

Abstract
Aims/Introduction
Mannose is a monosaccharide constituent of glycoproteins and glycolipids. Experiments in rats have shown previously that the plasma mannose level decreases after glucose load, but does not decrease in diabetic rats, and that hepatic glycogenolysis is a source of this plasma mannose; however, these results are not fully elucidated in humans. Plasma mannose levels before/after glucose loading in humans with various degrees of glucose intolerance were examined to analyze their association with clinical factors.
Materials and Methods
The 75‐g oral glucose tolerance test was carried out in Japanese individuals not taking diabetes medications. Participants were classified into normal glucose tolerance, impaired glucose metabolism and diabetes mellitus groups. Insulinogenic index as an index of insulin secretion, and Matsuda Index as an index of insulin sensitivity were calculated. Mannose was assayed by the established method using high‐performance liquid chromatography after labeling.
Results
After glucose load, the plasma mannose level decreased gradually in the normal glucose tolerance group, but did not decrease in the diabetes mellitus group. Plasma mannose changes during 120 min from baseline (M120‐M0) were significantly different among the three groups (normal glucose tolerance: −16.7 ± 1.7; impaired glucose metabolism: −9.0 ± 1.9; diabetes mellitus: −1.4 ± 1.8 μmol/L [n = 25 in each group], P < 0.0001). Plasma glucose 120 min after glucose loading (R 2 = 0.412) or loge‐insulinogenic index, loge‐Matsuda Index and age (R 2 = 0.230) were determinants of M120‐M0 in multiple regression analyses.
Conclusions
We clarified the relationship between plasma mannose level and glucose tolerance in humans. The present results are compatible with those using rats, in which mannose derived from glycogenolysis plays an important role in the alteration of mannose levels after glucose loading.
Keywords: Glucose tolerance, Glycogenolysis, Mannose
Introduction
Mannose is a monosaccharide constituent of glycoproteins and glycolipids. The mammalian serum mannose concentration ranges from approximately 50 to 100 μmol/L1. In humans, the mean concentrations of mannose in plasma determined by enzymatic methods were reported previously to be approximately 501, 202 and 40 μmol/L3. In a previous study3 of patients with diabetes, the plasma concentration of mannose was found to be higher than that in participants with normal glucose tolerance, and plasma mannose and glucose concentrations were positively correlated in participants with various severities of glucose intolerance. However, the mechanism of alteration of plasma mannose levels after glucose load was not elucidated in that study.
Taguchi et al.4 has shown using rats that hepatic glycogen is a source of plasma mannose. Oral administration of glucose increased the plasma glucose level, but decreased the plasma mannose level, showing that plasma glucose was not the direct source of plasma mannose. Interestingly, oral administration of glucose was found not to decrease plasma mannose levels in diabetic Goto‐Kakizaki rats, in which the plasma insulin response to glucose is impaired4. The authors proposed that part of the plasma mannose is supplied by breakdown of glycogen in the liver by the route of glycogen → glucose 1‐phosphate → fructose 6‐phosphate → mannose 6‐phosphate → mannose.
It is generally accepted that endogenous glucose production (EGP), which is the sum of gluconeogensis and glyogenolysis, is increased, and results in an elevated level of fasting plasma glucose in patients with diabetes. However, the relative and absolute contribution of glycogenolysis to increased EGP is controversial5. One of the reasons for this is the limited number of subjects available for such studies because of the complicated and expensive methods for using glucose isomer. A more convenient biomarker of glycogenolysis is necessary.
In the present study, to explore the possibility that the plasma mannose level is an indicator of glycogenolysis in humans, plasma mannose levels before and after glucose loading in participants with various degrees of glucose intolerance were examined and analyzed to clarify the association with clinical factors.
Materials and Methods
Participants
Japanese outpatients who visited Fukuda Clinic, Kochi, Japan, for hypertension and/or lipid metabolism disorders during the period of April 2012 through April 2014, not diagnosed with glucose intolerance previously, and took no diabetes medications were recruited for the study. Participants were successively recruited until the number of each group reached 25. Informed consent was obtained from each patient. Mannose levels in plasma samples were determined at Meijo University. Analysis of data was carried out at Kochi Medical School. The study protocol was approved by local ethical review boards of Kochi Medical School, Meijo University and Fukuda Clinic.
Laboratory examination
The 75‐g oral glucose tolerance test (OGTT) was carried out in the morning after an overnight fast. Samples were drawn just before, and 30, 60, 90 and 120 min after ingestion of glucose. Plasma glucose was measured by the glucose oxidase method. Plasma immunoreactive insulin (IRI) was measured using sandwich enzyme‐linked immunosorbent assay (Access® Ultrasensitive Insulin; Beckman Coulter, Brea, California, USA). Mannose was assayed using an established method4. Briefly, after labeling with 4‐aminobenzoyl ethyl ester, the concentration of mannose was determined using high‐performance liquid chromatography (HPLC)6. The glucose contained in the samples was confirmed not to affect the assay. To determine the time‐course of mannose levels during OGTT, samples of five participants among 25 participants belonging to each group were randomly chosen, and mannose levels at 30, 60 and 90 min after glucose load were measured. Mannose levels before and at 120 min after glucose loading were measured in all participants. Glycated hemoglobin was measured by HPLC7. Using the 2006 World Health Organization criteria and Japan Diabetes Society criteria8, 9, participants were categorized as having normal glucose tolerance (NGT; fasting plasma glucose [FPG] <6.1 mmol/L and 2‐h plasma glucose [2‐h PG] <7.8 mmol/L), impaired glucose metabolism (IGM; either impaired fasting glucose [IFG]: FPG ≥6.1 and <7.0 mmol/L, and/or impaired glucose tolerance [IGT]: 2‐h PG ≥7.8 and <11.1 mmol/L) or diabetes (FPG ≥7.0 mmol/L and/or 2‐h PG ≥11.1 mmol/L). Anti‐glutamic acid decarboxylase antibody was not measured in the participants.
Indices of insulin secretion and insulin sensitivity/resistance
In the 75‐g OGTT, glucose (G), mannose (M) and IRI (I) levels in plasma were determined at fasting (G0, M0, I0), 30 (G30, M30, I30), 60 (G60, M60, I60), 90 (G90, M90, I90) and 120 min (G120, M120, I120) after glucose loading, respectively. Gm and Im were calculated by dividing the area under the curve of G and IRI during OGTT by 120 min, respectively. As insulin secretion indices, insulinogenic index (IGI) and homeostasis model assessment of β‐cell function (HOMA‐β) were calculated using the following formula10, 11: IGI: (I30 ‐ I0[pmol/L])/(G30 ‐ G0[mmol/L]); HOMA‐β: I0(μU/mL) × 20 / (G0[mmol/L] − 3.5). As an insulin resistance index, homeostasis model assessment of insulin resistance (HOMA‐IR) was calculated using the following formula11: I0(μU/mL) × G0(mmol/L) / 22.5. As insulin sensitivity indices, the Quantitative Insulin Sensitivity Check Index (QUICKI) and Matsuda Index were calculated using the following formula12, 13: QUICKI: 1 / (log10 G0[mg/dL] + log10 I0[μU/mL]); Matsuda Index: 10,000 / (G0[mg/dL] × I0[μU/mL] × Gm[mg/dL] × Im[μU/mL])0.5. The oral disposition index (DIO) was calculated using the following formula14: .
Statistical analysis
Statistical analysis was carried out with the StatView 5.0 system (SAS Institute Inc., Cary, North Carolina, USA). Normally distributed continuous data are presented as mean ± standard error, and non‐normally distributed continuous data are presented as median value, 25th percentile value and 75th percentile value. Differences among more than three groups were determined by analysis of variance (anova) for normally distributed continuous data, and by Kruskal–Wallis tests for non‐normally distributed continuous data. Scheffe's test was carried out as post‐hoc analysis. The relationships between the parametric data and between non‐parametric data were determined by Pearson's analysis and by Spearman's analysis, respectively. As dependent variables, M0 and M0‐M120 were used. As independent variables, normally‐distributed loge‐transformed HOMA‐β, IGI, Matsuda Index and DIO were used. In multiple regression analyses, to reduce the number of independent variables matching with the total number of participants, and to avoid confounding factors, factors with >0.7 coefficient in simple correlation among independent variables were omitted (Table S1). As loge‐HOMA‐β and QUICKI are strongly correlated with G0 (multiple correlation coefficient 0.873), and loge‐IGI and loge‐MI are strongly correlated with G120 (multiple correlation coefficient 0.720), these variables were not included in the same analysis to avoid confounding results. Independent variables were selected by the forward stepwise selection method, according to the significance level for the addition of variables <0.10. P‐values <0.05 were considered statistically significant.
Results
Clinical and biochemical profiles
Clinical and biochemical profiles among the NGT, IGM and diabetes groups are shown in Table 1. Glycated hemoglobin, plasma glucose during OGTT and plasma IRI 120 min after glucose loading were significantly different among groups. Indices of insulin secretion, except HOMA‐β and insulin resistance/sensitivity, were significantly different among groups.
Table 1.
Clinical and biochemical profiles of participants with normal glucose tolerance, impaired glucose metabolism and diabetes
| NGT | IGM | DM | P‐value | |
|---|---|---|---|---|
| n | 25 | 25 | 25 | |
| Age | 60.0 ± 1.8 | 65.4 ± 2.6 | 65.6 ± 2.2 | 0.1364 |
| Sex (male/female) | 12/13 | 11/14 | 11/14 | 0.9476 |
| BMI (kg/m2) | 24.0 ± 0.7 | 25.5 ± 0.7 | 25.3 ± 0.7 | 0.3117 |
| WC (cm) | 89.0 ± 1.9 | 90.2 ± 2.2 | 91.7 ± 2.0 | 0.6400 |
| HbA1c (%) | 5.70 ± 0.06 | 5.79 ± 0.09 | 6.28 ± 0.11*,** | <0.0001 |
| G0 (mmol/L) | 5.35 ± 0.08 | 5.68 ± 0.14 | 6.48 ± 0.20*,** | <0.0001 |
| G120(mmol/L) | 6.37 ± 0.22 | 9.17 ± 0.23* | 13.53 ± 0.41*,** | <0.0001 |
| Gm (mmol/L) | 7.68 ± 0.23 | 9.91 ± 0.31* | 12.60 ± 0.27*,** | <0.0001 |
| I0 (pmol/L) | 35.5 ± 4.6 | 44.3 ± 4.6 | 49.9 ± 5.7 | 0.1291 |
| I120 (pmol/L) | 264 ± 43 | 538 ± 112* | 494 ± 61* | 0.0319 |
| Im (pmol/L) | 293 ± 42 | 369 ± 50 | 306 ± 34 | 0.4005 |
| IGI (pmol/mmol) | 57.0 (36.3,96.5) | 39.9 (23.1,65.6) | 22.4* (13.2,41.7) | 0.0005 |
| HOMA‐β | 51.2 (41.0,86.6) | 61.9 (41.9,91.4) | 47.7 (37.7,80.2) | 0.5153 |
| HOMA‐IR | 1.21 (0.78,1.68) | 1.53 (1.07,2.48) | 1.97* (1.42,2.40) | 0.0064 |
| QUICKI | 0.375 ± 0.006 | 0.357 ± 0.006 | 0.344 ± 0.006* | 0.0025 |
| Matsuda Index | 6.55 (3.71,9.56) | 4.36* (2.73,5.53) | 3.61* (2.20,5.10) | 0.0026 |
| DIO (mmol/L) | 1.87 (1.30,2.60) | 0.91 (0.72,1.58) | 0.54* (0.40,0.81) | <0.0001 |
*P < 0.05 vs normal glucose tolerance (NGT); **P < 0.05 vs impaired glucose metabolism (IGM). G0, G120 and Gm are plasma glucose at 0 (fasting) and 120 min after glucose loading, and mean plasma level in 75‐g oral glucose tolerance test, respectively. I0, I120 and Im are plasma immunoreactive insulin at 0 (fasting) and 120 min after glucose loading and mean immunoreactive insulin in 75‐g oral glucose tolerance test, respectively. Gm and Im are calculated by dividing the area under the curve of plasma glucose and plasma immunoreactive insulin during oral glucose tolerance test by 120 min, respectively. BMI, body mass index; DIO, oral disposition index; DM, diabetes; HbA1c, glycated hemoglobin; HOMA‐β, homeostasis model assessment of β‐cell function; HOMA‐IR, homeostasis model assessment of insulin resistance; IGI, insulinogenic index; QUICKI, Quantitative Insulin Sensitivity Check Index; WC, waist circumference.
Time‐course of mannose levels during OGTT
Time‐courses of mannose levels during OGTT were determined in five randomly‐selected participants of each group. Plasma glucose was elevated after glucose loading in all groups, and were higher at 60, 90 and 120 min, and at 120 min after glucose loading in diabetes and IGM compared with those in NGT, respectively (Figure 1a). Plasma IRI was elevated after glucose loading in all groups, but did not differ among the three groups at any time‐point during OGTT (Figure 1b). Plasma mannose levels gradually decreased after glucose loading in NGT and reached plateau after 90 min, but the decrease was blunted in IGM, and was not observed in diabetes. Plasma mannose levels in NGT compared with those in diabetes were lower at 60, 90 and 120 min after glucose loading (Figure 1c,d).
Figure 1.
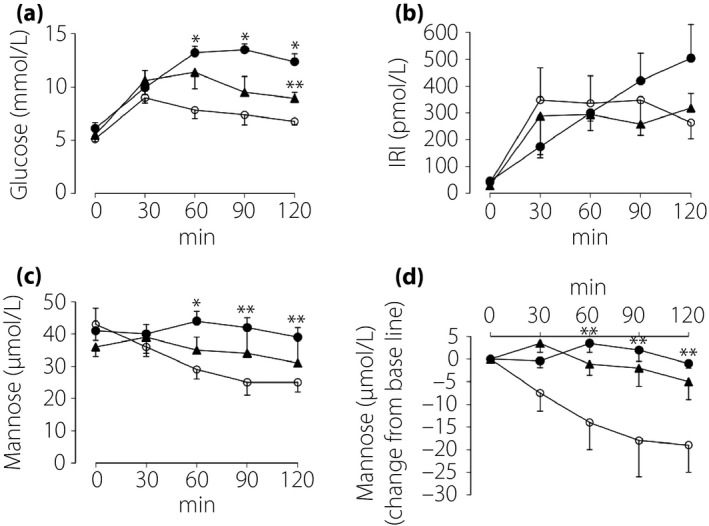
Time‐courses of (a) glucose, (b) immunoreactive insulin (IRI) and (c,d) mannose levels in plasma during the 75‐g oral glucose tolerance test in participants with normal glucose tolerance (NGT; open circle, n = 5), impaired glucose metabolism (IGM; closed triangle, n = 5) and diabetes (DM; closed circle, n = 5). Samples of five participants among 25 participants belonging to each group were randomly chosen. *P < 0.01 vs NGT; **P < 0.05 vs NGT.
Plasma mannose levels before and after 120‐min glucose loading
Fasting plasma mannose levels (M0) and plasma mannose levels after 120‐min glucose loading (M120) were determined in all participants in the NGT, IGM and diabetes groups (n of each group = 25). M0 did not differ among the three groups (Table 2). M120 in the NGT group was lower compared with those in the IGM and diabetes groups, and M120 in the IGM group was lower compared with that in the diabetes group. The decrease in mannose levels during 120 min after glucose loading from baseline in the NGT group was greater compared with those in the IGM and diabetes groups, and that in the IGM group was larger compared with that in the diabetes group.
Table 2.
Plasma mannose levels of participants with normal glucose tolerance, impaired glucose metabolism and diabetes
| n | NGT | IGM | DM | P‐value |
|---|---|---|---|---|
| 25 | 25 | 25 | ||
| M0 (μmol/L) | 40.8 ± 2.0 | 43.8 ± 2.2 | 45.0 ± 2.4 | 0.3977 |
| M120 (μmol/L) | 24.1 ± 1.4 | 34.7 ± 1.9* | 43.6 ± 2.8*,** | <0.0001 |
| M120‐M0 (μmol/L) | −16.7 ± 1.7 | −9.0 ± 1.9* | −1.4 ± 1.8*,** | <0.0001 |
*P < 0.01 vs normal glucose tolerance (NGT); **P<0.01 vs impaired glucose metabolism (IGM). M0 and M120 are plasma mannose levels at 0 (fasting) and 120 min after glucose loading. M120‐M0 is the alteration of mannose levels during 120‐min glucose loading (M120 minus M0). DM, diabetes.
Simple correlation between plasma mannose levels and clinical factors
M0 was correlated with body mass index, fasting plasma glucose (G0), fasting insulin (I0) and QUICKI; M0 was not significantly correlated with loge‐HOMA‐β (Table 3, Figure 2a,b). M0 was significantly correlated in the diabetes group, tended to be correlated in the IGM group and was not correlated with G0 (NGT: R = −0.183, P = 0.380; IGM: R = 0.370, P = 0.068; diabetes: R = 0.371, P = 0.001). The alteration of plasma mannose levels after glucose loading (M120‐M0) was correlated with plasma glucose at 120 min after glucose loading (G120), Gm, loge‐IGI, QUICKI and loge‐DIO (Table 3, Figure 2c,d).
Table 3.
Simple correlation between plasma mannose levels and clinical factors before and after glucose loading
| Before loading | ||
|---|---|---|
| Independent variables | Dependent variables M0 | |
| R | P‐value | |
| Age | −0.213 | 0.0664 |
| Sex | 0.098 | 0.4016 |
| BMI | 0.253 | 0.0283 |
| WC | 0.205 | 0.0778 |
| G0 | 0.371 | 0.0010 |
| loge‐HOMA‐β | 0.022 | 0.8487 |
| I0 | 0.295 | 0.0102 |
| QUICKI | −0.278 | 0.0156 |
| After loading | ||
|---|---|---|
| Independent variables |
Dependent variables M120−M0 |
|
| R | P‐value | |
| Age | 0.147 | 0.2086 |
| Sex | 0.122 | 0.2957 |
| BMI | 0.043 | 0.7131 |
| WC | 0.018 | 0.8758 |
| G120 | 0.642 | <0.0001 |
| Gm | 0.544 | <0.0001 |
| Im | −0.127 | 0.2794 |
| loge‐IGI | −0.334 | 0.0034 |
| loge‐MI | −0.192 | 0.0991 |
| QUICKI | −0.237 | 0.0410 |
| loge‐DIO | −0.431 | 0.0001 |
Sex: male = 1, female = 0. G0, G120 and Gm are plasma glucose at 0 (fasting) and 120 min after glucose loading, and mean PG in 75‐g oral glucose tolerance test, respectively. I0 and Im are plasma immunoreactive insulin at 0 (fasting) and mean immunoreactive insulin in 75‐g oral glucose tolerance test, respectively. BMI, body mass index; loge‐DIO, loge‐transformed oral disposition index; loge‐HOMA‐β, loge‐transformed homeostasis model assessment of β‐cell function; loge‐IGI, loge‐transformed insulinogenic index; loge‐MI, loge‐transformed Matsuda Index; QUICKI, Quantitative Insulin Sensitivity Check Index; WC, waist circumference.
Figure 2.
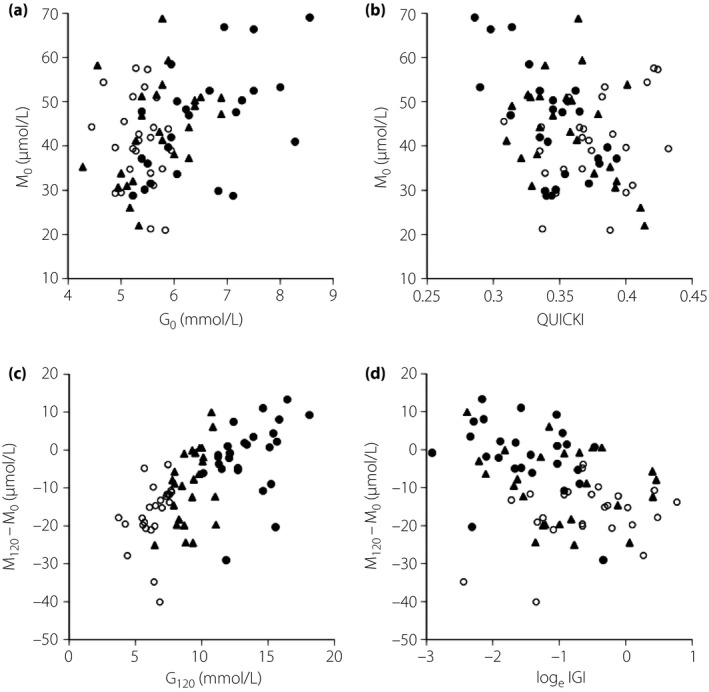
Relationships between (a) glucose at 0 min (G0) and mannose at 0 min (M0), (b) Quantitative Insulin Sensitivity Check Index (QUICKI) and M0, (c) glucose at 120 min (G120) and plasma mannose changes during 120 min from baseline (M120‐M0), and (d) loge insulinogenic index (IGI) and M120‐M0 in participants with normal glucose tolerance (open circle, n = 25), impaired glucose metabolism (closed triangle, n = 25) and diabetes (closed circle, n = 25).
Multiple regression analyses for plasma mannose levels
G0 and I0 were determinants of M0 in a model (R 2 = 0.172, P = 0.001) and QUICKI and loge‐HOMA‐β were determinants of M0 in another model (R 2 = 0.118, P = 0.010; Table 4a,b). G120 was a determinant of M120‐M0 in a model (R 2 = 0.412, P < 0.001), and loge‐IGI, loge‐MI and age were determinants of M120‐M0 in another model (R 2 = 0.230, P < 0.001; Table 4c,d).
Table 4.
Multiple regression analyses for the determinants of mannose levels
| (a) | ||
|---|---|---|
| Independent variables | Dependent variable: M0 | |
| β | P | |
| G0 | 0.308 | 0.008 |
| I0 | 0.195 | 0.090 |
| R 2 | 0.172 | |
| P | 0.001 | |
| (b) | ||
|---|---|---|
| Independent variables | Dependent variable: M0 | |
| β | P | |
| QUICKI | −0.444 | 0.003 |
| loge‐HOMA‐β | −0.260 | 0.074 |
| R 2 | 0.118 | |
| P | 0.010 | |
| (c) | ||
|---|---|---|
| Independent variables | Dependent variable: M120 − M0 | |
| β | P | |
| G120 | 0.642 | <0.001 |
| R 2 | 0.412 | |
| P | <0.001 | |
| (d) | ||
|---|---|---|
| Independent variables | Dependent variable: M120 − M0 | |
| β | P | |
| loge‐IGI | −0.396 | <0.001 |
| loge‐MI | −0.348 | 0.003 |
| Age | 0.212 | 0.055 |
| R 2 | 0.230 | |
| P | <0.001 | |
Variables were selected by stepwise selection method following candidate variables: (a) age, sex, body mass index (BMI), glucose at 0 min (G0) and insulin at 0 min (I0); (b) age, sex, BMI, loge‐transformed homeostasis model assessment of β‐cell function (loge‐HOMA‐β) and Quantitative Insulin Sensitivity Check Index (QUICKI); (c) age, sex, BMI, glucose at 120 min (G120) and plasma mean immunoreactive insulin in 75‐g oral glucose tolerance test (Im); (d) age, sex, BMI, loge‐transformed insulinogenic index (loge‐IGI) and loge‐transformed Matsuda Index (loge‐MI).
Discussion
We measured plasma mannose levels by HPLC after glucose loading, and confirmed that mannose levels decreased in participants with normal glucose tolerance, but did not decrease in patients with diabetes. These results are compatible with the results using normal and diabetic Goto‐Kakizaki rats in which mannose derived from glycogenolysis was found to play an important role in the alteration of mannose levels after glucose loading.
To precisely analyze the correlation between plasma glucose and mannose levels in participants, mannose should be measured without affection of coexisting glucose in samples. Mannose, a C‐2 epimer of glucose, is potentially metabolized by enzymes that metabolize glucose. In enzymatic methods, ~100‐fold excess of glucose in plasma samples potentially interferes with the assay in a glucose concentration‐dependent manner. A strength of the present study was that measurement bias was excluded by using a method of HPLC not affected by coexisting glucose in plasma samples6.
In dogs, portal infusion of catecholamine augments EGP by selectively stimulating glycogenolysis15, showing a more important role of glycogenolysis over gluconeogenesis in the short‐term regulation of glucose efflux from the liver. Intravenous administration of insulin decreased the plasma mannose level; in contrast, intravenous administration of epinephrine to fed rats increased plasma mannose. In addition, the increase in plasma mannose by epinephrine was canceled by fasting or by administration of glycogen phosphorylase inhibitor4. Furthermore, administration of lactate and alanine, which are gluconeogenic substrates to fasted rats, increased plasma glucose but did not increase plasma mannose4. These animal studies show the important roles of glycogenolysis in the short‐term regulation of glucose efflux from the liver that are tightly linked to mannose efflux from the liver.
Fasting plasma mannose levels (M0) were not significantly different among the NGT, IGM and diabetes groups, but the mannose levels after glucose loading differed. An implication of these results is that the FPG level overlaps largely among the three groups (Figure 2a); 68% of participants in the present study were classified to IGM and diabetes due to criteria for 2 h‐PG, not because of the criteria for FPG in this study. Therefore, the correlation between M0 and fasting glucose (G0) was analyzed in a combined population of the three groups. Simple and multiple regression analyses showed that M0 was significantly correlated with G0 (Tables 3 and 4).
In multiple regression analysis, QUICKI, an insulin sensitivity index, was an important determinant of fasting mannose level. In patients with type 2 diabetes, EGP, the sum of glycogenolysis and gluconeogenesis, is increased in the fasting state as a result of hepatic insulin resistance, which leads to fasting hyperglycemia16. The relative role of glycogenolysis and gluconeogenesis in increased EGP in type 2 diabetes is controversial. However, an increase in glycogenolysis might play some role in increased EGP, as metformin decreases EGP without affecting gluconeogenesis17.
In multiple regression analysis, the glucose level at 120 min after glucose loading, which is an index of glucose tolerance, was an important determinant of the decrease in mannose level after glucose loading. In addition, the insulinogenic index and Matsuda Index, indices of insulin secretion and sensitivity, respectively, were important determinants of the decrease in mannose level after glucose loading. These results show the relationship between plasma mannose level and glucose tolerance. Acute suppression in EGP in response to insulin elevation by nutrient intake is much more dependent on glycogenolysis relative to gluconeogenesis18, 19, 20. Taken together, our present results are compatible with the possibility of mannose as an indicator of gycogenolysis in humans.
The present study had limitations. First, insulin sensitivity was not measured by hyperinsulinemic euglycemic clamp, and EGP also was not measured. Second, as this study was cross‐sectional, causal association could not be evaluated. Third, glycogenolysis in vivo was not evaluated by methods using a glucose isomer. Fourth, hyperglucagonemia and reduced insulin/glucagon ratio after glucose loading were observed in patients with diabetes21, which might contribute to the increase in glycogenolysis. However, glucagon levels were not evaluated in the present study.
In conclusion, we clarified the relationship between plasma mannose levels and glucose tolerance in humans. Further investigation regarding plasma mannose levels as an indicator of glycogenolysis in human is required.
Disclosure
The authors declare no conflicts of interest.
Supporting information
Table S1 | Simple correlation coefficients between independent variables.
Acknowledgments
The authors thank Kaori Ikeda MD PhD, Department of Diabetes, Endocrinology and Nutrition, Kyoto University, for useful discussion.
J Diabetes Investig 2017; 8: 489–495
References
- 1. Alton G, Hasilik M, Niehues R, et al Direct utilization of mannose for mammalian glycoprotein biosynthesis. Glycobiology 1998; 8: 285–295. [DOI] [PubMed] [Google Scholar]
- 2. Soyama K. Enzymatic determination of D‐mannose in serum. Clin Chem 1984; 30: 293–294. [PubMed] [Google Scholar]
- 3. Sone H, Shimano H, Ebinuma H, et al Physiological changes in circulating mannose levels in normal, glucose‐intolerant, and diabetic subjects. Metabolism 2003; 52: 1019–1027. [DOI] [PubMed] [Google Scholar]
- 4. Taguchi T, Yamashita E, Mizutani T, et al Hepatic glycogen breakdown is implicated in the maintenance of plasma mannose concentration. Am J Physiol Endocrinol Metab 2005; 288: E534–E540. [DOI] [PubMed] [Google Scholar]
- 5. Treadway JL, Mendys P, Hoover DJ. Glycogen phosphorylase inhibitors for treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs 2001; 10: 439–454. [DOI] [PubMed] [Google Scholar]
- 6. Miwa I, Taguchi T. A simple HPLC assay for plasma D‐mannose. Clin Chim Acta 2013; 422: 42–43. [DOI] [PubMed] [Google Scholar]
- 7. Committee on the Standardization of Diabetes Mellitus‐Related Laboratory Testing of Japan Diabetes Society . International clinical harmonization of glycated hemoglobin in Japan: from Japan Diabetes Society to National Glycohemoglobin Standardization Program values. J Diabetes Investig 2012; 3: 39–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. World Health Organization . Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia: Report of a World Health Organization/IDF Consultation. Geneva: World Health Organization, 2006. [Google Scholar]
- 9. Committee of the Japan Diabetes Society on the Diagnostic Criteria of Diabetes Mellitus . Report of the committee on the classification and diagnostic criteria of diabetes mellitus. J Diabetes Investig 2010; 1: 212–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seltzer HS, Allen EW, Herron AL Jr, et al Insulin secretion in response to glycemic stimulus: relation of delayed initial release to carbohydrate intolerance in mild diabetes mellitus. J Clin Invest 1967; 46: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matthews DR, Hosker JP, Rudenski AS, et al Homeostasis model assessment: insulin resistance and β‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412–419. [DOI] [PubMed] [Google Scholar]
- 12. Katz A, Nambi SS, Mather K, et al Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 2000; 85: 2402–2410. [DOI] [PubMed] [Google Scholar]
- 13. Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 1999; 22: 1462–1470. [DOI] [PubMed] [Google Scholar]
- 14. Utzschneider KM, Prigeon RL, Faulenbach MV, et al Oral disposition index predicts the development of future diabetes above and beyond fasting and 2‐h glucose levels. Diabetes Care 2009; 32: 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chu CA, Sindelar DK, Neal DW, et al Direct effects of catecholamines on hepatic glucose production in conscious dog are due to glycogenolysis. Am J Physiol Endocrinol Metab 1996; 271: E127–E137. [DOI] [PubMed] [Google Scholar]
- 16. DeFronzo RA. The triumvirate: β‐cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667–687. [DOI] [PubMed] [Google Scholar]
- 17. Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin‐dependent diabetes mellitus. J Clin Endocrinol Metab 1996; 81: 4059–4067. [DOI] [PubMed] [Google Scholar]
- 18. Sindelar DK, Balcom JH, Chu CA, et al A comparison of the effects of selective increases in peripheral or portal insulin on hepatic glucose production in the conscious dog. Diabetes 1996; 45: 1594–1604. [DOI] [PubMed] [Google Scholar]
- 19. Sindelar DK, Chu CA, Venson P, et al Basal hepatic glucose production is regulated by the portal vein insulin concentration. Diabetes 1998; 47: 523–529. [DOI] [PubMed] [Google Scholar]
- 20. Balasubramanyam A, McKay S, Nadkarni P, et al Ethnicity affects the postprandial regulation of glycogenolysis. Am J Physio Endocrinol Metab 1999; 277: E905–E914. [DOI] [PubMed] [Google Scholar]
- 21. Yabe D, Kuroe A, Watanabe K, et al Early phase glucagon and insulin secretory abnormalities, but not incretin secretion, are similarly responsible for hyperglycemia after ingestion of nutrients. J Diabetes Complications 2015; 29: 413–421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Simple correlation coefficients between independent variables.