

Abstract
Blood formation is believed to occur through step-wise progression of haematopoietic stem cells (HSCs) following a tree-like hierarchy of oligo-, bi- and unipotent progenitors. However, this model is based on the analysis of predefined flow-sorted cell populations. Here we integrated flow cytometric, transcriptomic and functional data at single-cell resolution to quantitatively map early differentiation of human HSCs towards lineage commitment. During homeostasis, individual HSCs gradually acquire lineage biases along multiple directions without passing through discrete hierarchically organized progenitor populations. Instead, unilineage-restricted cells emerge directly from a “Continuum of LOw primed UnDifferentiated hematopoietic stem- and progenitor cells” (CLOUD-HSPCs). Distinct gene expression modules operate in a combinatorial manner to control stemness, early lineage priming and the subsequent progression into all major branches of haematopoiesis. These data reveal a continuous landscape of human steady state haematopoiesis downstream of HSCs and provide a basis for the understanding of hematopoietic malignancies.
INTRODUCTION
All mature blood and immune cells are thought to derive from self-renewing and multipotent HSCs. According to the current model, initiation of differentiation is associated with the loss of self-renewal and generation of discrete multipotent, oligopotent and subsequently unipotent progenitor cell stages1,2. These lineage-restricted progenitors are thought to be generated in a stepwise manner by several subsequent binary branching decisions leading to the classical hierarchical tree-like model of haematopoiesis1-6. However, this model is mainly based on analyses of FACS-purified cell populations. Even if followed up by single cell assays3,4,7, such analyses derive average properties of predefined cell populations and thereby miss both quantitative changes within gates as well as transition states falling between often subjectively set gates.
Moreover, the lineage contribution associated with each population is typically determined by assays such as colony formation or transplantation. While these assays read out lineage potential, the actual cell fate during homeostasis in vivo may be different8,9. Depending on the assays and markers used, partly conflicting branching points and hierarchies have been proposed10-14.
Recent studies based on novel single-cell approaches have challenged more fundamental aspects of this classical model. For instance, unipotent progenitors can derive directly from HSCs without proceeding through oligopotent progenitors14,15 and lineage commitment was observed in progenitors proposed to be oligopotent 7,10,16. However, many of these studies focused on more differentiated compartments7,10,16 or used predefined subpopulations to investigate single-cell heterogeneity7,17, impeding the characterization of transitions between cell stages. Therefore, it remains unclear how individual HSCs enter lineage commitment during homeostasis in vivo. To establish a comprehensive model of haematopoiesis that can reconcile previous findings, a combined view of transcriptomic and functional changes along the developmental progression of individual cells is required. Here we developed an approach that integrates the reconstruction of developmental trajectories18,19 with the quantitative linkage between transcriptomic and functional single cell data17 and thus provides a detailed view on lineage commitment of individual haematopoietic stem and progenitor cells (HSPCs) into all major branches of human haematopoiesis.
RESULTS
Healthy human bone marrow cells were labelled with a panel of up to 11 FACS surface markers commonly used to characterize human HSPCs5,6 (see Methods, Supplementary Table 1). All HSPCs, defined by the absence of lineage markers (Supplementary Table 1) and expression of CD34 (Lin-CD34+), were individually sorted and enriched for immature cells (see supplementary methods). The surface marker fluorescence intensities of all markers were recorded to retrospectively reconstruct immunophenotypes (CD10, CD38, CD45RA, CD90, CD135, and depending on the experiment CD2, CD7, CD49f, CD71, CD130, FCER1A, ITGA5 and KEL, Supplementary Fig. 1a). Such index-sorted HSPCs derived from the bone marrow of two healthy individuals were subjected to RNA-Seq analysis (index-omics, 1034 and 379 single cells; see Supplementary Fig. 1b for the distribution of cells within classically defined gates5,6 and Supplementary Fig. 2 for quality metrics of single cell RNA-Seq) to determine their transcriptomes or individually cultured ex vivo (“index-culture”, 2038 single cells) to quantify megakaryocytic, erythroid and myeloid lineage potential. Subsequently, the functional and transcriptomic data sets were integrated by regression models using commonly indexed surface marker expression to identify the molecular and cellular events associated with the differentiation of human HSCs at the single cell level (Fig. 1). To make this data type accessible, we developed indeXplorer, a web-based platform that combines features of FACS software (e.g. custom gating) with tools for single-cell transcriptomics data analysis (e.g. differential expression analysis, clustering, principal component analysis) in a single graphical user interface (Supplementary Fig. 3 and http://steinmetzlab.embl.de/shiny/indexplorer/?launch=yes).
Fig. 1. Experimental strategy.
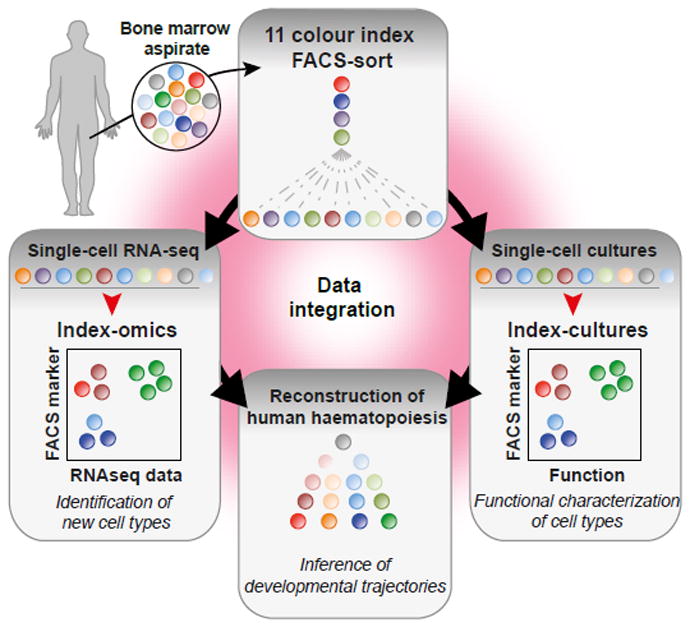
Adult human HSPCs were stained with antibodies against up to 11 surface markers and individually sorted for either single-cell RNA-seq or single-cell cultures. Data from the two experiments were then integrated based on surface marker expression to reconstruct developmental trajectories of haematopoiesis.
Early haematopoiesis is a continuous process
HSCs and their immediate progeny, such as multipotent progenitors (MPPs) or multilymphoid progenitors (MLPs), are located in the Lin-CD34+CD38- compartment, whereas more differentiated progenitors reside in the Lin-CD34+CD38+ compartment5,7. Global gene expression analysis of single cells within these two compartments revealed fundamentally different transcriptomic structures. In both individuals, the Lin-CD34+CD38+ progenitors could be separated into clusters corresponding to distinct progenitor cell types of all major branches of haematopoiesis (Fig. 2a and see below). In contrast, clustering within the Lin-CD34+CD38- compartment was largely unstable, as demonstrated by cluster stability analysis (Supplementary Fig. 4a), the absence of clusters according to Gap statistics (Supplementary Fig. 4b), and a recently published algorithm for the clustering of single cells20 (Supplementary Fig. 4c). A simulated series of random steps from an individual cell to one of its nearest neighbours (see methods) revealed that the majority of Lin-CD34+CD38- cells were highly interconnected, contrasting the disconnected cell types from the Lin-CD34+CD38+ compartment (Fig. 2b). Unsupervised visualization of all individual cells irrespective of FACS markers by t-SNE confirmed that Lin-CD34+CD38- cells formed a single continuously connected entity. In contrast, Lin-CD34+CD38+ cells emerged into locally clustered cell populations, with the exception of some phenotypic CMPs and CD10+ MLPs, suggesting that the classification based on differential CD38 expression is excellent, but not absolute (Fig. 2c).
Fig. 2. A stem and progenitor cell continuum precedes the establishment of discrete lineages at the CD34+CD38+ stage.
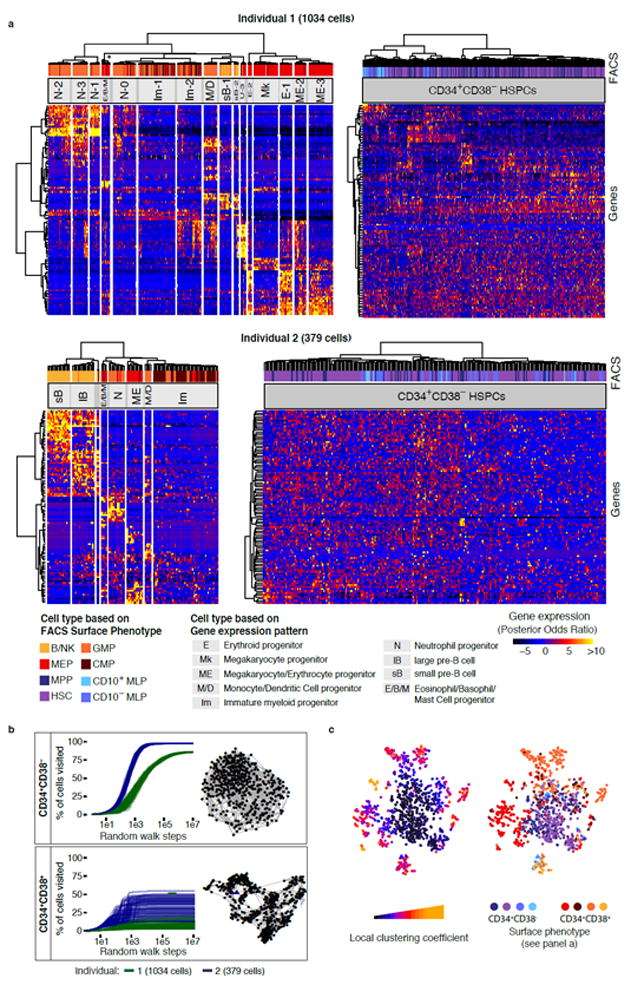
(a) Hierarchical clustering of Lin-CD34+CD38- (Individual 1: 467 cells, Individual 2: 261 cells) and Lin-CD34+CD38+ (I1: 567 cells, I2: 118 cells) compartments for both individuals. Clustering was performed on the most variable 1000 genes of each population. The most variable 100 genes were displayed in the heatmap. The asterisk indicates that 3 putative Eosinophil/Basophil/Mast cell progenitor subclusters of <5 cells were merged. (b) Random walk analysis of Lin-CD34+CD38- and Lin-CD34+CD38+ compartments for both individuals. 100 random walks, i.e. series of random steps from one cell to any of its 5 nearest neighbours in correlation distance space, were simulated and the number of cells reached was evaluated in relation to the total number of cells. 5-Nearest-neighbour networks are depicted on the right. (c) t-SNE visualization of all cells (individual 1) highlighting the degree to which cells are associated with local clusters (left panel, see also methods) and the immunophenotype (right panel).
Notably, the absence of hierarchical structures in the primitive Lin-CD34+CD38- compartment was due to the gradual nature of differences between cells in that compartment, and not due to insufficient data quality or a lack of transcriptomic heterogeneity: A principal component analysis of Lin-CD34+CD38- cells resolved more than 10 distinct, variable biological processes in this compartment, such as cell cycle activation and lineage priming (Supplementary Fig. 4d-f). These processes are tightly correlated to surface marker expression (Supplementary Fig. 4g).
Collectively, these observations are incompatible with the classical model of early haematopoiesis, which assumes a hierarchical tree-like structure of discrete progenitors downstream of HSCs. In contrast, our data suggest that HSCs and their immediate progeny are initially part of a Continuum of LOw-primed UnDifferentiated (“CLOUD”)-HSPCs within the Lin-CD34+CD38- compartment (see also below). Discrete populations are only established when differentiation has progressed to the level of restricted progenitors typically associated with the up-regulation of CD38.
Lineage-restriction downstream of the HSPC continuum
To characterize the discrete populations in the Lin-CD34+CD38+ compartment, we performed gene expression and cell surface marker analyses as well as functional validations at the single cell level. Our analyses revealed that these populations correspond to lineage-restricted progenitors of all major branches of bone marrow haematopoiesis, including B-cell progenitors of distinct stages, megakaryocyte/erythrocyte committed progenitors (ME, Ery, Mk), neutrophil-primed progenitors (Neutro), monocyte/dendritic cell (Mono/DC) progenitors, and eosinophil/basophil/mast cell progenitors (Eo/Baso/Mast), as well as immature myeloid progenitors (Fig. 3a, Supplementary Table 2). Importantly, populations cluster by cell type and not by individual in a cross-individual comparison (Fig. 3b). The comparison of the surface marker expression of these populations to the commonly applied gating scheme5 using our indexed data set showed that immunophenotypically defined oligopotent progenitor populations (megakaryocyte-erythroid progenitors, MEPs; granulocyte-monocyte progenitors, GMPs; B cell–NK cell progenitors, B-NKPs) were mainly comprised of cell types with unilineage-specific gene expression profiles (Fig. 3c) and functional unipotency (Fig. 4a,b).
Fig. 3. The Lin-CD34+CD38+ compartment consists of distinct lineage-restricted progenitors.
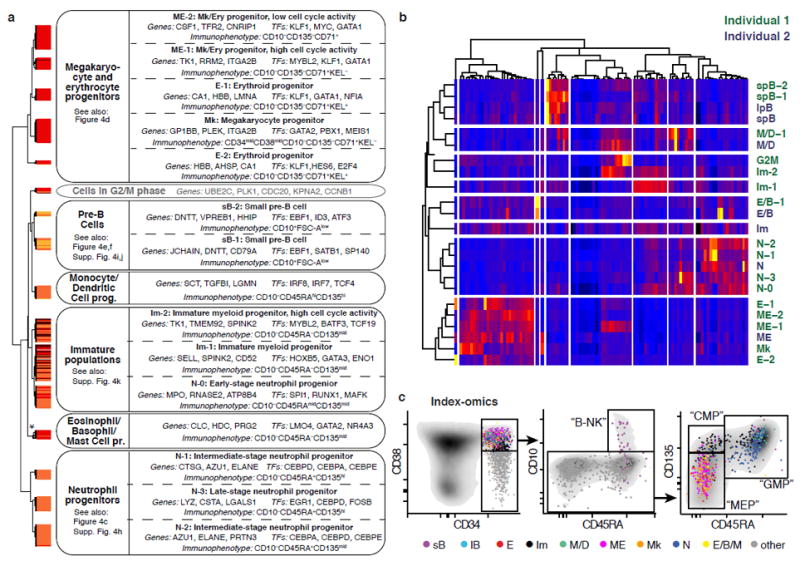
(a) Overview of putative cell types in individual 1 (see panel b for a comparison between individuals). Classes obtained from hierarchical clustering of the Lin-CD34+CD38+ compartment (Fig. 2a) were assigned to putative cell types based on analyses of gene- and surface marker expression. The asterisk indicates that 3 putative Eosinophil/Basophil/Mast cell progenitor subclusters of <5 cells were merged for this analyses. (b) Averaged gene expression profiles for cell types from both individuals defined in Fig. 2a were clustered based on the 1000 most variable genes. Only the most variable 100 genes are shown in the heatmap. (c) Index-omics display of Lin-CD34+CD38+ progenitors. Sequenced single Lin-CD34+CD38+ cells were arranged according to their cell surface marker expression in classical FACS gating strategies to identify B- and NK cell progenitors (“B-NK”), Megakaryocytic-Erythroid Progenitors (“MEP”), Common Myeloid Progenitors (“CMP”) and Granulocyte-Monocyte Progenitors (“GMP”). Cells were colour-coded based on their cell type identity from Fig. 3a.
Fig. 4. Characterization of Lin-CD34+CD38+ lineage restricted progenitors.
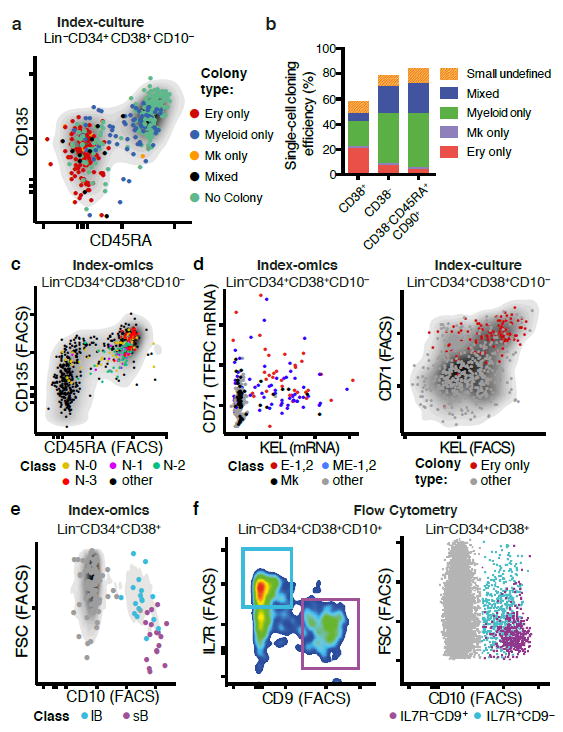
(a) Index-culture display of Lin-CD34+CD38+ HSPCs. Single HSPCs were cultured for 3 weeks and the resulting colony type was plotted in relation to CD45RA and CD135. (b) Single cells from the ex vivo culture assay were scored as unipotent (gave rise to one lineage) or mixed (gave rise to more than one lineage). (c) Neutrophil-primed subpopulations in relation to CD45RA and CD135 surface marker expression. (d) Megakaryocytic/Erythroid primed subpopulations in relation to TFRC (CD71) mRNA and KEL mRNA expression (left panel) and erythroid colony output in relation to CD71 and KEL surface marker expression (right panel). (e) Pre B-cell subpopulations from individual 2 in relation to CD10 surface expression and forward scatter (FSC). (f) Prospective isolation of B-cell subpopulations sB and lB using classical flow cytometry. FACS markers for IL7R and CD9 permit the separation of two populations with forward scatter (FSC)/CD10 profiles corresponding to sB and lB, as suggested from gene expression data.
Cells within the classic GMP compartment were separated into several neutrophil-primed progenitors (N0-N3), as well as into monocyte/dendritic cell progenitors (Mono/DC). The distinct neutrophil-primed progenitors likely represent progenitors at different developmental stages and granule composition (Fig. 4c, Supplementary Fig. 4h)21,22. Immunophenotypically, all neutrophil- primed progenitors express the surface markers CD135 and CD45RA, which are progressively upregulated during maturation (Fig. 4c). In contrast to neutrophil-primed progenitors, Eo/Baso/Mast progenitors did not fall into the classical GMP gate but displayed a Lin-CD34+CD38+CD10-CD45RA-CD135mid immunophenotpye (Fig. 3c), and expressed transcription factors important for early MEP commitment (GATA2 and TAL1) supporting a recent study suggesting that granulocyte subtypes might derive from distinct hematopoietic lineages12.
The MEP gate consisted of megakaryocytic (Mk) progenitors expressing typical Mk genes, of erythroid-committed (E1, E2) progenitors of distinct developmental stages, differing in haemoglobin and GATA1 expression, as well as of subpopulations showing combined expression of megakaryocytic and erythroid genes (M/E). Our single-cell transcriptome data suggested CD71 (TRFC) and the red blood cell antigen KEL to be highly indicative for erythroid fate, which was confirmed by single-cell culture assays using CD71 and KEL as indexing antibodies (Fig. 4d).
For individual 2, two CD10+ B-cell progenitor clusters (small pre-B cells, sB and large pre-B cells, lB) were observed. sB was characterized by high CD9 mRNA expression, high CD10 surface expression and small cell size (FSC), whereas lB showed high expression of interleukin-7 receptor (IL7RA) mRNA, intermediate CD10 surface levels, expression of cell cycle related genes and large cell size (Fig. 4e, Supplementary Fig. 4i, Supplementary Table 2). This suggests that sB corresponds to small pre-B cells, and lB to large pre-B cells, progenitor populations which have been well characterized in the murine system, but to a lesser extent in the human system23. To validate and prospectively isolate large pre-B-cells and small pre-B-cells, we used IL7RA and CD9 FACS markers, which allowed us to recapitulate the levels of CD10 surface expression, cell size and cell cycle activity as predicted from the index-omics data (Fig. 4f, Supplementary Fig. 4j). In contrast to individual 2, in individual 1, only small pre-B-cells were observed (Fig. 3b).
For both individuals, we also observed CD38-positive HSPCs with a gene expression profile of rather immature cells (Im) (Fig 3a). These clustered globally with the Lin-CD34+CD38- compartment in t-SNE analyses, and expressed lower levels of CD38 (Supplementary Fig. 4k). Most of these cells displayed an immunophenotype typical for CMPs (Lin-CD34+CD38+CD45RA-CD135+), however the composition of the cell types present in the CMP gate depends strongly on the exact gating strategy applied (see below, Supplementary Fig. 5h, i).
Based on these analyses, we provide markers and gating strategies for the prospective isolation of several of these newly defined populations using standard flow cytometry.
Developmental trajectories of early human haematopoiesis
To obtain a detailed view on the transition from stem cells to lineage-restricted progenitors in the continuous HSPC landscape, we developed STEMNET, a new dimensionality reduction algorithm. STEMNET identifies genes specific to the six Lin-CD34+CD38+ restricted progenitor populations defined above (Neutro, Eo/Baso/Mast, B-cell, Mono/DC, Ery and Mk; see Supplementary Table 3 for a list of genes used by STEMNET) and then computes the probability that each primitive (“CLOUD”) HSPC can be assigned to any of these classes. STEMNET thereby places the six developmental endpoints on the corners of a simplex. This resulted in the arrangement of the least primed HSCs, such as CD49f+ HSCs, to the centre, and the remaining HSPCs localizing in between according to their degree of priming (Fig. 5a, and see Supplementary Fig. 5a, b for individual 2). To describe the position of each cell we computed the predominant direction of priming d as the developmental endpoint closest to the cell and the degree of lineage priming Srel as the (Kullback-Leibler) distance from the least-primed cell.
Fig. 5. Visualization of the HSPC continuum.
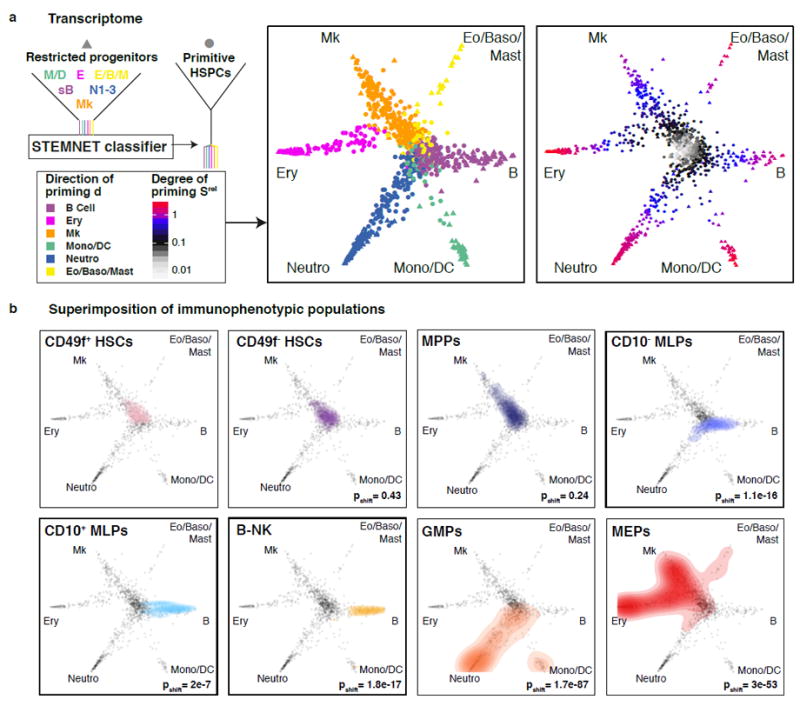
(a) The similarity of every cell to each of the progenitor classes was computed by STEMNET (see methods), projected on a unit circle, and used to quantify the degree and direction of transcriptomic priming. Data from individual 1 is shown, for individual 2 see Supplementary Fig. 5a, b. (b) Immunophenotypic populations5,6 were highlighted on the HSPC continuum. P-values were calculated by kernel-density based tests comparing each population to CD49f+ HSCs. For CMPs, see Supplementary Fig. 5h,i. For CD49f+ HSCs, n=101 single cells; CD49f- HSCs, n=117; MPPs, n=176; CD10- MLPs, n=52; CD10+MLPs, n=16; B-NKs, n=26; GMPs, n=244; MEPs, n=231
This analysis suggests that HSCs located in the centre of the “CLOUD” gradually acquired continuous lineage priming into either of the major branches. While lympho/myeloid and megakaryocytic/erythroid priming formed major points of attraction, a clear separation into single lineages was not present at this stage (Fig. 5a). In contrast, lineages were clearly separated at the level of Lin-CD34+CD38+ progenitors, without further sub-branching in this compartment (Fig. 5a, see Supplementary Fig. 5c for CD38 expression). Importantly, these results are not due to limitations of the bioinformatics method, as STEMNET is able to detect both subsequent branching points and discrete intermediate populations on simulated data (Supplementary Fig. 6a-d). Moreover, applying DPT, a different recently published method for the inference of developmental trajectories24 to our data confirmed the absence of subsequent binary branch points and the direct lineage commitment from CLOUD-HSPCs along continuous trajectories (Supplementary Fig. 6e).
Within the differentiation continuum, STEMNET analysis located previously defined immunophenotypic populations according to their known lineage potential5 (Fig. 5b, see Supplementary Fig. 5b for individual 2). For example, GMPs were distributed to the neutrophil and monocytic/dendritic cell branches while MEPs located to the megakaryocytic and erythroid branches (notice that the localization of CMPs critically depends on the exact CD38 and CD135 gating strategy, Supplementary Fig. 5h, i). In contrast, immunophenotypic MLPs located close to the separation of lymphoid, neutrophil and monocytic/dendritic cell lineages (Fig. 5b, Supplementary Fig. 5b), with individual cells already primed towards specific lineages, in line with frequent functional commitment to single lineages in mouse LMPPs15. Together, these analyses suggest that developmental stages immediately downstream of HSCs such as MLPs and MPPs do not represent discrete cell types located at defined branching points, but should rather be considered as transitory states within the HSPC continuum with higher probability for commitment to particular lineages.
While undergoing lineage commitment only very few cells acquired a transcriptomic state of dual-lineage priming (Supplementary Fig. 5d, e), in accordance with a recent single-cell transcriptomic study on mouse GMPs20. However, our analyses suggest that a direct transition from a primed multi-lineage towards a uni-lineage transcriptomic state represents the main route of lineage commitment, whereas dual-lineages states (such as Gfi1+Irf8+ GMPs, Supplementary Fig. 5f) exist, but represent rare exceptions. Importantly, both transcriptomic and functional (Supplementary Fig. 5g) lineage-combinations of bipotent cells were not restricted to the combinations predicted by the classical model, conflicting with a strictly ordered hierarchy of branching events. Along these lines, co-expression of opposing pairs of transcription factors, such as IRF8 and PU.1 (SPI1) that have been thought to establish an oligopotent state, occurred at much lower frequency than previously expected (see Fig. 8aviii, xi)25.
Fig. 8. Lineage commitment is a layered multi-step process.
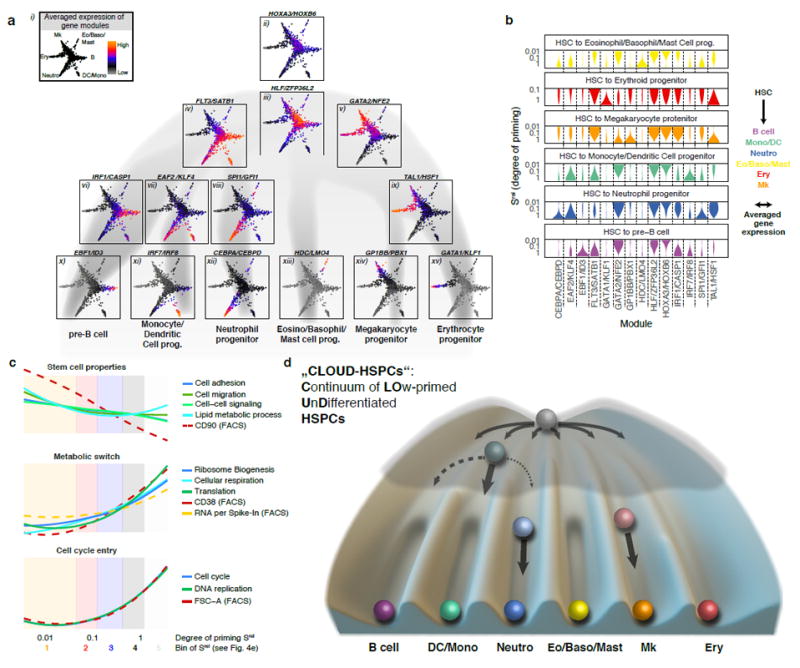
(a, b) Activity of gene modules associated with developmental progression of HSPCs. Genes depending on the degree and/or direction of priming were identified and clustered into modules displaying similar expression patterns (see methods). Averaged gene expression of selected modules from individual 1 was highlighted in the HSPC differentiation continuum (a) or smoothened and plotted against the degree of lineage-specific priming (b). For a complete list of modules and individual 2, see Supplementary Fig. 8 and Supplementary Table 4. (c) Gene ontology and FACS marker changes along the early priming of HSPCs (Srel < 0.4). During later stages of priming, GO activity and FACS marker expression additionally depend on the direction of priming (not shown). (d) Graphical summary of a continuum-based model of bone marrow haematopoiesis. Due to the interactions of gene regulatory networks, some cell states and transitions are more likely than others, represented by a lower elevation within a Waddington landscape. During early lineage commitment, small barriers between lineages arise early, thereby creating lineage biases in HSCs. At the progenitor stage these barriers are already more pronounced, making the oligopotent stage less likely. Note that T- and NK-cell development predominantly occurs outside the bone marrow42.
Transcriptomic priming mediates lineage commitment
Single-cell RNAseq protocols require cell lysis and therefore prohibit subsequent functional interrogation of the same single cell. However, the use of indexed FACS surface markers common to both single-cell ex vivo culture data and single-cell RNAseq data allowed us to quantitatively link the amount and direction of transcriptomic priming to functional properties such as lineage potential and proliferative capacity. For example, the STEMNET-predicted dominant direction of transcriptional priming into the lympho/myeloid versus the megakaryocytic/erythroid direction was strongly correlated to the surface marker expression of CD135 and CD45RA (Fig. 6ai, ii), which could be used to qualitatively predict the predominant cell type in colonies of our single-cell cultures (note that lymphoid progenitors do not grow in these conditions, and that myeloid sublineages are not resolved) (Fig. 6aii). Utilizing all recorded surface markers for linear models on the single-cell RNAseq data allowed us to quantitatively predict the dominant cell type present in the single-cell cultures for the Lin-CD34+CD38+ (p = 3.7e-23) and the Lin-CD34+CD38- compartment (p = 3.7e-22, Fig. 6aiii and Supplementary Fig. 7a for the full specification of regression models). Moreover, predicting erythroid and megakaryocytic priming individually revealed that the amount of lineage specific priming was linked to functional lineage commitment (Fig. 6b, c, Supplementary Fig. 7b, c). However, colonies derived from Mk-primed cells were frequently dominated by other cell types due to their lower proliferative capacity ex vivo (Supplementary Fig. 7b). STEMNET further predicted Lin-CD34+CD38-CD45RA-CD90-CD135- cells to be primed towards megakaryocytic differentiation (Fig. 6d, left panel). To functionally validate this prediction in vivo, we FACS-sorted these cells, transplanted them into sublethally irradiated NSG mice and quantified their lineage output 14 days post transplantation. As predicted, these cells, which we termed Mk-primed MPPs, predominantly generated thrombocytes if compared to MLPs and HSCs (Fig. 6d, right panel). Together, these analyses revealed that transcriptomic priming is linked to the restriction of lineage potential at an early stage in vitro and in vivo.
Fig. 6. The direction of transcriptomic priming is quantitatively linked to functional lineage potential.
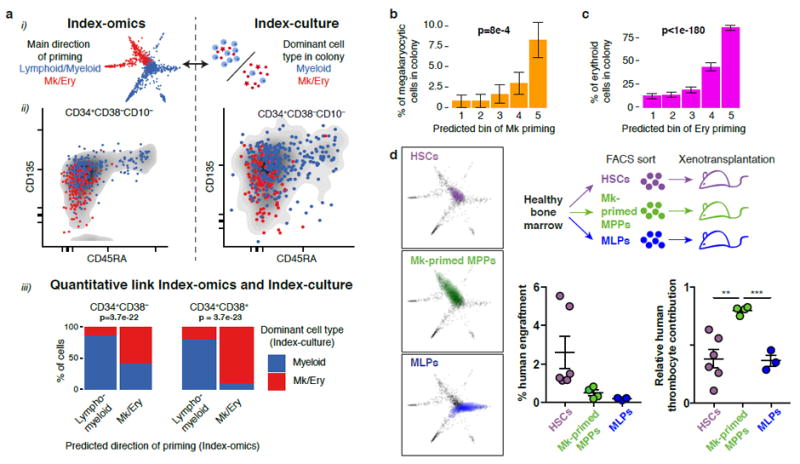
(a) Comparison of the predominant direction of priming d (lympho/myeloid versus megakaryocyte/erythroid) obtained from single-cell transcriptomics to the dominant cell type observed in colonies from single-cell culture. (i) Illustration. (ii) Qualitative comparison of the two quantities with respect to CD45RA and CD135 surface marker expression. (iii) Quantitative link. The most likely dominant direction of priming was estimated for each founder cell from index-culture based on regression models constructed on all surface markers and compared to the observed colony composition (see Supplementary Fig. 7a). p values are from a Fisher test with n=434 cells (left panel) and n=193 cells (right panel). (b) Comparison between inferred amount of transcriptomic Mk-priming and the percentage of CD41+ Mk-cells per colony. Errors bars denote S.E.M. p-value is from a Pearson product moment correlation test with n=627 single cells that formed colonies. See also Supplementary Fig. 7c. (c) Comparison between inferred amount of transcriptomic erythroid-priming and the percentage of CD235+ erythroid cells per colony. See also Supplementary Fig. 7c. Errors bars denote S.E.M. p-value is from a Pearson product moment correlation test with n=627 single cells that formed colonies. (d) Xenotransplantation validating a Mk-primed MPP population identified by STEMNET. HSCs, MLPs, and a population of putatively Mk-primed MPPs (Lin-CD34+CD38-CD45RA-CD90-CD135-) were sorted, transplanted into immunocompromised mice and chimerism of human lymphomyeloid cells (CD45+), thrombocytes and erythrocytes was determined 2 weeks post transplantation. Experimental setup (top right panel), localization of populations in STEMNET (left panels), and human engraftment (right panels, error bars denote SEM) are indicated. Relative contribution of thrombocytes was significantly higher in MK-primed MPPs compared to HSC (p=0.0031) and MLPs (p=0.0002, two-tailed unpaired t test, n=6 HSCs, n=4 Mk-primed MPPs, n=3 MLPs)
We next estimated the degree of transcriptomic lineage priming Srel for individual cells from the culture experiments (Fig. 7a, b). As expected, committed progenitors with a high degree of inferred transcriptomic lineage priming formed small colonies (Fig. 7a) of a single cell type (Fig. 7b). In contrast, primitive HSPCs (low inferred Srel) frequently displayed multi- or bilineage potential (Fig. 7b) and generated much larger colonies (Fig. 7a). However, not all of the primitive HSPCs displayed multipotency, but frequently appeared to be lineage-restricted while typically retaining a high proliferative capacity comparable to their multipotent counterparts (Fig. 7c). These data suggest that proliferative capacity and lineage potency are not obligatorily linked.
Fig. 7. The degree of transcriptomic priming is quantitatively linked to multipotency and proliferative capacity.
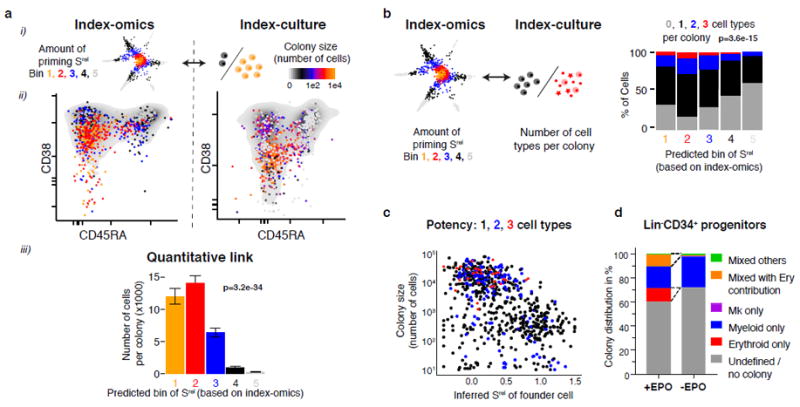
(a) Comparison between the inferred amount of transcriptomic priming Srel of the founder cell and the resulting colony size (cell number). (i) illustration, (ii) qualitative link and (iii) quantitative link. Errors bars denote S.E.M. p-value is from a Pearson product moment correlation test with n=1031 single cells. (b) Comparison between the inferred amount of priming Srel of the founder cell and the number of cell types in the colony. p-value is from a Pearson product moment correlation test with n=1031 single cells. (c) Inferred transcriptomic degree of priming Srel (x-axis) in relation to the colony size (y-axis) and the number of cell types per colony (colour-code). (d) Distribution of colony types in relation to the presence or absence of erythropoietin (EPO) in the culture medium.
In order to investigate the ability of cells with various amounts of priming to switch lineage potential, we cultured HSPCs in the absence and presence of erythropoietin (EPO). Progenitors that formed exclusively erythroid colonies in the presence of EPO were unable to give rise to alternative lineages in the absence of EPO (Fig. 7d). Moreover, we cultured single HSPCs for one week, split the colonies in four and determined the lineage outcome of the daughter colonies two weeks later. In line with the predictions of our model, the degree of transcriptomic priming was anticorrelated to the propensity of cells to generate daughters with variable lineage composition (Supplementary Fig. 7d, e). Together, these results support the hypothesis that early lineage priming of primitive HSPCs coincides with a loss of functional plasticity.
Molecular processes underlying HSC commitment
To characterize stemness, early lineage priming and transcriptional cell type manifestation on the molecular level, we identified co-expressed gene modules whose activities were associated with the direction and/or the degree of priming. We visualized the activity of these gene modules on the differentiation landscape established above (Fig. 8ai) and along the progression from HSCs to each of the six lineages (Fig. 8b, Supplementary Fig. 8a, b and Supplementary Table 4 for a complete list). Importantly, data from both individuals yielded highly comparable results (Supplementary Fig. 8). To gain additional information about biological processes associated with HSC differentiation, we determined the mean expression of genes for each gene ontology (GO) term, and selected representative examples that changed significantly during early lineage priming (Fig. 8c). Together, these analyses provide insights into the global molecular and cell biological processes HSCs encounter while undergoing continuous lineage priming, unilineage commitment and subsequent differentiation.
The least primed state was characterized by expression of the HOXA3/PRDM16/HOXB6 module26-28 (Fig. 8aii, 8b, Supplementary Table 4) and associated with typical stem cell properties such as cell cycle quiescence, low expression of the entire gene expression machinery, low total RNA content (measured by mRNA reads per in vitro spike in RNA read), low cellular respiration29, low CD38 and high CD90 surface expression5 (Fig. 8c). The expression of the HLF/ZFP36L2 module (which also contains the transcription factors MECOM/EVI1, HFL, GATA3) was highest in immature HSCs, but present in the entire “CLOUD” (Fig. 8aiii, 8b, Supplementary Table 4)30-32.
Intriguingly, stem cells also expressed genes from the earliest priming modules from both the lympho/myeloid (FLT3/SATB1 module) and the megakaryocyte/erythrocyte (GATA2/NFE2 module)33 lineages in a non-exclusive manner (Fig. 8aiv-v). These data suggest that the first transcriptional priming events into the predominantly lympho/myeloid or the megakaryocyte/erythrocyte direction are already present in most primitive HSCs, coinciding with the occurrence of first functional lineage biases already at this stage (Fig. 6a, b, 7a Srel bin 1 and 2). A number of additional gene modules was activated in a combinatorial fashion between lineages, similar to previous observations from bulk RNA Seq34 (Fig. 8, Supplementary Fig. 8a, Supplementary Table 4).
Upon acquisition of lineage priming, HSCs up-regulate their gene expression machinery, mRNA and protein biosynthesis, and respiration29,35, while cell cycle activity increases only marginally (Fig. 8c). At this stage, cells start to express lineage-specific gene modules, for example the SPI1/GFI1 module for the neutrophil lineage (Fig. 8aviii) or the IRF1/CASP1 module33 for the B-cell lineage (Fig. 8avi). Other modules active at this stage, however, are shared between lineages; for example, the TAL1/HFS1 module is shared between the erythroid and the megakaryocytic lineage, whereas the EAF2/KLF4 module is shared between the neutrophil and the monocyte lineage. This coincides with the observation that most progenitors at this stage display narrow restriction in their developmental potential, whereas some progenitor cells remain oligopotent15 (Fig. 7b, Srel bin 3).
Manifestation of lineage-specific differentiation is accomplished by activation of gene modules such as the CEBPA/CEPBD module for the neutrophil lineage, the EBF1/ID3 module for the B-cell lineage, the IRF8 module for the monocytic/dendritic lineage, the GPI1BB/PBX1 module for the megakaryocytic lineage and the GATA1/KLF1 module for the erythroid lineage33,36,37 (Fig. 8ax-xv, b). In all cases, this step is accompanied by cell cycle activation, CD38 surface marker up-regulation (Fig. 8c) and unipotency (Fig. 7b, Srel bin 4 and 5).
Together, our data suggest that HSCs are characterized by the expression of specific stem cell modules in combination with early, probably antagonizing priming modules. During the continuous priming and differentiation process the stem cell modules and certain (but not all) early priming modules already expressed in HSCs are turned off, while specific lineage modules become reinforced to drive differentiation towards lineage commitment and manifestation (Fig. 8a, b). Transcription factors from upstream modules may trigger expression of downstream modules, as in case of GATA2, TAL1 and GATA133. In contrast, transcription factors from mutually exclusive downstream modules may inhibit each other, for example IRF8 is known to repress CEBPA38. Such inhibitory interactions may render oligopotent progenitors unstable7,10,15, and thus less abundant than previously anticipated (Fig. 7b). In contrast, in cells with low amount of priming, expression levels of mutually exclusive modules are sufficiently small to allow uni-, oligo- or multipotency.
DISCUSSION
In summary, we provide a global view of the early human haematopoiesis during homeostasis. Our dataset combines both information on the lineage potential of HSCs (index-culture) and insights into the unperturbed lineage commitment of HSCs during human haematopoiesis (reconstruction of developmental trajectories from static single cell expression data), where lineage tracing approaches8,9 are not possible. Here, we rely on single-cell culture data and xenotransplantation for functional validation, which unlike gene expression or cellular barcoding measure developmental potential, not fate.
Our results are incompatible with fundamental aspects of the differentiation-tree model, in which HSCs are required to pass through discrete and definable intermediate progenitor cell stages by subsequent binary cell fate decisions made on branching points. Instead, we propose that early haematopoiesis is represented by a cellular Continuum of LOw-primed UnDifferentiated (“CLOUD”)-HSPCs. This HSPC continuum contains phenotypic MPPs and MLPs, which do not constitute discrete progenitor cell types, but rather transitory states. CLOUD-HSPCs gradually acquire transcriptomic lineage priming in a combination of multiple directions, with some cell state transitions and lineage combinations more likely to occur than others. Distinct lineages emerge directly from CLOUD-HSPCs, earlier than previously anticipated and without passing through a series of discrete, stable progenitors. Our data suggest a multidimensional molecular and cellular landscape of steady state human haematopoiesis defined by a continuous flow of differentiation and emergence of lineage trajectories independent of each other. This landscape can be visualized by using the classical Waddington’s landscape as a blueprint39-41, which more appropriately reflects the continuous nature of haematopoiesis than a “cell type tree” (Fig. 8d). Haematopoietic stem cells reside in a flat valley at the top. Barriers separating individual lineages emerge early and deepen gradually, illustrating the acquisition of lineage biases driven by small differences in gene expression of early fate mediators. When barriers become insurmountable, cell type manifestation and lineage commitment are established.
While our study provides detailed insight into lineage commitment from HSCs into all branches of human bone marrow haematopoiesis, it does not cover lineage decisions occurring further downstream or outside the bone marrow, such as T-cell development. Given the low frequency of Eosinophil/Basophil/Mast cell and Monocyte/Dendritic Cell progenitors within the CD34+ bone marrow compartment, our study cannot fully resolve the separation and maturation of these lineages.
Together, our data determine a comprehensive continuum-based model of early human haematopoiesis, which will likely have important implications for the aetiology of haematologic disorders and which may serve as a paradigm for other adult stem cell systems.
Methods
Bone marrow aspirations
Bone marrow aspirates from healthy individuals between 25 and 39 years of age were obtained at the University clinics in Heidelberg and Mannheim after written informed consent. The use of human samples for RNA-Sequencing and functional studies was approved by the local ethics committees in accordance with the Declaration of Helsinki. Bone marrow mononuclear cells were isolated by gradient centrifugation using Histopaque-1077 (Sigma).
Flow Cytometry
Bone marrow mononuclear cells were stained with surface markers for 30 minutes on ice according to standard protocols. For FACS-sorting BD FACS Aria II/III or Fusion flow cytometers (BD Bioscience) equipped with 405nm, 488nm, 561nm and 633nm (Aria) / 642nm (Fusion) lasers were used. For flow cytometric analyses LSRII and LSRFortessa flow cytometers (BD Biosciences) equipped with 350nm, 405nm, 488nm, 561nm, and 640nm lasers were used. For Ki67-Hoechst cell cycle analysis, surface staining was performed as described43. Subsequently, cells were fixed and permeabilized using cytofix-cytoperm buffer (BD Bioscience), and incubated with Ki67 antibody overnight at 4°C. Cells were stained with 2μg/mL Hoechst 33342 (Invitrogen) and analyzed. Data were analyzed using FlowJo (TreeStar), indeXplorer or R.
Single-cell liquid cultures (“index-cultures”)
Fresh human bone marrow mononuclear cells were stained as described above with fluorescence labeled antibodies against CD2, CD34, CD38, CD45RA, CD71, CD90, CD130, CD135, CD238 (KEL), Fc$\epsilon$RI and a lineage cocktail consisting of CD4, CD8, CD11b, CD14, CD19, CD20, CD56, CD235a plus CD10. Single Lin-CD34+CD38+CD10- and Lin-CD34+CD38-CD10-HSPCs were sorted into ultra-low attachment 96 well-plates (Corning) containing 100μL StemSpan SFEM media (Stem Cell Technologies), L-glutamine (100ng/mL), penicillin/streptomycin (100ng/mL) and the following human cytokines: SCF (20ng/mL, Peprotech), Flt3-L (20ng/mL, Peprotech), TPO (50ng/mL, Peprotech), IL-3 (20ng/mL, Peprotech), IL-6 (20ng/mL, Peprotech), G-CSF (20ng/mL, Peprotech), IL-5 (20ng/mL, Peprotech), M-CSF (20ng/mL, Peprotech), GM-CSF (20ng/mL, Peprotech) and EPO (4U/mL, R&D). For the experiment displayed in Fig. 7d, Epo was left out from the medium. Note that the CD38+ and CD38- gates were set to touch (see also Supplementary Fig. 1a).
Fluorescence intensities were recorded for every channel for each sorted cell and used to retrospectively reconstruct immunophenotypic populations. Cells were cultured for 21 days at 5% CO2 and 37°C. To characterize clonal progeny, colonies were imaged by microscopy and subsequently analyzed for CD15, CD33, CD41a and CD235a expression by flow cytometry. Note that under these conditions, only myeloid (CD33), erythroid (CD235a) and megakaryocytic (CD41a) colonies are efficiently generated. Colonies were judged based on their visual morphology and expression of surface markers. Colony size and lineage-output were based on flow cytometry and confirmed by microscopy. A colony was determined to be positive for a particular lineage if ≥10 cells of the respective cell type were detected.
For the `split-in-four’ experiment (Supplementary Fig. 7d, e), colonies were evaluated 7 days after seeding of single cells and colonies with more than 50 cells were equally split into 4 wells and cultured for additional 14 days before colony-size and lineage output were scored.
Mouse experiments
NSG mice were bred and housed under specific pathogen-free conditions at the central animal facility of the German Cancer Research Center. All animal experiments were approved by the Regierungspräsidium Karlsruhe under Tierversuchsantrag numbers G108/12 and G210/12. 17,000 FACS-sorted HSCs (Lin-CD34+CD38-CD90+CD45RA-), MLPs (Lin-CD34+CD38-CD45RA+) or MK-primed MPPs (Lin-CD34+CD38-CD90-CD135-) from healthy bone marrow were injected into the femoral bone marrow cavity of female mice 15 weeks of age that had been sublethally irradiated (200 cGy) 24 hours before injection.
2 weeks after xenotransplantation lineage specific human engraftment in the injected femur was evaluated by flow cytometry using anti-human-CD45-PE, anti-human-CD235a-APC and anti-human-CD41a-FITC antibodies.
Single-cell transcriptome sequencing (“index-omics”)
A 25-year old male donor (Individual 1) and a 29-year old female donor (Individual 2) were selected for single-cell RNA Sequencing. Fresh bone marrow mononuclear cells were stained as described above with fluorescence-labeled antibodies against CD34, CD38, CD45RA, CD90, CD49f, CD135, CD10, CD7 and a lineage cocktail consisting of CD4, CD8, CD11b, CD14, CD19, CD20, CD56 and CD235a. Fluorescence intensities were recorded for every channel for each sorted cell and used to reconstruct immunophenotypic populations subsequently.
While the frequently used smart-seq2 protocol44 failed to amplify transcriptomes from bone marrow derived human HSPCs, both the QUARTZ-seq protocol45 and a modified smart-seq2 protocol (see below) yielded good-quality cDNA (Supplementary Fig. 2a). To avoid method-specific biases, data were generated using both QUARTZ-seq (Individual 2) and smart-seq2.HSC (Individual 1), and all findings were systematically compared between individuals (Fig. 2, 3b, Supplementary Figures 4a, b, 5a,b, 8c).
For individual 1, eight plates of Lin-CD34+CD38- and six plates of Lin-CD34+CD38+ HSPCs were sorted and whole transcriptome amplification was performed using the smart-seq2 protocol44, but using 5μL of a modified RT buffer containing 1x SMART First Strand Buffer (Clontech), 1mM DTT (Clontech), 1μM template switching oligo (Exiqon), 10U/μL SMARTScribe (Clontech) and 1U/μL RNASin (Promega). ERCC spike-ins were included at a final dilution of 1:1,000,000. Libraries were constructed using a home-made Tn5 transposase (based on ref. 46). Note that the CD38+ and CD38-gates were set to touch (see also Supplementary Figure 1a).
For individual 2, eight plates of Lin-CD34+CD38-, one plate of Lin-CD34+CD38-CD90+CD45RA- and four plates of Lin-CD34+CD38+ HSPCs were sorted and whole transcriptome amplification was performed using the QUARTZ-Seq protocol45. ERCC spike-ins were included into the lysis buffer at a final dilution of 1:2,000,000. Libraries were constructed using Nextera Tn5 (Illumina) following the protocol provided, but using 1/4 of all volumes. Libraries were then sequenced on an Illumina HiSeq 2500 platform.
Raw data processing and quality control
Reads were demultiplexed and, where applicable, the remaining poly-A tail of the mRNA was trimmed off. Reads were then aligned to the Homo Sapiens genome (build 37.68, also containing the ERCC spike in sequences) using GSNAP47, with the expected paired-end length set to 400bp and the allowable deviation from the expected paired-end length set to 100bp. Reads overlapping uniquely with mRNA genes were counted using HTSeq48. As a first filtering step, we retained all cells in which we observed more than 750 genes at a minimum of 10 reads each, and a total of at least 150,000 reads. We removed all genes from the dataset that were not observed by at least 10 reads in at least 5 cells. Statistics on these filtering steps are displayed in Supplementary Fig. 2.
We then fitted error models49 to the readcount data (see also below). In 35 cells of individual 2 and 1 cell of individual 1, we observed an extreme overdispersion of the genes classified as non-dropout events. These cells were removed. In Individual 1, we further excluded 13 cells with an abnormal CD38-CD90high immunophenotype (Supplementary Fig. 1a). These cells were clear outliers also with regard to gene expression, as they mostly expressed genes associated with various types of mature immune cells (not shown).
Data normalization using Posterior Odds Ratio
We designed a normalization method to address the following two challenges:
Single-cell transcriptomics has large technical variability
Human hematopoietic stem and progenitor cells largely differ in RNA content (Supplementary Fig. 2h).
While lowly expressed genes are sometimes observed in cells with high total RNA content, they are almost never seen in cells with low total RNA content (Supplementary Fig. 2i). As this effect is the same for all genes of low expression level, it will induce some correlation structure on the data. In our data set, the first principal component was correlated to the library size and mRNA content, which may dominate over the effects of developmental transitions (Supplementary Fig. 2j, panel i). Normalization through division by total library size or harmonic mean estimator does not resolve this issue, as lowly expressed genes are still unobserved (zero) in cells of low mRNA content (Supplementary Fig. 2i,j panel ii). We and others have therefore used hierarchical models which assume that molecule counts are created by sampling from the true amount of mRNA molecules with cell-specific sampling efficiencies50,51. To adapt these approaches to the case where no molecular barcodes were used, we here use the error model of Kharchenko et al.49, which describes the posterior probability of a gene expression level x in a cell c as
where pd is the probability of a dropout event at gene expression x, pNB is the probability of observing rc reads in case of no dropout and pPoisson(x) is the the probability of observing rc spurious reads in case of a dropout. Ωc is a vector of cell-specific and numerically optimized parameters:
The slope and intercept of pd as a function of rc
The slope and intercept of x as a function of rc
The dispersion of the negative binomial distribution pNB(x|rc)
The background frequency λ of the Poisson distribution, which was fixed to 0.1
The maximum posterior average expression across all cells is then given by
While the mean of Πc p(x∣rc, Ωc) describes the expression magnitude of a gene in a given cell, its spread describes the uncertainty due to technical noise. To obtain a single number that weighs expression magnituide by confidence level, we compute a Posterior Odds Ratio (POR):
POR can be interpreted as the evidence (in bits) that a specific gene in a specific cell is expressed more highly (or lowly) than in the average cell. The use of POR scores in principal component analysis solved the problems associated with the above-mentioned normalization strategies (Supplementary Fig. 2j, panel iii) POR scores were used as the measure of gene expression for all analyses.
Clustering
For hierarchical clustering, we selected the 1000 most variable genes of each population. We then used Ward linkage on euclidean distances. Gap-statistics was computed on the same hierarchical clustering function using the R package cluster. Random walk analysis52 was performed by constructing a 5-nearest neighbor graph on correlation distances, initializing at a random node, and then simulating a series of random steps on the 5-connected graph. The local clustering coefficient of a node in such a graph quantifies the extent to which the neighbors of two connected cells are themselves connected to each other. It was computed using the transitivity function of the igraph package53
STEMNET
Basic setup
To identify processes associated with the transition of HSCs to progenitor cell types, we sought a lower-dimensional representation of the HSPC data that reflects lineage priming. We therefore trained an elastic-net regularized generalized linear model (GLMNET) of the multinomial family on the most mature populations (N1-3, EBM, MD, spB1/2, E1/2 and Mk from Fig. 2a for individual 1, or lpB, EBM, N, ME and MD for individual 2), using class membership as the response variable. During this step, a number of population-specific genes was identified (Supplementary Table 3). The classifier then used the expression of these genes in all cells to estimate the probability pij that a cell i belongs to class j. From these probabilities, we compute the Kullback-Leibler distance from the average HSPC, which can be interpreted as the amount of lineage information a given cell has acquired:
whre p̅j is the average probability of a cell to belong to class j. We further assign each cell a predominant direction of priming as
For displaying the six-dimensional vector pi in two dimensions, the developmental endpoints are arranged on the edge of a circle and all cells are placed in between. Each endpoint k is assigned with an angle αk. The class probabilties pik are then transformed to cartesian coordinates by
and
To find the optimal arrangement of the developmental endpoints on the circle, lineages with common precursor stages are placed next to each other. The proximity between lineages l and k is computed by
All arrangements are tested and the arrangement with the highest proximity is chosen. This approach is based on a method termed ’circular a posteriori projection’ (CAP)51.
Data simulation
To test the ability of the STEMNET method to uncover binary branching events and discrete sub-populations, we quantitatively specified alternative models of cell fate specification and reshuffled our original data according to these models (Supplementary Fig. 6). In particular, we assumed that each cell is located on a binary tree, where nodes represent branching points and edges between nodes represent developmental trajectories. Each node Vi is specified by a tuple (E1, E2, p1, p2, h) with E1,2 pointing to the left and right child, p1,2 giving the probability that a cell adapts the fate associated with the left and right child (p1 + p2 = 1), and h ∈ (0, 1) giving the height of the node (for developmental endpoints, h=1, and for the root, h=0). A cell is then defined by the tuple (h, E), where E points to the next node downstream of the cell.
For the scenario depicted in Supplementary Fig. 6a, cells were generated by randomly drawing values h from a Beta distribution with parameters (2,3). E was assigned by moving down a distance of h from the root and randomly choosing a branch according to p1,2 at each node that was passed. For the scenario depicted in Supplementary Fig. 6d, cells were then scattered around the nearest node assuming an average distance of 0.01. The developmental distance D(ci,Vj) between a cell ci and a node Vj is then computed by traversing through the tree and summing all distances h that are passed along the way. For example, the distance between two developmental endpoints that diverge at a node with h=0.6 is 0.8. To generate synthetic data from these cell fate specification models, we extracted the coefficients of the STEMNET classifer (Supplementary Table 3), and for each developmental endpoint j compiled lists of genes with nonzero coefficient. Gene expression values for these genes were then reordered across cells i to follow the developmental distance D(ci,Vj) (i.e. assuming that gene expression of lineage specific genes was entirely determined by developmental distance, Supplementary Fig. 6a). Alternatively, gene expression values were randomly reshuffled such that the correlation between developmental distance from Vj and gene expression equals the empirically observed correlation between gene expression and pj from the STEMNET classifier (Supplementary Fig. 6b-d).
Quantitative link between single-cell transcriptomics and single-cell culture
To quantitatively link single-cell transcriptomic properties (such as the amount or direction of priming) to single-cell functional properites, we made use of FACS markers used in both experiments. In particular, for each transcriptomic property, we constructed a regression model with logicle transformed flow cytometry markers as explanatory variables and the property as response variable. To achieve greater robustness than in standard linear regression, we applied GLMNET models of the normal family for this task, and used 10-fold cross validation to determine the regularization parameter λ. The regression coefficients of these models are shown in Supplementary Fig. 7a, together with the R2 these models achieve in 10-fold cross validation if applied to the single cell transcriptomic data. We then applied these classifiers to logicle transformed flow cytometry data from the single-cell culture experiment to estimate the magnitude of single-cell transcriptomic properties in that experiment. To further improve the classifier, we also included rank-transformed mRNA expression levels of TFRC (CD71) and KEL in the the training data, and rank-transformed flow cytometry data of CD71 and KEL in the single-cell culture experiment.
Identification of gene clusters associated with lineage priming
We then identified genes whose expression depends on Srel, d, or both, by separately fitting four different linear models to the expression data of each gene:
The first model describes gene expression as a function of the predominant direction d, which is a categorial variable. It best fits to genes that are up- or downregulated early during developmental progression in a certain direction and stay unchanged until the end.
The second model describes gene expression as a function of a 3rd degree polynomial through log10 Srel. It best fits to genes that are up- or downregulated at a specific stage of developmental progression, independent of the developmental direction.
The third model describes gene expression as a function of d, a 3rd degree polynomial through log10 Srel and the interaction of d and log10 Srel. It best fits to genes that are up- or downregulated at a specific stage of development in a specific direction.
The fourth model describes gene expression as a constant. It best fits to genes that do not change systematically during acquisition of lineage fate.
For each gene, we identified the optimal model by comparing the models’ Bayesian Information Criteria (BIC). For each class of genes (dependent on log10 Srel, d or both) separately, we identified subgroups of genes that display similar dependencies on log10 Srel and d by performing hierarchical clustering using correlation distance and complete linkage on the fitted values from the preferred model.
Code availability
Most analyses were performed in indeXplorer, a custom made software for the analysis of single cell index-sorting/transcriptomic datasets. indeXplorer was written in R and relies on the package shiny; code is available from https://git.embl.de/velten/indeXplorer/
For analyses that were not performed in indeXplorer directly, we provide an R package containing all code at https://git.embl.de/velten/STEMNET
Statistics and Reproducibility
Single cell RNA-Seq was performed on two different individuals. 1034 (for I1) and 379 cells (for I2) were included into the study. Single cell culture was performed for 2038 cells. As indicated in the figure legends, p-values are computed from Pearson product moment correlation test, Kernel density based global two sample comparison test or two-tailed unpaired t-test.
For animal experiments, no statistical method was used to predetermine sample size. The experiments were not randomized. The Investigators were not blinded to animal allocation during experiments and outcome assessment.
Data availability
RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE75478. Processed data are available at http://steinmetzlab.embl.de/shiny/indexplorer/?launch=yes for browsing. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank Christian Drumm for help with 3D graphics, Klaus Hexel, Steffen Schmitt, Claudia Felbinger and Marcus Eich from the DKFZ flow cytometry facility for flow cytometry support, the EMBL Genomics Core Facility for sequencing and Raeka Aiyar, Allan Jones, Michael Milsom and all members of HI-STEM and the Steinmetz group for helpful discussions on the manuscript as well as Timm Schroeder and Dirk Löffler for initial discussions. This work was supported by the SFB873 funded by the Deutsche Forschungsgemeinschaft (DFG) (to C.L., M.A.G.E. and A.T.), the Dietmar Hopp Foundation (to M.A.G.E. and A.T.) and the US National Institutes of Health (P01 HG000205 to L.M.S.).
Footnotes
Author Contributions
S.F.H., S.R., L.V., S.B and C.H. performed the experiments. L.V. analysed the data, with conceptual input from S.F.H., S.R., L.M.S., M.A.G.E. and A.T., and analytical advice from W.H.. S.I. and B.P.H. optimized genomics methods. C.L., E.C.B., D.N., T.B., W.K.H. and A.D.H. obtained bone marrow aspirates. L.V., S.F.H., S.R., M.A.G.E., L.M.S. and A.T. jointly conceived and designed the study, and wrote the manuscript.
Author information
The authors declare no competing financial interests.
References
- 1.Chao MP, Seita J, Weissman IL. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb Symp Quant Biol. 2008;73:439–449. doi: 10.1101/sqb.2008.73.031. [DOI] [PubMed] [Google Scholar]
- 2.Morrison S, Uchida N, Weissman I. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol. 1995;11:35–71. doi: 10.1146/annurev.cb.11.110195.000343. [DOI] [PubMed] [Google Scholar]
- 3.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 4.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 5.Doulatov S, et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nat Immunol. 2010;11:585–93. doi: 10.1038/ni.1889. [DOI] [PubMed] [Google Scholar]
- 6.Notta F, et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–21. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 7.Notta F, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science (80-) 2016;351:aab2116–aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busch K, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- 10.Perié L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-Myeloid Differentiation. Cell. 2015;163:1655–1662. doi: 10.1016/j.cell.2015.11.059. [DOI] [PubMed] [Google Scholar]
- 11.Haas S, et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell. 2015;17:422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Görgens A, et al. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 2013;3:1539–52. doi: 10.1016/j.celrep.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 13.Adolfsson J, et al. Identification of Flt3+ Lympho-Myeloid Stem Cells Lacking Erythro-Megakaryocytic Potential. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto R, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–26. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Naik SH, et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature. 2013;496:229–32. doi: 10.1038/nature12013. [DOI] [PubMed] [Google Scholar]
- 16.Paul F, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Wilson NK, et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell. 2015;16:712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin J, et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsson A, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537:698–702. doi: 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theilgaard-Monch K. The transcriptional program of terminal granulocytic differentiation. Blood. 2005;105:1785–1796. doi: 10.1182/blood-2004-08-3346. [DOI] [PubMed] [Google Scholar]
- 22.Borregaard N. Neutrophils, from Marrow to Microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2013;14:69–80. doi: 10.1038/nri3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haghverdi L, Büttner M, Wolf FA, Buettner F, Theis FJ. Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods. 2016;13:845–848. doi: 10.1038/nmeth.3971. [DOI] [PubMed] [Google Scholar]
- 25.Hoppe P, et al. Early myeloid lineage choice is not initiated by random PU.1 to GATA1 protein ratios. Nature. 2016;535:299–302. doi: 10.1038/nature18320. [DOI] [PubMed] [Google Scholar]
- 26.Fischbach NA, et al. HOXB6 overexpression in murine bone marrow immortalizes a myelomonocytic precursor in vitro and causes hematopoietic stem cell expansion and acute myeloid leukemia in vivo. Blood. 2005;105:1456–66. doi: 10.1182/blood-2004-04-1583. [DOI] [PubMed] [Google Scholar]
- 27.Iacovino M, et al. HoxA3 is an apical regulator of haemogenic endothelium. Nat Cell Biol. 2011;13:72–78. doi: 10.1038/ncb2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12:999–1006. doi: 10.1038/ncb2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–56. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shojaei F, et al. Hierarchical and ontogenic positions serve to define the molecular basis of human hematopoietic stem cell behavior. Dev Cell. 2005;8:651–663. doi: 10.1016/j.devcel.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Kataoka K, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208:2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frelin C, et al. GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat Immunol. 2013;14:1037–44. doi: 10.1038/ni.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–6269. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Signer RAJ, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509:49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- 37.Hystad ME, et al. Characterization of Early Stages of Human B Cell Development by Gene Expression Profiling. J Immunol. 2007;179:3662–3671. doi: 10.4049/jimmunol.179.6.3662. [DOI] [PubMed] [Google Scholar]
- 38.Kurotaki D, et al. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun. 2014;5:4978. doi: 10.1038/ncomms5978. [DOI] [PubMed] [Google Scholar]
- 39.Waddington CH. The Strategy of the Genes. Routledge; 1957. [Google Scholar]
- 40.Brock A, Chang H, Huang S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10:336–42. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- 41.Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–62. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 43.Essers MAG, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 44.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 45.Sasagawa Y, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA-Seq reveals non-genetic gene expression heterogeneity. Genome Biol. 2013;14:R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picelli S, et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014;24:2033–2040. doi: 10.1101/gr.177881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Velten L, et al. Single-cell polyadenylation site mapping reveals 3’ isoform choice variability. Mol Syst Biol. 2015;11:812. doi: 10.15252/msb.20156198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaitin DA, et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science (80-) 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Dongen S. Graph Clustering Via a Discrete Uncoupling Process. SIAM J Matrix Anal Appl. 2008;30:121–141. [Google Scholar]
- 53.Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal Complex Sy. 2006:1695. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE75478. Processed data are available at http://steinmetzlab.embl.de/shiny/indexplorer/?launch=yes for browsing. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.