

Summary
The Bartonella gene transfer agent (BaGTA) is an archetypical example for domestication of a phage-derived element to permit high-frequency genetic exchange in bacterial populations. Here we used multiplexed transposon sequencing (TnSeq) and single-cell reporters to globally define the core components and transfer dynamics of BaGTA. Our systems-level analysis has identified inner- and outer-circle components of the BaGTA system, including 55 regulatory components, as well as an additional 74 and 107 components mediating donor transfer and recipient uptake functions. We show that the stringent response signal guanosine-tetraphosphate (ppGpp) restricts BaGTA induction to a subset of fast-growing cells, whereas BaGTA particle uptake depends on a functional Tol-Pal trans-envelope complex that mediates outer-membrane invagination upon cell division. Our findings suggest that Bartonella evolved an efficient strategy to promote genetic exchange within the fittest subpopulation while disfavoring exchange of deleterious genetic information, thereby facilitating genome integrity and rapid host adaptation.
Keywords: TnSeq, systems biology, Bartonella, gene transfer agent, horizontal gene transfer, ppGpp
Graphical Abstract
Highlights
-
•
Comparative TnSeq deciphers BaGTA donor and recipient activities
-
•
The entire chromosome is a substrate for high-frequency recombination via BaGTA
-
•
BaGTA induction in the fittest subpopulation is ppGpp dependent
-
•
BaGTA uptake is linked to the Tol/Pal trans-envelope complex
Donor and recipient activities of a gene transfer agent mediating highly efficient genome-wide DNA exchange are described using comparative TnSeq analysis and follow-up experiments.
Introduction
Clonal populations typically deteriorate due to gradual loss of fitness and eventual extinction caused by accumulation of slightly deleterious mutations via genetic drift (Charlesworth et al., 1993, Muller, 1964). This evolutionary mechanism, known as Muller's ratchet, appears to be the typical fate of pathogens that thrive on complex infection cycles, including intracellular parasitism (Iranzo et al., 2016, Moran, 1996), that experience frequent population bottlenecks. One route of actual escape from Muller's ratchet appears to be gene acquisition via horizontal gene transfer (HGT), resulting in either repair of a deleteriously mutated gene by a functional copy or acquisition of new genes that offsets the deleterious effects of accumulating mutations (Takeuchi et al., 2014).
A prominent source of HGT in bacterial communities is gene transfer agents (GTAs). GTAs are chromosomally encoded DNA transfer systems that promote genetic exchanges between individual cells through cell lysis and release of bacteriophage-like intermediates. The current view is that a fraction of a bacterial population sacrifices itself to serve as GTA donors, producing phage-like particles that are packed with a random portion of their chromosomal DNA. The remaining part of the population may act as recipient, taking up the particles and incorporating the contained DNA into their chromosome (Figure 1A and for review, see Lang et al., 2012). Considering the limited DNA packing size of GTAs (less than the size of the locus encoding the GTA function) and the apparent lack of sequence specificity, GTAs are considered to be strictly vertically inherited. This is in contrast to bacteriophages, which largely promote exchange of phage-specific genes and are therefore self-transmissible. In this respect, GTAs can be considered as a form of “domesticated” prophage—that is, ancestrally derived from a bacteriophage genome but altered by the host to confer an adaptive benefit—and thus represent one of many phage-derived adaptive functions observed in bacterial genomes (Bobay et al., 2014, Hynes et al., 2016). It is, however, unclear how maintenance of a system that can induce the lysis of individual cells may still be beneficial to the entire population.
Figure 1.

Chromosomal Marker Exchanges during B. henselae Co-culture
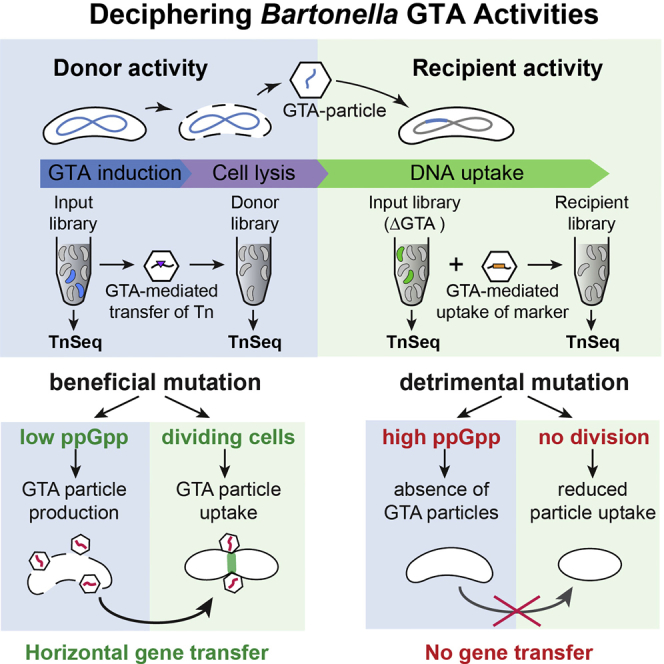
(A) Schematic model for BaGTA-mediated genetic exchange in Bartonella. In a three-stage process, induction of BaGTA in donor cells (blue) leads to host cell lysis (purple) and release of BaGTA particles that are taken up by recipient cells (green).
(B) Induction dynamics of the BaGTA locus as detected by FACS analysis of gfp-bgtC reporter strain upon cultivation of Bartonella in M199. A subpopulation of wild-type cells synchronously induces BaGTA locus (open circles) leading to early and late peaks in GTA induction. BaGTA induction is absent in cells expressing the constitutive active ppGpp synthase gene RelA1–455 from E. coli, suggesting that elevated levels of ppGpp repress induction of BaGTA. The mean values from a biological triplicate and their associated SEs are displayed.
(C) Cell density upon cultivation in M199 is shown as a spline fit (dotted line, purple) to the experimental growth data as determined by CFU counts. A temporal decrease in viable cells around 10 and 30 hr indicates BaGTA-induced cell lysis (early and late lysis). The mean values and associated SD from a biological triplicate are displayed.
(D) Relative transduction dynamics as a function of the co-culturing time are shown. The experimentally determined DNA uptake rate (transduction frequency, open circles) follows a time-delayed function (black line) proportional to the integrated GFP-expression rate of the first BaGTA induction peak (blue dashed line), while the integrated GFP-expression rate of the second BaGTA induction peak (cyan dashed line) does not contribute to detectable DNA uptake. The mean values from a biological triplicate and their associated SEs are displayed.
The alpha-proteobacterial genus Bartonella comprises an expanding number of arthropod-borne pathogens (e.g., lice, fleas, or keds) that share the ability to cause long-lasting intraerythrocytic infections in their mammalian reservoir hosts (Harms and Dehio, 2012). Genomic sequence analysis has revealed that all bacteria of the genus Bartonella are characterized by the presence of a Bartonella-specific GTA (BaGTA), which shares no homologies to previously described GTA systems. Interestingly, BaGTA is encoded upstream from an origin of run-off replication (ROR), another conserved feature of Bartonella genomes which has been suggested to be linked to the activity of BaGTA. Phylogenetic analyses have identified BaGTA as a key innovation associated with the spectacular adaptive radiation that characterizes these zoonotic bacterial pathogens (Guy et al., 2013). Although BaGTA has not been directly linked to Bartonella pathogenicity, it has been proposed to drive the exchange and the diversification of host-interaction factors within Bartonella communities such as the well-characterized VirB type IV secretion system (T4SS) and its cognate Bartonella effector proteins (Engel et al., 2012, Guy et al., 2012). Maintenance of BaGTA is likely driven by selection to increase the likelihood of genetic exchange and facilitates rapid adaptation to host-specific defense systems during infection (Guy et al., 2013). In addition, BaGTA may help Bartonella avoid Muller's ratchet. As a facultative intracellular pathogen Bartonella experiences major transmission bottlenecks throughout its infection cycle, both within the arthropod and while breaching the mammalian host barriers during invasion of erythrocytes. Exposure to host defense mechanisms such as reactive oxygen species likely results in the accumulation of mutations within the pathogen’s chromosome. Most acquired mutations are neutral, only very few evolve new functions and possibly show beneficial effects, while a sizable fraction of mutations will have deleterious effects resulting in fitness losses.
Despite clear genomics-based arguments pointing to a central role for BaGTA in Bartonella biology, direct experimental evidence for its activity are scarce and the molecular mechanisms underlying its activity and regulation have remained fully elusive. Here, we have taken an experimental systems biology approach based on multiplexed transposon sequencing (TnSeq) measurements and single-cell reporter assays to globally define the core components and transfer dynamics of the Bartonella GTA system. Our analysis has identified inner- and outer-circle components of BaGTA mediating donor transfer and recipient uptake functions. Moreover, rather than being a random process, we found that BaGTA specifically promotes genetic exchange between the fittest subpopulation of cells, as the stringent response signal guanosine-tetraphosphate (ppGpp) restricts BaGTA induction to a subset of fast-growing cells.
Results
The BaGTA System Is Transiently Induced within a Subpopulation of Cells
Because the BaGTA locus is broadly conserved across Bartonella species (Guy et al., 2012), we hypothesized that the regulation of BaGTA is likely connected to the infection cycle of these arthropod-borne pathogens. To characterize BaGTA behavior across time in a population of Bartonella cells, we constructed a reporter strain carrying a transcriptional gfp fusion to the phage-related tail collar gene (BH13960, renamed bgtC) located at the BaGTA locus. We monitored single-cell BaGTA expression dynamics upon growth in medium M199/10% fetal calf serum (hereafter M199), a host cell-free system commonly used to study the temporal progression of the Bartonella virulence program (Québatte et al., 2013). Time-course analysis using flow cytometry (fluorescence-activated cell sorting [FACS]) revealed that the gfp-bgtC reporter was heterogeneously expressed within a subpopulation of cells. While the majority displayed no detectable fluorescence (off-state), we observed a distinct subpopulation, corresponding to 6% of all cells, with induced GFP reporter (on-state) (Figure 1B). This observation is in accord with reports on the GTA systems in other species. For example, similar heterogeneous induction of the prototypical GTA system encoded by Rhodobacter capsulatus (RcGTA) system within 0.1%–3% of all cells has been observed in R. capsulatus (Fogg et al., 2012, Hynes et al., 2012). When we followed bgtC expression dynamics as a function of the M199 culturing time, we found that the BaGTA locus displayed a bimodal expression pattern with a dominant early peak of expression followed by a second less prominent peak at late time points (Figure 1B). Taken together, these findings indicate that upon M199 cultivation, BaGTA is induced in a synchronized manner within a subpopulation of cells.
To gain quantitative insights into BaGTA induction, we modeled the bgtC expression with an empirical function that captures the observed trend of a latent induction phase leading to an exponential increase in GFP reporter followed by an exponential decay (STAR Methods) (Holder and Beauchemin, 2011). Maximal BaGTA induction occurred 6.62 ± 0.44 hr post inoculation with 14.1% ± 0.61% of all cells displaying GFP expression (Figure 1B). BaGTA expression was followed by a sharp decay in GFP signal intensity with a half-life of 66.8 min (+12.1, −8.8) min. These kinetics are in agreement with previously published transcription profiling of Bartonella henselae genes during human endothelial cell infection (Québatte et al., 2010).
Furthermore, we noticed that the decay in early peak expression of the BaGTA locus coincides with a short time window when, after an initial phase of rapid cell proliferation, cell number temporally declines (Figure 1C), suggesting that induction of BaGTA is interlinked with cell growth. To test whether the observed decrease in GFP-positive cells could be caused by cell lysis, we recovered cells in the GFP on- and off-state using FACS and assessed their viability using a colony formation assay (STAR Methods). BaGTA-induced cells showed a ∼5-fold decrease in viability with less than 21% of all cells maintaining the ability to form colonies. This suggests that induction of BaGTA produces an irreversible state change in many cells that commits them to lysis. Accordingly, the second induction peak at time point 24 hr post co-cultivation also resulted in cell lysis, as suggested by a drop in colony-forming unit (CFU) counts at the 30 hr time point (Figure 1C). However, cell lysis associated with the second induction peak did not result in any detectable DNA uptake (Figure 1D) as would be expected if the BaGTA particles were taken up by recipients. This suggests that, while a small subpopulation of cells is still capable of BaGTA induction and cell lysis, the majority of the recipient cells fail to take up transducing particles after the initial fast-growing state observed in the culture.
The BaGTA System Mediates High-Frequency Chromosomal Exchange
The experimental system described above allowed us to observe the expression of key genes involved in BaGTA induction across time, but it does not provide direct evidence of BaGTA function, that is, evidence of genetic exchange between individual bacteria. To directly observe BaGTA function, we co-cultured two B. henselae strains chromosomally tagged with either kanamycin or gentamycin antibiotic resistance markers and measured the frequency of double-resistant clones using a colony-forming assay (STAR Methods). After 36 hr of co-culturing in the presence of the wild-type (WT) BaGTA system, one out of 10,000 cells acquired both resistance markers (frequency <10−4). A similar frequency was obtained when a WT strain was cultivated in the presence of a BaGTA deletion mutant (frequency <10−4). However, when co-culturing two BaGTA deletion mutants, only background levels of double-resistant bacteria were detected (frequency <10−7). Similarly, co-culturing a WT strain tagged with a plasmid-encoded resistance together with a BaGTA deletion mutant only resulted in background levels of double-resistant bacteria (frequency <10−7). Absence of genetic exchange for plasmid encoded resistance markers indicated that BaGTA selectively transduces chromosomal loci and excludes episomal DNA. We therefore reasoned that an intact BaGTA is a prerequisite for high-frequency genetic exchange of Bartonella and that genetic exchange via BaGTA is restricted to chromosomal rather than plasmid DNA.
Next, we determined the temporal dynamics of BaGTA-mediated HGT by following acquisition of antibiotic markers by recipient cells (STAR Methods). After 12 hr of co-cultivation, exchange of chromosomal antibiotic marker reached saturation. We modeled BaGTA transduction data as a time-delayed function proportional to the integrated early BaGTA induction peak (STAR Methods). The measured mean delay time between peak expression and appearance of cells resistant to both antibiotics was 162 (±21) min (Figure 1D and STAR Methods). Thus, transduction of chromosomal loci occurs within a time frame similar to the replication time of Bartonella (about 330 min [Riess et al., 2008]). In sum, these results demonstrate that BaGTA mediates high frequency of chromosomal DNA exchange. Furthermore, induction of BaGTA is restricted to a synchronized subpopulation of cells that promote genetic exchange during a short period with fast cell proliferation.
The ppGpp Signaling Network Couples Induction of the BaGTA System to Unrestricted Cell Growth
To define the cellular processes driving BaGTA expression, we designed a series of multiplexed transposon sequencing (TnSeq) experiments. TnSeq couples hypersaturated transposon (Tn) mutagenesis with next-generation sequencing to profile changes in genome-wide insertion patterns occurring upon selection of large mutant pools (Christen et al., 2011, Christen et al., 2016). We constructed a himar-based Tn probe and generated large input mutant libraries encompassing several hundreds of thousands of insertion events (STAR Methods). In total, we performed four TnSeq selection analyses under single and co-culturing conditions (STAR Methods and Tables S1–S7). For each selection (detailed in Table S1), we recovered between 35,321 and 88,565 unique Tn insertion sites across the 1.9 Mb B. henselae genome (median insertion resolution: 6–15 bp). Genome-wide Tn insertions statistics for all four selection experiments are listed in Table S2. Cumulatively, we retrieved insertion events for more than 46% (120,099 out of 256,618) of all possible himar target sites displaying the obligate TA dinucleotide motif.
We reasoned that having a prophage-like element such as the BaGTA locus integrated in the bacterial genome requires a sophisticated mechanism to repress lysis in most cells. In that case, Tn insertions disrupting genes required for BaGTA repression would cause its constitutive activation and lead to elevated rates of lysis. Consequently, this class of mutants would be quickly outcompeted during outgrowth in M199 and lost. However, in the absence of a functional BaGTA these mutants would persist, as the deleterious phenotype of a constitutive activated BaGTA would be alleviated. Using this rationale, we performed multiplexed TnSeq outgrowth experiments with WT and ΔBaGTA mutant cells (ΔbgtCD, STAR Methods). Across the entire genome, 56 out of 1,612 protein-coding sequences (3.47%) were essential in the WT background (Table S2) but specifically became dispensable in absence of a functional BaGTA (Tables S3 and S6; STAR Methods). We hypothesized that these genes build the regulatory network that restricts activation of BaGTA to a subpopulation of cells during distinct stages of the Bartonella infection cycle.
The most predominant difference in Tn insertions was observed within spoT (79 in ΔBaGTA compared with 7 in WT, Figure 2A and Table S3). The spoT gene encodes for an Rhs guanosine-tetraphosphate synthase enzyme (ppGpp synthase). SpoT consists of an N-terminal synthetase domain that catalyzes the formation of the stringent response alarmone ppGpp (Québatte et al., 2013). The alarmone ppGpp functions by binding to target proteins, leading to bacterial cells shutting down active growth and entering a dormant state that promotes survival (Steinchen and Bange, 2016). The regulatory TGS/CC/ACT domains at the C terminus of SpoT inhibit ppGpp synthesis, resulting in low ppGpp levels during phases of optimal cell growth. In the ΔBaGTA background, the 79 Tn insertions within spoT were scattered throughout the entire open reading frame (ORF), whereas the seven Tn insertions recovered in the WT were restricted to the TGS domain, leaving the entire synthetase domain intact (Figure 2A). The essentiality of the SpoT ppGpp synthetase domain in WT but not in the ΔBaGTA background suggested that ppGpp acts as a repressor of BaGTA. Indeed, increase in ppGpp levels using a constitutively active E. coli ppGpp synthase (RelA1–455 [Schreiber et al., 1991]) allele causes strong repression of BaGTA induction (Figure 1B). Furthermore, we reasoned that a marked drop in cellular ppGpp levels should be observed coinciding with induction of BaGTA. High-performance liquid chromatography-mass spectrometry measurements of the cellular nucleotide pool confirmed that ppGpp levels are lowest at the time window of BaGTA induction in M199 (Figure 2B).
Figure 2.

Global Identification of Negative Regulators of BaGTA
(A) Differences in Tn insertions between a wild-type (wt) (blue marks) and a ΔBaGTA strain (gray marks) are shown for a subset of the identified negative regulators of BaGTA.
(B) Correlating with the induction of BaGTA, cellular ppGpp levels are lowest at 6 hr post inoculation in M199. The mean values from a biological triplicate and their associated SEs are displayed.
(C) Genome-wide HGT mapped by TnSeq from the donor output library. The global Tn insertion density fitted as bidirectional exponential decay functions (red line, see STAR Methods) reveals two maxima of transfer: a narrow peak centered on the Bartonella prophage (BAP) and a broad peak centered at the origin of run of replication (OriROR). See also Figure S1.
(D) Identification of the OriROR based on the Tn insertion ratio between input and output libraries within intergenic sequences. The genome annotation track with Tn insertions recovered from the control experiment (gray bars) and the donor experiment (blue bars) are indicated.
If ppGpp acts as a master repressor of BaGTA induction, additional components of the ppGpp signaling network should show a marked increase in Tn insertion in ΔBaGTA, similar to the spoT gene. Indeed, we found 63 Tn insertions within the guanosine pentaphosphate (pppGpp) hydrolase gene gppA (BH09810) compared with 17 for the WT control (Figure 2A and Table S2). In E. coli disruption of gppA abrogates the hydrolysis of pppGpp, resulting in low levels of ppGpp (Mechold et al., 2013). In addition, we found that the nitrogen regulatory ptsN gene (BH00180) and the enolase gene (BH05720) catalyzing the interconversion of 2-phosphoglycerate into phosphoenolpyruvate are exclusively dispensable in ΔBaGTA (Figure 2A) The ptsN and enolase genes showed a more than 5-fold increase in himar insertions in the ΔBaGTA background compared with WT (3 versus 17, and 7 versus 41 insertions) (Table S2). Similarly, fruB (BH01410), which encodes for a sugar phosphotransferase system (PTS) involved in the regulation of carbohydrate uptake, showed a more than 4-fold increase in Tn insertions in ΔBaGTA compared with WT (133 versus 32 insertions) (Table S2). In Pseudomonas putida, fruB is interlinked with the PTSNtr system, leading to a sensor-actuator device that integrates nitrogen and carbon availability (Pflüger-Grau and de Lorenzo, 2014). In Caulobacter crescentus, it has been recently shown that upon nitrogen starvation, the PTSNtr system inhibits the hydrolase activity of SpoT leading to elevated ppGpp production (Ronneau et al., 2016). Similarly, our data suggest that the B. henselae PTSNtr-SpoT-ppGpp signaling system (Table S3) senses the cellular metabolic state and represses BaGTA under nitrogen and carbon starvation conditions.
In addition, we found that the vceAB genes (BH12700, BH12710) annotated as multidrug efflux system and the phaAD pH-adaptation genes (BH16460, BH16440) showed reduced Tn insertion rates in WT compared with the ΔBaGTA strain. This suggests that BaGTA perceives additional signaling inputs in response to changes in pH homeostasis and stress cues. Similarly, we found the outer membrane protein BH10650, the prophage integrase BH09580, as well as six hypothetical proteins (BH04560, BH09950, BH00080, BH03800, BH15430, and BH08580) to be essential in the WT but dispensable in the ΔBaGTA background. Whether these genes encode components at the periphery of the ppGpp signaling network or alternative pathways repressing the transcriptional activation of the BaGTA locus remains to be further investigated.
The BaGTA System Promotes Genome-wide DNA Transfer
To obtain a quantitative view on the transduction efficiency of BaGTA, we performed a second set of multiplexed TnSeq experiments. We generated a high-complexity himar donor library and used the Tn-associated Gmr resistance cassette as selection marker to quantify genome-wide DNA transfer into recipient cells. To follow unidirectional DNA transfer, we used a recipient strain tagged with a plasmid-encoded Kmr resistance cassette, as this plasmid is not transferred by BaGTA (see above and STAR Methods). Out of 67,681 unique Tn insertions from the donor library, we recovered 59,488 Tn mutants in the double-resistant recipient library (Table S1). Within the donor library, Tn insertions were uniformly distributed across the genome. However, after BaGTA-dependent transduction, Tn insertions frequency peaked in proximity to the BaGTA gene cluster. We compared global insertion density between control and donor output libraries using a 1-kb sliding window (Figure 2C) and fitted the genome-wide transduction data with a bidirectional exponential decay function (STAR Methods). The global transduction maximum of BaGTA is located at genome position 1,680,170 ± 6,500 bp corresponding to the region of the Bartonella ROR origin (Figure 2C and Table 1). Across the entire genome, we measured an average transduction efficiency of 58.72% and a minimal efficiency of 40.38% at the genome locations opposite to the region surrounding the ROR origin (genome coordinate 714,646 ± 6,500 bp). Furthermore, we observed only a mild decrease in transduction efficiency when moving away from the ROR region as illustrated by a decay constant of 1.61 × 10−6 per base pair. Therefore, the DNA replication forks initiated at the ROR are highly processive, covering on average 1.24 Mb ± 23.8 kb of chromosomal DNA.
Table 1.
DNA Transfer Characteristics of BaGTA and BAP Locus
| BaGTA Locus | BAP Locus | |
|---|---|---|
| Genome position of OriROR (kb) | 1,680,170 ± 6,500 | 386,016 |
| Genome-wide transduction efficiency (%) | 58.72 | 0.38 |
| Maximal efficiency (%) | 100 | 21.16 |
| Minimal efficiency (%) | 40.38 | <0.5 |
| Mean BaGTA particles produced | 88 ± 1 | NA |
| Half-maximal distance from OriROR (kb) | 619.5 ± 11.9 | 14.6 |
OriROR, run-off replication origin; BaGTA, Bartonella gene transfer agent; BAP, Bartonellaprophage; NA, not applicable.
These findings demonstrate that any part of the B. henselae chromosome transduces with a high frequency into recipient cells. Moreover, correlation between the transduction efficiency and the position relative to the ROR origin indicate that ROR is active in BaGTA-expressing cells (Figure 2C and Table 1). In addition, we observed a second, less prominent local transduction maximum at genome coordinate 386 kb corresponding to the Bartonella prophage (BAP) locus. However, in contrast to the global transduction coverage of the ROR, the transduction peak centered at the BAP site was indicative of a localized transfer, with a packing range of 29.2 kb, which agrees well with the predicted size of BAP (Alsmark et al., 2004). This suggests that both the BAP and BaGTA are capable of promoting genetic exchange, but only BaGTA promotes generalized transduction of the Bartonella genome.
To fine-map the location of the run-off replication origin, we searched for genomic regions showing specific loss of Tn insertion in the donor output library as compared with the outgrowth control experiment. We reasoned that any Tn insertion targeting the ROR locus will abrogate initiation of DNA replication and, thus, will not be transduced into recipient cells. The most dramatic loss of insertions occurred within an 893-bp-long region (genome position 1,656,090 to 1,656,983, Figure S1) for which we recovered 45 unique Tn hits in the control library but not a single insertion event in the donor output library (Figure 2D). This region encompasses a 555 bp-long intergenic sequence as well as a 338-bp-long segment spanning the 5′ portion of the gene BH14490 (renamed brrC, for Bartonella ROR) (Table S6). Previous estimates on the ROR location based on sequence conservation have also linked brrC with the site of ROR replication initiation (Berglund et al., 2009). brrC is positioned within a putative operon that includes BH14520 (renamed brrF) (Table S7), which encodes for a TOPRIM domain, a conserved catalytic domain found in type IA and II topoisomerases, and DnaG-type primases (Aravind et al., 1998). The gene upstream (BH14480, renamed brrB) (Table S7) to the mapped ROR region lacked Tn insertions in both the donor and control TnSeq experiments, pinpointing to an essential negative regulator of ROR replication initiation. In agreement with this hypothesis, brrB possesses an HTH DNA binding domain and has homology to the TraY protein. In E. coli, TraY binds and regulates the oriT functions of the conjugative F plasmid (Frost et al., 1994). By a similar mechanism, BrrB from B. henselae might bind and repress activity of the neighboring ROR.
The functional link between the ROR activity and the frequency of BaGTA-mediated transfer constitute a remarkable difference from the RcGTA system, which functions without known ROR and exhibits no significant packaging bias (Hynes et al., 2012).
Core Components of the BaGTA System Are Clustered within an 80 kb-Long Genome Segment
Within the donor output library, we expected depletion of Tn insertions associated with genes required for production and release of BaGTA particles from donor cells. To globally identify such inner-circle components of BaGTA (core components), we calculated a DNA transfer Z-score for each protein-coding sequence by comparing insertion frequency in control and output libraries (Tables S4 and S6). As genes within close linkage to the BaGTA locus display a higher likelihood for transduction than genes positioned further apart, we used the experimentally determined global transduction function to correct for position-specific insertion bias within the output library. For each of the 1,612 Bartonella ORFs, we calculated a Z-score metric and identified 74 BaGTA donor genes that exhibit a significantly lower (Z-score < −3.5, p = 0.0002, Table S4) and 29 genes displaying significant higher Tn insertion coverage (Z-score > 3.0, p = 0.0013) (Figure 3A and STAR Methods).
Figure 3.

Identification of Bartonella Genes Involved in HGT
(A) Donor transfer Z-score calculated by comparative TnSeq analysis reveals a prominent multi-gene cluster (red dots) spanning the BaGTA locus and the ROR segment. In addition, components of the branched-chain amino acid transport system (livGFHJM, blue dots) cause impairment of BaGTA. Negative values in the donor transfer Z score indicate a decrease in BaGTA transduction efficiency while positive values show an increase.
(B) Tn insertions recovered from the control (gray bars) and the donor experiment (blue bars) are shown above and below the genome annotation track of the BaGTA cluster, with identified BaGTA genes highlighted in red and essential genes of BaGTA shown in purple.
(C) Selected BaGTA donor genes identified by TnSeq were validated by co-cultivation experiments using individual Tn mutants. Analysis of single Tn mutants within bgtEHIJ and brrE demonstrate complete abrogation of HGT. The control specifies a wild-type strain with an antibiotic marker at a neutral locus. The averaged log10 transformed frequency of double resistant bacteria (log recipient frequency) obtained after 24 hr co-culture and associated SDs are displayed (n=3). See also Figures S1 and S2.
Among the top-ranked BaGTA donor genes, we identified a 78.8-kb multi-gene locus (Figures 3A and S2), which we termed the bgt gene cluster (for Bartonella GTA). This region was flanked on both sides by cryptic prophage integrases and cumulatively encompassed 66 annotated protein-coding genes including 29 genes required for BaGTA-mediated gene transfer. At the 5′ end of the BaGTA genome region, we found a cluster composed of 11 genes (named bgtA to bgtK) (Table S7) that belonged to the bona fide GTA locus (Figure 3B) as previously inferred by sequence conservation analysis (Guy et al., 2013). In addition, we found seven genes grouped around the fine-mapped ROR segment (Figure 3B), which we termed brrA to brrG (for Bartonella run-off replication) as well as 11 orphan BaGTA genes named bgtL to bgtO, bgtPQR, and bgtS to bgtV (Figures 3B and Table S7). In addition to numerous hypothetical proteins, the identified BaGTA genes encompass structural phage components for capsid (bgtG), base-plate and tail fibers (bgtC), and phage endolysins such as endosialidases (bgtD), highlighting the phage-derived origin of BaGTA (Table S7). The gene organization of the bgt locus and corresponding himar insertions recovered from the control and donor output TnSeq experiments are detailed in Figure 3B.
Interspersed within the bgt multi-gene locus, we also found ten ORFs that lacked himar insertions in both TnSeq measurements and, thus, likely encode essential proteins. Four ORFs corresponded to genes involved in purine and folate metabolism (purEK, BH14400) and peptidoglycan synthesis (murA), while six of the essential ORFs are annotated as hypothetical protein but likely encode components that repress BaGTA induction or regulate activity of the ROR (BH14150 bgtL, BH14200 bgtM, BH14240 bgtO, BH14330 bgtR, BH14480 brrB, and BH14500 brrD). In support of this hypothesis, bgtLMO and bgtR, while essential in the WT background, became specifically dispensable in the ΔBaGTA background, indicating that these components are likely repressors of BaGTA. Thus, in addition to essential host genes, additional essential factors of BaGTA ensure genetic stability of the gta locus.
To validate our findings from the TnSeq analysis, we used a panel of individual Tn insertion mutants for a subset of the identified bgt genes (Québatte et al., 2013) as donor. Subsequent quantification of transduction efficiencies revealed that insertions within bgtEHIJ and brrS completely abrogates genetic exchange (frequency <10−7.5, Figure 3C) confirming the requirement of these genes for a functional BaGTA. A Tn mutant in bgtA encoding for a lysozyme displayed a 36-fold reduction in transduction efficiency but did not abrogate GTA activity completely (Figure 3C). This suggests that bgtA is likely required for efficient lysis and release of phage-like particles but that alternative lysis mechanisms may also exist. We conclude that at least 24 BaGTA donor genes distributed within the 80 kb-long genome region form the core components of the BaGTA transduction machinery.
To define outer circle (or peripheral) components required for efficient BaGTA particle production, we searched for BaGTA donor genes genetically unlinked to the gta locus. Among factors that caused strong impairment of BaGTA when mutated, we identified five genes livFGHJM (BH08250–BH08290) encoding for a high-affinity branched-chain amino acid transport system as well as a putative proline permease gene (BH06520, proY). In agreement with these observations, we found that a Tn mutant of proY showed more than 64-fold reduction in BaGTA transfer efficiency (Figure 3C and Table S2). We identified several additional genes involved in amino acid metabolism including proA, proC, cysQ, and tyrC genes. These findings suggest that imbalances in intracellular levels of multiple amino acids inhibit proper functioning of BaGTA, possibly by increasing cellular ppGpp levels. Accordingly, in addition to nitrogen and carbon starvation conditions, spoT is also induced upon ribosome stalling. We identified the 16S rRNA methyltransferase gene gidB, the 23S rRNA pseudouridine synthase gene rsmG (BH02610), as well as rplY and rpmE encoding the 50S ribosomal protein L25 and I31 as additional critical determinants required for BaGTA activity. Taken together, these data suggest that the amino acid metabolism and the fidelity of ribosomes both serve as inputs that repress the activity of BaGTA, possibly in a ppGpp-dependent manner.
In total, our data suggest that BaGTA donor activity does not only rely on the genes encoding the structural components that likely compose the released phage-like particles, but also depends on an extended set of accessory genes, possibly of phage origin, scattered in a ∼80-kb region around the previously identified BaGTA locus. These components constitute what we defined as the inner-circle components of BaGTA. Among these are found components of the ROR, suggesting that a functional ROR activity is linked to the production of BaGTA. Furthermore, our analysis implies a regulatory role for components that directly act on the metabolic state of the bacteria, supporting the view that a low ppGpp concentration is a prerequisite for BaGTA activation.
BaGTA Particle Uptake Preferentially Targets Cells with Active Division in a Competence-Related Mechanism
To understand the fate of the released BaGTA particles and identify genome-wide determinants required for BaGTA uptake and chromosomal integration of the cargo DNA, we performed a third set of multiplexed TnSeq experiments. We reasoned that Tn insertions disrupting genes required for recipient functions are lost upon selection for cells that underwent GTA-mediated transduction. We thus generated a gentamycin-resistant himar library (complexity >106) in a ΔBaGTA strain (ΔbgtCD) and selected for acquisition of a chromosomal kanamycin resistance marker from a BaGTA-competent donor strain (STAR Methods). Cells resistant to both antibiotics were recovered to yield a recipient output Tn mutant library (Table S8). As control, we subjected the input Tn library in ΔBaGTA background to an outgrowth in M199 (Table S6). For each Bartonella protein-coding sequence, we calculated the DNA uptake Z-score and compared Tn insertion frequencies in both recipient output and control libraries (Table S4 and STAR Methods). Cumulatively, we identified 107 BaGTA susceptibility genes with significant decrease in Tn insertion rate (Z-score >3, p < 0.0013, Table S5). BaGTA susceptibility genes are distributed along the entire B. henselae chromosome (Figure 4A).
Figure 4.

Identification of Bartonella Genes Involved in BaGTA Uptake
(A) DNA uptake Z-score calculated by comparative TnSeq analysis reveals genes involved in the uptake and incorporation of BaGTA DNA in recipient bacteria. Negative values indicate a decrease of BaGTA uptake and positive values, an increase. Highlighted are components of the Tol-Pal trans-envelope module (green) belonging to the divisome machinery and the identified competence and recombination pathway (brown). Metabolic components that presumably impair cell division such as the putative proline-specific permease proY or three components of the Pts (phosphate transport system) encoded by pstA, B, and C are highlighted in purple.
(B) Genome annotation track for selected genes with himar insertions recovered from control (gray bars) and recipient experiment (green bars).
(C) Scheme of the identified components of the Tol-Pal trans-envelope module involved in BaGTA particle uptake. OM, outer membrane; PP, periplasm; IM, inner membrane.
(D) Validation of the TnSeq results by co-cultivation experiments using individual Tn insertion mutants. Displayed are the averaged log10 transformed frequency of double resistant bacteria (log recipient frequency) obtained after 24 hr co-culture and associated SDs (n = 3). Mutations in the competence genes dprA, comEC, and comM and the outer circle components proY and pstA, B, C affecting cell-growth (blue) show reduced DNA uptake rates compared with the wild-type control. The control specifies a wild-type strain with an antibiotic marker at a neutral locus.
(E) Scheme of the identified competence module required for BaGTA DNA uptake. See also Figure S3.
DNA uptake by recipient cells is a multistep process, requiring docking of BaGTA particles to a surface receptor, translocation of the cargo DNA over the outer and inner membranes, and subsequent integration into the host chromosome. To gain mechanistic insights into the DNA uptake process, we classified the 107 BaGTA susceptibility genes according to their subcellular localization using our previously curated genome-wide B. henselae protein localization atlas (Stekhoven et al., 2014). As candidates for BaGTA particle docking and translocation sites, we identified two cell surface-exposed outer membrane proteins including the hbpC gene (BH02550) that encodes for the hemin-binding protein C and BH10650 that encodes for a TolC-family type-I secretion protein. In E. coli, the TolC protein is involved in maintaining outer membrane integrity and serves as the outer membrane receptor for entry of the bacteriophage TLS (German and Misra, 2001). Several bacteriophages are able to translocate the outer membrane by a two-receptor system, whereby one receptor is used for initial binding and the second for translocation. Our data suggest that BaGTA translocation may involve a similar two-receptor system with HbpC and TolC as likely candidates.
Among the top-ranking BaGTA susceptibility components, we discovered a module composed of seven genes that encode for a trans-envelope spanning machinery coordinating the invagination of the multi-layered cell envelope at the division site (Figure S3 and Table S5). Among them, we found BH05630 (renamed dipM) coding for an outer membrane-associated peptidoglycan endopeptidase, the peptidoglycan carboxypeptidase DacA1 (BH06160) as well as an AmiC-like hydrolytic amidase (BH08710, renamed amiC). Homologs of these periplasmic proteins are widespread, and have been shown to localize to the division site to permit septal peptidoglycan remodeling and facilitate cell separation (Goley et al., 2010, Möll et al., 2010, Uehara et al., 2010). In addition, among the top-ranked BaGTA susceptibility genes, we identified tolQRA and the pal gene encoding the peptidoglycan-associated outer membrane lipoprotein Pal (Figures 4B and 4C). In E. coli, tolQRA and pal are components of the Tol-Pal trans-envelope system that enable outer membrane constriction during active cell division (Gerding et al., 2007). The E. coli Tol-Pal system is important for maintaining outer membrane integrity, and its role in facilitating phage infection has been reported (Bernadac et al., 1998).
Based on these observations, we speculated that cells with an active division status, a functional Tol-Pal system, and a septal peptidoglycan remodeling apparatus are preferential targets for BaGTA particle attachment and DNA uptake. Indeed, DNA uptake rates of a marked tolQ disruption mutant were 70-fold reduced compared with WT controls (Figure 4D and STAR Methods). However, Tn insertions within spoT were not significantly enriched the recipient experiment. Therefore, in contrast to BaGTA particle formation, low ppGpp levels do not activate DNA-uptake functions. Rather, our analysis suggests that BaGTA preferentially targets actively replicating cells as binding of the particles requires divisome-specific cell surface markers (Figure 5A).
Figure 5.

BaGTA Promotes Genetic Exchange among the Fittest Subpopulation of Cells
(A) Replication cycle of BaGTA mediating horizontal gene transfer in B. henselae. BaGTA is induced during optimal growth and low cellular levels of ppGpp. BaGTA particles are released upon cell lysis and adsorb to the divisome complex of dividing recipient cells, where DNA uptake and integration is mediated by the competence and recombination machinery of the bacterial cell.
(B) Scheme explaining how BaGTA may promote evolvability among the fittest subpopulation during B. henselae infection cycle.
Furthermore, we identified comEC and comF as components that translocate BaGTA DNA across the inner membrane. The trans-membrane protein ComEC is involved in translocation of single-stranded DNA (ssDNA) into the cytoplasm while the ComF protein is thought to assist ComEC in transporting DNA. This suggests that the broadly conserved competence genes are responsible for BaGTA DNA uptake across the inner membrane into the cytoplasm (Figure 4E). Out of the 107 GTA susceptibility genes, we identified 38 cytoplasmic proteins. Among the top cytosolic hits involved in BaGTA uptake were the DNA repair gene radA, recombination gene recA, the Holliday junction helicase recG, the DNA processing factor dprA, the excision repair nuclease uvrB, the DNA topoisomerase IV parE, and the competence factor comM. Using individual Tn dprA, comEC, and comM mutants as recipients, we found a more than 1,000-fold reduction in marker uptake (Figure 4D). We propose that these seven genes, commonly associated with the integration of ssDNA in other organism, constitute the recombination machinery that catalyzes the successful integration of BaGTA DNA into the chromosome of recipient cells, with DprA targeting the ssDNA delivered by ComEC directly to RecA, promoting its loading and the subsequent integration by homologous recombination.
Discussion
We combined quantitative single-cell FACS reporter studies with multiplexed TnSeq analyses to define the regulatory networks and molecular determinants of BaGTA that mediate DNA transfer from donor to recipient cells. Using a GFP-based FACS reporter assay, we found that BaGTA is heterogeneously induced during a short time window within a subpopulation of rapidly proliferating cells, possibly at the onset of exponential growth.
Our systems-genetic experiments derived from TnSeq measurements support two further key features for BaGTA (Figure 5). First, because the alarmone ppGpp is a key repressor of BaGTA induction, only healthy cells can serve as DNA donors within the population. Second, uptake of donor DNA is likely restricted to actively dividing cells, as a plausible consequence of BaGTA tropism toward components of the divisome. Considering population dynamics, these findings clearly support the logical argument that an individual bacterium with a high fitness cost mutation is less likely to take up transduced DNA as the result of its dilution in a competing set-up. While our multiplexed TnSeq analysis reveals a broad panel of inner- and outer-circle components of BaGTA, additional essential genes that contribute to the functioning of BaGTA likely exist, and further studies will be required to decipher the precise involvement of the individual components identified to the global processes described.
Taken together, our findings suggest that both the induction of BaGTA particle formation and uptake preferentially occur within a cell subpopulation that experience rapid cell proliferation. In evolutionary terms, this suggests that BaGTA is tuned toward achieving high rates of DNA exchange between the fittest bacteria within a population. This would maintain the species' overall genetic stability and promote the propagation of beneficial traits among the fittest subpopulations of cells (Figure 5A). Combination of ppGpp-dependent BaGTA induction with cell division-dependent particle adsorption (Figure 5B) provides an excellent strategy to promote evolvability and to circumvent Muller's ratchet in isolated bacterial communities. Furthermore, such genome-wide HGT would preferentially occur when the bacteria reach a favorable environment, such as their replicative niche in the mammalian host or the midgut of their arthropod vector.
Thus, the wide spread of BaGTA among Bartonella species appears to present strong evidence of evolvability within these ubiquitous zoonotic pathogens, which increases their capacity to generate descendants with increased fitness and acquire new adaptive characteristics for efficient host infection. Moreover, the strict conservation of BaGTA observed across the bartonellae suggests that it supports an essential function during the infection cycle of these bacteria, where a high selection pressure could avoid the emergence of cheaters. Further studies should determine whether GTA-mediated HGT takes place in the mammalian host, where it would provide an excellent strategy to circumvent Muller's ratchet in isolated bacterial communities. An alternative, but not exclusive, possibility is that this process happens in the arthropod vector. Despite constituting a separate transmission bottleneck, the lumen of the arthropod gut also constitutes a potential melting pot for genetically diverse genotypes with distinct successful infection history.
Besides the RcGTA system, only few other GTA systems have been characterized to date. Our study indicates that despite a distinct origin, the later steps of DNA uptake and integration into recipient cells seem conserved between BaGTA and RcGTA (Brimacombe et al., 2014, Brimacombe et al., 2015), suggesting a shared uptake mechanism between all GTA systems. This also supports the emerging view that GTA-mediated DNA transfer may generally be considered as a form of transformation rather than transduction or conjugation (Redfield, 2001, Brimacombe et al., 2015, Takeuchi et al., 2014, Hynes et al., 2016).
It is very likely that similar examples of phage-derived systems will emerge as functional analysis of sequenced genomes progress. In this context, our study also provides a methodological framework that can be applied to analyze and decipher the functionality of any gene transfer machinery at the systems level and should contribute toward extending our understanding of the molecular mechanisms underlying HGT in bacterial communities.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Columbia agar | Oxoid | Cat. # CM0331 |
| Sheep blood, defibrinated | Oxoid | Cat. # SB055 |
| Medium 199 (HEPES, L-Glutamine) | Gibco | Cat. # 22340-020 |
| Fetal Calf Serum | Amimed | Cat. # 2-01F30G |
| meso-2,6-diaminopimelic acid (DAP) | Sigma | Cat. # 07036 |
| Restriction enzymes used in this study | New England Biolabs | N/A |
| Critical Commercial Assays | ||
| DNeasy 96 Blood and Tissue kit | Qiagen | Cat. # 69582 |
| Rapid DNA dephosphorylation and ligation kit | Roche | Cat. # 04 898 117 001 |
| Taq polymerase BioMix Red | Bioline | Cat. # BIO-25006 |
| Illumina sequencing chemistry version v4 | Illumina | HiSeq SBS Kit v4 |
| iProof DNA polymerase | Biorad | Cat. # 72-5301 |
| Experimental Models: Organisms/Strains | ||
| Escherichia coli MG1655 | Lab stock | N/A |
| Escherichia coli MFDpir | Ferrières et al., 2010 | JKE201 |
| Bartonella henselae ATCC 49882T Smr | Schulein and Dehio, 2002 | RSE247 |
| Bartonella henselae ATCC 49882T Smr -PvirB-gfp | Québatte et al., 2013 | MQB528 |
| Bartonella henselae 49882T SmrbgtC-gfp | This work | MQB1366 |
| Bartonella henselae 49882T Smr ΔbgtCD | This work | MQB759 |
| Bartonella henselae 49882T Smr ΔbgtCD BH15040::himarI (Kmr) | This work | MQB1439 |
| Bartonella henselae ATCC 49882T Smr -PvirB-gfp ΔbgtCD | This work | MQB1028 |
| Bartonella henselae 49882T SmrbgtC-gfp carrying pMQ165.1 | This work | MQB1633 |
| Bartonella henselae ATCC 49882T Smr -PvirB-gfp x::himarI (Kmr)(Pool) | This work | MQB1380 |
| Bartonella henselae ATCC 49882T Smr -PvirB-gfp bepG::himarI (Kmr) | This work | MQB1562 |
| Bartonella henselae ATCC 49882T Smr -PvirB::himarI:gfp (Gmr) | Québatte et al., 2013 | MQT222 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bgtE::himarI (Gmr) | Québatte et al., 2013 | MQT226 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bepG::himarI (Kmr) | This work | MQB1562 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp pstB::himarI (Gmr) | Québatte et al., 2013 | MQT269 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp proY::himarI (Gmr) | Québatte et al., 2013 | MQT215 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bgtA::himarI (Gmr) | Québatte et al., 2013 | MQT265 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bgtH::himarI (Gmr) | Québatte et al., 2013 | MQT260 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bgtI::himarI (Gmr) | Québatte et al., 2013 | MQT274 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp bgtJ::himarI (Gmr) | Québatte et al., 2013 | MQT211 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp brrE::himarI (Gmr) | Québatte et al., 2013 | MQT313 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp pstC::himarI (Gmr) | Québatte et al., 2013 | MQT224 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp pstA::himarI (Gmr) | Québatte et al., 2013 | MQT216 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp pstB::himarI (Gmr) | Québatte et al., 2013 | MQT269 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp dprA::himarI (Gmr) | Québatte et al., 2013 | MQT220 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp comEC::himarI (Gmr) | Québatte et al., 2013 | MQT311 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp comM::himarI (Gmr) | Québatte et al., 2013 | MQT212 |
| Bartonella henselae ATCC 49882T Smr PvirB-gfp ΔbgtCD tolQ:pMQ169_O1 | This work | MQB1662 |
| Oligonucleotides | ||
| Primer used in this study | This paper | Table S8 |
| Recombinant DNA | ||
| pHS006tag004 (Kmr himarI transposon) | Saenz et al., 2007 | N/A |
| pML001 (Gmr himarI transposon) | Québatte et al., 2013 | N/A |
| pMQ999 (Gmr himarI transposon for TnSeq libraries) | This paper | N/A |
| pAH184 | This paper | N/A |
| pBG01 | Schulein and Dehio, 2002 | N/A |
| pBZ485a | Harms, A. | N/A |
| pCD366 | Dehio et al., 1998 | N/A |
| pCD353 | Dehio et al., 1998 | N/A |
| pMQ075 | This paper | N/A |
| pMQ149 | This paper | N/A |
| pMQ165.1 | This paper | N/A |
| pMQ169_O1 | This paper | N/A |
| pTR1000 | Schulein and Dehio, 2002 | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo LLT | N/A |
| Matlab | MathWorks | N/A |
| OLB genome analyzer software suite OLB | Illumina | N/A |
| Biopython | Cock et al., 2009 | N/A |
| Burrows-Wheeler Aligner (bwa) | Li and Durbin, 2010 | N/A |
Contact for Reagent and Resource Sharing
Further information may be obtained from the Lead Contact Christoph Dehio (christoph.dehio@unibas.ch).
Experimental Model and Subject Details
Bacterial Strains and Growth Condition
All bacterial strains and plasmids used in this study are listed in the key resource table. B. henselae strains were grown at 35°C for 2 days on Columbia agar supplemented with 5% sheep blood (CBA agar) in a humidified atmosphere containing 5% CO2. E. coli strains were cultured in either liquid or solid (1.5% agar) Luria-Bertani (LB) medium at 37°C and 200 rpm. Plasmids were introduced into B. henselae by conjugation from a derivative of the E. coli MFDpir strain (Ferrières et al., 2010) as described previously (Dehio and Meyer, 1997). Unless otherwise indicated, antibiotics and other supplements were used at the following concentrations: (i) B. henselae: 10 μg ml-1 gentamycin (Gm), 30 μg ml-1 kanamycin (Km), 100 μg ml-1 streptomycin (Sm) or 50 μg ml-1 spectinomycin (Sp), 500 μg ml-1 isopropyl-β-D-thiogalactopyranosid (IPTG); (ii) E. coli: 200 μg ml-1 ampicillin (Ap), 20 μg ml-1 Cm, 20 μg ml-1 Gm, 50 μg ml-1 Km, 50 μg ml-1 Sp or 1 mM 2,6-diaminopimelic acid (DAP).
Authentication of Cell Lines Used
The Bartonella strains used in this study are all derivative of the spontaneous streptomycin resistant variant of Bartonella henselae ATCC 49882T (Schulein and Dehio, 2002). The strain was authentified by DNA sequencing of multiple PCR-amplified regions and comparison to the reference sequence (Alsmark et al., 2004).
Construction of Himar Delivery Plasmid
To perform TnSeq selection experiments, we constructed a himarI Tn delivery plasmid. To this end, a himar transposable cassette composed of an outward-pointing Ptac/lac+ promoter, a gentamycin resistance gene and an illumina adapter sequence linker flanked by himar repeat sequences was constructed and ligated into XbaI digested pHS006tag004 (Saenz et al., 2007). To generate stable himarI insertions, the himarI Tn delivery plasmid contains a himar transposase gene that resides outside of the transposable cassette. The transposable cassette contains a marker that upon genomic integration confers resistance to gentamicin to the host. The transposase gene itself is not transferred to the host genome ensuring stability of insertion mutations. The himarI transposon cassette was constructed as follows: a synthetic DNA template carrying the illumine adapter linker sequence was re-amplified using primers prMQ1736 and prMQ1730. The gentamicin resistance cassette was amplified from pML001 (Québatte et al., 2013) using prMQ1735 and prMQ1735 with the high fidelity iProof DNA polymerase (Biorad). The Ptac/lac promoter was amplified from a derivative of pCD353 (Dehio et al., 1998) using prMQ1706a and prMQ1777. The 3 fragments were assembled by SOEing PCR using prMQ1736 x prMQ1777, the resulting DNA fragment purified and digested by AvrII and ligated into XbaI digested pHS006tag004 (Saenz et al., 2007) yielding pMQ999. Primer sequences are provided in Table S8.
Construction of Targeted Chromosomal Mutations
Chromosomal insertions or deletions of B. henselae were generated by a two-step gene replacement procedure as described in (Schulein and Dehio, 2002). In brief, integration of the suicide plasmid into the Smr recipient was selected by plating the conjugation mixtures on CBA plates containing 30 μg ml-1 Km for 7-10 days. Independent clones were expanded on CBA-Km plates. Negative selection was performed on CBA plates containing 100 μg ml-1 Sm and 100 μg ml-1 IPGT plates to select for the loss of the integrated plasmid and clones harboring the expected insertion or deletion were identified by overspanning PCR. Gene disruption by incomplete duplication was performed as described in (Schulein and Dehio, 2002). In brief, integration of the suicide plasmid into the recipient was selected by plating the conjugation mixtures on CBA plates containing 30 μg ml-1 Km plates for 7-10 days. Plasmid recombination was confirmed by inside-out PCR.
BaGTA Reporter Strain
The plasmid to generate the chromosomal gfp-bgtC translational fusion was constructed as follows: flanking homologies regions were amplified from B. henselae RSE247 DNA using primers prMQ1717 x prMQ1718 and prMQ1721 x prMQ1722. The gfpmut2 was amplified from pCD366 using prMQ1719 and prMQ1720). The 3 fragments were assembled by SOEing PCR using prMQ1717 and prMQ1722. The resulting fragment was purified, digested by SalI and ligated into SalI-digested pTR1000 (Schulein and Dehio, 2002) yielding pMQ149. Conjugation of pMQ149 into B. henselae strain RSE247 followed by positive and negative selection resulted in B. henselae strain MQB1366.
Deletion of bgtC-bgtD
the plasmid to generate the in-frame deletion of bgtC-bgtD (BH13960-BH13970) was constructed by amplifying flanking homologies regions from B. henselae RSE247 using primers prMQ1357 x prMQ1358 and prMQ1355 x prMQ1356. Both fragments were assembled using prMQ1355 x prMQ1357, digested with XbaI and ligated into XbaI digested pTR1000 yielding pMQ075. Conjugation of pMQ075 into B. henselae strain MQB528 followed by positive and negative selection resulted in B. henselae strain MQB1028.
Disruption of tolQ
The plasmid used to disrupt tolQ by incomplete gene duplication was constructed as follows: a 524 bp fragment comprising a 499 bp internal tolQ fragment of flanked by two stop codons and two EcoRI sites (gBLOCKs-tolQ) was digested by EcoRI and cloned into the corresponding sites of pBG01 (Schulein and Dehio, 2002), resulting in pMQ169_O1. Conjugation of pMQ169_O1 into B. henselae strain MQB1028 resulted in B. henselae strain MQB1662.
E. coli RelA1-455 Expression Construct
A 1411 bp fragment encoding a constitutive active allele of E. coli relA (amino acids 1-445, (Schreiber et al., 1991)) was amplified from MG1655 boiled colonies using primers prMQ1889 x prMQ1890. The resulting fragment was purified, digested by NotI and EcoRI and ligated into the corresponding site of pBZ485a. Sequencing of the resulting plasmid revealed different mutations upstream from the start codon for all tested clones, suggesting toxicity of the construct for E. coli in that plasmid backbone. Plasmid pMQ165.1 (carrying the mutation CCCGGGGATC instead of CAAG---ATC within prMQ1889 sequence) was conjugated into B. henselae strain MQB1366 resulting in MQB1633. Functionality of the construct was confirmed by the experimental read-out.
Method Details
Flow Cytometry
Determination of BaGTA transcriptional activity by flow cytometry was performed as follows. B. henselae strains were grown on CBA plates supplemented with appropriated antibiotics and grown in a humidified atmosphere at 35°C and 5% CO2 for three days followed by re-streaking on fresh CBA plates and growth for 48 h. The bacteria were resuspended in medium M199 supplemented with 10% FCS (M199 hereafter) at a final OD600 nm of 0.008 and incubated in 48 well plates in a humidified atmosphere at 35°C and 5% CO2. Expression of the chromosomal gfp reporter was measured as GFP fluorescence using a FACSCalibur flow cytometer (BD Biosciences) with an excitation at 488 nm. The effect of E. coli RelA1-455 overexpression was performed as described above in the presence of 0.5 M IPTG as inducer. Data analysis was performed using the FlowJo software and geometric mean of the GFP fluorescence intensities were used for calculation of promoter activity. All experiments were performed as biological triplicates.
To determine the relative survival between off-state and GTA expressing cells, B. henselae was induced for 6 hM199 and subjected to cell sorting using an Aria III (BD Biosciences). At least 50’000 GFP-positive and GFP-negative cells were sorted from triplicated samples. Sorted cells were resuspended in M199, subjected to serial dilution followed by plating on CBA. After 5-7 days, CFU were determined and regrowth rate was determined for each condition as the ratio of observed CFU / sorted events. The relative recovery rate was obtained by the recovery rate ratio between GFP positive and GFP negative cells. This experiment was performed as biological triplicate.
GFP-Reporter Analysis by FACS and Data Modeling
The experimental data obtained from the FACS time course analysis of the bgtC-gfp reporter strains was baseline corrected and then fitted with an empirical function capturing the characteristics of a latent phase followed by an exponential signal increase followed by an exponential decay according to:
with Fpeak corresponding to the peak value of GFP positive cells, λi and λd corresponding to the induction rate and decay rates and τ specifying the delay time when maximal titers of GFP positive cells are reached. The data was fitted using Levenberg-Marquart algorithm (adjusted R-square 0.9831). We measure for the delay time τ1 and τ2 6.62 ± 0.44 h and 27.1 ± 4.0 h respectively and for Fpeak1 and Fpeak2 14.11± 0.61% and 3.68 ±2.44%, induction constants λi1 of 0.53 ± 0.26 and for λi2 of 0.06 ± 0.06, and decay constants λd1 of 0.62 ± 0.19 and for λd2 of 0.25 ± 0.48.
Co-cultivation Assay
Bacteria were grown for 2 days on CBA with appropriate antibiotics, resuspended in medium 199 supplemented with 10% FCS (M199 hereafter) and diluted to a final OD600 of 0.008 per strain. Co-cultivation assays were performed using two B. henselae strains carrying distinct resistance marker in M199. For each tested condition 2 x 1 ml M199 were prepared, placed in 24 well plates (Corning) and incubated for 0 to 56 h at 35°C and 5% CO2 in a water-saturated atmosphere. For donor assays, selection of double resistant bacteria (DRB) and frequency determination was performed as follows. Bacteria were resuspended by pipetting and transferred to 2 ml Eppendorf tubes. 10 μl of each culture was used to perform serial dilution and spotted on CBA containing 100 μg ml-1 Sm or 100 μg ml-1 Sm plus 10 μg ml-1 Gm and/or 30 μg ml-1Km. The rest of the culture was centrifuged for 5 min at 5000 x g, resuspended in 50 μl M199 and plated on CBA containing 100 μg ml-1 Sm, 10 μg ml-1 Gm and 30 μg ml-1Km. CFU were counted after 5-7 days growth at 35°C and 5% CO2. CFU/ml was determined for each growth condition and DRB frequency was calculated as the ratio of DRB (Kmr, Gmr) / total bacteria (Smr) or DRB / Gmr bacteria when testing individual Tn mutants. To ensure directionality in donor assays using individual Gmr Tn mutants, co-cultures were performed with the wild-type strain RSE247 harboring plasmid pCD366 conferring Kmr as recipient, since episomal DNA is not a substrate for BaGTA (results section).
In order to ensure directionality when testing individual Gmr himar mutants in recipient assays, we needed to establish a new selection marker applicable for Bartonella selection. To this end, a derivative of pBZ485a carrying a spectinomycin (Sp) resistance cassette was generated. The aminoglycoside adenylyltransferase aadA cassette was amplified from pGB2 (Churchward et al., 1984) using prAH1111 and prAH1112, digested with MluI and EcoRV and ligated into MluI and NaeI digested pBZ485a yielding pAH184. Primers sequence can be found in Table S8. Conjugation of pAH184 into B. henselae effectively conferred Spr. Therefore, pAH184 was introduced in all Gmr himar mutants tested in recipient assays. Co-cultures were performed using the WT Kmr donor MQB1562. 10 μl of each culture was used to perform serial dilution and spotted on CBA containing 100 μg ml-1 Sm or 50 μg ml-1 Sp plus 10 μg ml-1 Gm and/or 30 μg ml-1 Km. The rest of the culture was centrifuged for 5 min at 5000 x g, resuspended in 50 μl M199 and plated on CBA containing 100 μg ml-1 Sp, 10 μg ml-1 Gm and 30 μg ml-1Km. CFU were counted after 5-7 days growth at 35°C and 5% CO2. CFU/ml was determined for each growth condition and recipient frequency was calculated as the ratio of (Kmr Gmr Spr) / (Gmr Spr) CFU, yielding the frequency of DNA transfer per recipient. Uptake frequency for the tolQ disruption mutant was normalized against total bacterial counts (Kmr Gmr Spr) / (Smr) CFU to account for the filamentous phenotype resulting from tolQ disruption, as recipient CFU would not accurately captures the total amounts of bacterial cells. Each DNA transfer experiment was performed as biological triplicate.
Transposon Mutagenesis and DNA Library Generation
Transposon Mutagenesis
Each himar Tn library was generated by conjugation of a pool of suicide plasmid (pMQ999), carrying a modified himar Tn using a derivative of the E. coli strain MFDpir as donor strain. Conjugations were performed as described above. After 5 day of selective growth on CBA, transconjugants were harvested in M199/10% FCS (M199) as 96 sub-pools of more than 10’000 CFU using 96 deep well plates (Eppendorf). For the generation of the donor library, each 96 sub-pools of transconjugants in the background of the WT MQB528 were co-cultivated with B. henselae MQB1379 as recipient in 4 x 1 ml M199 at a final OD600 of 0.008 per strain. After 24 h of co-cultivation, bacteria were collected into 2 x 96 deep well plates and centrifuged for 5 min at 5000 x g. Each pellet was resuspended in 50 μl M199 and plated on CBA containing 100 μg ml-1 Sm, 10 μg ml-1 Gm and 30 μg ml-1Km. After 5 days, DRB from each 96 co-cultures (c.a. 10’000) were collected in M199. OD600 was determined and the equivalent of 1 ml OD600 = 0.75 was transferred into a new 96 deep well plate, centrifuged for 5 min at 5000 x g. Pellets were stored at -80°C until proceeding with DNA extraction. The generation of the recipient library was performed similarly, with the following modifications: pMQ999 was introduced into B. henselae strain MQB1028 (ΔbgtC-D, ΔBaGTA hereafter). The resulting 96 sub-libraries were used as recipient in co-culture for 24 h with the donor B. henselae strain MQB1380. The input (control) libraries in WT and ΔBaGTA backgrounds were generated by conjugation of pMQ999 into MQB1379 and MQB1028 carrying plasmid pCD366, respectively. For both library, 96 x 10’000 mutants were harvested and incubated for 24 h in M199 before plating, to match the treatment of the output libraries.
DNA Extraction and TnSeq DNA Library Generation
DNA extraction was performed using the DNeasy 96 Blood and Tissue kit (Qiagen) according to manufacturer’s instruction. Using a two-step arbitrary PCR based strategy, Tn junctions were amplified in parallel from genomic DNA samples as previously reported (Christen et al., 2011). First and second round of PCR amplification was performed in 10 μl reaction volume in 384 well PCR plates on thermocycler instruments (C1000 touch, BioRad, Cressier, Switzerland) using Taq polymerase mix (BioMix Red, Bioline, USA). To each PCR reaction 20ng genomic DNA or 1.5 μl of the first round PCR product was added as a template. For the first round of PCR a Tn specific primer (TGTAAAACGACGGCCAGT) was used with arbitrary PCR primers as previously described (Christen et al., 2016) with the following modifications. To adjust the annealing probability of the semi-arbitrary primers to the low GC content of the Bartonella genome, different arbitrary penta-nucleotide sequences were chosen (ACGCC, ACATC, AAATC, ATTTC). In addition, the arbitrary primers carried short barcode sequences for DNA library multiplexing. The first PCR amplification was performed according to following thermocycling program: (1) 94°C for 3 min, (2) 94°C for 30 s, (3) 42°C for 30 s, slope -1°C/cycle, (4) 72°C for 1min, (5) go to step 2, 6 times, (6) 94°C for 30 s, (7) 58°C for 30 s, (8) 72°C for 1 min, (9) go to step 6, 25 times, (10) 72°C for 3 min, (11) 12°C hold. Products of the first round of PCR were further amplified in a second nested PCR step, using, Illumina paired-end primers PE1.0 and PE2.0: (1) 94°C for 3 min, (2) 94°C for 30 s, (3) 64°C for 30 s, (4) 72°C for 1 min, (5) go to step 2, 30 times, (6) 72°C for 3 min, (7) 12°C hold. PCR products derived from each Tn mutant library were separately pooled. PCR products corresponding to amplified Tn junctions from each selection experiment were separated on a 2.5% agarose gel and DNA molecules between 200-700 bp were size selected and purified over a silica column (Machery-Nagel, Switzerland). DNA sample concentration was quantified on a Nanodrop ND-1000 spectrometer and equimolar amount were pooled prior high-throughput sequencing.
Sequencing and Sequence Analysis
Transposon Sequencing
DNA samples were paired-end sequenced (2 x125 bases) on HiSeq Illumina instruments using Illumina sequencing chemistry version v4 and sequencing primers PE1.0 and PE2.0. The DNA samples were quantified by qPCR for library viability and titrated to produce approximately 2x108 clusters per HiSeq flow cell lane. Standard base-calling from raw images was performed using a phiX reference spike-in according to the genome analyzer software suite OLB (Illumina). Sequencing was performed at the Functional Genomics Center Zürich, Zürich, Switzerland.
Sequence Analysis and Mapping of Transposon Insertion Sites
Raw sequencing data processing and read alignment was performed using a custom sequence analysis pipeline based on Python, Biopython (Cock et al., 2009), bwa (Li and Durbin, 2010) and Matlab routines as previously described (M. Christen et al., 2016). Low quality reads (low phred scores) were discarded and Tn reads with at least a 15 bp long perfect match to the Himar transposon end sequence [‘TATCAGCCAACCTGT’] were selected. In the wild-type B. henselae genome (BX897699.1) the k-mer [‘TATCAGCCAACCTGT’] does not occur. The genomic insert size from each paired end read was inferred by testing for overlapping read sequences between mate reads. Only read pairs with genomic inserts larger than 15 base pairs were selected. Sequencing strings corresponding to the illumina adapters, Tn end or arbitrary PCR primers sequences were trimmed. Demultiplexing into the different TnSeq selection experiments was performed according to a defined barcode sequence tag internal to the arbitrary primer sequence (see above). Using bwa-07.12 (Li and Durbin, 2010), filtered Tn reads were aligned onto the Bartonella henselae Houston-1 NCBI reference genome (BX897699.1). Reads with correct paired-end alignment, no mismatches within the first 15 bases of both paired-end reads, and with an insert size smaller than 500 bases, were selected for subsequent analysis. The himar1 transposition generates a two base pair long duplication upon integration, which was taken into account for subsequent insertion occurrence analysis. The location of a given Tn insertion was defined as the genome position of the first reference base detected adjacent to the Tn end sequence. A custom Matlab script was used to analyze global insertion statistics and calculate Tn insertion occurrence and distributions within each annotated GenBank feature of the B. henselae genome. Metrics and routines applied for this analysis have been previously described (B. Christen et al., 2011; M. Christen et al., 2016). See the Quantification and Statistical Analysis section for a description of the TnSeq analysis.
Quantification of ppGpp by LC-MS/MS
Quantification of cellular concentrations of ppGpp by LC-MS/MS was performed as previously described in (Québatte et al., 2013). In brief, B. henselae WT was inoculated in M199/10% FCS at a final concentration of OD600 = 0.075 and incubated for 48 h. Cultures were rapidly cooled on iced-water and harvested by centrifugation. Pellets were washed with PBS and aliquots were taken for protein quantification. Pellets were resuspended in 300 μl of extraction solution (acetonitrile/methanol/ddH2O, 2/2/1, v/v/v) and extraction was performed as described for cyclic-di-GMP (Spangler et al., 2010). Levels of ppGpp were measured by liquid chromatography-tandem mass spectrometry on a QTRAP 5500 mass spectrometer (AB SCIEX) coupled with a Series 200 HPLC System (Perkin Elmer Instruments). The ppGpp was detected in positive ionization mode via selected reaction monitoring (SRM). Liquid chromatography separation was achieved on a porous graphitic carbon (Hypercarb) column. A linear gradient from 96% solvent A (10 mM ammonium acetate, pH 10) and 4% solvent B (acetonitrile) to 60% solvent B over 8 min at a flow rate of 0.6 ml/min was applied. The column was re-equilibrated for further 4 min to reach the starting condition again. To determine the amount of protein in each sample, 800 μl of 0.1 M NaOH was added to the bacteria culture aliquot, and the protein concentrations were determined with the Protein Assay kit (BioRad) after heating the sample at 95°C for 15 min. Measurements were repeated in triplicate and values were expressed as ppGpp per mg of protein.
Quantification and Statistical Analysis
Fitting of Growth and Transduction Data
The growth curve for the co-cultivation assay was fitted with Matlab’s built-in curve-fitting tool (cftool) using an empirical smoothing spline fit based on a piecewise polynomial function (Smoothing parameter p = 0.99990086, goodness of fit: SSE: 0.02028, R-square: 0.9774, adjusted R-square: 0.943 and RMSE: 0.05187). To obtain quantitative insights into the transduction kinetics, we fitted the observed transduction frequency as a time-delayed function proportional to the integrated GFP-expression rate of the first BaGTA induction peak. The second GFP expression peak occurred after the transduction plateaued and, thus was not considered for fitting. The GFP-expression rate was numerically integrated and normalized to obtain relative transduction frequencies between 0 and 1. The time delay was calculated as the time difference between the half-maximal GFP expression of the integrated first GFP-expression peak and the half maximal transduction frequency observed in the experimental data.
Comparative TnSeq Analysis
Calculation of Global himar1 Insertion Densities and Fitting of Genome Transfer Function
For the GTA donor TnSeq dataset, global insertion densities across the 1.9 Mb Bartonella genome, were calculated using a window of 1000 bp and a step size corresponding to the insertion distance between consecutive insertions. To calculate a genome-transfer function for the GTA system, insertion densities across the genome were fitted by Levenberg-Marquardt using a symmetrical two-sided exponential decay function that progresses bidirectional along the circular Bartonella genome:
Where x corresponds to the relative genome position, θ corresponds to the relative genome position of the run-off replication origin (ROR), λ corresponds to the decay constant, α corresponds to the ratio between the maximum and minimal BaGTA efficiency measured across the genome and mod() specifying the modulus function with a divisor of 1.
Genomic insertion densities were then normalized according to the fitted genome transfer function for BaGTA. To define the transfer characteristics of the second Bartonella phage locus (BAP) Tn insertion densities were fitted by Levenberg-Marquardt in a second iteration to obtain a genome transfer function for the Bap locus.
To define the precise location of the run-off replication (ROR) origin of BaGTA, the insertion ratio between the input library and the BaGTA transferred library was determined for each intergenic region of the Bartonella genome using a 400 bp window. Minimum insertion number for intergenic regions in the BaGTA transferred library was set to 1. A single maximum for the insertion ratio was obtained for the intergenic region between BH14480 and BH14490 pinpointing to the ROR origin of the GTA system.
Essentiality Analysis and Calculation of Donor Transfer Z-score and Recipient Uptake Z-score
The metrics and statistical analysis for classification of protein coding sequences (CDS) into essential, promoting fitness, and non-essential categories have been previously described (Christen et al., 2016). CDS were classified as essential if their non-disruptable 5’ portion covers more than 60% of the total CDS. In order to identify essential CDS with potential misannotated translational start codons, CDS were classified as essential if the Tn insertion density was less than six-times the average insertion density measured across the genome and has either a single Tn insertion gap covering more than 60% of the CDS or had two internal Tn gaps covering more than 80% of the CDS. If the average Tn density was less than four-times the average insertion density across the genome but no essentiality criterion was satisfied, CDS were classified as promoting fitness.
To derive quantitative gene fitness values and correct for the observed bias in himar transfer efficiency as a function of the distance to the GTA or BAP locus, the insertion density of each gene was adjusted using the fitted transfer function for the GTA and the BAP locus. For each Bartonella gene a donor transfer score was calculated based on a z-score metric comparing input Tn insertion frequencies with the frequencies for the donor transfer selection experiment after normalizing for the positional bias of the GTA and BAP system. Similarly, a recipient uptake score was calculated based on the mean z-score comparing the insertion frequency of the M199 media selection for WT and a BaGTA deficient background against the recipient transfer frequency. Genes involved in repression of the BaGTA locus, were identified according to following criteria. Genes were classified as essential in the WT background but were dispensable when BaGTA was inactivated by a site-specific deletion. Furthermore, genes were required to exhibit a z-scores calculated based on the insertion differences observed that was greater than 2. The genome-wide results for each TnSeq experiments are provided in Table S6.
Data Availability
The raw TnSeq insertion data is available as Supplemental Information.
Author Contributions
M.Q., B.C., M.C., and C.D. designed the research. M.Q., A.H., and J.K. performed experimental work. M.C. and B.C. performed TnSeq and related data analysis. M.C. and B.C. performed the data modeling of the single-cell FACS data. M.C., B.C., M.Q., and C.D. wrote the manuscript.
Acknowledgments
We are thankful to Volkhard Kaever and Annette Garbe from the Research Core Unit Metabolomics of the Hannover Medical School, Germany, for ppGpp measurements. We are also thankful to Janine Bögli and Anna Sproll from the FACS core facility of the Biozentrum, Basel, Switzerland, for cell sorting. This work was supported by grants 31003A_166476 to B.C. and 310030B_149886 to C.D. from the Swiss National Science Foundation (SNSF, www.snf.ch), advanced grant 340330 to C.D. (FicModFun) from the European Research Council (ERC), grant 51RTP0_151029 to C.D. for the Research and Technology Development (RTD) project TargetInfectX in the frame of SystemsX.ch (www.systemX.ch), the Swiss Initiative for System Biology, and by the Eidgenössische Technische Hochschule (ETH) Zürich (ETH-08 16-1) to B.C.
Published: June 14, 2017
Footnotes
Supplemental Information includes three figures, eight tables, and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2017.05.011.
Contributor Information
Beat Christen, Email: beat.christen@imsb.biol.ethz.ch.
Christoph Dehio, Email: christoph.dehio@unibas.ch.
Supplemental Information
References
- Aravind L., Leipe D.D., Koonin E.V. Toprim--a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsmark C.M., Frank A.C., Karlberg E.O., Legault B.-A., Ardell D.H., Canbäck B., Eriksson A.-S., Näslund A.K., Handley S.A., Huvet M. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc. Natl. Acad. Sci. USA. 2004;101:9716–9721. doi: 10.1073/pnas.0305659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund E.C., Frank A.C., Calteau A., Vinnere Pettersson O., Granberg F., Eriksson A.-S., Näslund K., Holmberg M., Lindroos H., Andersson S.G.E. Run-off replication of host-adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet. 2009;5:e1000546. doi: 10.1371/journal.pgen.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadac A., Gavioli M., Lazzaroni J.C., Raina S., Lloubès R. Escherichia coli tol-pal mutants form outer membrane vesicles. J. Bacteriol. 1998;180:4872–4878. doi: 10.1128/jb.180.18.4872-4878.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay L.M., Touchon M., Rocha E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA. 2014;111:12127–12132. doi: 10.1073/pnas.1405336111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe C.A., Ding H., Beatty J.T. Rhodobacter capsulatus DprA is essential for RecA-mediated gene transfer agent (RcGTA) recipient capability regulated by quorum-sensing and the CtrA response regulator. Mol. Microbiol. 2014;92:1260–1278. doi: 10.1111/mmi.12628. [DOI] [PubMed] [Google Scholar]
- Brimacombe C.A., Ding H., Johnson J.A., Beatty J.T. Homologues of genetic transformation DNA import genes are required for Rhodobacter capsulatus Gene Transfer Agent recipient capability regulated by the response regulator CtrA. J. Bacteriol. 2015;197:2653–2663. doi: 10.1128/JB.00332-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Morgan M.T., Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen B., Abeliuk E., Collier J.M., Kalogeraki V.S., Passarelli B., Coller J.A., Fero M.J., McAdams H.H., Shapiro L. The essential genome of a bacterium. Mol. Syst. Biol. 2011;7:528. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M., Beusch C., Bösch Y., Cerletti D., Flores-Tinoco C.E., Del Medico L., Tschan F., Christen B. Quantitative selection analysis of bacteriophage φCbK susceptibility in Caulobacter crescentus. J. Mol. Biol. 2016;428:419–430. doi: 10.1016/j.jmb.2015.11.018. [DOI] [PubMed] [Google Scholar]
- Churchward G., Belin D., Nagamine Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene. 1984;31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M.J.L. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio C., Meyer M. Maintenance of broad-host-range incompatibility group P and group Q plasmids and transposition of Tn5 in Bartonella henselae following conjugal plasmid transfer from Escherichia coli. J. Bacteriol. 1997;179:538–540. doi: 10.1128/jb.179.2.538-540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio M., Knorre A., Lanz C., Dehio C. Construction of versatile high-level expression vectors for Bartonella henselae and the use of green fluorescent protein as a new expression marker. Gene. 1998;215:223–229. doi: 10.1016/s0378-1119(98)00319-9. [DOI] [PubMed] [Google Scholar]
- Engel P., Goepfert A., Stanger F.V., Harms A., Schmidt A., Schirmer T., Dehio C. Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature. 2012;482:107–110. doi: 10.1038/nature10729. [DOI] [PubMed] [Google Scholar]
- Ferrières L., Hémery G., Nham T., Guérout A.M., Mazel D., Beloin C., Ghigo G.M. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J. Bacteriol. 2010;192:6418–6427. doi: 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg P.C.M., Westbye A.B., Beatty J.T. One for all or all for one: heterogeneous expression and host cell lysis are key to gene transfer agent activity in Rhodobacter capsulatus. PLoS One. 2012;7:e43772. doi: 10.1371/journal.pone.0043772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost L.S., Ippen-Ihler K., Skurray R.A. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 1994;58:162–210. doi: 10.1128/mr.58.2.162-210.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding M.A., Ogata Y., Pecora N.D., Niki H., de Boer P.A.J. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol. Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German G.J., Misra R. The TolC protein of Escherichia coli serves as a cell-surface receptor for the newly characterized TLS bacteriophage. J. Mol. Biol. 2001;308:579–585. doi: 10.1006/jmbi.2001.4578. [DOI] [PubMed] [Google Scholar]
- Goley E.D., Comolli L.R., Fero K.E., Downing K.H., Shapiro L. DipM links peptidoglycan remodeling to outer membrane organization in Caulobacter. Mol. Microbiol. 2010;77:56–73. doi: 10.1111/j.1365-2958.2010.07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L., Nystedt B., Sun Y., Näslund K., Berglund E.C., Andersson S.G.E. A genome-wide study of recombination rate variation in Bartonella henselae. BMC Evol. Biol. 2012;12:65. doi: 10.1186/1471-2148-12-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L., Nystedt B., Toft C., Zaremba-Niedzwiedzka K., Berglund E.C., Granberg F., Näslund K., Eriksson A.-S., Andersson S.G.E. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet. 2013;9:e1003393. doi: 10.1371/journal.pgen.1003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A., Dehio C. Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin. Microbiol. Rev. 2012;25:42–78. doi: 10.1128/CMR.05009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder B.P., Beauchemin C.A.A. Exploring the effect of biological delays in kinetic models of influenza within a host or cell culture. BMC Public Health. 2011;11(Suppl 1):S10. doi: 10.1186/1471-2458-11-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes A.P., Mercer R.G., Watton D.E., Buckley C.B., Lang A.S. DNA packaging bias and differential expression of gene transfer agent genes within a population during production and release of the Rhodobacter capsulatus gene transfer agent, RcGTA. Mol. Microbiol. 2012;85:314–325. doi: 10.1111/j.1365-2958.2012.08113.x. [DOI] [PubMed] [Google Scholar]
- Hynes A.P., Shakya M., Mercer R.G., Grüll M.P., Bown L., Davidson F., Steffen E., Matchem H., Peach M.E., Berger T. Functional and evolutionary characterization of a gene transfer agent's multilocus “genome”. Mol. Biol. Evol. 2016 doi: 10.1093/molbev/msw125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo J., Puigbò P., Lobkovsky A.E., Wolf Y.I., Koonin E.V. Inevitability of genetic parasites. Genome Biol. Evol. 2016 doi: 10.1093/gbe/evw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang A.S., Zhaxybayeva O., Beatty J.T. Gene transfer agents: phage-like elements of genetic exchange. Nat. Rev. Microbiol. 2012;10:472–482. doi: 10.1038/nrmicro2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechold U., Potrykus K., Murphy H., Murakami K.S., Cashel M. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 2013;41:6175–6189. doi: 10.1093/nar/gkt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möll A., Schlimpert S., Briegel A., Jensen G.J., Thanbichler M. DipM, a new factor required for peptidoglycan remodeling during cell division in Caulobacter crescentus. Mol. Microbiol. 2010;77:90–107. doi: 10.1111/j.1365-2958.2010.07224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N.A. Accelerated evolution and Muller's ratchet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H.J. The relation of recombination to mutational advance. Mutat. Res. 1964;106:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Pflüger-Grau K., de Lorenzo V. From the phosphoenolpyruvate phosphotransferase system to selfish metabolism: a story retraced in Pseudomonas putida. FEMS Microbiol. Lett. 2014;356:144–153. doi: 10.1111/1574-6968.12459. [DOI] [PubMed] [Google Scholar]
- Québatte M., Dehio M., Tropel D., Basler A., Toller I., Raddatz G., Engel P., Huser S., Schein H., Lindroos H.L. The BatR/BatS two-component regulatory system controls the adaptive response of Bartonella henselae during human endothelial cell infection. J. Bacteriol. 2010;192:3352–3367. doi: 10.1128/JB.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Québatte M., Dick M.S., Kaever V., Schmidt A., Dehio C. Dual input control: activation of the Bartonella henselae VirB/D4 type IV secretion system by the stringent sigma factor RpoH1 and the BatR/BatS two-component system. Mol. Microbiol. 2013;90:756–775. doi: 10.1111/mmi.12396. [DOI] [PubMed] [Google Scholar]
- Redfield R.J. Do bacteria have sex? Nat. Rev. Genet. 2001;2:634–639. doi: 10.1038/35084593. [DOI] [PubMed] [Google Scholar]
- Riess T., Dietrich F., Schmidt K.V., Kaiser P.O., Schwarz H., Schäfer A., Kempf V.A.J. Analysis of a novel insect cell culture medium-based growth medium for Bartonella species. Appl. Environ. Microbiol. 2008;74:5224–5227. doi: 10.1128/AEM.00621-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneau S., Petit K., De Bolle X., Hallez R. Phosphotransferase-dependent accumulation of (p)ppGpp in response to glutamine deprivation in Caulobacter crescentus. Nat. Commun. 2016;7:11423. doi: 10.1038/ncomms11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz H.L., Engel P., Stoeckli M.C., Lanz C., Raddatz G., Vayssier-Taussat M., Birtles R., Schuster S.C., Dehio C. Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat. Genet. 2007;39:1469–1476. doi: 10.1038/ng.2007.38. [DOI] [PubMed] [Google Scholar]
- Schreiber G., Metzger S., Aizenman E., Roza S., Cashel M., Glaser G. Overexpression of the relA gene in Escherichia coli. J. Biol. Chem. 1991;266:3760–3767. [PubMed] [Google Scholar]
- Schulein R., Dehio C. The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol. Microbiol. 2002;46:1053–1067. doi: 10.1046/j.1365-2958.2002.03208.x. [DOI] [PubMed] [Google Scholar]
- Spangler C., Böhm A., Jenal U., Seifert R., Kaever V. A liquid chromatography-coupled tandem mass spectrometry method for quantitation of cyclic di-guanosine monophosphate. J. Microbiol. Methods. 2010;81:226–231. doi: 10.1016/j.mimet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Steinchen W., Bange G. The magic dance of the alarmones (p)ppGpp. Mol. Microbiol. 2016;101:531–544. doi: 10.1111/mmi.13412. [DOI] [PubMed] [Google Scholar]
- Stekhoven D.J., Omasits U., Québatte M., Dehio C., Ahrens C.H. Proteome-wide identification of predominant subcellular protein localizations in a bacterial model organism. J. Proteomics. 2014;99:123–137. doi: 10.1016/j.jprot.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Takeuchi N., Kaneko K., Koonin E.V. Horizontal gene transfer can rescue prokaryotes from Muller's ratchet: benefit of DNA from dead cells and population subdivision. G3 (Bethesda) 2014;4:325–339. doi: 10.1534/g3.113.009845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T., Parzych K.R., Dinh T., Bernhardt T.G. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 2010;29:1412–1422. doi: 10.1038/emboj.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw TnSeq insertion data is available as Supplemental Information.