

Abstract
Several intrauterine environmental factors can increase the future risk of type 2 diabetes. The microbiome can influence the balance between health and disease. However, the influence of the maternal gut microbiome on the future risk of diabetes in the fetus is unknown. The present study investigated the associations between maternal gut microbiome and differentially methylated regions of diabetes‐associated genes in umbilical cord samples. The present study included 10 pregnant participants from a birth cohort study. 16S ribosomal ribonucleic acid metagenome analysis of maternal stool samples and deoxyribonucleic acid methylation assays of umbilical cord samples were carried out. The present study found that changes in the UBE2E2 and KCNQ1 methylation rates in umbilical cord samples were associated with the proportion of Firmicutes in the maternal gut, albeit with marginal correlations after adjustment for age and body mass index. These findings suggest a link between the methylation of diabetes‐associated genes in fetuses and maternal microbiota components during pregnancy.
Keywords: Deoxyribonucleic methylation, Microbiome, Umbilical cord
Introduction
Epigenetic alternations of diabetes‐associated genes have been reported to increase the risk of diabetes1, 2. Although, low birthweight and maternal malnutrition, which are considered as fetal environmental factors, can cause epigenetic alternations of diabetes‐associated genes3, few studies have investigated the epigenetic alternations in type 2 diabetes‐associated genes of fetal tissues.
The gut microbiome has been shown to be a novel environmental factor that directly affects metabolism4. Perturbations in the gut microbiome have been implicated to cause metabolic syndrome5, and the role of the gut microbiome in pregnancy has become the subject of considerable interest6.
Although a change in the exposure to maternal microbial diversity in fetuses remains one of the leading explanations for epigenetic changes and an increase in the risk of non‐communicable diseases7, the association between the maternal gut microbiome and fetal epigenetic changes of diabetes‐associated genes has not been elucidated.
The umbilical cord is fetal tissue, and it can be obtained from infants non‐invasively. Several studies have reported that alternations of umbilical cord deoxyribonucleic acid (DNA) methylation are associated with some phenotypes of children8. In the present study, we investigated associations between changes in the maternal gut microbiome and differentially methylated regions of diabetes‐associated genes in umbilical cord samples.
Materials and Methods
Participants
We recruited 10 pregnant women who were participants in the Chiba Study of Mother and Children's Health, which included collection of stool, blood and umbilical cord samples; assessment of dietary records; and administration of questionnaires9. This observational study was carried out according to the guidelines of the Declaration of Helsinki, and study protocol was approved by the Biomedical Research Ethics Committee of the Graduate School of Medicine, Chiba University. Additionally, written informed consent was obtained from the participants.
Clinical data were obtained from the medical records, and food consumption data were obtained using a brief self‐administered diet history questionnaire10. The serum levels of biochemical parameters were measured using enzyme‐colorimetric automated methods (SRL Inc., Tokyo, Japan).
16S ribosomal ribonucleic acid metagenome analysis
Stool samples were collected for analysis of the gut microbiome composition at the third trimester of pregnancy. Stool samples were transferred to Chiba University CPMS Biobank and kept frozen at −80°C.
Total DNA from the stool samples was extracted, sequenced and analyzed by quantitative metagenomics at Hokkaido System Science (Sapporo, Japan). A detailed description of the metagenomic analysis has been reported previously11. In each sample, bacterial 16S ribosomal ribonucleic acid gene sequences were polymerase chain reaction‐amplified using a primer for the V3‐V4 hypervariable region of the 16S ribosomal ribonucleic acid gene with overhang adapters attached. Pair‐ended sequencing with read lengths of 300 bp was carried out using an Illumina sequencing system (Illumina, San Diego, CA, USA). Analysis was carried out using the open‐source software package Quantitative Insights Into Microbial Ecology (QIIME, qiime.org). Sequences were clustered as operational taxonomic units based on 0.97 similarity using UCLUST (drive5.com).
DNA methylation assays
Umbilical cord samples were obtained at delivery and stored at −80°C until analysis. Genomic DNA was extracted from each sample using the NucleoSpin Tissue Kit (TaKaRa Bio, Shiga, Japan). The DNA methylation profiles of the umbilical cords were determined using the Infinium HumanMethylation450 BeadChip (Illumina). The methylation levels at each cytosine‐phosphate‐guanine dinucleotide quantified with average β‐values were calculated using GenomeStudio 2011.1 (Module M Version 1.9.0; Illumina). We selected the candidate region using following criteria: (i) standard deviation of β‐values >0.05; (ii) presence of a differentially methylated regions tag, according to the data sheet provided by the manufacturer12; and (iii) location near or in diabetes‐associated genes13, 14.
Statistical analysis
Clinical baseline characteristics are presented as mean ± SD. Spearman's rank correlation coefficient analysis was carried out. All statistical analyses were carried out using spss version 22 (IBM Corp., Armonk NY, USA). Statistical significance was defined as a P‐value <0.05.
Results
Health characteristics and diets of the participants
The clinical baseline characteristics of the participants are presented in Table 1. None of the participants had gestational diabetes.
Table 1.
Characteristics of the participants at the first trimester of pregnancy
| Characteristic | ||
| Age (years) | 34.1 ± 3.0 | |
| Prepregnancy body mass index (kg/m2) | 21.2 ± 1.9 | |
| Prepregnancy body weight (kg) | 52.3 ± 5.8 | |
| Bodyweight gain during pregnancy (kg) | 9.75 ± 2.47 | |
| Food consumption | ||
| Energy intake (kJ/day) | 6532.4 ± 1854.7 | |
| Carbohydrate intake (%energy) | 57.0 ± 4.6 | |
| Fat intake (%energy) | 27.0 ± 4.7 | |
| Protein intake (%energy) | 15.1 ± 2.1 | |
| Fiber (g/10 MJ) | 16.5 ± 5.2 | |
| Biochemical parameters | Reference value | |
| Glycoalbumin (%) | 13.8 ± 0.9 | (12.4–16.3) |
| Total cholesterol (mg/dL) | 186.2 ± 18.0 | (150–219) |
| HDL‐cholesterol (mg/dL) | 76.0 ± 8.2 | (40–96) |
| Triglyceride (mg/dL) | 114.4 ± 42.8 | (50–149) |
| Folic acid (ng/mL) | 15.2 ± 11.8 | (≥4.0) |
| Vitamin B12 (pg/mL) | 280.7 ± 75.7 | (180–914) |
| Total homocysteine (nmol/mL) | 4.69 ± 0.88 | (3.7–13.5) |
Values are presented as mean ± SD. Food consumption data were obtained using a brief self‐administered diet history questionnaire. The density method was used to compute the amount of each nutrient consumed daily, as a percentage of daily energy intakes for energy‐containing nutrients or per 10 MJ of daily energy intake for non‐energy‐containing nutrient. HDL, high‐density lipoprotein.
Relative abundances of gut bacterial phyla
The four major bacterial phyla in the third trimester were Firmicutes (71.8 ± 7.8%), Actinobacteria (16.7 ± 8.0%), Bacteroidetes (7.3 ± 4.8%) and Proteobacteria (1.7 ± 2.5%).
Correlation between the microbiome composition and host parameters
According to previous reports, we selected Firmicutes to assess the correlation between the microbiome composition and host parameters15. We found no significant correlation between the proportion of Firmicutes and their anthropometric or nutritional parameters.
Umbilical cord DNA methylation of the diabetes‐associated genes
The number of genes with standard deviation of the β‐value >0.05 was 72,848 in the chip without sex chromosomes. Of these genes, 3,097 had differentially methylated regions tags. The candidates that fulfilled our criteria were the following three regions: UBE2E2 (cg13447539, cg10632094) and KCNQ1 (cg08376310). The locations of the identified regions are shown in Figure 1. We investigated the relationships of these three regions with the proportion of Firmicutes. There was a significant positive correlation between the UBE2E2 (cg13447539) β‐value and the proportion of Firmicutes, and a significant negative correlation between the KCNQ1 β‐value and the proportion of Firmicutes in the third trimester (Rs = 0.685, P = 0.040; Figure 2a and Rs = −0.661, P = 0.048; Figure 2b, respectively). We also carried out multiple regression analysis using age and body mass index. After adjustment, the UBE2E2 (cg13447539) and KCNQ1 β‐values, and the proportion of Firmicutes tended to show a marginal correlation (Rs = 0.641, P = 0.087; Rs = −0.606, P = 0.111, respectively). Additionally, there was a moderate positive correlation between the β‐value of the UBE2E2 cytosine‐phosphate‐guanine dinucleotide site (cg10632094) and the proportion of Firmicutes (Rs = 0.617, P = 0.080).
Figure 1.
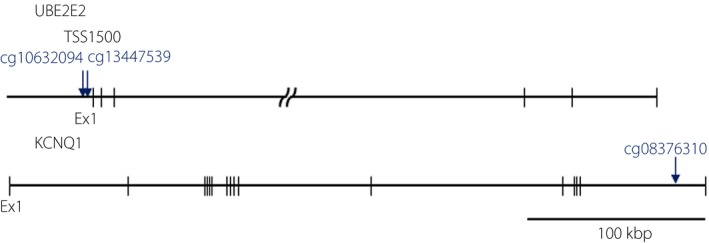
The location of the identified cytosine‐phosphate‐guanine dinucleotide sites in the UBE2E2 and KCNQ1 genes. The three regions that fulfilled our criteria were UBE2E2 (cg13447539 and cg10632094) and KCNQ1 (cg08376310). Each arrow indicates the position of each probe in the BeadChip.
Figure 2.
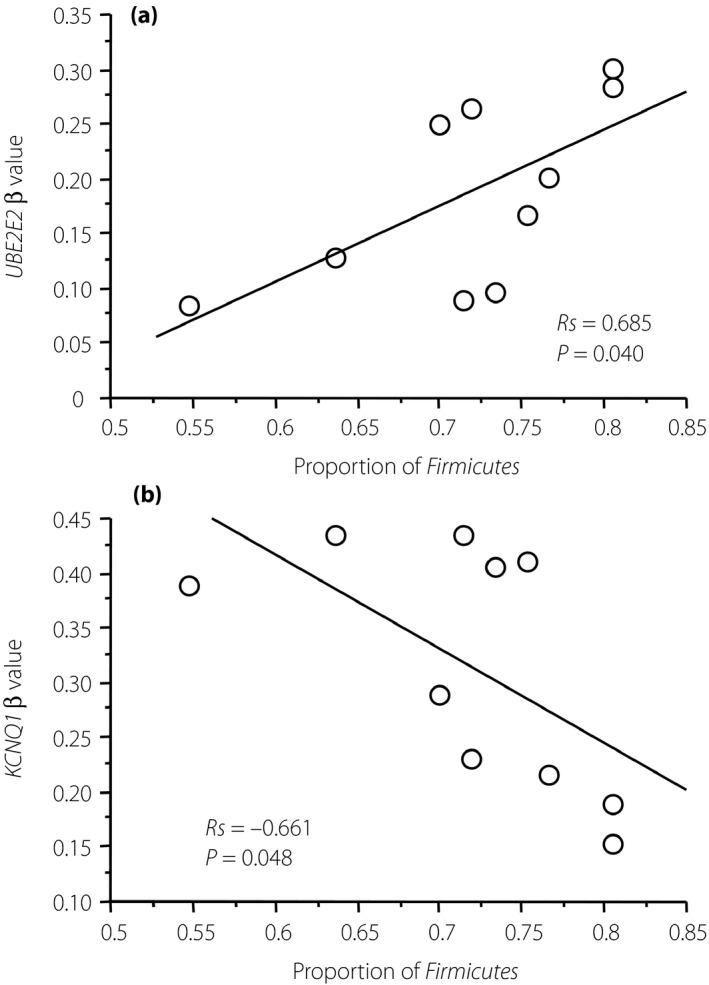
Correlation between maternal gut microbiome and deoxyribonucleic acid methylation of diabetes‐associated genes in the umbilical cord. (a) Scatter plot of the proportion of Firmicutes in the maternal gut (x‐axis) and the umbilical cord UBE2E2 (cg13447539) β‐value (y‐axis) in the third trimester among the 10 participants. (b) Scatter plot of the proportion of Firmicutes in the maternal gut (x‐axis) and the umbilical cord KCNQ1 (cg08376310) β‐value (y‐axis) in the third trimester among the 10 participants. The methylation levels at each cytosine‐phosphate‐guanine dinucleotide that was quantified with average β‐values, where 1 corresponds to complete methylation and 0 to no methylation, are shown.
Discussion
Alternations in the levels of DNA methylation have been shown to be present in numerous candidate genes for type 2 diabetes16. Furthermore, epigenetic variations were shown to be present before disease development and were found to be risk factors2. UBE2E2 encodes the ubiquitin‐conjugating enzyme E2‐E2, and has been reported to play an important role in insulin secretion. Single‐nucleotide polymorphisms in UBE2E2 were shown to be associated with type 2 diabetes in individuals of East Asian descent14. Similarly, the association of KCNQ1 with type 2 diabetes was shown in non‐European populations17. Furthermore, epigenetic regulation of the KCNQ1 gene has been reported to contribute to the onset of type 2 diabetes in mice18 and humans19. We found that the UBE2E2 and KCNQ1 methylation rates in umbilical cord samples were associated with the maternal gut microbiome composition. DNA methylation is known to regulate gene expression in a tissue‐specific manner20. Further studies are required to investigate whether the methylation rates in umbilical cord samples reflect the methylation rates in other tissues.
Changes in the gut microbiome in late pregnancy have been shown to be associated with known changes in insulin resistance and inflammation6. However little is known about the impact of this dysbiosis on maternal and fetal metabolism4. It has been reported that Firmicutes‐dominant microbiota is associated with the development of obesity and metabolic syndrome21. Furthermore, Firmicutes might contribute to epigenetic changes by producing folate and butylate15. The present study provides possibilities for the interactions between the maternal gut microbiome and the epigenetic changes of diabetes‐associated genes in fetal tissue.
The results of the present study are limited by the small sample size. Further studies with a larger sample size and long‐term follow up are required to confirm the present findings.
In conclusion, a link is assumed to be present between changes in the methylation of type 2 diabetes‐associated genes in fetuses and the microbiota components in mothers during pregnancy.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
The authors acknowledge the Chiba Study of Mother and Children's Health participants and staff for their dedication. This study was supported by JSPS KAKENHI grants (grant numbers 20241016 and 15H06087), and grants from the Chiba Foundation for Health Promotion & Disease Prevention. We thank Editage (www.editage.jp) for English language editing.
J Diabetes Investig 2017; 8: 550–553
References
- 1. Kwak SH, Park KS. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp Mol Med 2016; 48: e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ronn T, Ling C. DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics. 2015; 7: 451–460. [DOI] [PubMed] [Google Scholar]
- 3. Lee HS. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients. 2015; 7: 9492–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Subramanian S, Blanton LV, Frese SA, et al Cultivating healthy growth and nutrition through the gut microbiota. Cell 2015; 161: 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ley RE, Backhed F, Turnbaugh P, et al Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005; 102: 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koren O, Goodrich JK, Cullender TC, et al Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012; 150: 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pfefferle PI, Prescott SL, Kopp M. Microbial influence on tolerance and opportunities for intervention with prebiotics/probiotics and bacterial lysates. J Allergy Clin Immunol. 2013; 131: 1453–1463, quiz 64. [DOI] [PubMed] [Google Scholar]
- 8. Godfrey KM, Sheppard A, Gluckman PD, et al Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 2011; 60: 1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakurai K, Miyaso H, Eguchi A, et al Chiba study of Mother and Children's Health (C‐MACH): cohort study with omics analyses. BMJ Open. 2016; 6: e010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi S, Murakami K, Sasaki S, et al Comparison of relative validity of food group intakes estimated by comprehensive and brief‐type self‐administered diet history questionnaires against 16 d dietary records in Japanese adults. Public Health Nutr. 2011; 14: 1200–1211. [DOI] [PubMed] [Google Scholar]
- 11. Takahashi S, Tomita J, Nishioka K, et al Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next‐generation sequencing. PLoS One 2014; 9: e105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Availabe from: http://support.illumina.com/downloads/humanmethylation450_15017482_v1-2_product_files.html Accessed July 17, 2015.
- 13. Miyake K, Yang W, Hara K, et al Construction of a prediction model for type 2 diabetes mellitus in the Japanese population based on 11 genes with strong evidence of the association. J Hum Genet 2009; 54: 236–241. [DOI] [PubMed] [Google Scholar]
- 14. Yamauchi T, Hara K, Maeda S, et al A genome‐wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A‐C2CD4B. Nat Genet 2010; 42: 864–868. [DOI] [PubMed] [Google Scholar]
- 15. Kumar H, Lund R, Laiho A, et al Gut microbiota as an epigenetic regulator: pilot study based on whole‐genome methylation analysis. MBio 2014; 5: e02113–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dayeh T, Volkov P, Salo S, et al Genome‐wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non‐diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 2014; 10: e1004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yasuda K, Miyake K, Horikawa Y, et al Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008; 40: 1092–1097. [DOI] [PubMed] [Google Scholar]
- 18. Asahara S, Etoh H, Inoue H, et al Paternal allelic mutation at the Kcnq1 locus reduces pancreatic beta‐cell mass by epigenetic modification of Cdkn1c. Proc Natl Acad Sci USA. 2015; 112: 8332–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kong A, Steinthorsdottir V, Masson G, et al Parental origin of sequence variants associated with complex diseases. Nature 2009; 462: 868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toperoff G, Aran D, Kark JD, et al Genome‐wide survey reveals predisposing diabetes type 2‐related DNA methylation variations in human peripheral blood. Hum Mol Genet 2012; 21: 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gohir W, Ratcliffe EM, Sloboda DM. Of the bugs that shape us: maternal obesity, the gut microbiome, and long‐term disease risk. Pediatr Res 2015; 77: 196–204. [DOI] [PubMed] [Google Scholar]