

Abstract
Aims/Introduction
Peroxisome proliferator‐activated receptor‐α (PPARα) is a therapeutic target for hyperlipidemia. K‐877 is a new selective PPARα modulator (SPPARMα) that activates PPARα transcriptional activity. The aim of the present study was to assess the effects of K‐877 on lipid metabolism in vitro and in vivo compared with those of classical PPARα agonists.
Materials and Methods
To compare the effects of K‐877 on PPARα transcriptional activity with those of the classical PPARα agonists Wy14643 (Wy) and fenofibrate (Feno), the cell‐based PPARα transactivation luciferase assay was carried out. WT and Ppara −/− mice were fed with a moderate‐fat (MF) diet for 6 days, and methionine–choline‐deficient (MCD) diet for 4 weeks containing Feno or K‐877.
Results
In luciferase assays, K‐877 activated PPARα transcriptional activity more efficiently than the classical PPARα agonists Feno and Wy. After being fed MF diet containing 0.001% K‐877 or 0.2% Feno for 6 days, mice in the K‐877 group showed significant increases in the expression of Ppara and its target genes, leading to marked reductions in plasma triglyceride levels compared with those observed in Feno‐treated animals. These K‐877 effects were blunted in Ppara −/− mice, confirming that K‐877 activates PPARα. In further experiments, K‐877 (0.00025%) and Feno (0.1%) equally improved the pathology of MCD diet‐induced non‐alcoholic fatty liver disease, with increased expression of hepatic fatty acid oxidation genes.
Conclusions
The present data show that K‐877 is an attractive PPARα‐modulating drug and can efficiently reduce plasma triglyceride levels, thereby alleviating the dysregulation of lipid metabolism.
Keywords: Lipid metabolism, Peroxisome proliferator‐activated receptor‐α, Selective peroxisome proliferator‐activated receptor‐α modulator
Introduction
Impaired nutrient homeostasis is a common characteristic of metabolic disorders, such as obesity, diabetes, cardiovascular diseases and fatty liver disease. Nutrient homeostasis is tightly regulated through the balance between energy‐producing pathways, such as ketogenesis, gluconeogenesis and lipid synthesis, and energy utilization pathways, such as lipid oxidation. Because of the emerging epidemic of obesity and diabetes, the factors determining progression of non‐alcoholic fatty liver disease (NAFLD) present a major clinical challenge. In particular, fatty liver can progress to non‐alcoholic steatohepatitis (NASH) after overwhelming of the adaptive mechanisms that mediate lipid partitioning and metabolism, and protect hepatocytes from lipotoxicity of excess fatty acids (FAs) and other lipids, resulting in inflammation and fibrosis.
Peroxisome proliferative‐activated receptors (PPARs) are members of the nuclear receptor superfamily, and include PPARα, PPARβ/δ and PPARγ. On ligand binding, PPARs form heterodimers with the retinoid X receptor, and interact with PPAR response elements to regulate target gene expression. PPARα is most prominently expressed in the liver, and is activated by hypolipidemic fibrate‐class drugs (fibrates). PPARα controls lipid flux in the liver by modulating FA transport and β‐oxidation, and improves plasma lipid profiles by decreasing triglyceride (TG) levels and increasing high‐density lipoprotein cholesterol levels. In addition, PPARα activation inhibits inflammatory genes that are induced by nuclear factor‐κB, and decreases the expression of acute‐phase response genes. Accordingly, PPARα deficiency increases susceptibility to NAFLD, NASH, hepatic inflammation and acute phase responses1. Fibrates, such as gemfibrozil, bezafibrate, and fenofibrate (Feno), decrease plasma TG levels, and increase high‐density lipoprotein cholesterol levels in patients with hyperlipidemia and type 2 diabetes, and can prevent coronary heart disease and stroke2, 3, 4, 5, 6. However, these drugs are weak agonists of PPARα, have poor substrate selectivity and require high clinical doses. Therefore, a potent and selective PPARα agonist is required for patients with metabolic syndrome. K‐877 is a novel selective PPARα modulator (SPPARMα) that enhances PPARα activity7; it also elicits higher PPARα activation than other fibrates, with lower EC50 values and higher PPAR subtype selectivity8.
In the present study, we compared the effects of K‐877 on lipid metabolism and hepatic gene expression related to NASH/NAFLD with the classical PPARα agonists, Feno and Wy14643 (Wy), in vitro and in vivo.
Materials and Methods
Reagents
K‐877 was kindly provided by Kowa Co. Ltd. Feno and Wy were purchased from Sigma‐Aldrich (St. Louis, Missouri, USA).
Animals
Eight‐week‐old male C57BL/6J (wild‐type; WT) mice were obtained from CLEA Japan (Tokyo, Japan). B6;129S4‐Ppara tm1Gonz/J (Ppara −/−) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). Methionine–choline‐deficient (MCD)‐diet analyses were carried out on 8‐week‐old male mice that were fed for 4 weeks and killed without fasting. All animal husbandry procedures and experiments were compliant with the University of Tsukuba's Regulations for Animal Experiments, and were approved by the Animal Experiment Committee at the University of Tsukuba.
Histological analysis
Harvested livers were fixed, embedded in paraffin, sectioned and stained with hematoxylin–eosin.
Plasmids
The expression vector for the Gal4‐PPARα (pM Gal4‐PPARα) fusion protein was generated by inserting a human PPARα fragment (166–468 aa) downstream of Gal4 in the pM vector (Clontech, Palo Alto, California, USA). The GAL4 UAS–LUC vector contains eight copies of the UAS Gal4‐binding site9.
Cell culture
Mouse AML12.2 hepatoma cells were maintained in Dulbecco's modified Eagle's medium/Ham's F12 media supplemented with ITS Liquid Media Supplement (Sigma‐Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin and 10% fetal bovine serum. Cells were incubated with Feno (50 μmol/L) or K‐877 (5 and 50 μmol/L) for 48 h.
Transfections and Luc assays
HepG2 cells were grown at 37°C in an atmosphere of 5% CO2 in Dulbecco's modified Eagle's medium containing 25 mmol/L glucose, 100 U/mL penicillin, 100 μg/mL streptomycin and 10% fetal bovine serum. Transfection studies were carried out in cells on 24‐well plates. Cells were transfected with pM Gal4‐PPARα DBD and GAL4 UAS luciferase vectors9, and a pRL‐SV40 plasmid as a reference (Promega, Madison, Wisconsin, USA) using X‐tremeGENE 9 (Roche, Basel, Switzerland). After 24 h of transfection, Wy (50 μmol/L), Feno (30 μmol/L) or K‐877 (50 nmol/L) were added to the medium. After additional 24‐h incubation, firefly luciferase activity was measured and normalized to that of Renilla.
Metabolic measurements
Plasma levels of TG, non‐esterified FA (NEFA), total cholesterol (TC), aspartate aminotransferase, alanine aminotransferase, and liver TG and TC levels were measured as described previously10.
Analysis of gene expression
Total ribonucleic acid from cells and tissues was prepared using a Trizol reagent (Invitrogen, Carlsbad, California, USA). Before real‐time polymerase chain reaction analyses, total ribonucleic acid was reversed transcribed into complementary deoxyribonucleic acid using a reverse transcriptase according to the manufacturer's instructions (Invitrogen). Real‐time polymerase chain reaction was carried out using an ABI Prism 7300 system (ABI, Carlsbad, California, USA) with TaqMan probes (Invitrogen) and a SYBR Green Master Mix (Roche)11. Primer sequences are available on request.
Statistical analysis
Comparisons of treatment groups were made using Tukey–Kramer post‐hoc tests, and differences were considered significant when P < 0.05. All data are expressed as mean ± standard error of the mean.
Results
K‐877 effectively activates PPARα transcriptional activity
To compare the effects of K‐877 on PPARα transcriptional activity with those of the classical PPARα agonists Wy and Feno, cell‐based transactivation assays were carried out using a Gal4‐luciferase assay system. After co‐transfecting human HepG2 hepatoma cells with pM‐GAL4 PPARα ligand‐binding domain and GAL4 UAS reporter vectors, cells were treated with K‐877 for 24 h. Although K‐877, Wy and Feno induced PPARα luciferase activation in a dose‐dependent manner, the dose–response curve of K‐877 was shifted to the left side compared with those of Wy14643 and Feno (Figure 1a). K‐877 is a highly potent agonist of PPARα transcriptional activity, with half‐maximal effective concentrations (EC50) of 0.49 nmol/L. The PPARα agonists, Wy (50 μmol/L), Feno (30 μmol/L) or K‐877 (50 nmol/L) were treated for 24 h. PPARα luciferase activation by K‐877 was comparable with that by Wy or Feno (Figure 1b). Considering the relative concentrations of these drugs, results show that K‐877 activates the PPARα transcription activity more effectively than the other agents. To determine the effects of K‐877 on fibroblast growth factor 21 (Fgf21) expression, a typical target gene of PPARα, in vitro, mouse AML12.2 hepatoma cells were incubated with the PPARα agonists, Feno (50 μmol/L) or K‐877 (5 and 50 μmol/L) for 48 h based on a previous report12. Both doses of K‐877 significantly increased Fgf21 expression, but the increase in expression by Feno was slight (Figure 1c). Thus, K‐877 can efficiently activate Fgf21 expression.
Figure 1.
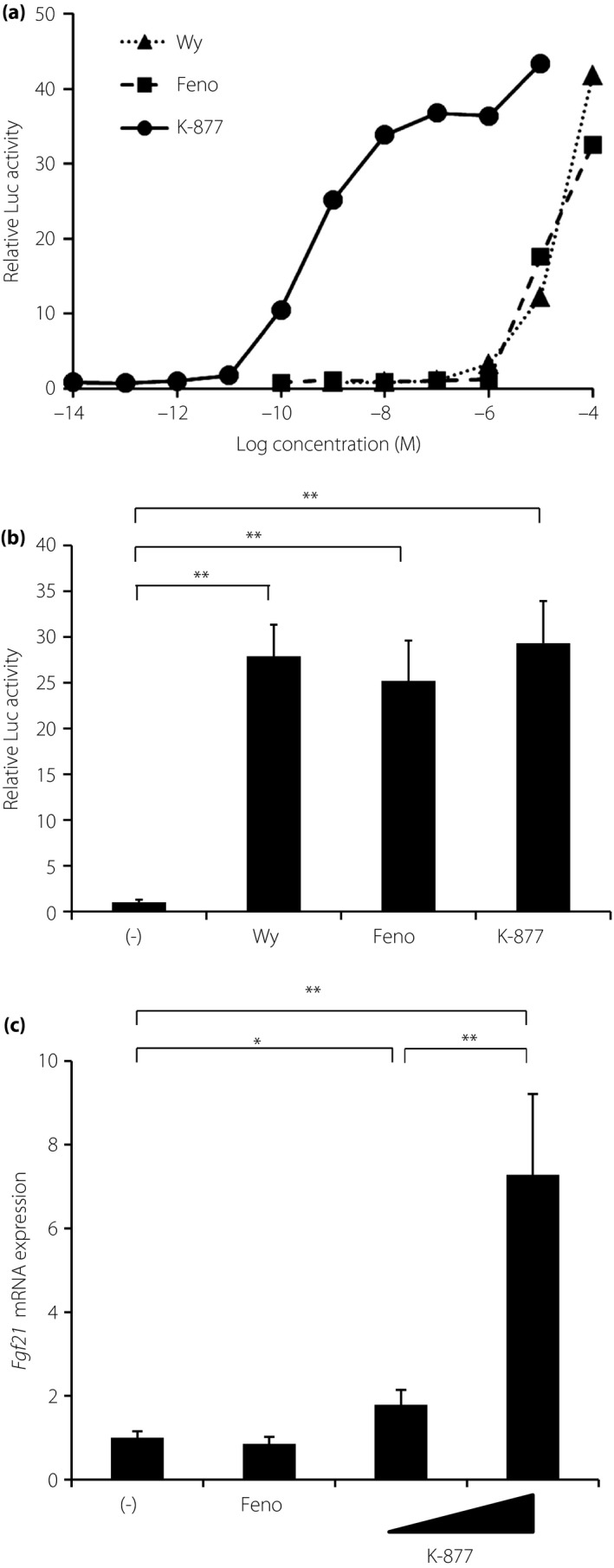
K‐877 activates the Peroxisome proliferator‐activated receptor‐α (PPARα) transcriptional activity. (a,b) HepG2 cells were co‐transfected with GAL4 UAS‐LUC and pM GAL4‐hPPARα ligand‐binding domain vectors, and with a pRL‐SV40 plasmid as a reference in 24‐well plates for 24 h. Cells were then treated with Wy, Feno or K‐877 for 48 h, and were harvested at 48 h after transfection. Luciferase activity was measured and normalized to that of Renilla luciferase activity. (a) Dose–response curves of PPARα transactivation. (b) Cells were then treated with Wy14643 (Wy; 50 μmol/L), fenofibrate (Feno; 30 μmol/L) or K‐877 (50 nmol/L); n = 8 per group. (c) AML12.2 cells were treated with Feno (50 μmol/L) or K‐877 (5 and 50 μmol/L) for 48 h. Fgf21 expression was determined by quantitative polymerase chain reaction; n = 9 per group, *P < 0.05 and **P < 0.01.
K‐877 decreased plasma lipid levels in WT mice
Based on a previous report7, 8‐week‐old male WT mice were fed a moderate‐fat (MF) diet containing 0.2% (w/w) Feno or 0.001% (w/w) K‐877 for 6 days. Although the dose of K‐877 was 200‐fold lower than that of Feno, compared with no treatment, K‐877 and Feno significantly reduced plasma TG levels, and tended to reduce TC and NEFA levels (Figure 2a), and the effects of both agonists were blunted in Ppara −/− mice (Figure 2a). These data show that considering the concentrations of agonists, K‐877 elicits greater PPARα‐mediated lipid‐lowering effects than Feno. Accordingly, hepatic gene expression of Ppara and its target genes, such as those encoding cyclic adenosine monophosphate responsive element binding protein 3‐like 3 (Creb3l3), Fgf21, acyl‐CoA oxidase 1, palmitoyl (Acox1) and carnitine palmitoyltransferase 1a, liver (Cpt1a), were significantly increased by both PPARα agonists (Figure 2b). These changes in gene expression were similar in the presence of K‐877 and Feno despite dose differences, showing that K‐877 has more powerful effects on PPARα activation than Feno. In addition, the effects of both agonists were abolished in Ppara −/− mice, confirming that these agonists target the PPARα pathway.
Figure 2.
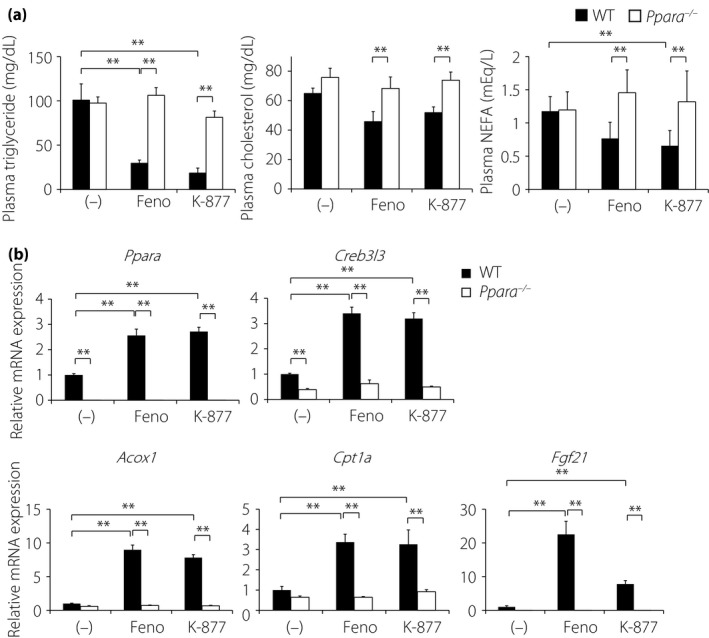
K‐877 reduces plasma lipid levels and increases the expression of hepatic fatty acid oxidation genes in wild type, but not in Ppara −/− mice. Eight‐week old male wild‐type (WT) and Ppara −/− mice were administrated with fenofibrate (Feno; 0.2%) or K‐877 (0.001%) for 6 days. (a) Plasma triglyceride, total cholesterol and non‐esterified fatty acid (NEFA) levels, and (b) hepatic gene expression in WT and Ppara −/− mice; n = 9–13 per group; *P < 0.05 and **P < 0.01. mRNA, messenger ribonucleic acid.
K‐877 suppresses MCD‐induced liver injury in normal mice, but not in Ppara −/− mice
To compare the effects of PPARα agonists on the progression of NAFLD, WT and Ppara −/− mice were fed an MCD diet containing 0.1% Feno or 0.00025% K‐877 for 4 weeks, and their optimum doses were determined according to previously reported methods7, 12. This diet has been used extensively to produce diet‐induced animal models of NASH that show similar histology to that of human NASH1. Histological analyses of hematoxylin–eosin‐stained liver sections from WT mice showed that the MCD diet led to slight lipid accumulation in hepatocytes (Figure 3a). The addition of Feno and K‐877 suppressed MCD‐induced lipid accumulation (Figure 3a). Furthermore, sections from MCD‐fed Ppara −/− mice showed greater lipid accumulation and macrophage invasion than those from MCD‐fed WT mice, but the addition of Feno and K‐877 could not improve them (Figure 3a). Hepatic TG and TC contents were significantly increased in MCD diet‐fed mice than in MF diet‐fed mice (Figure 3b). The addition of K‐877 and Feno into MCD diets tended to reduce liver TG and TC levels, and Ppara −/− mice showed more severe liver lipid accumulation than WT mice (Figure 3b). However, PPARα agonists did not improve these phenotypes significantly (Figure 3b). In further analyses, increased plasma alanine aminotransferase and aspartate aminotransferase levels were observed in MCD diet‐fed WT mice, compared with those in MF diet‐fed WT mice (Figure 3c). The administration of K‐877 or Feno tended to decrease MCD diet‐induced alanine aminotransferase and aspartate aminotransferase levels. However, both agonists failed to suppress these increases in Ppara −/− mice (Figure 3c). Taken together, these data suggest that K‐877 and Feno ameliorate MCD diet‐induced fatty liver progression.
Figure 3.
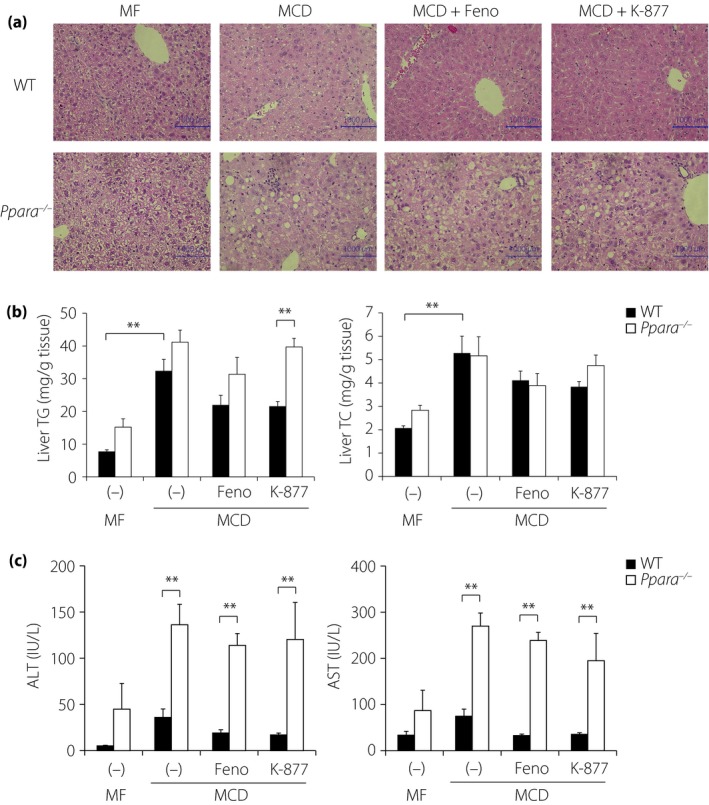
K‐877 suppresses methionine–choline‐deficient (MCD) diet‐induced non‐alcoholic fatty liver disease in wild‐type (WT), but not in Ppara −/− mice. Eight‐week‐old male WT and Ppara −/− mice were fed moderate‐fat (MF) or MCD diets containing fenofibrate (Feno; 0.1%) or K‐877 (0.00025%) for 4 weeks. Hematoxylin–eosin staining in the (a) liver, (b) hepatic lipid contents, and (c) plasma aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were determined in WT and Ppara −/− mice; n = 5–10 per group, *P < 0.05 and **P < 0.01. TC, total cholesterol; TG, triglyceride.
K‐877 activates PPARα target gene expression and reduces Xbp1s expression in the liver of MCD‐fed mice
Hepatic gene expression was determined in WT and Ppara −/− mice after feeding on a MCD diet containing PPARα agonists for 4 weeks. Consistent with the expression levels of Ppara and its target genes Creb3l3 in normal mice, Acox1 and Cpt1a were significantly increased to similar levels in the presence of PPARα agonists (Figure 4). However, Fgf21 expression was decreased in the presence of PPARα agonists. Furthermore, no apparent differences in inflammatory and macrophage hepatic gene expression were observed between MCD diet‐fed WT mouse groups. The administration of PPARα agonists on Ppara −/− mice showed increased Cd68 expression, supporting the increase of macrophages including Kupffer cells. However, the MCD diet induced endoplasmic reticulum (ER) stress markers, such as X‐box binding protein 1s (Xbp1s) in WT mice. In addition, the effects of K‐877 on Xbp1s expression were abolished in Ppara −/− mice. In subsequent experiments, K‐877 suppressed MCD diet‐induced Xbp1s expression more efficiently than Feno, thereby ameliorating ER stress in MCD diet‐fed WT mice. In contrast, neither PPARα agonist suppressed ER stress‐related gene expression in Ppara −/− mice. These data show that K‐877 increases PPARα target genes that are related to FA oxidation and reduces Xbp1s expression, leading to decreased liver injury.
Figure 4.
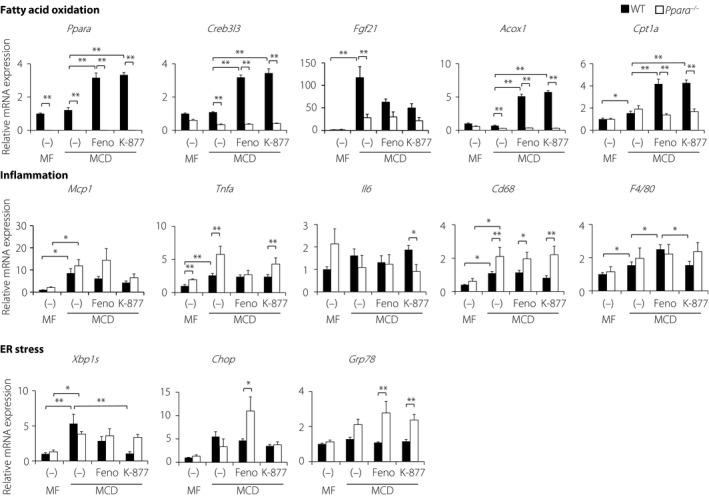
Hepatic gene expression in wild‐type (WT) and Ppara −/− mice fed the methionine–choline‐deficient (MCD) diet with K‐877 or fenofibrate (Feno) for 4 weeks. Eight‐week‐old male WT and Ppara −/− mice were fed moderate‐fat (MF) or MCD diets containing Feno (0.1%) or K‐877 (0.00025%) for 4 weeks. Hepatic gene expression profiles of WT and Ppara −/− mice; n = 5–10 per group; *P < 0.05 and **P < 0.01. mRNA, messenger ribonucleic acid.
Discussion
In the present study, we showed that K‐877 specifically and efficiently activates PPARα transactivation activity in vitro and in vivo. Furthermore, K‐877 had greater lipid‐lowering effects than the classical PPARα agonists in mice, and significantly induced PPARα target genes. These effects reduced MCD‐induced liver injury by increasing PPARα target gene expression and decreasing ER stress in the liver. Therefore, K‐877 might be an effective drug for hyperlipidemia.
Fibrates are widely used to ameliorate the macro‐ and microvascular risks associated with metabolic syndrome. However, these agents are weak PPARα agonists, and have limited efficacy due to dose‐related adverse events. To address this problem, a new generation of PPARα‐specific agonists, known as SPPARMαs, has been developed to maximize receptor‐mediated effects and diminish side effects. In luciferase analyses, we confirmed that K‐877 activates PPARα at 1,000‐fold lower doses than Feno and Wy. K‐877 increases Fgf21 expression, one of the typical target genes for PPARα in AML12.2 cells. FGF21 is known to ameliorate obesity, diabetes and hyperlipidemia by inducing lipid catabolism13, 14. The in vivo studies showed that K‐877 and Feno induced the PPARα target genes, Fgf21, Acox1 and Cpt1a, in the liver of MF‐fed WT mice, suggesting that these compounds activate hepatic FA oxidation. These effects were similar in the presence of low doses of K‐877, or high doses of Feno, suggesting superiority of K‐877 as a clinical agent. As a result, the administration of K‐877 and Feno in MF‐fed WT mice reduced plasma lipids, including TG, TC and NEFA. Previous reports have shown that some PPARα agonists prevent the progression of NAFLD15, 16. Accordingly, K‐877 efficiently increased the PPARα transcriptional activity and subsequently transactivated FA oxidation genes, including Acox1 and Cpt1a, resulting in reduced hepatic lipid accumulation in MCD‐fed mice. However, Fgf21 expression was decreased in MCD‐fed WT mice in the presence of PPARα agonist compared with that in MCD‐fed WT mice without agonists. Fgf21 expression was induced in both WT and Ppara −/− mice in response to MCD, suggesting that a PPARα independent mechanism underlies MCD‐induced Fgf21 expression.
ER stress is associated with an accumulation of misfolded and unfolded proteins in the ER lumen. ER plays an essential role in controlling lipid metabolism. XBP1s induces the expression of multiple inflammatory cytokines17, suggesting important roles of ER stress in the pathology of NAFLD. K‐877 reduced hepatic Xbp1s expression, indicating potential roles in the management of ER stress. Furthermore, reduced lipid accumulation in the presence of K‐877 might lead to decreases in the ER stress‐related gene expression. Finally, the effects of K‐877 in Ppara −/− mice were blunted, confirming that this agonist specifically targets PPARα.
Selective activation of PPARα by K‐877 was associated with beneficial changes in liver disease markers, suggesting the potential of this novel agent in the treatment of NASH/NAFLD might relate to PPARα pathway activation and reduced ER stress. However, the mechanisms of K‐877‐mediated PPARα activation remain unknown, warranting further studies to characterize the specificity of K‐877 for PPARα activation, recruitment of cofactors, and downstream gene expression.
Disclosure
The authors declare no conflict of interest.
Acknowledgment
This work was supported by JSPS KAKENHI, grant number 16H03253. This manuscript was edited by Enago English language editors.
J Diabetes Investig 2017; 8: 446–452
These authors contributed equally to this work.
References
- 1. Ip E, Farrell GC, Robertson G, et al Central role of PPARalpha‐dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003; 38: 123–132. [DOI] [PubMed] [Google Scholar]
- 2. Diabetes Atherosclerosis Intervention Study Investigators . Effect of fenofibrate on progression of coronary‐artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet 2001; 357: 905–910. [PubMed] [Google Scholar]
- 3. Bloomfield Rubins H, Davenport J, Babikian V, et al Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: the Veterans Affairs HDL Intervention Trial (VA‐HIT). Circulation 2001; 103: 2828–2833. [DOI] [PubMed] [Google Scholar]
- 4. Rubins HB, Robins SJ, Collins D, et al Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high‐density lipoprotein cholesterol. Veterans affairs high‐density lipoprotein cholesterol intervention trial study group. N Engl J Med 1999; 341: 410–418. [DOI] [PubMed] [Google Scholar]
- 5. Tanne D, Koren‐Morag N, Graff E, et al Blood lipids and first‐ever ischemic stroke/transient ischemic attack in the Bezafibrate Infarction Prevention (BIP) Registry: high triglycerides constitute an independent risk factor. Circulation 2001; 104: 2892–2897. [DOI] [PubMed] [Google Scholar]
- 6. The Bezafibrate Infarction Prevention(BIP) Study . Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000; 102: 21–27. [DOI] [PubMed] [Google Scholar]
- 7. Raza‐Iqbal S, Tanaka T, Anai M, et al Transcriptome Analysis of K‐877 (a Novel Selective PPARalpha Modulator (SPPARMalpha))‐Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J Atheroscler Thromb 2015; 22: 754–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fruchart JC. Selective peroxisome proliferator‐activated receptor alpha modulators (SPPARMalpha): the next generation of peroxisome proliferator‐activated receptor alpha‐agonists. Cardiovasc Diabetol 2013; 12: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamamoto T, Shimano H, Nakagawa Y, et al SREBP‐1 interacts with hepatocyte nuclear factor‐4 alpha and interferes with PGC‐1 recruitment to suppress hepatic gluconeogenic genes. J Biol Chem 2004; 279: 12027–12035. [DOI] [PubMed] [Google Scholar]
- 10. Nakagawa Y, Shimano H, Yoshikawa T, et al TFE3 transcriptionally activates hepatic IRS‐2, participates in insulin signaling and ameliorates diabetes. Nat Med 2006; 12: 107–113. [DOI] [PubMed] [Google Scholar]
- 11. Fujimoto Y, Nakagawa Y, Satoh A, et al TFE3 controls lipid metabolism in adipose tissue of male mice by suppressing lipolysis and thermogenesis. Endocrinology 2013; 154: 3577–3588. [DOI] [PubMed] [Google Scholar]
- 12. Hennuyer N, Duplan I, Paquet C, et al The novel selective PPARalpha modulator (SPPARMalpha) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016; 249: 200–208. [DOI] [PubMed] [Google Scholar]
- 13. Badman MK, Pissios P, Kennedy AR, et al Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 2007; 5: 426–437. [DOI] [PubMed] [Google Scholar]
- 14. Inagaki T, Dutchak P, Zhao G, et al Endocrine regulation of the fasting response by PPARalpha‐mediated induction of fibroblast growth factor 21. Cell Metab 2007; 5: 415–425. [DOI] [PubMed] [Google Scholar]
- 15. Jha P, Claudel T, Baghdasaryan A, et al Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology 2014; 59: 858–869. [DOI] [PubMed] [Google Scholar]
- 16. Chanda D, Lee CH, Kim YH, et al Fenofibrate differentially regulates plasminogen activator inhibitor‐1 gene expression via adenosine monophosphate‐activated protein kinase‐dependent induction of orphan nuclear receptor small heterodimer partner. Hepatology 2009; 50: 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinon F, Chen X, Lee AH, et al TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 2010; 11: 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]