

Abstract
Activation of G protein-gated inwardly rectifying potassium (GIRK) channels leads to a hyperpolarization of the neuron’s membrane potential, providing an important component of inhibition in the brain. In addition to the canonical G protein-activation pathway, GIRK channels are activated by small molecules but less is known about the underlying gating mechanisms. One drawback to previous studies has been the inability to control intrinsic and extrinsic factors. Here we used a reconstitution strategy with highly purified mammalian GIRK2 channels incorporated into liposomes and demonstrate that cholesterol or intoxicating concentrations of ethanol, i.e., >20 mM, each activate GIRK2 channels directly, in the absence of G proteins. Notably, both activators require the membrane phospholipid PIP2 but appear to interact independently with different regions of the channel. Elucidating the mechanisms underlying G protein-independent pathways of activating GIRK channels provides a unique strategy for developing new types of neuronal excitability modulators.
Introduction
Many modulatory neurotransmitters in the brain, such as dopamine, acetylcholine, serotonin and GABA, inhibit neuronal activity by stimulating G protein-coupled receptors (GPCRs) that couple to G protein-gated inwardly rectifying (GIRK, also referred to as Kir3) channels1. The activation of GIRK channels hyperpolarizes the neuron’s membrane potential, and thereby reduces action potential firing1, 2. GIRK channels are widely expressed in the brain, existing as predominantly heterotetramers of three different GIRK channel subunits, GIRK1, GIRK2 and/or GIRK3, or as homotetramers of the GIRK2 subunit1, 2. GIRK channels have been implicated in the pathophysiology of several human neurological disorders3. Genome-wide association studies (GWAS) of people with schizophrenia have identified single nucleotide polymorphisms (SNPs) in the Kcnj3 (GIRK1) gene4. SNPs in Kcnj6 (GIRK2) have been linked to alcohol and nicotine dependence, reduced opioid withdrawal and increased opioid requirement for analgesia5–8. Recently, a mutation in Kcnj6 has been proposed to contribute to Keppen-Lubinsky syndrome, a severe developmental disorder with cognitive deficits9.
Prior work on GIRK channels has focused on delineating the G protein-dependent pathway for activation of GIRK channels. Following stimulation of GPCRs that couple to pertussis toxin-sensitive Gi/o G proteins, the G protein Gβγ subunits bind directly to the channel, and induce a conformational change that opens the channel in a manner that depends on the membrane phospholipid PI(4,5)P2 (referred to as PIP2)10–14. Recently, this interaction was confirmed in an atomic resolution structure based on x-ray crystallography of GIRK2 in complex with Gβγ15. Activation of GIRK channels through so-called ‘G protein-independent’ pathways, however, are poorly understood. For example, GIRK channels are activated by alcohol16–18 and, similar to Gβγ activation, requires PIP2 19. However, while previous studies with heterologous expression systems and neurons suggested that alcohol directly activates GIRK channels16–19, the role of endogenous Gβγ subunits could not be unequivocally dismissed. Furthermore, alternative mechanisms through which alcohol could exert its effects were possible, such as increasing the fluidity of the plasma membrane20, 21. Another example of a putative G protein-independent modulator is cholesterol. Cholesterol, which accounts for up to 50% of the total lipid membrane content22, has been shown to modulate the activity of potassium channels23–28, including cardiac GIRK channels, i.e., GIRK1/GIRK4 subunits23, 27. However, whether cholesterol affects brain GIRK channels, comprised mostly of GIRK2-containing tetramers, requires Gβγ subunits, and interacts with alcohol-dependent activation is unknown1.
In previous studies using heterologous expression systems, e.g., Xenopus oocytes, HEK-293 cells, or neurons, the contribution of endogenous proteins could not be entirely eliminated or controlled29, 30. For example, cholesterol concentration in plasma membranes is affected by numerous factors and varies between expression systems31. To elucidate more definitively the mechanisms of GIRK activation, a different approach is needed in which the lipid components, G proteins and activators can be all controlled experimentally. In the current study, we implemented a methodology in which we purified GIRK2 channels, reconstituted the channels into liposomes with different lipid compositions and then probed channel function with exogenous ligands. We demonstrate that alcohol and cholesterol directly activate neuronal GIRK2 channels, in the absence of G protein Gβγ subunits but requiring PIP2. Interestingly, although PIP2 has been considered a co-factor that is necessary, but not sufficient, for channel activation, here we demonstrate that PIP2 is sufficient to activate GIRK2 channels in the presence of Na+. Further, we discovered that alcohol and cholesterol appear to interact structurally with different regions of the channel. Determining the mechanism of activation of GIRK channels by modulators that bypass G protein-gating could provide a new therapeutic strategy for treating a variety of human diseases.
Results
PIP2 is necessary and sufficient to activate GIRK2 channels
Mouse GIRK2 channels were expressed in Pichia pastoris, purified, and reconstituted into liposomes of defined compositions (Fig. 1a), following a protocol described previously15, 32. GIRK2 channels express efficiently in Pichia pastoris and retain their activation by Gβγ subunits15. To study K+ flux through GIRK2 channels, we used a fluorescence-based K+ flux assay15 (Fig. 1b) with a plate-reader equipped with a fluidic system to add compounds acutely. In GIRK2-containing liposomes loaded with K+ and incubated with the membrane permeable pH-sensitive ACMA dye, the addition of the proton ionophore CCCP results in quenching of the ACMA dye if GIRK channels are open, i.e., H+ enter via CCCP if K+ can exit the proteoliposome (Fig. 1b). Thus the decrease in fluorescence, i.e., quenching, provides a measure of the relative K+ flux that correlates directly with GIRK2 channel activity.
Figure 1.
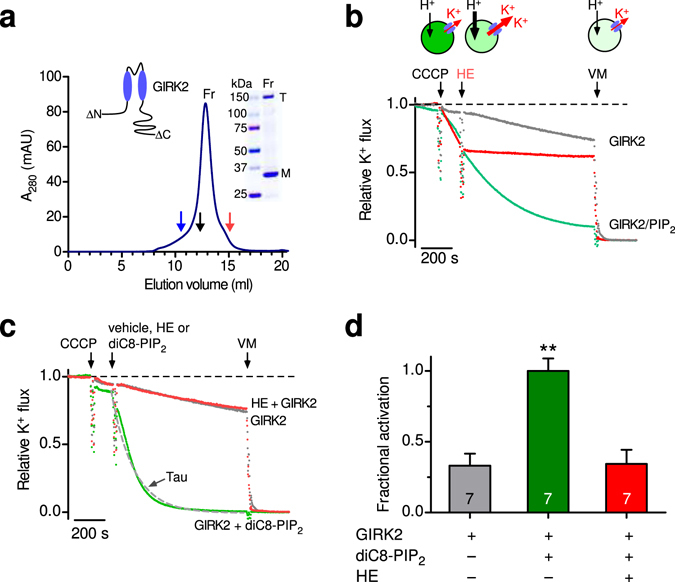
Brain PIP2 and soluble PIP2 activate reconstituted brain GIRK2 channels in the absence of other proteins. (a) Graph shows purification of mouse GIRK2 from Pichia pastoris. The absorbance is plotted as a function of elution volume, with molecular weight (MW) standards; Blue - 440 kDa, Black - 158 kDa, Red - 75 kDa (arrows). The major peak elutes at a volume consistent with the size of a GIRK2 tetramer (T). Inset, Coomassie blue staining of a gradient SDS-PAGE protein gel shows two bands, one for tetramer (T) and one for monomer (M). Inset, wild-type GIRK2 was truncated at the amino- and carboxy-termini (see methods). (b) Example of K+ flux assay with GIRK2 reconstituted into PE/PG liposomes in the absence or presence of brain PIP2 (1%) (see methods). The fluorescence is normalized and plotted as a function of time (‘Relative K+ flux’). Arrows indicate addition of the proton ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP), the GIRK2 channel blocker MTS-HE (HE, 100 μM) and valinomycin (VM). Note the quenching of fluorescence for GIRK2 with PIP2 (green) upon addition of CCCP, which is attenuated with addition of MTS-HE. Dashed line represents no flux. (c) Representative fluorescent traces of GIRK2 alone (grey), GIRK2 and MTS-HE (red), and GIRK2 with acute application of 30 μM diC8-PIP2 (green). The decay was fit with a single exponential to determine that rate constant (1/τ, s-1). (d) Bar graph shows the average ( ± SEM) fractional activation with diC8-PIP2 based on the change in steady-state fluorescence (green bar vs. grey bar). MTS-HE (red bar) significantly inhibits diC8-PIP2-activated GIRK2 channels (n = 7, P < 0.0001).
Purified GIRK2 channels, containing amino acids 52 - 380, were reconstituted into liposomes containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG) at a 3:1 ratio15. PE:PG lipids were found previously to support both Gβγ and Na+-dependent activation15. We first examined the requirement of PIP2 for channel activation12, 14. PIP2 is found in the plasma membrane at concentrations up to 1%33 and has been shown previously to be required for GIRK activity12, 14, 34. We measured the relative K+ flux through GIRK2 channels reconstituted in either the absence or presence of 1% brain PIP2, under conditions that saturate the cytoplasmic Na+ binding site in GIRK235, 36. GIRK2-containing liposomes containing brain PIP2 exhibited a robust quenching of fluorescence upon addition of CCCP (‘GIRK2 /PIP2’, Fig. 1b). Subsequent addition of a small molecular inhibitor of GIRK2 channels, MTS-HE (100 μM)37, abruptly slowed the rate of quenching, indicating closure of GIRK2 channels (‘GIRK2/PIP2 + HE’, Fig. 1b). Similarly, pre-incubating GIRK2-containing liposomes with the K channel inhibitor BaCl2 (2 mM) prevented the CCCP-dependent quenching (data not shown). In the absence of PIP2, however, there was no significant change in fluorescence, i.e., no quenching, upon CCCP addition (‘GIRK2’, Fig. 1b). Thus, under these defined conditions of PE/PG lipids and cytoplasmic Na+, brain PIP2 is necessary and sufficient for robust activation of GIRK2 channels12, 14, 35, 36.
We next examined whether GIRK2 channels could be directly activated by PIP2 applied in real-time. Previous studies demonstrated that analogs of PIP2 with shorter tails, e.g., diC8-PIP2, can also activate GIRK channels38. With GIRK2-containing liposomes, the acute addition of diC8-PIP2 (30 μM) revealed potent activation, i.e., increase in the rate of quenching (Fig. 1c). To quantify the change in K+ flux, we measured both the steady state fluorescence at the end of 15 minutes and the rate of quenching following the addition of diC8-PIP2 (see methods for details). Using both analyses, diC8-PIP2 clearly activates GIRK2 channels (Figs 1d and 2a,b).
Figure 2.

Potency of PIP2 activation of GIRK2 channels. (a) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2 following addition of the indicated concentration of diC8-PIP2 or MTS-HE (100 μM) (n = 9). (b) Plot of the rate of K+ flux as a function of diC8-PIP2 concentration. Line shows best fit using the Hill equation with apparent EC50 of 25.1 ± 3.3 μM and Hill coefficient of 1.7 ± 0.1 (n = 10). (c) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 1% brain PIP2 upon addition of the indicated concentration of neomycin, a competitive inhibitor of PIP2 (n = 4). (d) Plot of the fractional inhibition of K+ flux as a function of neomycin concentration. Fraction inhibition was calculated using the apparent steady-state K+ flux measurement. Line shows best fit using the Hill equation with IC50 for neomycin inhibition 11.1 ± 2.4 μM and with a Hill coefficient of 1.3 ± 0.3 (n = 4).
To determine the PIP2 sensitivity, we examined the effect of applying different concentrations of diC8-PIP2 to GIRK2-containing liposomes (Fig. 2a). Note the progressively faster rate of quenching with higher diC8-PIP2 concentrations. Plotting the rate of K+ flux (1/tau) as a function of the diC8-PIP2 concentration reveals a concentration-dependent increase in the rate of quenching. The best-fit with the Hill equation indicates an apparent EC50 of 25.1 ± 3.3 μM and a Hill coefficient of 1.7 ± 0.1 (n = 10) (Fig. 2b). The EC50 with diC8-PIP2 is higher than for that for GIRK2 reconstituted in bilayers in the presence of Gβγ (~15 μM)34 and lower than that for GIRK1/GIRK4 channels expressed heterologously (~ 45 μM)39.
To probe the sensitivity of GIRK2 channel activation by brain PIP2, we examined the effect of neomycin, which is a competitive inhibitor of PIP2 40, on the K+ flux with GIRK2 channels. Increasing concentrations of neomycin applied acutely to GIRK2-containing liposomes with brain PIP2 attenuated the rate of quenching (Fig. 2c). The IC50 for neomycin inhibition is 11.1 ± 2.4 μM (n = 4), with a Hill coefficient of 1.3 ± 0.3 (Fig. 2d). Taken together, these experiments demonstrate that both brain PIP2 and soluble diC8-PIP2 activate GIRK2 channels in a dose-dependent manner, in the absence of G proteins or other ligands, indicating that GIRK2 channels are directly gated by PIP2 in the presence of Na+.
Direct action of alcohol on brain GIRK2 channels
In addition to G proteins, GIRK channels are activated by alcohols16–18. In order to address whether this property is retained in the purified protein, we examined the effect of directly applying ethanol to GIRK2-containing liposomes with brain PIP2. Ethanol, added at increasing concentrations, produced a dose-dependent increase in the relative rate of K+ flux (Fig. 3a). Similarly, increasing concentrations of propanol also enhanced the relative rate of K+ flux (Fig. 3b). We calculated the change in the rate of K+ flux, relative to 0 mM alcohol, and plotted the normalized rate as a function of alcohol concentration (Fig. 3c). Note the increase in the normalized rate of K+ flux with concentrations greater than 10 mM, a rank order of activation with propanol greater than ethanol, and no apparent saturation at 200 mM. These properties of alcohol activation are remarkably similar to those with GIRK2 channels expressed heterologously in HEK293 cells16–18. Importantly, alcohol-dependent activation required brain PIP2 (Fig. 3d). Thus, we show for the first time that alcohol-dependent activation of GIRK channels requires only PIP2, and no other proteins, e.g., Gβγ subunits.
Figure 3.
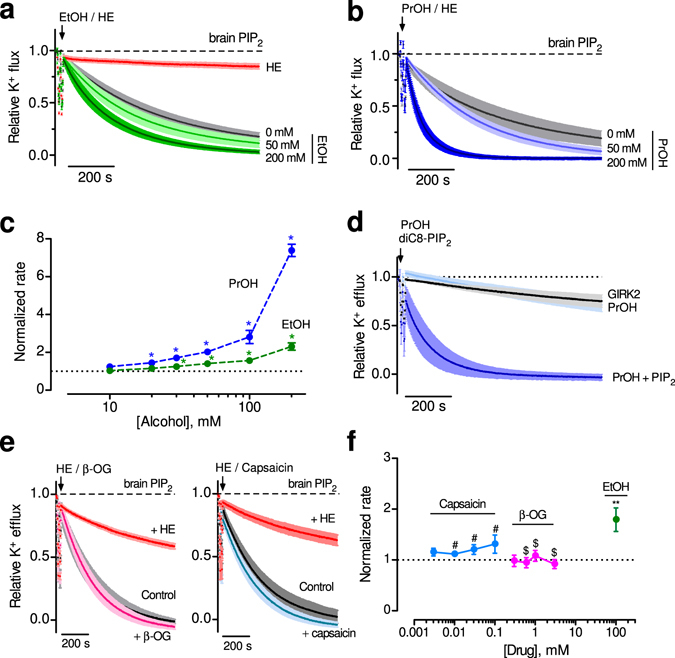
Alcohol directly activates GIRK2 channels in absence of G proteins. (a,b) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 1% brain PIP2 and increasing concentrations of ethanol (EtOH) (a) (n = 12) (green traces) or propanol (PrOH) (b) (n = 4–6) (blue traces). (c) The normalized rate of K+ flux is plotted as a function of ethanol (green) and propanol (blue) concentration. Inset, zoom of the dose-response curves over the physiological alcohol concentrations. * p < 0.05 vs. 10 mM using one-way ANOVA and Dunnett’s post hoc test (n = 5–6 for PrOH and 11–12 for EtOH). (d) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2 in the presence of 100 mM PrOH in the absence (light blue, n = 3) or presence of 50 μM diC8-PIP2 (dark blue, n = 4). GIRK2 in the absence of both PrOH and diC8-PIP2 is shown for comparison (black, n = 4). (e) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 1% brain PIP2 and 1 mM β-OG (left) (n = 4, magenta) or 30 μM capsaicin (right) (n = 4, blue). MTS-HE inhibition is shown for comparison (red). (f) The normalized rate of K+ flux is plotted as a function of different β-OG (pink) or capsaicin (blue) concentrations, and is compared to 100 mM EtOH (green). #not significant vs. 3 μM Capsaicin, $not significant vs. 0.3 mM β-OG, ** P < 0.05 vs. 3 μM Capsaicin or 0.3 mM β-OG using one-way ANOVA and Dunnett’s post hoc test.
Using the lipid-reconstitution system, we could now investigate the possibility that changes in lipid membrane dynamics could affect GIRK2 function20, 21. We tested the effect of compounds demonstrated previously to increase membrane elasticity, e.g., β-octyl glucoside (β-OG) and capsaicin41, 42. Direct application of β-OG (1 mM) or capsaicin (30 μM) did not alter the rate of K+ flux for GIRK2 channels reconstituted into liposomes with brain PIP2 (Fig. 3e). There was no significant change in the rate of K+ flux with increasing concentrations of β-OG or capsaicin, in contrast to the change in K+ flux with ethanol (Fig. 3f). Taken together, these experiments demonstrate that known lipid-membrane disruptors have little effect on GIRK2 channel activity, providing additional support for the direct activation of GIRK2 by ethanol.
Direct action of cholesterol on brain GIRK2 channels
Cholesterol inhibits constitutively open inwardly-rectifying potassium channels25, 26 but appears to activate cardiac (GIRK1/GIRK4) and brain (GIRK2) channels23, 27, 43. Whether cholesterol acts directly on brain GIRK2 channels in the absence of Gβγ is unknown. Using GIRK2-containing liposomes that have brain PIP2 (1%), we examined the effect of cholesterol on the relative K+ flux. The basal rate of K+ flux through GIRK2 channels was significantly enhanced in membranes containing 5% cholesterol (Fig. 4a). Both 5% and 10% cholesterol significantly increased the relative rate of K+ flux, while 1% was ineffective (Fig. 4b ). Interestingly, the relative rate of K+ flux was similar for 5% and 10% cholesterol, suggesting near saturation for this response.
Figure 4.
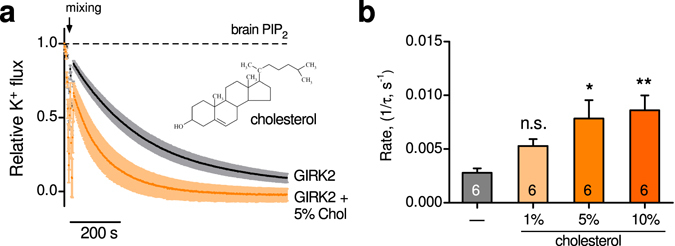
Cholesterol directly activates GIRK2 channels. (a) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 1% brain PIP2 in the absence (black) or presence of 5% cholesterol (orange) (n = 6). Inset, chemical structure of cholesterol is shown. (b) Bar graph shows the increase in the rate of K+ flux with different cholesterol concentrations (1%, 5%, and 10%). Statistical significance * p < 0.05, ** p < 0.01 (n = 6).
GIRK2 channels reconstituted in cholesterol-containing liposomes (5%) lacking brain PIP2 exhibited no basal K+ flux, indicating that PIP2 is required for cholesterol activation, similar to alcohol activation. We hypothesized that cholesterol may enhance GIRK2 activity by altering the sensitivity to PIP2. To test this, we measured the rate of K+ flux in GIRK2-containing liposomes containing 5% cholesterol that were directly exposed to different concentrations of diC8-PIP2 (Fig. 5a). The dose-response curve for diC8-PIP2 in the presence of cholesterol shifted to lower concentrations, compared to that without cholesterol (Fig. 5b). The EC50 for PIP2 activation of GIRK2 in the presence of cholesterol was 12.2 ± 2.5 μM (n = 6), compared to an EC50 of 25.1 ± 3.3 μM without cholesterol. Thus, the apparent affinity for PIP2 increases nearly 2-fold in the presence of cholesterol. In GIRK2 proteoliposomes containing both brain PIP2 (1%) and 5% cholesterol, acute application of neomycin decreased the rate of K+ flux, with an IC50 of 15.0 ± 4.6 μM (Fig. 5c,d). The IC50 for neomycin inhibition is indistinguishable from that measured in the absence of cholesterol (Fig. 2d), suggesting little difference in the off-rate of PIP2. Thus, in addition to alcohol, cholesterol appears to directly activate GIRK2 channels through enhancement of the interaction with PIP2.
Figure 5.

Cholesterol enhances sensitivity of GIRK2 channels to PIP2. (a) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 5% cholesterol in response to increasing concentrations of diC8-PIP2 (green) or MTS-HE (100 μM, red). Note little change in K+ flux in the absence of PIP2 (black) similar to that of liposomes without cholesterol (n = 7). (b) The rate of K+ flux is plotted as a function of different diC8-PIP2 concentrations for GIRK2 in the presence of 5% cholesterol (mustard circles). Line shows best fit using the Hill equation with EC50 of 12.2 ± 2.5 μM and a Hill coefficient of 1.5 ± 0.2 (n = 6). For reference, dose-response without cholesterol from Fig. 2B is shown (green circles). (c) Normalized fluorescent traces (mean ± SEM) show K+ flux for GIRK2-containing liposomes with 1% brain PIP2 / 5% cholesterol with different concentrations of neomycin (n = 5). (d) The rate of K+ flux for GIRK2/brain PIP2/5% cholesterol is plotted as a function of different neomycin concentrations. Line shows best fit using the Hill equation with an IC50 15.0 ± 4.6 μM and Hill coefficient of 1.2 ± 0.3 (n = 5).
Cholesterol does not affect the alcohol sensitivity of GIRK2 channels
Both alcohol and cholesterol activate GIRK2 channels by increasing the apparent affinity for PIP2. We next investigated the interaction between alcohol and cholesterol on GIRK2 activation. With GIRK2-containing liposomes in 5% cholesterol (a saturating concentration in our experiments), ethanol enhanced the relative K+ flux to a greater extent than for cholesterol alone or ethanol alone (Fig. 6a). The rate of K+ flux with 5% cholesterol increased over a range of ethanol concentrations (Fig. 6b). However, the dose-response curves completely overlap over the physiological range of activation, e.g., 10–100 mM, after adjusting for the choleseterol-dependent increase in the basal K+ flux (Fig. 4b), (Fig. 6b , inset). These results indicate that the effects of alcohol and cholesterol on GIRK channel are additive, suggesting these two modulators increase channel activity via separate structural sites in the channel.
Figure 6.
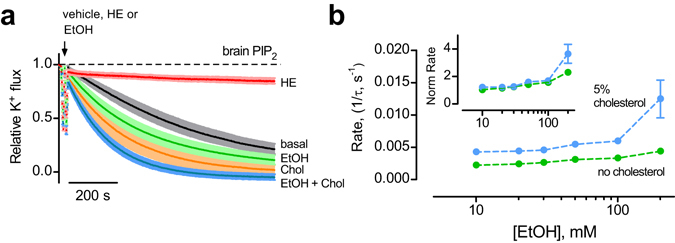
Cholesterol does not affect the ethanol sensitivity of GIRK2 channels. (a) Normalized fluorescent traces (mean ± SEM) show the K+ flux for GIRK2-containing liposomes with 1% brain PIP2 (black trace), and either 100 mM EtOH (green), 5% cholesterol (orange), or both 100 mM EtOH and 5% cholesterol (blue trace) (n = 4–12) (b) The rate of K+ flux for GIRK2 with PIP2 and 5% cholesterol (blue) or PIP2 alone (green) is plotted as a function of different EtOH concentrations. Inset, the normalized rate of K+ flux is plotted as a function of EtOH concentration.
Discussion
GIRK channels provide a major pathway for inhibition in the brain that is important in both normal and diseased states. The canonical pathway for GIRK channel activation is mediated by G protein Gβγ subunits, which occurs following stimulation of a Gi/o-coupled GPCRs, such as GABAB, dopamine D2 or mu opioid receptors1, 2. Recent crystal structures of GIRK2 in complex with Gβγ and PIP2 have provided structural and mechanistic details of how Gβγ associates with GIRK channels15, 32. It has become increasingly evident that G protein-independent pathways also exist for modulating GIRK channels; however, much less is known about the mechanism of action. A majority of the identified compounds that modulate GIRK channels independently of G proteins appear to inhibit GIRK channel activity44–48. Conversely, the number of drugs that directly activate GIRK channels is small16, 17, 49, 50. Here, we demonstrate for the first time that ethanol and cholesterol directly activate GIRK2 channels via changes in the interaction with PIP2, and do not require the presence of G proteins or other endogenous proteins.
In previous studies, extracellular application of alcohol enhanced GIRK channel activity, both with heterologously expressed channels, or with natively expressed channels in cerebellar neurons16, 17. Alcohol activation was determined to be insensitive to pertussis toxin (PTX) or antibodies to the G protein β1 subunit, suggesting that G proteins were not involved17. Further, deletion studies16, the identification of an alcohol pocket in x-ray crystallographic structures18, 51 and alcohol-tagging studies19 all indicated that alcohol activation was likely mediated by the direct interaction of alcohol with the channel. However, the involvement of endogenous proteins, e.g. Gβγ subunits, could not be ruled out with heterologous expression systems, such as oocytes or HEK293 cells, or native cell systems, i.e., neurons. In addition, a component of ethanol activation could include an alteration in the lipid bilayer fluidity21, since this was not directly tested. By reconstituting purified GIRK2 channels in liposomes, controlling the components of the bilayer, and adding alcohol in isolation of any other proteins, we could overcome these previous limitations and demonstrate that intoxicating concentrations of ethanol, i.e., >20 mM, directly activate GIRK2 channels in the presence of PIP2. For reference, a blood alcohol level of 0.08% is ~ 17 mM, binge-drinking levels can reach 50 mM, and the anesthetic concentration is around 190 mM52. On the other hand, we find that two types of lipid disruptive amphiphiles, e.g., β-octyl glucoside (β-OG) and capsaicin, do not activate GIRK2 channels. Thus, the mechanism of ethanol activation of GIRK2 channels is unlikely to involve an indirect component from an alcohol-dependent changes in the membrane lipids or from Gβγ subunits.
Using mammalian GIRK2 channels reconstituted in defined lipid membranes, we also determined that cholesterol directly activates GIRK2 channels. By contrast, cholesterol inhibits the activity of many different types of ion channels, including other members of the inwardly rectifying potassium channel family (Kir)24, 26, 53, 54. Like alcohol, cholesterol activation of GIRK2 depends on the presence of PIP2. Both activators appear to increase the apparent affinity of the channel for PIP2, similar to Gβγ activation34, suggesting that a tighter association of PIP2 with the channel may contribute to the increase in channel activity. In addition, we demonstrate that PIP2 is not only necessary, but is sufficient to activate GIRK2 channels with cytoplasmic Na+ in the absence of Gβγ subunits. As such, PIP2 could be considered an ‘agonist’ for GIRK channels. Moreover, molecules typically described as channel activators, i.e., Gβγ subunits and alcohol, could be mechanistically referred to as positive allosteric modulators (PAMs). PAMs are molecules that cannot activate the channel on their own but increase the efficacy or potency of an agonist, i.e., PIP2. For example, benzodiazepine is a PAM for GABAA receptors; it does not activate GABAA receptors but shifts the EC50 for agonist, i.e., GABA, activation55. However, the endogenous levels of PIP2 are low relative to the sensitivity of GIRK channels, resulting in a small basal channel activity. GIRK channels are considered to have a relatively low affinity for PIP2, compared to constitutively active Kir2.1 channels56. Consistent with this, the EC50 for activation of GIRK2 with diC8-PIP2 is ~25 μM, which compares to a 10-fold lower EC50 of ~2 μM for Kir2.157. Interestingly, the GIRK channel activators, i.e., Gβγ, alcohol and cholesterol, do not alter the levels of PIP2 but instead change the apparent affinity of the channel for PIP2. The shift in apparent PIP2 affinity with these activators likely occurs through allosteric changes in the channel that alter the rates of association and dissociation of PIP2. On the other hand, stimulation of GPCRs that couple to Gq G proteins can reversibly lower the levels of membrane PIP2, leading to a decrease in GIRK channel activity58.
Similar to its effects on brain GIRK2 channels, cholesterol enhances acetylcholine activated potassium (KACh) channels in the heart, which are composed of GIRK1 and GIRK4 subunits23, 27. Interestingly, this effect was proposed to be independent of Gβγ but with little change in the apparent PIP2 affinity23. In these experiments, the relative association of PIP2 with GIRK1/GIRK4 heteromeric or GIRK4* homomeric channels was inferred from measuring the rate of current inhibition following activation of a PIP2-depleting phosphatase23. The rate was similar in the absence and presence of cholesterol23. By contrast, we find that cholesterol activates the neuronal GIRK2 channel activity by increasing the apparent association with PIP2 in the absence of Gβγ subunits. Whether this difference in PIP2 dependence is unique to GIRK2 remains to be determined.
A high-resolution structural view of the binding site for cholesterol in GIRK2 is not available. By contrast, a structural pocket has been identified in GIRK2 channels for alcohol activation18, 51. The alcohol pocket is located at the interface of the cytoplasmic domain of two adjacent channel subunits, and is formed by hydrophobic amino acids in the N-terminal domain, the βL-βM strands, and the βD-βE strands18. This binding pocket overlaps with the region involved in Gβγ subunits activation15, 59. Molecular dynamics (MD) simulations combined with functional studies have suggested cholesterol may be coordinated by residues in the transmembrane spanning regions60, 61. Recently, Bukiya et al. (2017) conducted cholesterol-docking experiments and identified four amino acids that potentially interact directly with cholesterol, V99, V101, L174 and L183. Interestingly, these sites are physically close to the binding pocket for PIP2 in the GIRK2 closed structure32, raising the possibility that cholesterol may enhance the access of PIP2 to the channel. Thus, the site for cholesterol modulation is unlikely to be the same as that for alcohol (i.e. membrane vs. cytoplasmic). In addition, we noted functional differences between alcohol and cholesterol activation. First, there was no apparent shift in ethanol sensitivity in the presence of cholesterol. A direct or allosteric effect of alcohol on cholesterol activation might be reflected in differences in the dose-response curves. Second, the basal rate of K+ flux through GIRK2 channels in the presence of alcohol and a saturating level of cholesterol was higher than the rate for alcohol alone. Together, these results suggest that alcohol and cholesterol interact structurally and mechanistically with different regions of the channel.
Cholesterol is ubiquitous in plasma membranes, accounting for up to 50% of the lipid mass22. Changes in brain cholesterol are implicated in numerous neurodegenerative diseases, such as Alzheimer’s, Huntington’s, Parkinson’s, and Niemann-Pick disease62. Cholesterol metabolism and utilization in the brain is significantly different than the rest of the body. Although the brain only makes up 2-5% of the body mass, almost 25% of the total body cholesterol resides in the brain63. Moreover, cholesterol degradation in the brain is low. An increase of cholesterol in the brain has been reported to decrease the firing rate in hippocampal neurons, suggesting it may reduce excitability64. Notably, some types of GIRK channels are localized in lipid rafts, which are enriched in cholesterol65, 66. Thus, changes in membrane cholesterol content may alter the basal activity of neuronal GIRK channels. Consistent with this, Bukiya et al. (2017) reported that cholesterol enrichment increased baclofen-induced, tertiapin-inhibited GIRK-like currents in hippocampal neurons. Similarly, cholesterol (33%) enhanced the basal activity of a constitutively-active mutant GIRK2 channel43.
GIRK channels are emerging as a potential drug target for treating alcoholism and other neurological disorders3. Interestingly, chronic alcohol exposure leads to increased cholesterol levels and more widespread distribution of cholesterol in the membrane leaflets67, 68. Moreover, increases in cholesterol have been also reported in cases of fetal alcohol exposure69. The potentiation of GIRK channel activity by cholesterol could amplify the effects of alcohol, perhaps contributing to a comorbidity of cholesterol and alcohol. With a better understanding of the molecular mechanism underlying the direct activation of GIRK channels by small compounds like ethanol and cholesterol, it should be possible to design new drugs that specifically modulate the activity of GIRK channels through these G protein-independent pathways.
Methods
Molecular Biology
We used a variant of GIRK2 shown previously to express efficiently in Pichia pastoris; mouse GIRK2 (containing amino acids 52–380) is linked in-frame with an HRV 3 C protease site, green fluorescent protein (GFP) and a decahistidine (HIS10) tag (a generous gift from R. MacKinnon, The Rockefeller University, New York, NY). GIRK2 clones in pPICZ were transformed into Pichia pastoris using the lithium chloride transformation method70 or electroporation (according to manufacturer protocols). Transformants were screened based upon Zeocin resistance (>500 μg/ml) and GFP emission. Highest expressing clones were selected for large-scale purification.
Protein purification and reconstitution
All GIRK channels were expressed and purified in P. pastoris as described previously15, 32. Briefly, the highest-expressing clone was grown in BMGY medium and induced in BMM medium containing 1% methanol. Cells were harvested, resuspended in buffer (50 mM HEPES, pH 7.4; 150 mM KCl; 1 mM TCEP; 1 mM AEBSF and Complete EDTA-free protease inhibitor tablets (Roche)), dripped into liquid nitrogen, and placed at −80 °C. Frozen cells were lysed in a Mixer Mill (Retsch) 5-times for 3 minutes at 25 Hz and solubilized in 50 mM HEPES, pH 7.35; 150 mM KCl; 1 mM TCEP; 1 mM AEBSF; 3% (w/v) n-Dodecyl-β-D-maltoside (DDM; Anatrace) and Complete ULTRA EDTA-free protease inhibitor tablets (Roche) with gentle stirring at 4 °C. Unsolubilized material was separated by centrifugation at 40,000 × g for 40 min at 4 °C. The supernatant was injected onto a HISTrap HP column (GE Healthcare) equilibrated in wash buffer (50 mM HEPES, pH 7.0; 150 mM KCl; 0.4% DDM; 20 mM imidazole) connected to an ÄKTA pure (GE Healthcare) chromatography system and eluted in buffer containing 300 mM imidazole. The HISTrap column eluate was pooled, exchanged into imidazole-free buffer and digested overnight at 4 °C with HRV 3 C protease, purified as described71 (a generous gift of Daniel Minor, UCSF, San Francisco, CA). Digested protein was concentrated and run on a Superdex-200 gel filtration column in 20 mM TRIS-HCl pH 7.5, 150 mM KCl, 0.1% (w/v) DDM (anagrade), 5 mM DTT, and 1 mM EDTA. Fractions eluting at a volume consistent with the GIRK channel tetramer were pooled, concentrated and examined by SDS-PAGE and Coomassie blue staining. We also analyzed the purified protein using tandem mass spectrometry and detected significant peptides for only GIRK2 and the mass spectrometry experimental artifacts trypsin and keratin. No other proteins were evident.
Purified GIRK2 channels were reconstituted into lipid vesicles as described previously15, 34. Briefly, a lipid mixture containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG), and L-α-phosphatidylinositol-4,5-bisphosphate (Brain, PI(4,5)P2 (Porcine)) at mass ratios of 3:1:0.04 (POPE:POPG:PIP2) or 3:1 (POPE:POPG) was prepared, reconstituted in vesicle buffer (20 mM K-HEPES, pH 7.4; 150 mM KCl; 0.5 mM EDTA containing 35 mM CHAPS) and incubated with protein in detergent at a 1:200 protein: lipid ratio unless otherwise indicated. Where indicated, cholesterol (Ovine wool) was added to vesicles at a mole percentage of 1%, 5% or 10%. Cholesterol could not be incorporated into liposomes at concentrations higher than 10% as noted previously25. Detergent was removed through sequential addition of Bio-beads SM-2 (Bio-rad). All phospholipids, cholesterol, and Brain PIP2 were purchased from Avanti Polar Lipids, Inc. Soluble PIP2 (diC8-PIP2) was purchased from Echelon Biosciences.
Flux assay
Liposomes were diluted 1:20 into flux buffer (20 mM Na-HEPES, pH 7.4; 150 mM NaCl; 0.5 mM EDTA) containing 5 μM of the H+ sensitive dye 9-Amino-6-chloro-2-methoxyacridine (ACMA). Fluorescence was measured using a Flexstation 3 microplate reader (Molecular Devices) with the following parameters: 410 nm excitation, 480 nm emission, 455 nm cutoff, medium PMT sensitivity, and sampling at 2 seconds. After a stable baseline fluorescence (150 s) was obtained, the H+ ionophore m-chlorophenyl hydrazone (CCCP) was automatically added (1 μM), then a second addition consisting of different compounds or vehicle was added 150 s later, followed 900 s later by a third addition with the K+ ionophore Valinomycin (100 nM), to determine the maximal K+ flux. GIRK2 channels are likely arranged in both orientations in the liposomes. However, we expect the channels oriented inside-out to support high K+ flux because of high Na+ in the flux buffer and high K+ in the liposome14, 35, 36.
Data Analysis
To compare between different flux assay runs, the fluorescence was either normalized to the baseline fluorescence before CCCP (F b) and the fluorescence following Valinomycin (F v), or in the case of acutely adding a compound, the fluorescence after CCCP (F b) and the fluorescence following Valinomycin (Fv). This normalized fluorescence (F N), defined as ‘relative K+ flux”, was calculated according to the equation (Eq. 1):
| 1 |
where F N (t) is the normalized fluorescence at time t and F(t) is the relative fluorescence unit (RFU) as a function of time.
The rate of K+ flux (1/τ) was determined by fitting the normalized fluorescence decay with a single exponential (Eq. 2):
| 2 |
where Fo is the amplitude, 1/τ is the rate, and C is a constant. In experiments where it was not possible to fit the decay, the amplitude of the fluorescence just prior to adding valinomycin (1190 s) was used to calculate the fractional inhibition of K+ flux.
Dose-response curves for diC8-PIP2/neomycin were fit to the Hill equation (Eq. 3):
| 3 |
where ymax is the maximal flux rate or fractional inhibition, [X] is the concentration, h is the Hill coefficient, and x 50 is the half-maximal effective concentration, i.e. EC50 for PIP2 or IC50 neomycin. Dose response curves for modulators, e.g., alcohol, β-OG, capsaicin, do not reach saturation and were normalized to the basal flux rate in the absence of ligand. All values are reported as mean ± S.E.M, with statistical significance (P < 0.05) determined using one-way ANOVA followed by Dunnett’s multiple comparison post hoc test unless otherwise noted. All data were analyzed using Excel (Microsoft) and Prism (GraphPad).
Acknowledgements
We thank Dr. Rod Mackinnon for providing GIRK2 cDNA and P. pastoris strain, Dr. Daniel Minor for the HRV 3C protease cDNA, Dr. Olaf Andersen for discussions on lipid disruptors, Dr. Alessio Accardi for helpful discussions, Dr. Iban Urbarretxena for technical assistance and sharing equipment, and the Slesinger lab for comments on the text. This work was supported by grants from the NIAAA (AA018734; PAS and F32AA024671; IWG) and the NIDA (DA037170; PAS).
Author Contributions
I.W.G. conducted the experiments and collected the data. P.A.S. and I.W.G. planned the research, analyzed the data, and prepared the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ian W. Glaaser, Email: ian.glaaser@mssm.edu
Paul A. Slesinger, Email: paul.slesinger@mssm.edu
References
- 1.Luscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci. 2010;11:301–15. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hibino H, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 3.Mayfield J, Blednov YA, Harris RA. Behavioral and Genetic Evidence for GIRK Channels in the CNS: Role in Physiology, Pathophysiology, and Drug Addiction. Int Rev Neurobiol. 2015;123:279–313. doi: 10.1016/bs.irn.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamada K, et al. Association study of the KCNJ3 gene as a susceptibility candidate for schizophrenia in the Chinese population. Hum Genet. 2012;131:443–51. doi: 10.1007/s00439-011-1089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishizawa D, et al. Association between KCNJ6 (GIRK2) gene polymorphism rs2835859 and post-operative analgesia, pain sensitivity, and nicotine dependence. J Pharmacol Sci. 2014;126:253–63. doi: 10.1254/jphs.14189FP. [DOI] [PubMed] [Google Scholar]
- 6.Clarke TK, et al. KCNJ6 is associated with adult alcohol dependence and involved in gene x early life stress interactions in adolescent alcohol drinking. Neuropsychopharmacology. 2011;36:1142–8. doi: 10.1038/npp.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saccone SF, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lotsch J, Pruss H, Veh RW, Doehring A. A KCNJ6 (Kir3.2, GIRK2) gene polymorphism modulates opioid effects on analgesia and addiction but not on pupil size. Pharmacogenet Genomics. 2010;20:291–7. doi: 10.1097/FPC.0b013e3283386bda. [DOI] [PubMed] [Google Scholar]
- 9.Masotti A, et al. Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am J Hum Genet. 2015;96:295–300. doi: 10.1016/j.ajhg.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wickman KD, et al. Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368:255–7. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 11.Reuveny E, et al. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–6. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- 12.Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2and its stabilization by Gβγ. Nature. 1998;391:803–6. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 13.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–6. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 14.Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA. 1998;95:1307–12. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whorton MR, MacKinnon R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature. 2013;498:190–7. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewohl JM, et al. G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci. 1999;2:1084–90. doi: 10.1038/16012. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi T, et al. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat Neurosci. 1999;2:1091–7. doi: 10.1038/16019. [DOI] [PubMed] [Google Scholar]
- 18.Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009;12:988–95. doi: 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodhinathan K, Slesinger PA. Molecular mechanism underlying ethanol activation of G-protein-gated inwardly rectifying potassium channels. Proc Natl Acad Sci U S A. 2013;110:18309–14. doi: 10.1073/pnas.1311406110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yevenes GE, et al. Molecular Requirements for Ethanol Differential Allosteric Modulation of Glycine Receptors Based on Selective Gβγ Modulation. Journal of Biological Chemistry. 2010;285:30203–30213. doi: 10.1074/jbc.M110.134676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingolfsson HI, Andersen OS. Alcohol’s effects on lipid bilayer properties. Biophys J. 2011;101:847–55. doi: 10.1016/j.bpj.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeagle PL. Modulation of membrane function by cholesterol. Biochimie. 1991;73:1303–10. doi: 10.1016/0300-9084(91)90093-G. [DOI] [PubMed] [Google Scholar]
- 23.Deng W, et al. Hypercholesterolemia induces up-regulation of KACh cardiac currents via a mechanism independent of phosphatidylinositol 4,5-bisphosphate and Gβγ. J Biol Chem. 2012;287:4925–35. doi: 10.1074/jbc.M111.306134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowley JJ, Treistman SN, Dopico AM. Cholesterol antagonizes ethanol potentiation of human brain BKCa channels reconstituted into phospholipid bilayers. Mol Pharmacol. 2003;64:365–72. doi: 10.1124/mol.64.2.365. [DOI] [PubMed] [Google Scholar]
- 25.D’Avanzo N, Hyrc K, Enkvetchakul D, Covey DF, Nichols CG. Enantioselective protein-sterol interactions mediate regulation of both prokaryotic and eukaryotic inward rectifier K+ channels by cholesterol. PLoS One. 2011;6:e19393. doi: 10.1371/journal.pone.0019393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romanenko VG, et al. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–61. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bukiya AN, et al. Cholesterol increases the open probability of cardiac KACh currents. Biochim Biophys Acta. 2015;1848:2406–13. doi: 10.1016/j.bbamem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Bukiya AN, Belani JD, Rychnovsky S, Dopico AM. Specificity of cholesterol and analogs to modulate BK channels points to direct sterol-channel protein interactions. J Gen Physiol. 2011;137:93–110. doi: 10.1085/jgp.201010519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber W. Ion currents of Xenopus laevis oocytes: state of the art. Biochim Biophys Acta. 1999;1421:213–33. doi: 10.1016/S0005-2736(99)00135-2. [DOI] [PubMed] [Google Scholar]
- 31.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. 2010;22:422–9. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–75. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 34.Wang W, Whorton MR, MacKinnon R. Quantitative analysis of mammalian GIRK2 channel regulation by G proteins, the signaling lipid PIP2 and Na+ in a reconstituted system. Elife. 2014;3:e03671. doi: 10.7554/eLife.03671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho IH, Murrell-Lagnado RD. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels. J Physiol. 1999;520(Pt 3):645–51. doi: 10.1111/j.1469-7793.1999.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho IH, Murrell-Lagnado RD. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J Biol Chem. 1999;274:8639–48. doi: 10.1074/jbc.274.13.8639. [DOI] [PubMed] [Google Scholar]
- 37.Guo Y, Waldron GJ, Murrell-Lagnado R. A role for the middle C terminus of G-protein-activated inward rectifier potassium channels in regulating gating. J Biol Chem. 2002;277:48289–94. doi: 10.1074/jbc.M207987200. [DOI] [PubMed] [Google Scholar]
- 38.Rohacs T, Chen J, Prestwich GD, Logothetis DE. Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J Biol Chem. 1999;274:36065–72. doi: 10.1074/jbc.274.51.36065. [DOI] [PubMed] [Google Scholar]
- 39.Lopes CM, et al. Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K+ channels. Channels (Austin) 2007;1:124–34. doi: 10.4161/chan.4322. [DOI] [PubMed] [Google Scholar]
- 40.Xie LH, John SA, Ribalet B, Weiss JN. Phosphatidylinositol-4,5-bisphosphate (PIP2) regulation of strong inward rectifier Kir2.1 channels: multilevel positive cooperativity. J Physiol. 2008;586:1833–48. doi: 10.1113/jphysiol.2007.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–30. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 42.Aranda FJ, Villalain J, Gomez-Fernandez JC. Capsaicin affects the structure and phase organization of phospholipid membranes. Biochim Biophys Acta. 1995;1234:225–34. doi: 10.1016/0005-2736(94)00293-X. [DOI] [PubMed] [Google Scholar]
- 43.Bukiya, A.N., Durdagi, S., Noskov, S. & Rosenhouse-Dantsker, A. Cholesterol Up-regulates Neuronal G Protein-Gated Inwardly Rectifying Potassium (GIRK) Channel Activity in the Hippocampus. J Biol Chem292, 6135–6147 (2017) [DOI] [PMC free article] [PubMed]
- 44.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by various antidepressant drugs. Neuropsychopharmacology. 2004;29:1841–51. doi: 10.1038/sj.npp.1300484. [DOI] [PubMed] [Google Scholar]
- 45.Zhou W, Arrabit C, Choe S, Slesinger PA. Mechanism underlying bupivacaine inhibition of G protein-gated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 2001;98:6482–7. doi: 10.1073/pnas.111447798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by different classes of antidepressants. PLoS One. 2011;6:e28208. doi: 10.1371/journal.pone.0028208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi T, Nishizawa D, Iwamura T, Ikeda K. Inhibition by cocaine of G protein-activated inwardly rectifying K+ channels expressed in Xenopus oocytes. Toxicol In Vitro. 2007;21:656–64. doi: 10.1016/j.tiv.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Kobayashi T, Ikeda K, Kumanishi T. Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K(+) (GIRK) channels expressed in xenopus oocytes. Br J Pharmacol. 2000;129:1716–22. doi: 10.1038/sj.bjp.0703224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaufmann K, et al. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Neurosci. 2013;4:1278–86. doi: 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su Z, Brown EC, Wang W, MacKinnon R. Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci U S A. 2016;113:5748–53. doi: 10.1073/pnas.1602815113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pegan S, Arrabit C, Slesinger PA, Choe S. Andersen’s syndrome mutation effects on the structure and assembly of the cytoplasmic domains of Kir2.1. Biochemistry. 2006;45:8599–606. doi: 10.1021/bi060653d. [DOI] [PubMed] [Google Scholar]
- 52.Harris, R.A., Trudell, J.R. & Mihic, S.J. Ethanol’s molecular targets. Sci Signal1, re7 (2008). [DOI] [PMC free article] [PubMed]
- 53.Hajdu P, Varga Z, Pieri C, Panyi G, Gaspar R., Jr. Cholesterol modifies the gating of Kv1.3 in human T lymphocytes. Pflugers Arch. 2003;445:674–82. doi: 10.1007/s00424-002-0974-y. [DOI] [PubMed] [Google Scholar]
- 54.Lundbaek JA, et al. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J Gen Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walters RJ, Hadley SH, Morris KD, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci. 2000;3:1274–81. doi: 10.1038/81800. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol. 1999;1:183–8. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 57.Tang QY, et al. Mutations in Nature Conferred a High Affinity Phosphatidylinositol 4,5-Bisphosphate-binding Site in Vertebrate Inwardly Rectifying Potassium Channels. J Biol Chem. 2015;290:16517–29. doi: 10.1074/jbc.M115.640409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobrinsky E, Mirshahi T, Zhang H, Jin T, Logothetis DE. Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat Cell Biol. 2000;2:507–14. doi: 10.1038/35019544. [DOI] [PubMed] [Google Scholar]
- 59.Glaaser IW, Slesinger PA. Structural Insights into GIRK Channel Function. Int Rev Neurobiol. 2015;123:117–60. doi: 10.1016/bs.irn.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 60.Furst O, Nichols CG, Lamoureux G, D’Avanzo N. Identification of a cholesterol-binding pocket in inward rectifier K(+) (Kir) channels. Biophys J. 2014;107:2786–96. doi: 10.1016/j.bpj.2014.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. 2013;288:31154–64. doi: 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. 2012;5:746–55. doi: 10.1242/dmm.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–12. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 64.Wang D, Schreurs BG. Dietary cholesterol modulates the excitability of rabbit hippocampal CA1 pyramidal neurons. Neurosci Lett. 2010;479:327–31. doi: 10.1016/j.neulet.2010.05.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koyrakh L, et al. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–78. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Delling M, et al. The neural cell adhesion molecule regulates cell-surface delivery of G-protein-activated inwardly rectifying potassium channels via lipid rafts. J Neurosci. 2002;22:7154–64. doi: 10.1523/JNEUROSCI.22-16-07154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wood WG, Schroeder F, Hogy L, Rao AM, Nemecz G. Asymmetric distribution of a fluorescent sterol in synaptic plasma membranes: effects of chronic ethanol consumption. Biochim Biophys Acta. 1990;1025:243–6. doi: 10.1016/0005-2736(90)90103-U. [DOI] [PubMed] [Google Scholar]
- 68.Omodeo-Sale F, Pitto M, Masserini M, Palestini P. Effects of chronic ethanol exposure on cultured cerebellar granule cells. Mol Chem Neuropathol. 1995;26:159–69. doi: 10.1007/BF02815010. [DOI] [PubMed] [Google Scholar]
- 69.Barcelo-Coblijn G, Wold LE, Ren J, Murphy EJ. Prenatal ethanol exposure increases brain cholesterol content in adult rats. Lipids. 2013;48:1059–68. doi: 10.1007/s11745-013-3821-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gietz RD, Schiestl RH. Transforming yeast with DNA. Methods in Molecular and Cellular Biology. 1995;5:255–269. [Google Scholar]
- 71.Shaya D, et al. Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc Natl Acad Sci USA. 2011;108:12313–8. doi: 10.1073/pnas.1106811108. [DOI] [PMC free article] [PubMed] [Google Scholar]