic tourniquet to the upper limb for a total of 5 minutes (38). Tissue hypoxia and pH changes in the area distal to the occlusion cause reactive vasodilation in the skin and muscle microcirculation immediately after release of the occlusion. This process causes a brief period of high blood flow and increased shear stress in the brachial artery that stimulates the endothelial production of NO and vasodilation that can be measured on high resolution ultrasound. “see Figure 5” Endothelium-independent vasodilatory function of the brachial artery can be assessed by intra-arterial or sublingual administration of NO donors such as nitroglycerin or nitroprusside.
Figure 5.
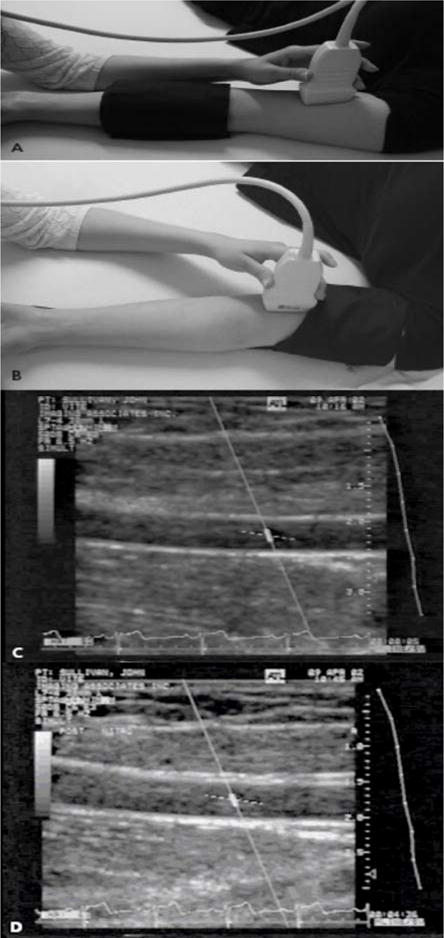
Assessment of FMD in the brachial artery. A 7.0-MHz or greater liner array transducer is used to image the brachial artery above the antecubital fossa in the longitudinal plane. A blood pressure cuff is employed to occlude the arterial blood flow and can be placed either at the forearm (a) or the upper arm level (b). Two-dimensional grayscale scans are taken, one at rest at rest, before the cuff inflation (c) and 1 minute after the cuff deflation that leads to arterial dilation (d). The percentage of the postocclusive artery diameter increase over the baseline represents the FMD. Figure adapted with permission from A. Veves (145).
In contrast, common carotid IMT identifies anatomic changes consistent with early atherosclerosis. Carotid artery intima-media thickness (IMT) is an ultrasound measure of the distance between the intima to the outer edge of the media. Increased intima-media thickness occurs early in the process of atherosclerotic plaque formation prior to luminal narrowing. IMT is associated with the presence of conventional atherosclerotic risk factors and can predict the development of cardiovascular events (39, 40).
5.2. Microcirculatory measurements
Microcirculatory vascular reactivity is most commonly assessed by LASER Doppler flowmetry to measure blood flow in the skin. Blood flow is estimated from the combination of number and velocity of moving red cells within arterioles, capillaries, and postcapillary venules. A LASER beam is delivered to the skin via a fiber optic light guide, and reflected light is gathered by a second set of photodetectors. Light reflected by moving objects, such as red blood cells, is reflected at a different frequency. The Doppler shifted fraction of the light signal and the mean Doppler frequency shift is calculated to generate a value in mV, which is proportional to the quantity and velocity of red blood cells with the measured superficial skin microcirculation (41).
The microcirculation can be studied without systemic side effects by using iontophoresis and microdialysis techniques that allow for precise, local delivery of vasoactive agents. Iontophoresis uses a small charge to facilitate transcutaneous delivery of charged substances into the skin without trauma or pain. The length of stimulation, strength of current used, and area of delivery determine the number of molecules transported. Endothelium-dependent vasodilation is assessed by delivery of acetylcholine using anodal current given its positive charge, whereas endothelium-independent vasodilation is assessed by the delivery of the anion sodium nitroprusside using cathodal current. Microdialysis can be used to deliver larger, water-soluble vasoactive agents that lack a charge. These techniques allow for non-invasive measurement of abnormal endothelial function prior to the development of overt clinical disease.
6. THERAPEUTIC INTERVENTIONS THAT MODIFY OXIDATIVE STRESS
In subsequent sections we will discuss how these measurements of vascular function, animal models, and larger clinical trials have been used to evaluate the efficacy of therapeutic agents in combating the increased oxidative stress in diabetes and subsequent ill effects on the vasculature. These agents include vitamins E, C, alpha-lipoic acid, statins, angiotensin converting enzyme inhibitors (ACE inhibitors), angiotensin II receptor blockers (ARBs) and thiazolinedones. Many other agents have been noted to have antioxidant properties, but have not been evaluated in human clinical trials, and are beyond the scope of this review. Therefore, we will limit our discussion to those compounds noted above.
6.1. Vitamin E
Vitamin E is a fat soluble vitamin with known anti-oxidant properties. It is frequently co-administered with vitamin C because its oxidized form is regenerated by vitamin C. Vitamin E and C supplementation improves markers of oxidative stress and endothelium-dependent vasodilation in experimental diabetic models (42–45). Acute administration of vitamin E has generally been shown to improve endothelial dependent brachial artery vasodilatation in both type 1 and 2 diabetes (46).
Chronic administration of vitamin E was found to be vasoconstrictive in patients with type 1 and 2 diabetes (47). Treatment with twelve months of vitamin E at doses of 1,800 IU per day is associated with no improvement in FMD, a deterioration in endothelial independent vasodilation and a trend towards increased systolic blood pressure. Vitamin E does not change microcirculatory responses to either acetylcholine and nitroprusside or progression of diabetic retinopathy after twelve months (47). Thus, chronic administration of vitamin E appears to worsen endothelial independent vasoreactivity and increase blood pressure in patients with diabetes.
Several prospective interventional trials have found that vitamin E decreases cardiovascular events in the general population and in patients with a high risk of cardiac disease, but without diabetes. In particular, vitamin E was reported to reduce the risk of non-fatal MI when administered at doses of 400–800 IU per day in patients with prior symptomatic coronary atherosclerosis (CHAOS trial) (48). However, it is also associated with a disturbing, non-significant trend towards an excess of cardiovascular deaths.
A lack of benefit with vitamin E supplementation in a subsequent series of randomized trials dampened enthusiasm for vitamin E’s utility as an antioxidant in diabetes. In fact, both cardiovascular outcomes and atherosclerosis progression by carotid IMT are not improved by vitamin E in a group of high risk patients with vascular disease or diabetes (HOPE study and SECURE trial) (49–52). The lack of benefit in regards to cardiovascular outcomes and nephropathy persisted after a subgroup analysis of patients with diabetes. In addition, there was no reduction in cardiovascular events or death in 1031 patients with diabetes after vitamin E supplementation in the PPP trial (53). In addition, an increased risk of adverse events with vitamin E supplementation raises further concerns about its use. Vitamin E supplementation appeared safe during the initial HOPE study with a follow-up period of 4.5 years, but extended followup for a total of 7 years was associated with an increased risk of heart failure. The excess heart failure risk was also evident after 3.5 years of follow-up of post-infarction patients supplemented with vitamin E in the GISSI-Prevenzione trial (54, 55). In fact, patients with left ventricular dysfunction (ejection fraction <50%) who are treated with vitamin E demonstratd a 50% increased risk of developing congestive heart failure (p=0.034).
In addition, higher doses are associated with increased mortality risk. In a meta-analysis of vitamin E trials prior to August 2004, low dose vitamin E (<150 IU per day) was associated with a non-significant decrease in all cause mortality (56). Doses larger that 150 IU per day are associated with a progressive increase in all cause mortality as dose increased. Furthermore, the benefical effect of low dose vitamin E was attenuated when adjustments were made for concomitant use of other vitamin supplements. Thus, the routine use of vitamin E supplementation with or without vitamin C cannot be recommended in patients with diabetes.
6.2. Vitamin C
Vitamin C, also called ascorbic acid, is a water soluble vitamin with many biological roles in addition to its function as an antioxidant. Vitamin C stabilizes eNOS cofactor BH4, leading to increases in NO production. It also prevents oxidation of LDL and regenerates oxidized vitamin E. Initial physiologic studies demonstrate improvement in endothelial function with acute infusion of vitamin C in patients with acute hyperglycemia, type I and II diabetes and hypertension (57–60). Longer term, orally delivered vitamin C has variable effectiveness in improving brachial artery reactivity in patients with type II diabetes (61, 62).
Epidemiologic data suggest that higher intake of vitamin C is associated with improvements in mortality, particularly from cardiovascular causes. The First National Health and Nutrition Examination Survey (NHANES I) studied a cohort of 11,348 adults for 10 years (63). In this cohort, increased vitamin C intake (approx 300mg per day) was associated with a 45 to 25% risk reduction in all cause mortality and mortality from cardiovascular causes in men and women respectively. Vitamin C supplement use was associated with a significantly lower risk (28%) of coronary disease (relative risk of 0.72) after controlling for other cardiovascular risk factors in an observational study of 85,118 female nurses followed for 16 years (4, 64). This benefit was noted again by researchers in the EPIC-Norfolk prospective population study (65). The highest quartile of ascorbic acid had an odds ratio for future coronary artery disease of 0.67 compared with those in the lowest quartile. There are no randomized, controlled studies addressing the cardiovascular benefits of vitamin C supplementation independent of other vitamin supplements. Therefore, at this time, the use of vitamin C for cardiovascular benefits cannot be recommended in diabetes or the general population.
6.3. Alpha-lipoic acid
Alpha-lipoic acid is a more potent antioxidant than either vitamin E or C, and a critical cofactor in aerobic metabolism. Alpha-Lipoic acid reduced to its conjugate base, dihydrolipoate, is able to regenerate other antioxidants such as vitamins E and C, as well as reduced glutathione. Thus, one might expect more potent vascular benefits.
Diabetic animal models demonstrate improvements in metabolic profile and the microvasculature after treatment with alpha-lipoic acid. Thus, blood glucose, plasma insulin, cholesterol, triglycerides and lipid peroxidation improvements are associated with increased antioxidant enzymatic activity (catalase and glutathione peroxidase activity) (66). These benefits are partly attributable to the recovery of insulin producing cells in the pancreas, and are significant enough to prevent atherosclerotic lesions (67). In the microvasculature of diabetic rats, alpha-lipoic acid reduces nitrotyrosine levels and prevents pathologic retinal vessel changes (68). In endothelial cell cultures, alpha-lipoic acid prevents AGE dependent depletion of reduced glutathione and ascorbic acid and subsequent activation of NF-kappa B (69). Thus, it appears alpha-lipoic acid supplementation reduces oxidative stress and thereby improves metabolic derangements and microvascular function in animal and in vitro models.
Alpha-lipoic acid has been mainly studied in randomized controlled human clinical trials for the treatment of diabetic polyneuropathy. Short term IV alpha-lipoic acid for 19 days appeared to improve symptoms, and longer term alpha-lipoic acid (IV infusions, followed by oral therapy for 2 years) were reported to improve objective peripheral nerve function (70, 71). Prolonged treatment of four years duration in the NATHAN 1 trial found improvements in only some neuropathic deficits and symptoms, but not objective nerve conduction in patients with mild to moderate distal symmetric neuropathy (72). In addition, there was a nonsignificant trend towards an increased rate of serious adverse events from 28% to 38% in the active treatment group. Thus, although there may be a possibility of improvements in neuropathy with short term IV infusion of alpha-lipoic acid, these improvements have not been sustained with long term oral therapy and are may increase the risk of serious adverse events.
The effects of alpha-lipoic acid on autonomic dysfunction and surrogate markers of macrovascular disease have been studied in only small numbers of patients. Alpha-lipoic acid treatment for four months slightly improves measures of heart rate variability, a measure of autonomic dysfunction, but does not change symptoms of autonomic dysfunction (73). Four weeks of therapy with oral alpha-lipoic acid improves endothelium-dependent vasorelaxation of the brachial artery by 44% compared to the placebo group and was accompanied by reductions in markers of endothelial activation, plasma interleukin-6 and plasminogen activator-1 (74). Thus, the impact of lipoic acid on clinical cardiovascular end-points is still unknown. Given this, and the increased risk of serious adverse events with long term use, the use of alpha-lipoic acid supplements cannot be recommended for patients with diabetes.
6.4. Statins
Statins inhibit the enzyme hydroxymethylglutaryl coenzyme A reductase (HMG CoA reductase) thereby improving serum lipid profile and lowering cardiovascular morbidity and mortality (75). These agents were initially thought to exert their beneficial effects on endothelial function secondary to their lipid lowering capacity. However, it appears that improvements in vascular function are only partly mediated by this mechanism (76). Statins also improve endothelial function by decreasing oxidative stress, inflammation and the thrombogenic response (77).
Statins achieve this enhanced vascular function by decreasing NADPH activity, reducing formation of reactive oxygen species and downregulating the renin angiotensin system. Statins reduce activity of the NADPH oxidase in endothelial cells, thus reducing the formation of ROS as well as oxidation of LDL (78–85). In addition, statins decrease uptake of oxidized LDL by monocytes that develop into foam cells in atherosclerotic lesions (86, 87). Atorvastatin is unique in that its hydroxymetabolites are present at usual doses and demonstrate free radical scavenging abilities (88). Statins downregulate AT1 receptor at the transcriptional level further improving measures of oxidative stress and vascular function (81).
All of the above mechanisms upregulate eNOS activity, which plays a central role in mediating the beneficial effects of statins in endothelial cells. Transcription of eNOS is reduced by the presence of oxidized LDL, but not native LDL. Statins prevent this inhibition of eNOS at the transcriptional level (89). This increased expression of eNOS is associated with an improvement in vascular function in animal models of type II diabetes and hypercholesteremia (90, 91). Statin mediated increases in eNOS function appear critical in vascular regeneration and restored myocardial vasorelaxation after experimentally induced myocardial infarction in the mouse model, as these benefits were not observed in eNOS −/− mice after statin treatment (92).
Statins unequivocally reduce the risk of major vascular events such as stroke, myocardial infarction, and coronary revascularization in patients with diabetes (93, 94). However, surrogate markers of such macrovascular events, endothelial dependent vasorelaxation, are not clearly improved with statins. In particular, vasoreactivity does not improve after statin treatment in patients with poorly controlled diabetes (95). Endothelial dependent vasodilation does improve independently of lipid lowering in patients with better glycemic and lipid control in both type 1 & type 2 diabetes (96–101). Statin use was also reported to ameliorate postprandial hypertriglyceridemic-and hyperglycemia-induced endothelial dysfunction and reduced serum nitrotyrosine levels in type II diabetes suggesting that its short term, lipid-independent vascular benefits are secondary to decreased oxidative and nitrosative stress (102).
Thus, statins improve endothelial function prior to reductions in LDL unless there is overwhelming oxidative stress related to factors such as hyperglycemia and hypercholesteremia in type 2 diabetes. A lack of response to statins may be related to elevated asymmetric dimethylarginine (ADMA) levels, a competitive inhibitor of eNOS. ADMA is elevated in by many cardiovascular risk factors and patients with elevated ADMA levels are less likely to have an improvement in vasoreactivity with statin use after 3 weeks (103).
6.5. ACE-inhibitors and Angiotensin II receptor blockers
Both ACE inhibitors and ARBs exert their clinical effects by decreasing the binding of angiotensin II to the AT1 receptor, by decreasing levels of angiotensin II and by inhibiting the interaction of angiotensin II to the AT1 receptor, respectively. It should be noted that ACE inhibitors reduce formation of angiotensin II by inhibiting ACE1, but have no effect on ACE2 or other angiotensin II forming enzymes. Angiotensin II opposes many of the actions of NO; it causes vasoconstriction, altered vascular smooth muscle function, increased inflammation via NF-kB and hypercoagulability by increased formation of PAI-1. In addition, inflammation itself may sustain endothelial dysfunction by activating the renin-angiotensin system, and subsequently increasing ROS formation and decreasing endothelial dependent vasoreactivity (104). Angiotensin II also induces vascular superoxide production by uncoupling eNOS upon loss of dihydrofolate reductase (DHFR), a BH4 salvage enzyme (105). Thus, ARBs and ACE inhibitors improve endothelium-dependent vasorelaxation by decreasing superoxide production, and increases NO bioavailability (105–108).
ACE inhibitors and ARBs improve vascular function and cardiovascular outcomes in type 2 diabetes. Both agents unequivocally improve endothelial function in patients with type 2 diabetes (109–112). Valsartan therapy improved resting forearm skin blood flow and resting brachial artery diameter after 12 weeks in patients with type 2 diabetes. However, their impact on endothelial function in patients with type 1 diabetes is less clear (113–116). ACE inhibitors and ARBs improve cardiovascular and all-cause mortality outcomes in patients with diabetes, in fact, to a greater degree than in non-diabetics as noted in subgroup analysis of the HOPE and LIFE studies (approx 38% and 19%, respectively, of subjects had diabetes) (117, 118). In addition, both of these agents appear to reduce the onset of type 2 diabetes in susceptible populations. Thus, it appears that ACE inhibitors and ARBs improve vascular outcomes in patients with diabetes.
As briefly mentioned earlier, LDL and the renin-angiotensin system modulate one another. The presence of native LDL increases AT1 receptor expression at least two fold in a sustained manner for 24 hours by stabilization of post-transcriptional mRNA (119). Increased AT1 receptor expression and activity is associated with increased production of superoxide and decreased endothelial-dependent vasodilatation (120). However, statins reduce the half life of AT1 receptor mRNA and thereby reduce angiotensin II induced production of reactive oxygen species (121). Native LDL, not oxidized LDL, increases angiotensin II-AT1 receptor induced vasoconstriction (122–124).
Conversely, angiotensin II binding of the AT1 receptor upregulates endothelial oxidized LDL receptor (LOX-1) in endothelial cells. This upregulation of LOX-1 receptor is prevented by ARBs and ACE inhibitors, thus limiting the potential diffusion of oxidized LDL from the blood into the vessel wall where it can result in plaque formation (125). Thus, co-administration of ACE-inhibitors/ARBs with a statin may decrease oxidative stress, vasoconstriction, decrease uptake of oxidized LDL and improve endothelial function.
6.6. Thiazolinediones
Thiazolinedones, also known as PPAR gamma agonists, include pioglitazone (Actos), rosiglitazone (Avandia), and troglitazone (Rezulin). These agonists bind nuclear PPAR-gamma receptors in adipocytes that function as transcription factors for genes important in adipocyte differentiation, lipid metabolism and insulin sensitivity. In addition, PPAR-gamma receptors are expressed in cells integral to the development of atherosclerosis: endothelial cells, vascular smooth muscle cells, monocytes/macrophages and T cells. Thus, one would expect all thiazolinedones to improve vascular function in a similar manner, just as they all improve insulin sensitivity. Indeed, all thiazolinediones have been demonstrated to enhance glycemic control, and improve surrogate measures of vascular disease. Thiazolinediones improve endothelial dependent vasoreactivity and measurements of carotid IMT in patients with diabetes (126–130). In addition, both rosiglitazone and pioglitazone have been reported to increase the regenerative capacity of endothelial progenitor cells in individuals with diabetes (131, 132). This improvement in vascular function is associated with reduced NADPH oxidase activity, decreased LDL oxidation and reduction in vascular inflammation (130, 133).
Despite these improvements in oxidative stress and vascular function, individual thiazolinediones appear to worsen clinical cardiovascular outcomes. Cardiovascular outcome data on troglitazone is limited because it was withdrawn from the market shortly after reports of severe, idiosyncratic, often fatal hepatic failure (134). However, rosiglitazone and pioglitazone have been reported to differentially affect the risk of myocardial infarction and both appear to increase the risk of heart failure.
Rosiglitazone was reported to elevate the risk of myocardial infarction and congestive heart failure. A meta-analysis by Nissen and Wolski reported a 43% increased risk of myocardial infarction (MI) and increased risk of death from cardiovascular causes (by 64%) with rosiglitazone therapy (135). These results were also supported by a meta-analysis of data from the manufacturer, GlaxoSmithKine, which reported a 31% increased risk of MI (136). In response to this data, the RECORD trial (which was sponsored by the manufacturer) performed an unplanned interim analysis which revealed no increased risk of MI or death from cardiovascular causes with rosiglitazone treatment in patients with type 2 diabetes (137). However, at the time of the interim analysis, the study lacked power to substantiate this negative finding. Recently, another new meta-analysis of long term (>12 months), randomized controlled trials in subjects that had impaired glucose tolerance or diabetes was performed because the Nissen and Wolski meta-analyiss included many small studies of short duration with heterogenous populations. This report again confirmed a similar increased risk of myocardial infarction (42% increase), but without the increase in cardiovascular mortality (138). Thus, the current consensus is that rosiglitazone may have detrimental effects in patients with previous heart disease and diabetes, and its use cannot be recommended in these patients.
Rosiglitazone has also been associated with an increased risk of congestive heart failure. In particular, rosiglitazone treatment was associated with an increased risk of heart failure (hazard ratio 2.15) in the interim analysis of the RECORD trial as well as in the most recent meta-analysis by Singh et. al (RR 2.09) (137, 138). It appears that this increased risk of heart failure can be mitigated by close attention to fluid status. Thus, rates of heart failure or worsening heart failure were not elevated in a group of patients with diabetes and mild heart failure (139). However, the rosiglitazone treated group suffered from significantly more cases of worsening edema (25.5% vs. 8.8%, P = 0.005), an increase in heart failure medication (33% vs. 18%), as well as, small, statistically significant, increases in brain natriutetic peptide levels. Despite the increased edema formation associated with rosiglitazone treatment, it was not reported to cause structural changes in left ventricular size or function in patients without a prior history of heart failure (140). Thus, rosiglitazone appears to worsen heart failure and its use cannot be recommended in patients with heart failure or those at risk of heart failure.
Unlike the first two thiazolinediones, larger clinical trials of pioglitzone in high-risk patients with type 2 diabetes and prior MI demonstrate an improvement in rates of myocardial infarction, but increased edema formation and heart failure remain concerns. After a median follow up of almost 3 years, pioglitazone was reported to reduce the risk of MI by 28%, acute coronary syndromes (ACS) by 37%, and the composite end point of nonfatal MI, coronary revascularization, ACS, and cardiac death by 19% (141). In addition, pioglitazone therapy has been reported to improve nocturnal blood pressure (142). The above findings were confirmed by a recent meta-analysis by Nissen et al which reported a slight decrease in the composite end point of death, myocardial infarctio, or stroke (hazard ration 0.82) and a slight increase in the risk of serious heart failure (hazard ratio 1.41) after approximately a year of pioglitazone therapy (143). Much of the differential benefit of pioglitazone is likely due to its favorable lipid profile. It improves total cholesterol, LDL, HDL and triglycerides, whereas rosiglitazone therapy increases in total cholesterol, LDL, and triglycerides (144).
7. SUMMARY
In summary, diabetes is a state associated with increased oxidative stress secondary to hyperglycemia and increased free fatty acid production. Agents such as vitamin E, C, and alpha lipoic acid that mitigate this oxidative stress by quenching already formed reactive oxygen species have had limited success in improving vascular function. Drugs which limit the production of superoxide and other reactive oxygen species such as statins, ACE inhibitors, ARBs and thiazolinediones are more successful in improving vascular outcomes in patients with diabetes. Their success is based on limiting the cascade of antioxidant production and subsequent vascular inflammation. In particular, the coadministration of statins and ACE inhibitors/ARBs will may lead to synergistic reductions in oxidant burden and vascular disease. Additionally, outcome studies are needed to confirm vascular benefits, as measurements of surrogate markers often do not predict longterm clinical outcomes.
Figure 6.
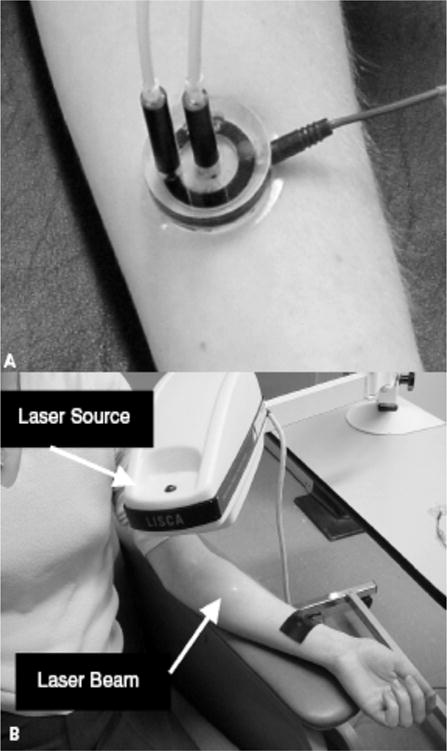
Measurement of skin microcirculation. (a) A small quantity of (<1ml) of 1% Ach chloride solution or 1% sodium nitroprusside solution is placed in the iontophoresis chamber. A constant current of 200 mA is applied for 60 seconds achieving a dose of 6 mC/cm2 between the iontophoresis chamber and a second nonactive electrode placed 10 to 15 cm proximal to the chamber (black strap around the wrist). This current causes a movement of solution to be delivered toward the skin. (b) Laser Doppler flowmetry: A helium-neon laser beam is emitted from the laser source to sequentially scan the circular hyperemic area produced by the iontophoresed vasoactive substance to a small area on the volar surface of the forearm. Figure adapted with permission from A. Veves (145).
Acknowledgments
Dr. Veves is funded by R01HL075678, R01NS046710, R01DK076937 and Dr. Malhotra is funded by NIH P50 HL060292-09, AG024837-01, RO1-HL73146. Dr. Yeh is funded by the National Sleep Foundation 2007 Pickwick Postdoctoral Fellowship.
Abbreviations
- TCA cycle
tricarboxylic acid cycle
- NO
nitric oxide
- eNOS
endothelial NO synthase
- BH4
(6R)-5,6,7,8-tetrahydro-L-biopterin
- PARP
poly(ADP-ribose) polymerase
- PKC
protein kinase C
- AGE
advanced glycation end products
- RAGE
receptor for advanced glycation end products
- FMD
flow-mediated dilation
- IMT
intima-media thickness
- ACE inhibitors
angiotensin converting enzyme inhibitors
- ARBs
angiotensin II receptor blockers
- LDL
low density lipoprotein
- HDL
high density lipoprotein
- ROS
reactive oxygen species
- AT1 receptor
angiotensin II type 1 receptor
- MI
myocardial infarction
- ACS
acute coronary syndromes
References
- 1.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 2.Schrauwen P, Hesselink MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–7. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz D, Malhotra A, Fink M. Cytopathic Hypoxia in Sepsis: An Overview. Sepsis. 1999;2:279–89. [Google Scholar]
- 4.Pacher P, Szabo C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr Opin Pharmacol. 2006;6:136–41. doi: 10.1016/j.coph.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res. 2001;34:541–81. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- 6.Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, White CR. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–8. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 7.Mihm MJ, Jing L, Bauer JA. Nitrotyrosine causes selective vascular endothelial dysfunction and DNA damage. J Cardiovasc Pharmacol. 2000;36:182–7. doi: 10.1097/00005344-200008000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mabley JG, Soriano FG. Role of nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic vascular dysfunction. Curr Vasc Pharmacol. 2005;3:247–52. doi: 10.2174/1570161054368571. [DOI] [PubMed] [Google Scholar]
- 10.Yim S, Malhotra A, Veves A. Antioxidants and CVD in diabetes: where do we stand now. Curr Diab Rep. 2007;7:8–13. doi: 10.1007/s11892-007-0003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–57. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brownlee M. A radical explanation for glucose-induced beta cell dysfunction. J Clin Invest. 2003;112:1788–90. doi: 10.1172/JCI20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szabo C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–6. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 14.Adaikalakoteswari A, Rema M, Mohan V, Balasubramanyam M. Oxidative DNA damage and augmentation of poly(ADP-ribose) polymerase/nuclear factor-kappa B signaling in patients with type 2 diabetes and microangiopathy. Int J Biochem Cell Biol. 2007;39:1673–84. doi: 10.1016/j.biocel.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 16.Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, Haller H, Cooper ME, Forbes JM. Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker ALT-711 via a protein kinase C-alpha-dependent pathway. Diabetes. 2004;53:2921–30. doi: 10.2337/diabetes.53.11.2921. [DOI] [PubMed] [Google Scholar]
- 17.Inoguchi T, Xia P, Kunisaki M, Higashi S, Feener EP, King GL. Insulin’s effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol. 1994;267:E369–79. doi: 10.1152/ajpendo.1994.267.3.E369. [DOI] [PubMed] [Google Scholar]
- 18.Xia P, Inoguchi T, Kern TS, Engerman RL, Oates PJ, King GL. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes. 1994;43:1122–9. doi: 10.2337/diab.43.9.1122. [DOI] [PubMed] [Google Scholar]
- 19.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–45. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 20.Cotter MA, Jack AM, Cameron NE. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 2002;103:311–21. doi: 10.1042/cs1030311. [DOI] [PubMed] [Google Scholar]
- 21.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 22.Aiello LP, Davis MD, Girach A, Kles KA, Milton RC, Sheetz MJ, Vignati L, Zhi XE. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006;113:2221–30. doi: 10.1016/j.ophtha.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 23.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 24.Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–4. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- 25.Berg TJ, Dahl-Jorgensen K, Torjesen PA, Hanssen KF. Increased serum levels of advanced glycation end products (AGEs) in children and adolescents with IDDM. Diabetes Care. 1997;20:1006–8. doi: 10.2337/diacare.20.6.1006. [DOI] [PubMed] [Google Scholar]
- 26.Berg TJ, Bangstad HJ, Torjesen PA, Osterby R, Bucala R, Hanssen KF. Advanced glycation end products in serum predict changes in the kidney morphology of patients with insulin-dependent diabetes mellitus. Metabolism. 1997;46:661–5. doi: 10.1016/s0026-0495(97)90010-x. [DOI] [PubMed] [Google Scholar]
- 27.Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A. 1991;88:11555–8. doi: 10.1073/pnas.88.24.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch Ophthalmol. 1990;108:1234–44. doi: 10.1001/archopht.1990.01070110050024. [DOI] [PubMed] [Google Scholar]
- 29.Greene DA, Arezzo JC, Brown MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology. 1999;53:580–91. doi: 10.1212/wnl.53.3.580. [DOI] [PubMed] [Google Scholar]
- 30.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–8. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akbari CM, Saouaf R, Barnhill DF, Newman PA, LoGerfo FW, Veves A. Endothelium-dependent vasodilatation is impaired in both microcirculation and macrocirculation during acute hyperglycemia. J Vasc Surg. 1998;28:687–94. doi: 10.1016/s0741-5214(98)70095-3. [DOI] [PubMed] [Google Scholar]
- 32.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 33.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- 34.Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial. Am J Cardiol. 1995;75:894–903. doi: 10.1016/s0002-9149(99)80683-3. [DOI] [PubMed] [Google Scholar]
- 35.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–53. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 37.UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. U.K. Prospective Diabetes Study Group. Diabetes Care. 1998;21:87–92. doi: 10.2337/diacare.21.1.87. [DOI] [PubMed] [Google Scholar]
- 38.Saouaf R, Arora S, Smakowski P, Caballero AE, Veves A. Reactive hyperemic response of the brachial artery: comparison of proximal and distal occlusion. Acad Radiol. 1998;5:556–60. doi: 10.1016/s1076-6332(98)80207-9. [DOI] [PubMed] [Google Scholar]
- 39.Heiss G, Sharrett AR, Barnes R, Chambless LE, Szklo M, Alzola C. Carotid atherosclerosis measured by B-mode ultrasound in populations: associations with cardiovascular risk factors in the ARIC study. Am J Epidemiol. 1991;134:250–6. doi: 10.1093/oxfordjournals.aje.a116078. [DOI] [PubMed] [Google Scholar]
- 40.O’Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK., Jr Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999;340:14–22. doi: 10.1056/NEJM199901073400103. [DOI] [PubMed] [Google Scholar]
- 41.Tooke JE, Ostergren J, Fagrell B. Synchronous assessment of human skin microcirculation by laser Doppler flowmetry and dynamic capillaroscopy. Int J Microcirc Clin Exp. 1983;2:277–84. [PubMed] [Google Scholar]
- 42.Cinar MG, Ulker S, Alper G, Evinc A. Effect of dietary vitamin E supplementation on vascular reactivity of thoracic aorta in streptozotocin-diabetic rats. Pharmacology. 2001;62:56–64. doi: 10.1159/000056072. [DOI] [PubMed] [Google Scholar]
- 43.Keegan A, Walbank H, Cotter MA, Cameron NE. Chronic vitamin E treatment prevents defective endothelium-dependent relaxation in diabetic rat aorta. Diabetologia. 1995;38:1475–8. doi: 10.1007/BF00400609. [DOI] [PubMed] [Google Scholar]
- 44.Rosen P, Ballhausen T, Bloch W, Addicks K. Endothelial relaxation is disturbed by oxidative stress in the diabetic rat heart: influence of tocopherol as antioxidant. Diabetologia. 1995;38:1157–68. doi: 10.1007/BF00422364. [DOI] [PubMed] [Google Scholar]
- 45.Alper G, Olukman M, Irer S, Caglayan O, Duman E, Yilmaz C, Ulker S. Effect of vitamin E and C supplementation combined with oral antidiabetic therapy on the endothelial dysfunction in the neonatally streptozotocin injected diabetic rat. Diabetes Metab Res Rev. 2006;22:190–7. doi: 10.1002/dmrr.586. [DOI] [PubMed] [Google Scholar]
- 46.Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Keaney JF, Jr, Creager MA. Oral antioxidant therapy improves endothelial function in Type 1 but not Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. 2003;285:H2392–8. doi: 10.1152/ajpheart.00403.2003. [DOI] [PubMed] [Google Scholar]
- 47.Economides PA, Khaodhiar L, Caselli A, Caballero AE, Keenan H, Bursell SE, King GL, Johnstone MT, Horton ES, Veves A. The effect of vitamin E on endothelial function of micro- and macrocirculation and left ventricular function in type 1 and type 2 diabetic patients. Diabetes. 2005;54:204–11. doi: 10.2337/diabetes.54.1.204. [DOI] [PubMed] [Google Scholar]
- 48.Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS) Lancet. 1996;347:781–6. doi: 10.1016/s0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 49.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:154–60. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 50.Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, Bosch J, Dagenais G, Mann JF, Gerstein HC. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care. 2002;25:1919–27. doi: 10.2337/diacare.25.11.1919. [DOI] [PubMed] [Google Scholar]
- 51.Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. Jama. 2005;293:1338–47. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 52.Lonn E, Yusuf S, Dzavik V, Doris C, Yi Q, Smith S, Moore-Cox A, Bosch J, Riley W, Teo K. Effects of ramipril and vitamin E on atherosclerosis: the study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE) Circulation. 2001;103:919–25. doi: 10.1161/01.cir.103.7.919. [DOI] [PubMed] [Google Scholar]
- 53.Sacco M, Pellegrini F, Roncaglioni MC, Avanzini F, Tognoni G, Nicolucci A. Primary prevention of cardiovascular events with low-dose aspirin and vitamin E in type 2 diabetic patients: results of the Primary Prevention Project (PPP) trial. Diabetes Care. 2003;26:3264–72. doi: 10.2337/diacare.26.12.3264. [DOI] [PubMed] [Google Scholar]
- 54.Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354:447–55. [PubMed] [Google Scholar]
- 55.Marchioli R, Levantesi G, Macchia A, Marfisi RM, Nicolosi GL, Tavazzi L, Tognoni G, Valagussa F. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J Cardiovasc Med (Hagerstown) 2006;7:347–50. doi: 10.2459/01.JCM.0000223257.09062.17. [DOI] [PubMed] [Google Scholar]
- 56.Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 57.Beckman JA, Goldfine AB, Gordon MB, Creager MA. Ascorbate restores endothelium-dependent vasodilation impaired by acute hyperglycemia in humans. Circulation. 2001;103:1618–23. doi: 10.1161/01.cir.103.12.1618. [DOI] [PubMed] [Google Scholar]
- 58.Timimi FK, Ting HH, Haley EA, Roddy MA, Ganz P, Creager MA. Vitamin C improves endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1998;31:552–7. doi: 10.1016/s0735-1097(97)00536-6. [DOI] [PubMed] [Google Scholar]
- 59.Ting HH, Timimi FK, Boles KS, Creager SJ, Ganz P, Creager MA. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1996;97:22–8. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider MP, Delles C, Schmidt BM, Oehmer S, Schwarz TK, Schmieder RE, John S. Superoxide scavenging effects of N-acetylcysteine and vitamin C in subjects with essential hypertension. Am J Hypertens. 2005;18:1111–7. doi: 10.1016/j.amjhyper.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 61.Tousoulis D, Antoniades C, Tountas C, Bosinakou E, Kotsopoulou M, Toutouzas P, Stefanadis C. Vitamin C affects thrombosis/fibrinolysis system and reactive hyperemia in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2003;26:2749–53. doi: 10.2337/diacare.26.10.2749. [DOI] [PubMed] [Google Scholar]
- 62.Chen H, Karne RJ, Hall G, Campia U, Panza JA, Cannon RO, 3rd, Wang Y, Katz A, Levine M, Quon MJ. High-dose oral vitamin C partially replenishes vitamin C levels in patients with Type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am J Physiol Heart Circ Physiol. 2006;290:H137–45. doi: 10.1152/ajpheart.00768.2005. [DOI] [PubMed] [Google Scholar]
- 63.Enstrom JE, Kanim LE, Klein MA. Vitamin C intake and mortality among a sample of the United States population. Epidemiology. 1992;3:194–202. doi: 10.1097/00001648-199205000-00003. [DOI] [PubMed] [Google Scholar]
- 64.Osganian SK, Stampfer MJ, Rimm E, Spiegelman D, Hu FB, Manson JE, Willett WC. Vitamin C and risk of coronary heart disease in women. J Am Coll Cardiol. 2003;42:246–52. doi: 10.1016/s0735-1097(03)00575-8. [DOI] [PubMed] [Google Scholar]
- 65.Boekholdt SM, Meuwese MC, Day NE, Luben R, Welch A, Wareham NJ, Khaw KT. Plasma concentrations of ascorbic acid and C-reactive protein, and risk of future coronary artery disease, in apparently healthy men and women: the EPIC-Norfolk prospective population study. Br J Nutr. 2006;96:516–22. [PubMed] [Google Scholar]
- 66.Kocak G, Aktan F, Canbolat O, Ozogul C, Elbeg S, Yildizoglu-Ari N, Karasu C. Alpha-lipoic acid treatment ameliorates metabolic parameters, blood pressure, vascular reactivity and morphology of vessels already damaged by streptozotocin-diabetes. Diabetes Nutr Metab. 2000;13:308–18. [PubMed] [Google Scholar]
- 67.Yi X, Maeda N. alpha-Lipoic acid prevents the increase in atherosclerosis induced by diabetes in apolipoprotein E-deficient mice fed high-fat/low-cholesterol diet. Diabetes. 2006;55:2238–44. doi: 10.2337/db06-0251. [DOI] [PubMed] [Google Scholar]
- 68.Kowluru RA, Odenbach S. Effect of long-term administration of alpha-lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes. 2004;53:3233–8. doi: 10.2337/diabetes.53.12.3233. [DOI] [PubMed] [Google Scholar]
- 69.Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T, Luther T, Berentshtein E, Tritschler H, Muller M, Wahl P, Ziegler R, Nawroth PP. Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes. 1997;46:1481–90. doi: 10.2337/diab.46.9.1481. [DOI] [PubMed] [Google Scholar]
- 70.Ziegler D, Hanefeld M, Ruhnau KJ, Meissner HP, Lobisch M, Schutte K, Gries FA. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized controlled trial (ALADIN Study) Diabetologia. 1995;38:1425–33. doi: 10.1007/BF00400603. [DOI] [PubMed] [Google Scholar]
- 71.Reljanovic M, Reichel G, Rett K, Lobisch M, Schuette K, Moller W, Tritschler HJ, Mehnert H. Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): a two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy. Free Radic Res. 1999;31:171–9. doi: 10.1080/10715769900300721. [DOI] [PubMed] [Google Scholar]
- 72.Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, Munzel U, Yakhno N, Raz I, Novosadova M, Maus J, Samigullin R. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29:2365–70. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- 73.Ziegler D, Schatz H, Conrad F, Gries FA, Ulrich H, Reichel G. Effects of treatment with the antioxidant alpha-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month randomized controlled multicenter trial (DEKAN Study). Deutsche Kardiale Autonome Neuropathie. Diabetes Care. 1997;20:369–73. doi: 10.2337/diacare.20.3.369. [DOI] [PubMed] [Google Scholar]
- 74.Sola S, Mir MQ, Cheema FA, Khan-Merchant N, Menon RG, Parthasarathy S, Khan BV. Irbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: results of the Irbesartan and Lipoic Acid in Endothelial Dysfunction (ISLAND) study. Circulation. 2005;111:343–8. doi: 10.1161/01.CIR.0000153272.48711.B9. [DOI] [PubMed] [Google Scholar]
- 75.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- 76.Lekakis JP, Papamichael CM, Barbaki P, Papaioannou TG, Stamatelopoulos KS, Dagre AG, Stamatelopoulos SF. Comparison of low-density lipoprotein cholesterol lowering by pravastatin to <100 mg/dl versus >100 mg/dl on brachial artery vasoreactivity in patients with severe hypercholesterolemia and previous atherosclerotic events or diabetes mellitus. Am J Cardiol. 2002;89:857–60. doi: 10.1016/s0002-9149(02)02200-2. [DOI] [PubMed] [Google Scholar]
- 77.Ceriello A, Assaloni R, Da Ros R, Maier A, Piconi L, Quagliaro L, Esposito K, Giugliano D. Effect of atorvastatin and irbesartan, alone and in combination, on postprandial endothelial dysfunction, oxidative stress, and inflammation in type 2 diabetic patients. Circulation. 2005;111:2518–24. doi: 10.1161/01.CIR.0000165070.46111.9F. [DOI] [PubMed] [Google Scholar]
- 78.Tsubouchi H, Inoguchi T, Sonta T, Sato N, Sekiguchi N, Kobayashi K, Sumimoto H, Utsumi H, Nawata H. Statin attenuates high glucose-induced and diabetes-induced oxidative stress in vitro and in vivo evaluated by electron spin resonance measurement. Free Radic Biol Med. 2005;39:444–52. doi: 10.1016/j.freeradbiomed.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 79.Ceylan A, Karasu C, Aktan F, Guven C, Can B, Ozansoy G. Effects of simvastatin treatment on oxidant/antioxidant state and ultrastructure of diabetic rat myocardium. Gen Physiol Biophys. 2003;22:535–47. [PubMed] [Google Scholar]
- 80.Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–9. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- 81.Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W, Itter G, Rosen R, Bohm M, Nickenig G. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. 2001;37:1450–7. doi: 10.1161/01.hyp.37.6.1450. [DOI] [PubMed] [Google Scholar]
- 82.Ceylan A, Karasu C, Aktan F, Ozansoy G. Simvastatin treatment restores vasoconstriction and the inhibitory effect of LPC on endothelial relaxation via affecting oxidizing metabolism in diabetic rats. Diabetes Nutr Metab. 2004;17:203–10. [PubMed] [Google Scholar]
- 83.Rosenson RS, Tangney CC, Levine DM, Parker TS, Gordon BR. Association between reduced low density lipoprotein oxidation and inhibition of monocyte chemoattractant protein-1 production in statin-treated subjects. J Lab Clin Med. 2005;145:83–7. doi: 10.1016/j.lab.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 84.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101:207–13. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 85.Morawietz H, Erbs S, Holtz J, Schubert A, Krekler M, Goettsch W, Kuss O, Adams V, Lenk K, Mohr FW, Schuler G, Hambrecht R. Endothelial Protection, AT1 blockade and Cholesterol-Dependent Oxidative Stress: the EPAS trial. Circulation. 2006;114:I296–301. doi: 10.1161/CIRCULATIONAHA.105.001313. [DOI] [PubMed] [Google Scholar]
- 86.Pietsch A, Erl W, Lorenz RL. Lovastatin reduces expression of the combined adhesion and scavenger receptor CD36 in human monocytic cells. Biochem Pharmacol. 1996;52:433–9. doi: 10.1016/0006-2952(96)00245-6. [DOI] [PubMed] [Google Scholar]
- 87.Umetani N, Kanayama Y, Okamura M, Negoro N, Takeda T. Lovastatin inhibits gene expression of type-I scavenger receptor in THP-1 human macrophages. Biochim Biophys Acta. 1996;1303:199–206. doi: 10.1016/0005-2760(96)00098-7. [DOI] [PubMed] [Google Scholar]
- 88.Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis. 1998;138:271–80. doi: 10.1016/s0021-9150(98)00032-x. [DOI] [PubMed] [Google Scholar]
- 89.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, Sanchez-Pascuala R, Hernandez G, Diaz C, Lamas S. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–9. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu Y, Ohmori K, Chen Y, Sato C, Kiyomoto H, Shinomiya K, Takeuchi H, Mizushige K, Kohno M. Effects of pravastatin on progression of glucose intolerance and cardiovascular remodeling in a type II diabetes model. J Am Coll Cardiol. 2004;44:904–13. doi: 10.1016/j.jacc.2004.04.050. [DOI] [PubMed] [Google Scholar]
- 91.Jimenez A, Arriero MM, Lopez-Blaya A, Gonzalez-Fernandez F, Garcia R, Fortes J, Millas I, Velasco S, Sanchez De Miguel L, Rico L, Farre J, Casado S, Lopez-Farre A. Regulation of endothelial nitric oxide synthase expression in the vascular wall and in mononuclear cells from hypercholesterolemic rabbits. Circulation. 2001;104:1822–30. doi: 10.1161/hc3901.095769. [DOI] [PubMed] [Google Scholar]
- 92.Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–9. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 93.Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, Thomason MJ, Mackness MI, Charlton-Menys V, Fuller JH. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 94.Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–16. doi: 10.1016/s0140-6736(03)13636-7. [DOI] [PubMed] [Google Scholar]
- 95.Mansourati J, Newman LG, Roman SH, Travis A, Rafey M, Phillips RA. Lipid lowering does not improve endothelial function in subjects with poorly controlled diabetes. Diabetes Care. 2001;24:2152–3. doi: 10.2337/diacare.24.12.2152. [DOI] [PubMed] [Google Scholar]
- 96.Dogra GK, Watts GF, Chan DC, Stanton K. Statin therapy improves brachial artery vasodilator function in patients with Type 1 diabetes and microalbuminuria. Diabet Med. 2005;22:239–42. doi: 10.1111/j.1464-5491.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 97.Mullen MJ, Wright D, Donald AE, Thorne S, Thomson H, Deanfield JE. Atorvastatin but not L-arginine improves endothelial function in type I diabetes mellitus: a double-blind study. J Am Coll Cardiol. 2000;36:410–6. doi: 10.1016/s0735-1097(00)00743-9. [DOI] [PubMed] [Google Scholar]
- 98.Tsunekawa T, Hayashi T, Kano H, Sumi D, Matsui-Hirai H, Thakur NK, Egashira K, Iguchi A. Cerivastatin, a hydroxymethylglutaryl coenzyme a reductase inhibitor, improves endothelial function in elderly diabetic patients within 3 days. Circulation. 2001;104:376–9. doi: 10.1161/hc2901.094094. [DOI] [PubMed] [Google Scholar]
- 99.Economides PA, Caselli A, Tiani E, Khaodhiar L, Horton ES, Veves A. The effects of atorvastatin on endothelial function in diabetic patients and subjects at risk for type 2 diabetes. J Clin Endocrinol Metab. 2004;89:740–7. doi: 10.1210/jc.2003-031116. [DOI] [PubMed] [Google Scholar]
- 100.Tan KC, Chow WS, Tam SC, Ai VH, Lam CH, Lam KS. Atorvastatin lowers C-reactive protein and improves endothelium-dependent vasodilation in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2002;87:563–8. doi: 10.1210/jcem.87.2.8249. [DOI] [PubMed] [Google Scholar]
- 101.Tousoulis D, Antoniades C, Vasiliadou C, Kourtellaris P, Koniari K, Marinou K, Charakida M, Ntarladimas I, Siasos G, Stefanadis C. Effects of atorvastatin and vitamin C on forearm hyperaemic blood flow, asymmentrical dimethylarginine levels and the inflammatory process in patients with type 2 diabetes mellitus. Heart. 2007;93:244–6. doi: 10.1136/hrt.2006.093112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ceriello A, Taboga C, Tonutti L, Quagliaro L, Piconi L, Bais B, Da Ros R, Motz E. Evidence for an independent and cumulative effect of postprandial hypertriglyceridemia and hyperglycemia on endothelial dysfunction and oxidative stress generation: effects of short- and long-term simvastatin treatment. Circulation. 2002;106:1211–8. doi: 10.1161/01.cir.0000027569.76671.a8. [DOI] [PubMed] [Google Scholar]
- 103.Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134:2842S–2847S. doi: 10.1093/jn/134.10.2842S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- 104.Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M, Nickenig G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. 2004;94:534–41. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- 105.Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes. 2007;56:118–26. doi: 10.2337/db06-0288. [DOI] [PubMed] [Google Scholar]
- 106.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–93. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 108.Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res. 2007;100:1016–25. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- 109.Cheetham C, O’Driscoll G, Stanton K, Taylor R, Green D. Losartan, an angiotensin type I receptor antagonist, improves conduit vessel endothelial function in Type II diabetes. Clin Sci (Lond) 2001;100:13–7. [PubMed] [Google Scholar]
- 110.Cheetham C, Collis J, O’Driscoll G, Stanton K, Taylor R, Green D. Losartan, an angiotensin type 1 receptor antagonist, improves endothelial function in non-insulin-dependent diabetes. J Am Coll Cardiol. 2000;36:1461–6. doi: 10.1016/s0735-1097(00)00933-5. [DOI] [PubMed] [Google Scholar]
- 111.O’Driscoll G, Green D, Maiorana A, Stanton K, Colreavy F, Taylor R. Improvement in endothelial function by angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1999;33:1506–11. doi: 10.1016/s0735-1097(99)00065-0. [DOI] [PubMed] [Google Scholar]
- 112.Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care. 1998;21:631–6. doi: 10.2337/diacare.21.4.631. [DOI] [PubMed] [Google Scholar]
- 113.Komers R, Simkova R, Kazdova L, Ruzickova J, Pelikanova T. Effects of ACE inhibition and AT1-receptor blockade on haemodynamic responses to L-arginine in Type 1 diabetes. J Renin Angiotensin Aldosterone Syst. 2004;5:33–8. doi: 10.3317/jraas.2004.006. [DOI] [PubMed] [Google Scholar]
- 114.Schalkwijk CG, Smulders RA, Lambert J, Donker AJ, Stehouwer CD. ACE-inhibition modulates some endothelial functions in healthy subjects and in normotensive type 1 diabetic patients. Eur J Clin Invest. 2000;30:853–60. doi: 10.1046/j.1365-2362.2000.00721.x. [DOI] [PubMed] [Google Scholar]
- 115.McFarlane R, McCredie RJ, Bonney MA, Molyneaux L, Zilkens R, Celermajer DS, Yue DK. Angiotensin converting enzyme inhibition and arterial endothelial function in adults with Type 1 diabetes mellitus. Diabet Med. 1999;16:62–6. doi: 10.1046/j.1464-5491.1999.00021.x. [DOI] [PubMed] [Google Scholar]
- 116.Mullen MJ, Clarkson P, Donald AE, Thomson H, Thorne SA, Powe AJ, Furuno T, Bull T, Deanfield JE. Effect of enalapril on endothelial function in young insulin-dependent diabetic patients: a randomized, double-blind study. J Am Coll Cardiol. 1998;31:1330–5. doi: 10.1016/s0735-1097(98)00099-0. [DOI] [PubMed] [Google Scholar]
- 117.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 118.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 119.Nickenig G, Sachinidis A, Michaelsen F, Bohm M, Seewald S, Vetter H. Upregulation of vascular angiotensin II receptor gene expression by low-density lipoprotein in vascular smooth muscle cells. Circulation. 1997;95:473–8. doi: 10.1161/01.cir.95.2.473. [DOI] [PubMed] [Google Scholar]
- 120.Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, Bohm M, Meinertz T, Munzel T. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–33. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 121.Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, Bohm M, Nickenig G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–54. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 122.Nickenig G, Jung O, Strehlow K, Zolk O, Linz W, Scholkens BA, Bohm M. Hypercholesterolemia is associated with enhanced angiotensin AT1-receptor expression. Am J Physiol. 1997;272:H2701–7. doi: 10.1152/ajpheart.1997.272.6.H2701. [DOI] [PubMed] [Google Scholar]
- 123.Nickenig G, Sachinidis A, Seewald S, Bohm M, Vetter H. Influence of oxidized low-density lipoprotein on vascular angiotensin II receptor expression. J Hypertens Suppl. 1997;15:S27–30. doi: 10.1097/00004872-199715066-00006. [DOI] [PubMed] [Google Scholar]
- 124.Strehlow K, Wassmann S, Bohm M, Nickenig G. Angiotensin AT1 receptor over-expression in hypercholesterolaemia. Ann Med. 2000;32:386–9. doi: 10.3109/07853890008995944. [DOI] [PubMed] [Google Scholar]
- 125.Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, Zerkowski HR, Sawamura T, Holtz J. Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation. 1999;100:899–902. doi: 10.1161/01.cir.100.9.899. [DOI] [PubMed] [Google Scholar]
- 126.Caballero AE, Saouaf R, Lim SC, Hamdy O, Abou-Elenin K, O’Connor C, Logerfo FW, Horton ES, Veves A. The effects of troglitazone, an insulin-sensitizing agent, on the endothelial function in early and late type 2 diabetes: a placebo-controlled randomized clinical trial. Metabolism. 2003;52:173–80. doi: 10.1053/meta.2003.50023. [DOI] [PubMed] [Google Scholar]
- 127.Mittermayer F, Schaller G, Pleiner J, Krzyzanowska K, Kapiotis S, Roden M, Wolzt M. Rosiglitazone prevents free fatty acid-induced vascular endothelial dysfunction. J Clin Endocrinol Metab. 2007;92:2574–80. doi: 10.1210/jc.2006-2130. [DOI] [PubMed] [Google Scholar]
- 128.Libby P, Plutzky J. Inflammation in diabetes mellitus: role of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma agonists. Am J Cardiol. 2007;99:27B–40B. doi: 10.1016/j.amjcard.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 129.Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB, Sr, Perez A, Provost JC, Haffner SM. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. Jama. 2006;296:2572–81. doi: 10.1001/jama.296.21.joc60158. [DOI] [PubMed] [Google Scholar]
- 130.Stocker DJ, Taylor AJ, Langley RW, Jezior MR, Vigersky RA. A randomized trial of the effects of rosiglitazone and metformin on inflammation and subclinical atherosclerosis in patients with type 2 diabetes. Am Heart J. 2007;153:445 e1–6. doi: 10.1016/j.ahj.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 131.Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, Doerries C, Horvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–73. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- 132.Wang CH, Ting MK, Verma S, Kuo LT, Yang NI, Hsieh IC, Wang SY, Hung A, Cherng WJ. Pioglitazone increases the numbers and improves the functional capacity of endothelial progenitor cells in patients with diabetes mellitus. Am Heart J. 2006;152:1051 e1–8. doi: 10.1016/j.ahj.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 133.Tao L, Liu HR, Gao E, Teng ZP, Lopez BL, Christopher TA, Ma XL, Batinic-Haberle I, Willette RN, Ohlstein EH, Yue TL. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-gamma agonist in hypercholesterolemia. Circulation. 2003;108:2805–11. doi: 10.1161/01.CIR.0000097003.49585.5E. [DOI] [PubMed] [Google Scholar]
- 134.Graham DJ, Green L, Senior JR, Nourjah P. Troglitazone-induced liver failure: a case study. Am J Med. 2003;114:299–306. doi: 10.1016/s0002-9343(02)01529-2. [DOI] [PubMed] [Google Scholar]
- 135.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 136.Couzin J. Drug safety. Heart attack risk overshadows a popular diabetes therapy. Science. 2007;316:1550–1. doi: 10.1126/science.316.5831.1550. [DOI] [PubMed] [Google Scholar]
- 137.Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, Komajda M, McMurray JJ. Rosiglitazone evaluated for cardiovascular outcomes–an interim analysis. N Engl J Med. 2007;357:28–38. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- 138.Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. Jama. 2007;298:1189–95. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- 139.Dargie HJ, Hildebrandt PR, Riegger GA, McMurray JJ, McMorn SO, Roberts JN, Zambanini A, Wilding JP. A randomized, placebo-controlled trial assessing the effects of rosiglitazone on echocardiographic function and cardiac status in type 2 diabetic patients with New York Heart Association Functional Class I or II Heart Failure. J Am Coll Cardiol. 2007;49:1696–704. doi: 10.1016/j.jacc.2006.10.077. [DOI] [PubMed] [Google Scholar]
- 140.Ghazzi MN, Perez JE, Antonucci TK, Driscoll JH, Huang SM, Faja BW, Whitcomb RW. Cardiac and glycemic benefits of troglitazone treatment in NIDDM. The Troglitazone Study Group. Diabetes. 1997;46:433–9. doi: 10.2337/diab.46.3.433. [DOI] [PubMed] [Google Scholar]
- 141.Erdmann E, Dormandy JA, Charbonnel B, Massi-Benedetti M, Moules IK, Skene AM. The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction: results from the PROactive (PROactive 05) Study. J Am Coll Cardiol. 2007;49:1772–80. doi: 10.1016/j.jacc.2006.12.048. [DOI] [PubMed] [Google Scholar]
- 142.Negro R, Dazzi D, Hassan H, Pezzarossa A. Pioglitazone reduces blood pressure in non-dipping diabetic patients. Minerva Endocrinol. 2004;29:11–7. [PubMed] [Google Scholar]
- 143.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. Jama. 2007;298:1180–8. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 144.Derosa G, Cicero AF, D’Angelo A, Gaddi A, Ciccarelli L, Piccinni MN, Salvadeo SA, Pricolo F, Ferrari I, Gravina A, Ragonesi PD. Effects of 1 year of treatment with pioglitazone or rosiglitazone added to glimepiride on lipoprotein (a) and homocysteine concentrations in patients with type 2 diabetes mellitus and metabolic syndrome: a multicenter, randomized, double-blind, controlled clinical trial. Clin Ther. 2006;28:679–88. doi: 10.1016/j.clinthera.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 145.Veves A, Freeman R. Diagnosing endothelial cell dysfunction. In: Aird WC, editor. Endothelial Biomedicine. Cambridge University Press; New York: 2007. [Google Scholar]
- 146.Danias P, Saouaf R. Noninvasive methods to assess vascular function and pathophysiology. In: Johnstone MT, Veves A, editors. Diabetes and Cardiovascular Disease. Humana Press; Totowa, NJ: 2005. [Google Scholar]