

Abstract
Aims/Introduction
Angiotensin‐(1–7) (Ang‐[1–7]), recognized as a new bioactive peptide in the renin–angiotensin system, shows biological and pharmacological properties in diabetic cardiovascular diseases. The leptin‐induced p38 mitogen‐activated protein kinase (MAPK) pathway has been reported to contribute to high glucose (HG)‐induced injury. In the present study, we showed the mechanism of how Ang‐(1–7) can protect against HG‐stimulated injuries in H9c2 cells.
Materials and Methods
H9c2 cells were treated with 35 mmol/L glucose (HG) for 24 h to establish a model of HG‐induced damage. Apoptotic cells were observed by Hoechst 33258 staining. Cell viability was analyzed by cell counter kit‐8. The expression of protein was detected by western blot. Reactive oxygen species was tested by 2′,7′‐dichlorodihydrofluorescein diacetate staining. Mitochondrial membrane potential was measured by 5,5′,6,6′‐Tetrachloro‐1,1′,3,3′‐tetraethyl‐imidacarbocyanine iodide staining.
Results
The present results showed that treating H9c2 cells with HG obviously enhanced the expressions of both the leptin and phosphorylated (p)‐MAPK pathway. However, the overexpression levels of leptin and p‐p38 MAPK/p‐extracellular signal‐regulated protein kinase 1/2 (ERK1/2), but not p‐c‐Jun N‐terminal kinase, were significantly suppressed by treatment of the cells with Ang‐(1–7). Additionally, leptin antagonist also markedly suppressed the overexpressions of p38 and ERK1/2 induced by HG, whereas leptin antagonist had no influence on the overexpression of c‐Jun N‐terminal kinase. More remarkable, Ang‐(1–7), leptin antagonist, SB203580 or SP600125, respectively, significantly inhibited the injuries induced by HG, such as the increased cell viability, decreased apoptotic rate, reduction of ROS production and increased mitochondrial membrane potential. Furthermore, the overexpressions of p38 MAPK, ERK1/2 and leptin were suppressed by N‐actyl‐L‐cystine.
Conclusions
The present findings show that Ang‐(1–7) protects from HG‐stimulated damage as an inhibitor of the reactive oxygen species–leptin–p38 MAPK/ERK1/2 pathways, but not the leptin–c‐Jun N‐terminal kinase pathway in vitro.
Keywords: Angiotensin‐(1–7), Cardiomyocytes, High glucose
Introduction
Diabetes mellitus is a common metabolic disease that can lead to hyperglycemia. Accumulative studies have shown that hyperglycemia can lead to the development of diabetic cardiomyopathy, which has become one of the major causes of heart failure in diabetes patients1, 2. Despite extensive studies, a thorough understanding of the pathogenesis has remained elusive, and there are no effective therapies for them.
Complex and highly diversified mechanisms are involved in the pathogenesis of hyperglycemia‐induced injuries in cardiomyocytes. A series of risk factors, such as leptin3, 4, activation of mitogen‐activated protein kinase (MAPK)5, 6, reactive oxygen species (ROS) production7, 8, diminution of the anti‐oxidant defense system9, 10, pro‐inflammatory cytokine11, 12 and mitochondrial dysfunction13, 14, have been reported to play key roles in hyperglycemia‐induced injuries. Sustained hyperglycemia could produce more ROS generation by enhancing oxidative phosphorylation, which results in oxidative stress and myocardial damage6. Furthermore, previous studies have shown that hyperglycemia significantly increases phosphorylation of the MAPK signaling pathway, mainly composed of p38, extracellular signal‐regulated protein kinase 1/2 (ERK1/2) and c‐Jun N‐terminal kinase (JNK), which are associated with the progression of diabetic heart disease15. Leptin, another crucial factor induced by HG, is able to regulate orexis and energy metabolism in vivo, which can modulate obesity or bodyweight16, 17. In contrast, a series of studies have shown the roles of leptin in inflammation, hematopoiesis, thermogenesis, reproduction and angiogenesis during physiological and pathological processes18. Then the activation of Janus tyrosine kinase (JAK) and signal transducers and activators of transcription are two main components in the leptin pathway19. Additionally, we also previously reported that inhibition of the leptin‐induced activation of the p38 MAPK pathway contributed to the protection of naringin against HG‐induced injury in cardiac cells3.
Angiotensin‐(1–7) (Ang‐[1–7]) is recognized as a new bioactive peptide in the renin–angiotensin system (RAS). Ang‐(1–7) is a counter‐regulatory mediator of Ang‐II, which appears to be protective against cardiovascular disease20. Recently, numerous scientific researchers have proved the extensive physiological and pathophysiological properties of Ang‐(1–7) on the progress of diabetes. Previous findings have shown that Ang‐(1–7) functions in reducing systematic hypertension, oxidation, fibrosis and inflammation21. However today, the beneficial effects of Ang‐(1–7), as a protection opposition to HG‐stimulated injuries, are still unclear. In the present study, we tried to demonstrate the hypothesis that Ang‐(1–7) was able to prevent H9c2 cells from HG‐induced damage by inhibiting the upregulation of the leptin–MAPKs pathway.
Materials and Methods
Cell culture and treatments
The H9c2 cells, a kind of rat cardiac myoblasts cell line, were acquired from Sun Yat‐sen University Experimental Animal Center (Guangzhou, China). The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum in a condition of 5% CO2 and 95% air at 37°C. The cells were treated with HG and 1 μmol/L Ang‐(1–7), or pre‐treated with leptin antagonist (LA), SB203580 (a selective inhibitor of p38 MAPK), SP600125 (a selective inhibitor of JNK) and N‐actyl‐L‐cystine (NAC; an inhibitor of ROS) for 60 min before exposure to HG for 24 h.
Western blotting
The H9c2 cardiac cells were seeded at a concentration of 1 × 104 cells/mL, and incubated at 37°C. The cells with indicated treatments were harvested and dissolved into lysis solution for carrying out western blot testing. Protein concentration was assessed by the bicinchoninic acid protein assay kit. The same amount of cell protein was loaded onto 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis for electroblotting, and then transferred onto a polyvinylidene fluoride membrane. After being blocked in 5% milk without fat, the membranes were incubated with anti‐p38 (1:1,000 dilution; Cell Signaling Technology, Massachusetts, Boston, USA), anti‐p‐p38 (1:1,000 dilution; Cell Signaling Technology), anti‐ERK1/2 (1:1,000 dilution; Cell Signaling Technology), anti‐p‐ERK1/2 (1:1,000 dilution; Cell Signaling Technology), anti‐p‐JNK, anti‐JNK antibody (1:1,000 dilution; Cell Signaling Technology) or anti‐leptin (1:1,000 dilution; Cell Signaling Technology) at 4°C overnight on a shaker. Then the horseradish peroxidase‐conjugated goat anti‐rabbit as a secondary antibody was used at room temperature for 2 h. The membranes were washed three times with phosphate‐buffered saline with Tween 20, and fluorescence was detected by enhanced chemiluminescence (Bio‐Rad Laboratories Inc., Hercules, California, USA) and exposed to X‐ray film.
Cell viability assay
H9c2 cells were seeded at a concentration of 1 × 104/mL in 96‐well plates, and the cell viability was assessed by the CCK‐8 assay. After being treated with the drugs, each well had 10 μL of working solution diluted 10 times added, and then incubated for 1.5 h. We detected the absorbance at 450 nm through a microplate reader (Molecular Devices, Sunnyvale, California, USA), and used the optical density of five wells to compute the cell viability based on the following formula: cell viability(%) = (optical densitytreatmentgroup/optical densitycontrolgroup)× 100%. The experiment was carried out three times.
Hoechst 33258 nuclear staining for measurement of apoptosis
The H9c2 cardiac cells were seeded at a concentration of 1 × 104 cells/mL, and incubated at 37°C. The apoptosis of H9c2 cells was evaluated by using the Hoechst 33258 staining method. The cells were pretreated with the drugs, and fixed with 4% paraformaldehyde and washed three times with phosphate‐buffered saline (PBS). After that, the nuclear deoxyribonucleic acid was stained with 5 mg/mL of working solution for 5 min, then washed three times again and visualized by using a fluorescence microscope (Olympus, Kanagawa, Japan). Viable H9c2 cell nuclei always presented consistent blue fluorescence and normal size, but apoptotic cells showed fractured or condensed nuclei. The experiment was repeated three times.
Assessment of ROS production
The H9c2 cardiac cells were seeded at a concentration of 1 × 104 cells/mL, and incubated at 37°C. The oxidative conversion of cell‐permeable oxidation of 2′,7′‐dichlorodihydrofluorescein diacetate to fluorescent 2′,7′‐dichlorodihydrofluorescein was used to assessed the generation of intracellular ROS. The cells were removed from the culture medium and washed with PBS twice. The cells were treated with 10 μmol/L working solution and then cultured in 37°C for 30 min. The cells were washed with PBS again, and the fluorescence product was assessed by a fluorescence microscope that was connected to an imaging system (Olympus). The mean fluorescence intensity, as an index of the amount of ROS, from five random fields was measured by IMAGEJ 1.47 i software (Sun Microsystems, Inc., Bethesda, Maryland, USA). The experiment was carried out three times.
Assessment of the mitochondrial membrane potential
The H9c2 cardiac cells were seeded at a concentration of 1 × 104 cells/mL, and incubated at 37°C. The mitochondrial membrane potential of H9c2 cells was detected by 5,5′,6,6′‐Tetrachloro‐1,1′,3,3′‐tetraethyl‐imidacarbocyanine iodide staining, a cell‐permeable fluorescent dye that preferentially goes into mitochondria ground on the highly negative mitochondrial membrane potential (MMP). The depolarization of MMP can lead to diminution of green fluorescence. The cells were cultured on slides with Dulbecco's modified Eagle's medium, and then washed three times with PBS after treatment with drugs. The cells were incubated with 1 mg/L 5,5′,6,6′‐Tetrachloro‐1,1′,3,3′‐tetraethyl‐imidacarbocyanine iodide at 37°C for 30 min, then washed three times with PBS and dried in air. Fluorescence was measured by a fluorescent microscope, which was connected to an imaging system (Olympus). The mean fluorescence intensity of Rh123 from five random fields, as an index of the MMP, was analyzed by the IMAGEJ 1.47 i software. The experiment was repeated three times.
Statistical analysis
All data are shown as the mean ± standard error of the mean. Differences between groups were analyzed by one‐way analysis of variance (anova) using spss software (SPSS version 19.0; IBM SPSS Inc. Chicago, Illinois, USA), and followed by the least significant difference post‐hoc comparison test. Statistical significance was set at P < 0.05.
Results
Ang‐(1–7) suppresses the HG‐induced activation of leptin and p‐38MAPK/ERK1/2 in H9c2 cells, but has no influence on overexpression of p‐JNK
We first examined expression levels of MAPK pathway phosphorylation and leptin in the condition of HG (35 mmol/L). As shown in Figures 1 and 2, the expression of p‐p38, p‐ERK1/2, p‐JNK and leptin were markedly upregulated when treated with HG. However, the total expression levels of p38, ERK1/2 and JNK had no obvious change.
Figure 1.
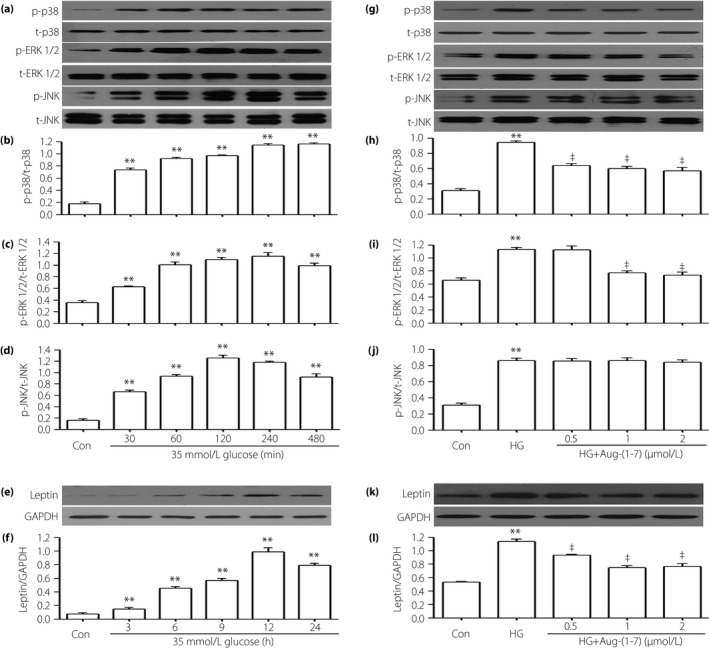
Different doses of angiotensin‐(1–7) (Ang‐[1–7]) suppresse the high glucose (HG)‐induced activation of leptin and p‐38 mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated protein kinase 1/2 (ERK1/2) in H9c2 cells, but have no influence on overexpression of phosphorylated (p)‐c‐Jun N‐terminal kinase (JNK). (a–f) H9c2 cells were exposed to 35 mmol/L glucose for the indicated times (30, 60, 120, 240 and 480 min, respectively) or (3, 6, 9, 12 and 24 h, respectively). (g–l) H9c2 cells were co‐treated with 35 mmol/L glucose and indicated concentrations of Ang‐(1–7) (0.5, 1 and 2 μmol/L, respectively) for 15,240 min or 24 h. The expression levels of (a,b,g,h) p38 MAPK, (a,c,g,i) ERK1/2, (a,d,g,j) JNK and (e,f,k,l) leptin were measured by western blot assay. (b,c,d,f,h,i,j,l) Densitometric analysis of the results from (a), (e), (g) and (k), respectively. Data are presented as the mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; p‐p38, phosphorylated‐p38; t‐p38, total p38.
Figure 2.
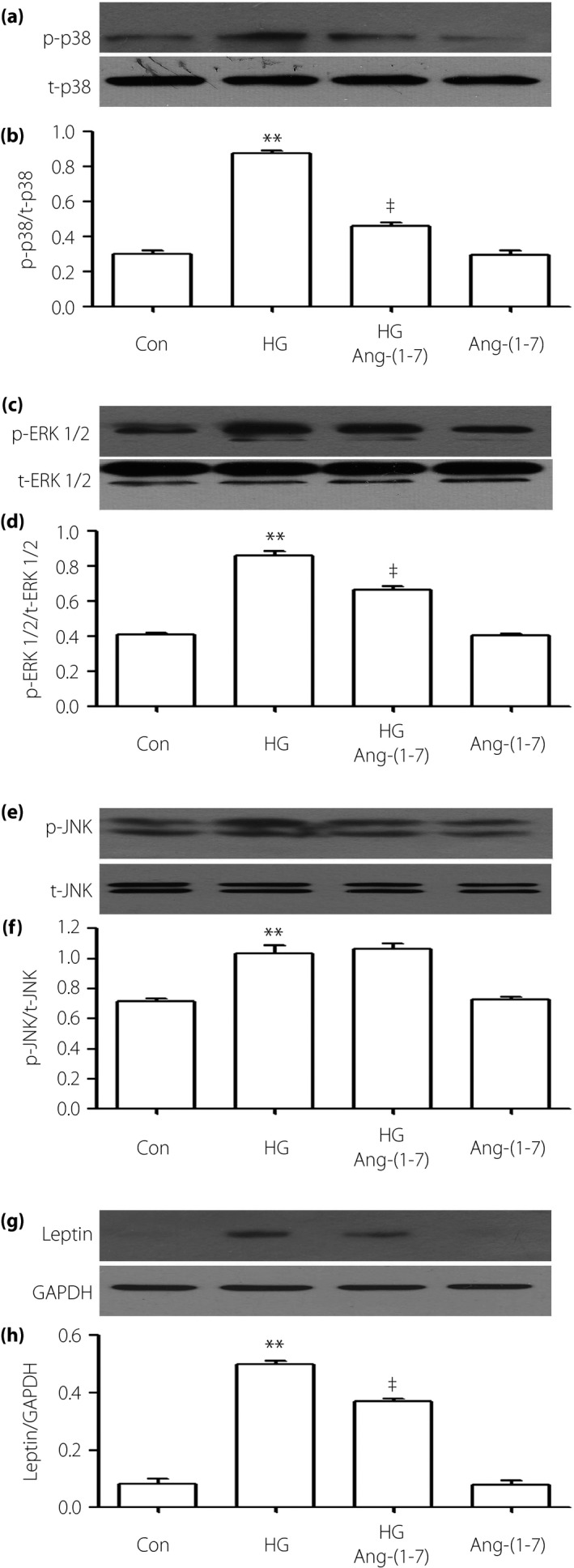
Angiotensin‐(1–7) (Ang‐[1–7]) suppresses the high glucose (HG)‐induced activation of leptin and p‐38 mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated protein kinase 1/2 (ERK1/2) in H9c2 cells, but has no influence on overexpression of phosphorylated (p)‐c‐Jun N‐terminal kinase (JNK). Cells were coconditioned with 1 μmol/L Ang‐(1–7) for 24 h with or without HG. (a,c,e,g) The expression of p38, ERK1/2, JNK and leptin were measured by western blot analysis. (b,d,f,h) Densitometric analysis of the related protein expression levels in (a,c,e,g), respectively. The data were quantified by densitometric analysis with IMAGEJ 1.47 i software. Data are shown as the mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; p‐p38, phosphorylatedp38; t‐p38, total p38.
To observe the effects of Ang‐(1–7), we co‐treated H9c2 cells with HG and Ang‐(1–7) for 24 h. As shown in Figures 1 and 2, the increased phosphorylation of MAPK (including p38 MAPK and ERK1/2) and leptin were decreased by the co‐treatment with HG and Ang‐(1–7), but the activation of p‐JNK was nearly unaffected.
Leptin antagonist decreases HG‐induced upregulation of p‐38MAPK/ERK1/2, but not overexpression of p‐JNK in H9c2 cells
According to the western blot analysis (Figure 3), treating the cells with LA can significantly suppress the upregulated expressions of p‐p38and p‐ERK1/2 induced by HG, but the overexpression of p‐JNK was almost unaffected.
Figure 3.
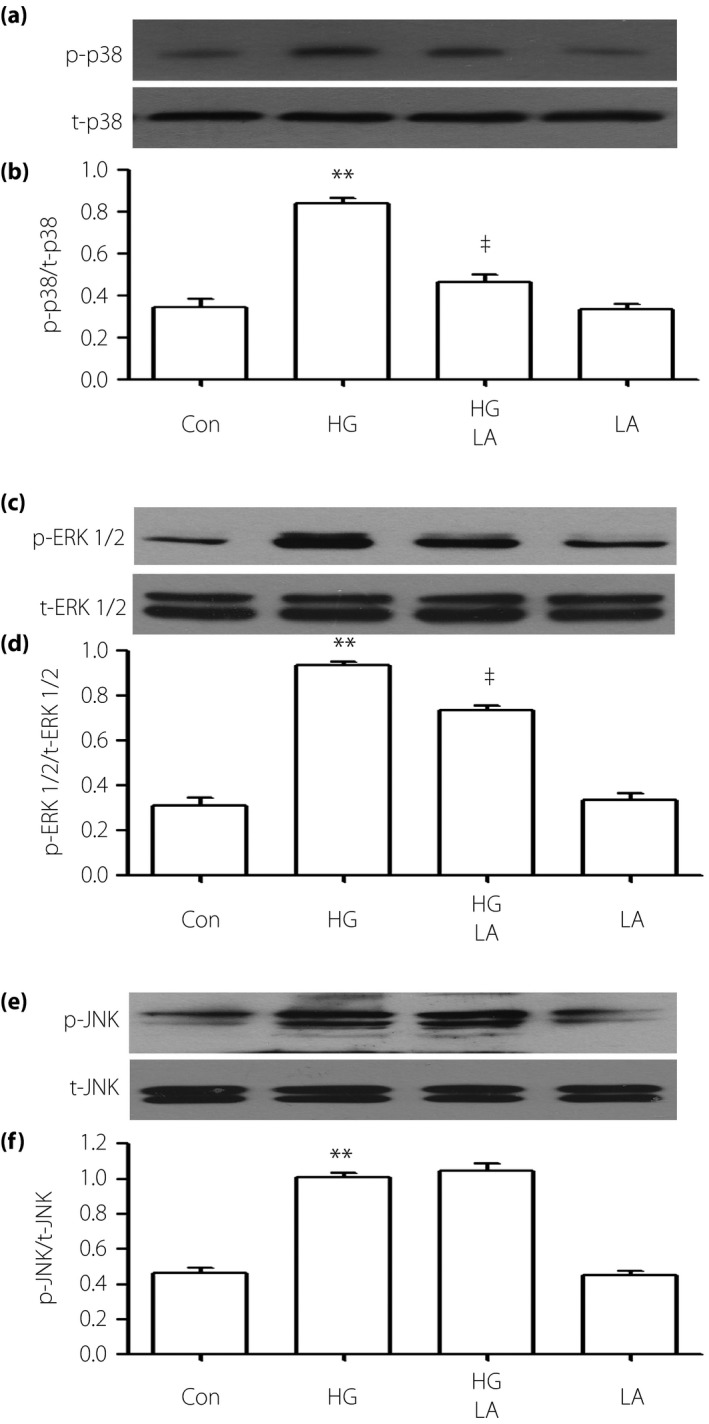
Leptin antagonist (LA) decreases the high glucose (HG)‐induced activation of p‐38 mitogen‐activated protein kinase (MAPK)/extracellular signal‐regulated protein kinase 1/2 (ERK1/2) in H9c2 cells, but has no influence on overexpression of phosphorylated (p)‐c‐Jun Nterminal kinase (JNK). Cells were preconditioned with 50 ng/mL LA for 24 h with or without HG for 24 h. (a,c,e) The expression levels of p38, ERK1/2 and JNK were assessed by western blot analysis. (b,d,f) Densitometric analysis of the related protein expression levels in (a,c,e), respectively. The data was quantified by densitometric analysis with IMAGEJ 1.47 i software. Data are shown as the mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group. p‐p38, phosphorylated‐p38; t‐p38, total p38.
Ang‐(1–7), LA, SB203580 and SP600125 relieved HG‐induced cytotoxicity in H9c2 cells
To further explore the effect of leptin‐p38/ERK1/2 on cytotoxicity induced by HG, we treated the cells with HG and 1 μmol/L Ang‐(1–7) at the same time. As shown in Figure 4, the cytotoxicity induced by HG was significantly relieved by Ang‐(1–7). Similarly, preconditioning with either 50 ng/mL LA or 3 μmol/L SB203580 or 10 μmol/L SP600125 for 60 min also markedly upregulated the cell viability, respectively.
Figure 4.
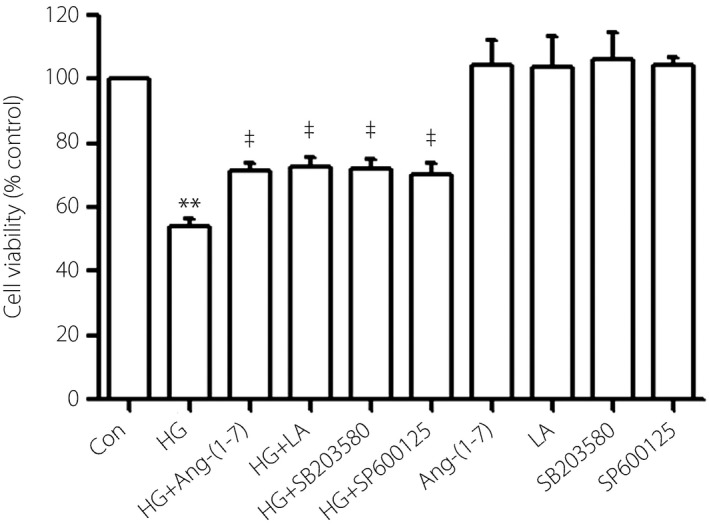
Angiotensin‐(1–7) (Ang‐[1–7]), leptin antagonist (LA), SB203580 (a selective inhibitor of p38 mitogen‐activated protein kinase) and SP600152 (a selective inhibitor of extracellular signal‐regulated protein kinase 1/2) relieved cytotoxicity induced by high glucose (HG) in H9c2 cells. Cell viability was tested by using the CCK‐8 assay. Cells were treated with 1 μmol/L Ang‐(1–7), 50 ng/mL LA, 3 μmol/L SB203580 or 10 μmol/L SP600125 with or without HG. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group.
Ang‐(1–7), LA, SB203580 and SP600125 decrease HG‐induced apoptosis in H9c2 cells
The amounts of apoptotic cells were obviously upregulated in the HG condition in H9c2 cells (Figure 5b), whereas co‐treatment of cells with HG and 1 μmol/L Ang‐(1–7) for 24 h can sharply diminish increased cell apoptosis (Figure 5c). To explore whether leptin, p38 and ERK1/2 function in cell apoptosis, we gave the cells priority treatment with 50 ng/mL LA, 3 μmol/L SB203580 or 10 μmol/L SP600125 for 60 min, the apoptosis induced by HG was obviously diminished, respectively (Figure 5d–f).
Figure 5.

Angiotensin‐(1–7) (Ang‐[1–7]), leptin antagonist (LA), SB203580 and SP600152 decrease apoptosis induced by high glucose (HG) in H9c2 cells. (a–j) Cells were treated with different drugs. Hoechst 33258 staining followed by photofluorography was used to assess the cellular apoptosis. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group.
Ang‐(1–7), LA, SB203580 and SP600125 alleviate the HG‐induced increased ROS generation in H9c2 cells
As shown in Figure 6b, the generation of ROS was obviously increased in the HG condition in H9c2 cells, whereas the co‐treatment of cells with HG and 1 μmol/L Ang‐(1–7) for 24 h can diminish the increased ROS generation significantly (Figure 6c). To explore whether leptin, p38 and ERK1/2 have an effect on HG‐induced oxidative stress, we gave the cells priority treatment with 50 ng/mL LA, 3 μmol/L SB203580 or 10 μmol/L SP600125 for 60 min, the HG‐stimulated ROS generation in H9c2 cells were obviously diminished, respectively (Figure 6d–f).
Figure 6.
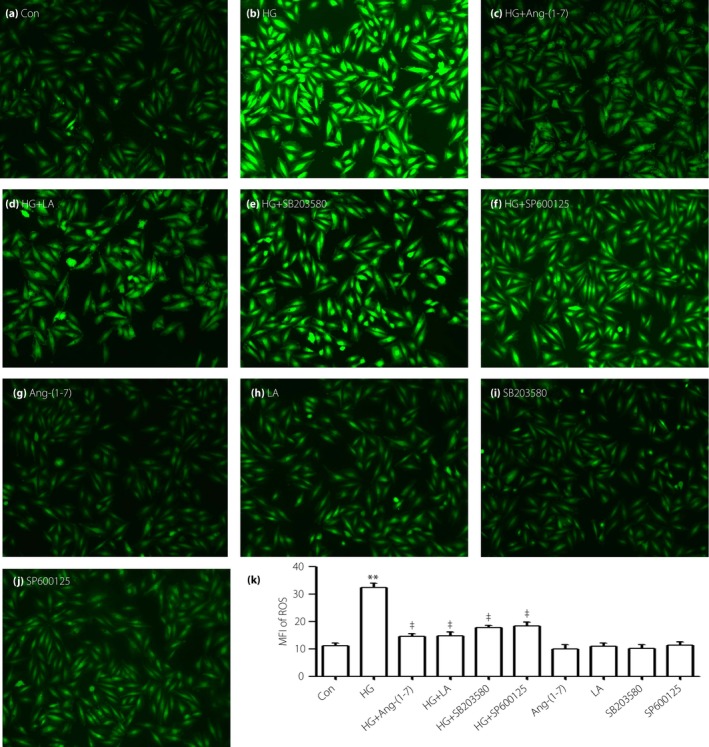
Angiotensin‐(1–7) (Ang‐[1–7]), leptin antagonist (LA), SB203580 and SP600152 alleviate the increased reactive oxygen species (ROS) generation induced by high glucose (HG) in H9c2 cells. (a–j) Cells were treated with different drugs, intracellular ROS level was assessed by 2′,7′‐dichlorodihydrofluorescein diacetate staining followed by photofluorography. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group.
Ang‐(1–7), LA, SB203580 and SP600125 block dissipation of HG‐induced MMP in H9c2 cells
To examine the effects of Ang‐(1–7) and other inhibitors on the loss of MMP stimulated by HG in vitro, we gave the cells co‐treatment with HG and 1 μmol/L Ang‐(1–7), or priority treatment with 50 ng/mL LA, 3 μmol/L SB203580 or 10 μmol/L SP600125. The HG‐stimulated dissipation of MMP was significantly attenuated, respectively (Figure 7c–e).
Figure 7.

Angiotensin‐(1–7) (Ang‐[1–7]), leptin antagonist (LA), SB203580 and SP600152 block the mitochondrial membrane potential (MMP) induced by high glucose (HG) in H9c2 cells. (a–j) Cells were treated with different drugs, and the MMP level was tested by 5,5′,6,6′‐Tetrachloro‐1,1′,3,3′‐tetraethyl‐imidacarbocyanine iodide staining followed by photofluorography. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group.
NAC reduces the HG‐induced activation of Leptin‐p38/ERK1/2 and decreased the H9c2 cell viability
To examine the effect of ROS on the leptin–p‐38/ERK1/2 pathway, 1,000 μmol/L NAC was added to H9c2 cells for 60 min before exposure to HG. According to the western blot analysis (Figure 8), preconditioning with NAC significantly reduced the HG‐stimulated overexpressions of leptin, p‐p38 and p‐ERK1/2. Additionally, NAC can also upregulate the cell viability suppressed by HG (Figure 9).
Figure 8.
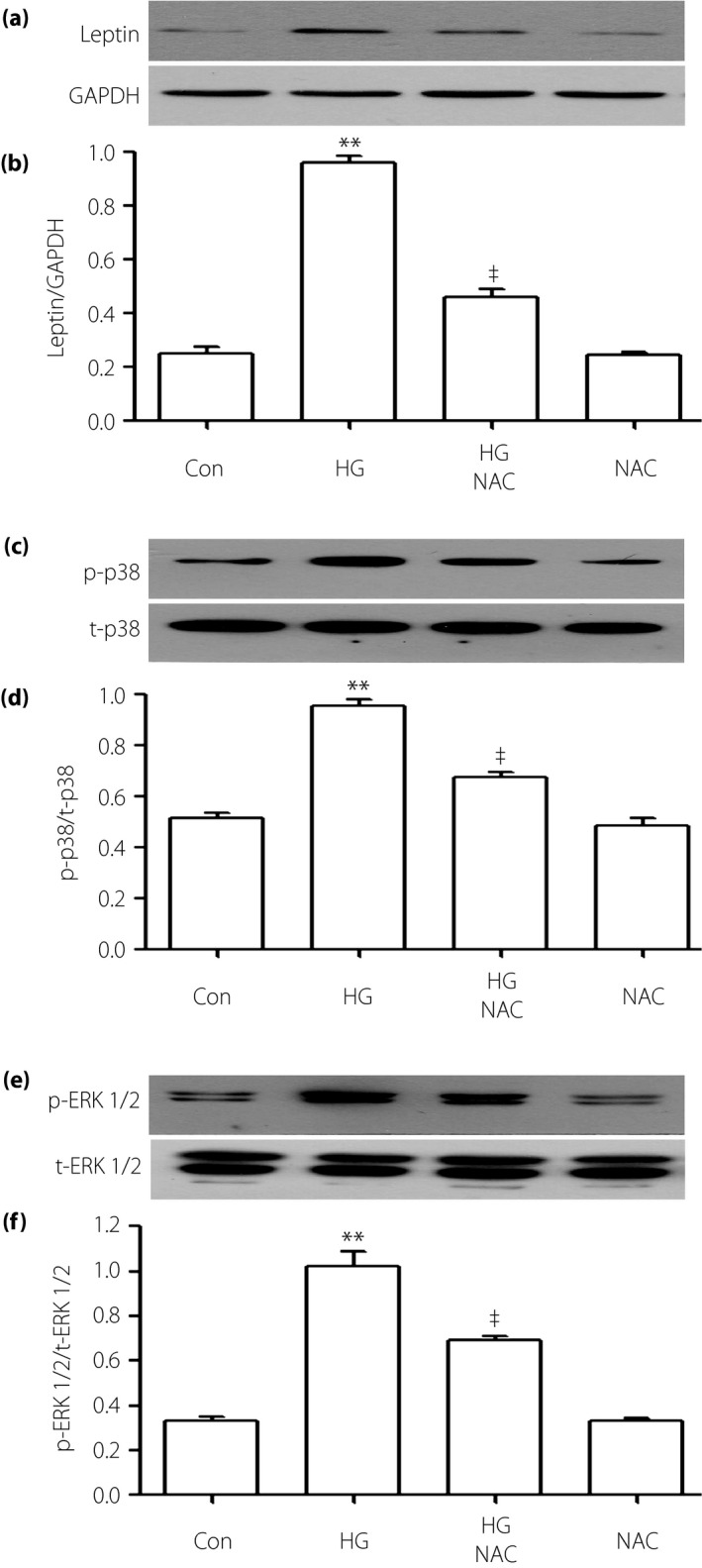
Reactive oxygen species (ROS) inhibitor suppresses the high glucose (HG)‐induced overexpressions of p38 mitogen‐activated protein kinase (MAPK), extracellular signal‐regulated protein kinase 1/2 (ERK1/2) and leptin in H9c2 cells. Cells were priority treated with 1,000 μmol/L N‐actyl‐L‐cystine for 60 min with or without HG. (a,c,e) The expressions of leptin, p38 and ERK1/2 were measured by western blot analysis. (b,d,f) Densitometric analysis of the related protein expression levels in (a,c,e), respectively. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; p‐p38, phosphorylated‐p38; t‐p38, total p38.
Figure 9.
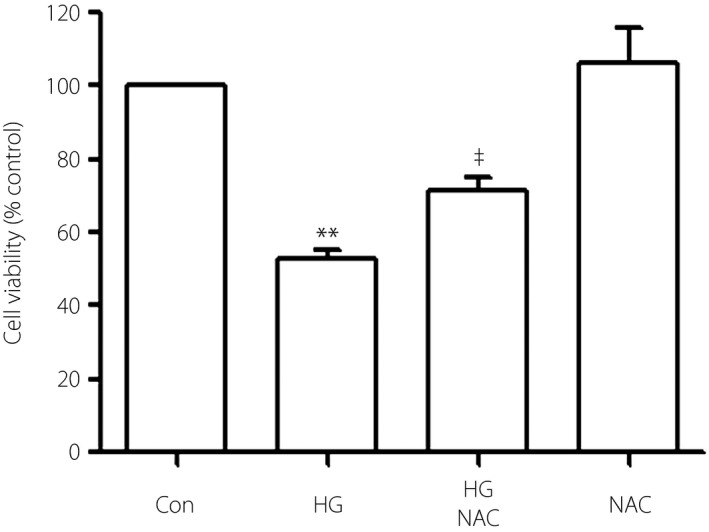
N‐actyl‐L‐cystine alleviates HG‐induced cytotoxicity in H9c2 cells. Cells were pretreated with 1,000 μmol/L NAC for 60 min with or without high glucose (HG). Cell viability was tested by using the CCK‐8 assay. Data are shown as mean ± standard error of the mean (n = 3). **P < 0.01 vs the control (Con) group; ‡ P < 0.01 vs the HG‐treated group.
Discussion
In the present study, we showed a novel finding of Ang‐(1–7) against injuries induced by HG and provided evidence to show its potential mechanisms in H9c2 cardiac cells. As the molecular mechanisms of Ang‐(1–7) are not fully understood in previous studies, we used HG to induce injuries in H9c2 cells for dissecting the protective function of Ang‐(1–7) against HG‐induced damage and its properties.
As our previous studies reported3, 4, 22, 23, 24, we showed that HG could have various degenerative effects, including cytotoxicity, apoptosis, oxidative stress and mitochondrial insult, as evidenced by the decreased cell viability, the increased apoptotic rate, the accumulation ROS generation and the loss of MMP. The ROS level is not only an oxidative stress indicator, but also one of the elements reflecting HG‐stimulated cardiomyocyte damage. Conclusive evidence has established that overgeneration of ROS is increased during ischemia/reperfusion and hypoxia/reperfusion in the heart and cardiomyocytes, which results in chemical hypoxia‐stimulated damage in the cardiac cells3. Analogously, the present data suggested that preconditioning with ROS inhibitor significantly diminished cell cytotoxicity and HG‐induced apoptosis, proving the participation of ROS in HG‐induced cardiomyocyte injury. Furthermore, we found that ROS scavengers obviously depressed the overexpressions of leptin, p38 and ERK1/2 in HG, which have been proven to be a result of the development of diabetic cardiomyopathy22, showing that ROS plays its role effectively though the leptin and p38 MAPK/ERK1/2 pathway.
Previous research has demonstrated that leptin had been shown to take part in HG‐induced cardiomyocyte hypertrophy4, which means that leptin might be associated with HG‐induced cardiovascular complications. Furthermore, we investigated whether leptin participates in other HG‐induced cardiac cells injury. In the present study, the data showed that the aforementioned HG‐induced H9c2 cells injuries were significantly suppressed by treatment with LA. Therefore, we can conclude that leptin is involved in HG‐induced injury, including apoptosis, cytotoxicity and ROS generation, as well as mitochondrial damage. The present findings confirm the data from our previous study3, and provide novel evidence of the molecular role of leptin in HG‐induced cardiac cells injury.
Additionally, MAPK, a family of serine/threonine kinase, presents important signal transduction machinery and regulates cell signaling in response to kinds of cellular stimulus, including growth, differentiation, apoptosis and inflammation. Previous studies have shown that the MAPK signaling pathway could be activated by HG in human retinal pigmented epithelial cells25 and rat mesangial cells26. In the present study, we showed a similar effect that HG obviously upregulated the expressions of the MAPK pathway, including p38, ERK1/2 and JNK, in cardiac cells. A variety of physical and chemical stresses, such as hypoxia/ischemia27, drugs28 and oxidative stress29, could activate the MAPK pathway. As far as we know, only a few studies have shown the relationship of MAPK with leptin in in vitro experiments. For instance, leptin has been shown to directly stimulate cellular hypertrophy through p38 MAPK in rat vascular smooth muscle cells30. However, it is still unclear whether the activation of the leptin–MAPK pathway contributes to cardiomyocyte damage induced by HG. Recently, our previous data showed that the activation of the lepin–p38 MAPK pathway could play a crucial role in the progress of HG‐induced injury in H9c2 cells3. Additionally, we found in the present study that the high expressions of p38 and ERK1/2 in HG were decreased by treatment with LA, testifying that leptin acts in the upstream of the p38–ERK1/2 pathway. However, LA has no influence on the overexpression of JNK induced by HG, showing that leptin does not function in activating the JNK pathway.
Previous research showed that Ang‐(1–7) appeared to be protective against cardiovascular disease20. Additional research found that Ang‐(1–7) exerted cardioprotective effects against various stimulates (Ang II, isoproterenol, vasopressin)‐induced heart failure, injuries, cardiac hypertrophy and fibrosis through its respective receptors (angiotensin type 2 receptor)31, 32. Ang‐(1–7) bound to its receptor, and then participated in various pathophysiological processes and affected the downstream signaling pathways. Importantly, in the present study we found that Ang‐(1–7) could markedly prevent injuries in cardiac cells, such as cytotoxicity, apoptosis, ROS generation and the loss of MMP induced by HG. Our hypothesis is also supported by the evidence that Ang‐(1–7) obviously inhibited the HG‐induced overexpressions of leptin‐p38 MAPK and ERK1/2, which are associated with the biological properties mentioned. Another mechanism of Ang‐(1–7) antihyperglycemic functions might be concerned with the inhibitory action, as Akhtar et al.33 reported that Ang‐(1–7), through its Mas receptor, acts as a epidermal growth factor receptor inhibitor to exert its beneficial effects in many disease states, including diabetes‐induced vascular complications. However, much deeper research should be carried out to explain the protective mechanism of Ang‐(1–7) in HG.
Notably, the present results showed that Ang‐(1–7) had no influence on the HG‐induced activation of the JNK pathway. However, some scientists reported that Ang‐(1–7) activates a MAPK phosphatase, and thereby reduces JNK phosphorylation to inhibit apoptosis and promote cell survival in the human A549 and mouse MLE12 AEC lines34, so the effects of Ang‐(1–7) on JNK are still controversial. Our explanation is that Ang‐(1–7) has different effects on different cells, and its inhibitory vs stimulatory effects on MAPK pathway might be complicated.
In conclusion, HG could induce various kinds of injuries in H9c2 cardiac cells. These effects might be prevented by Ang‐(1–7), leading to decreased expressions of ROS, leptin, p38 and ERK1/2. In diabetes mellitus, the findings provide novel insight into a unified concept, and identify Ang‐(1–7) as a protective factor and antihyperglycemia injury target. The deeper mechanism of Ang‐(1–7) in H9c2 cells is still unclear, and needs to be further investigated.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This study was supported by Technology Planning Project (201544‐01) of Huangpu District, Science and Technology Planning Project of Guangdong Province (2011B031600068), Medical Scientific Research Foundation of Guangdong Province (A2015287, A2014217) and Guangdong Province Natural Science Foundation (2015A030313690).
J Diabetes Investig 2017; 8: 434–445
References
- 1. Liu Q, Wang S, Cai L. Diabetic cardiomyopathy and its mechanisms: role of oxidative stress and damage. J Diabetes Investig 2014; 5: 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu H, Li GN, Xie J, et al Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF‐beta/periostin pathway in STZ‐induced diabetic mice. BMC Cardiovasc Disord 2016; 16: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen J, Mo H, Guo R, et al Inhibition of the leptin‐induced activation of the p38 MAPK pathway contributes to the protective effects of naringin against high glucose‐induced injury in H9c2 cardiac cells. Int J Mol Med 2014; 33: 605–612. [DOI] [PubMed] [Google Scholar]
- 4. Majumdar P, Chen S, George B, et al Leptin and endothelin‐1 mediated increased extracellular matrix protein production and cardiomyocyte hypertrophy in diabetic heart disease. Diabetes Metab Res Rev 2009; 25: 452–463. [DOI] [PubMed] [Google Scholar]
- 5. Evans JL, Goldfine ID, Maddux BA, et al Oxidative stress and stress‐activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 2002; 23: 599–622. [DOI] [PubMed] [Google Scholar]
- 6. Soetikno V1, Sari FR, Sukumaran V, et al Curcumin prevents diabetic cardiomyopathy in streptozotocin‐induced diabetic rats: possible involvement of PKC‐MAPK signaling pathway. Eur J Pharm Sci 2012; 47: 604–614. [DOI] [PubMed] [Google Scholar]
- 7. Murali R, Karthikeyan A, Saravanan R. Protective effects of D‐limonene on lipid peroxidation and antioxidant enzymes in streptozotocin‐induced diabetic rats. Basic Clin Pharmacol Toxicol 2013; 112: 175–181. [DOI] [PubMed] [Google Scholar]
- 8. Peake BF1, Nicholson CK, Lambert JP, et al Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia‐reperfusion injury by activating Nrf2 signaling in an Erk‐dependent manner. Am J Physiol Heart Circ Physiol 2013; 304: H1215–H1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ashour M1, Al‐Kattan K, Rafay MA, et al Current surgical therapy for bronchiectasis. World J Surg 1999; 23: 1096–1104. [DOI] [PubMed] [Google Scholar]
- 10. Maritim AC, Sanders RA, Watkins JB 3rd. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol 2003; 17: 24–38. [DOI] [PubMed] [Google Scholar]
- 11. Di Filippo C, Marfella R, Cuzzocrea S, et al Hyperglycemia in streptozotocin‐induced diabetic rat increases infarct size associated with low levels of myocardial HO‐1 during ischemia/reperfusion. Diabetes 2005; 54: 803–810. [DOI] [PubMed] [Google Scholar]
- 12. Venkatachalam K, Mummidi S, Cortez DM, et al Resveratrol inhibits high glucose‐induced PI3K/Akt/ERK‐dependent interleukin‐17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol 2008; 294: H2078–H2087. [DOI] [PubMed] [Google Scholar]
- 13. Ceriello A. Cardiovascular effects of acute hyperglycaemia: pathophysiological underpinnings. Diab Vasc Dis Res 2008; 5: 260–268. [DOI] [PubMed] [Google Scholar]
- 14. Boudina S, Sena S, Theobald H, et al Mitochondrial energetics in the heart in obesity‐related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007; 56: 2457–2466. [DOI] [PubMed] [Google Scholar]
- 15. Igarashi M, Wakasaki H, Takahara N, et al Glucose or diabetes activates p38 mitogen‐activated protein kinase via different pathways. J Clin Invest 1999; 103: 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tartaglia LA. The leptin receptor. J Biol Chem 1997; 272: 6093–6096. [DOI] [PubMed] [Google Scholar]
- 17. Murad A, Nath AK, Cha ST, et al Leptin is an autocrine/paracrine regulator of wound healing. FASEB J 2003; 17: 1895–1897. [DOI] [PubMed] [Google Scholar]
- 18. La Cava A, Alviggi C, Matarese G. Unraveling the multiple roles of leptin in inflammation and autoimmunity. J Mol Med (Berl) 2004; 82: 4–11. [DOI] [PubMed] [Google Scholar]
- 19. Bjorbaek C, Uotani S, da Silva B, et al Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 1997; 272: 32686–32695. [DOI] [PubMed] [Google Scholar]
- 20. Zhang F, Liu J, Li SF, et al Angiotensin‐(1‐7): new perspectives in atherosclerosis treatment. J Geriatr Cardiol 2015; 12: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Padda RS, Shi Y, Lo CS, et al Angiotensin‐(1‐7): A novel peptide to treat hypertension and nephropathy in diabetes? J Diabetes Metab 2015; 6: doi:10.4172/2155‐6156.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J, Guo R, Yan H, et al Naringin inhibits ROS‐activated MAPK pathway in high glucose‐induced injuries in H9c2 cardiac cells. Basic Clin Pharmacol Toxicol 2014; 114: 293–304. [DOI] [PubMed] [Google Scholar]
- 23. Xu W, Chen J, Lin J, et al Exogenous hydrogen sulfide protects H9c2 cardiac cells against high glucose‐induced injury by inhibiting the activities of the p38 MAPK and ERK1/2 pathways. Int J Mol Med 2013; 32: 917–925. [DOI] [PubMed] [Google Scholar]
- 24. You Q, Wu Z, Wu B, et al Naringin protects cardiomyocytes against hyperglycemia‐induced injuries in vitro and in vivo . J Endocrinol 2016; 230: 197–214. [DOI] [PubMed] [Google Scholar]
- 25. Liang W, Chen J, Mo L, et al ATP‐sensitive K+ channels contribute to the protective effects of exogenous hydrogen sulfide against high glucose‐induced injury in H9c2 cardiac cells. Int J Mol Med 2016; 37: 763–772. [DOI] [PubMed] [Google Scholar]
- 26. Dong XB, Yang CT, Zheng DD, et al Inhibition of ROS‐activated ERK1/2 pathway contributes to the protection of H2S against chemical hypoxia‐induced injury in H9c2 cells. Mol Cell Biochem 2012; 362: 149–157. [DOI] [PubMed] [Google Scholar]
- 27. Yuan Z, Feng W, Hong J, et al p38MAPK and ERK promote nitric oxide production in cultured human retinal pigmented epithelial cells induced by high concentration glucose. Nitric Oxide 2009; 20: 9–15. [DOI] [PubMed] [Google Scholar]
- 28. Fang S, Jin Y, Zheng H, et al High glucose condition upregulated Txnip expression level in rat mesangial cells through ROS/MEK/MAPK pathway. Mol Cell Biochem 2011; 347: 175–182. [DOI] [PubMed] [Google Scholar]
- 29. Liu AL, Wang XW, Liu AH, et al JNK and p38 were involved in hypoxia and reoxygenation‐induced apoptosis of cultured rat cerebellar granule neurons. Exp Toxicol Pathol 2009; 61: 137–143. [DOI] [PubMed] [Google Scholar]
- 30. Guo R, Lin J, Xu W, et al Hydrogen sulfide attenuates doxorubicin‐induced cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells. Int J Mol Med 2013; 31: 644–650. [DOI] [PubMed] [Google Scholar]
- 31. Marques FD, Ferreira AJ, Sinisterra RD, et al An oral formulation of angiotensin‐(1‐7) produces cardioprotective effects in infarcted and isoproterenol‐treated rats. Hypertension 2011; 57: 477–483. [DOI] [PubMed] [Google Scholar]
- 32. Flores‐Muñoz M1, Godinho BM, Almalik A, et al Adenoviral delivery of angiotensin‐(1‐7) or angiotensin‐(1‐9) inhibits cardiomyocyte hypertrophy via the masor angiotensin type 2 receptor. PLoS ONE 2012; 7: e45564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akhtar S, Chandrasekhar B, Attur S, et al Transactivation of ErbB Family of Receptor Tyrosine Kinases Is Inhibited by Angiotensin‐(1‐7) via Its Mas Receptor. PLoS ONE 2015; 10: e0141657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gopallawa I, Uhal BD. Angiotensin‐(1‐7)/mas inhibits apoptosis in alveolar epithelial cells through upregulation of MAP kinase phosphatase‐2. Am J Physiol Lung Cell Mol Physiol 2016; 310: L240–L248. [DOI] [PMC free article] [PubMed] [Google Scholar]