

Abstract
We describe in this Minireview the synthesis, properties, and applications of artificial genetic sets built from base pairs that are larger than the natural Watson—Crick architecture. Such designed systems are being explored by several research groups to investigate basic chemical questions regarding the functions of the genetic information storage systems and thus of the origin and evolution of life. For example, is the terrestrial DNA structure the only viable one, or can other architectures function as well? Working outside the constraints of purine—pyrimidine geometry provides more chemical flexibility in design, and the added size confers useful properties such as high binding affinity and helix stability as well as fluorescence. These features are useful for the investigation of fundamental biochemical questions as well as in the development of new biotechnological, biomedical, and nanostructural tools and methods.
Keywords: non-natural nucleobases, nucleic acids, replication, synthetic biology, Watson—Crick base pairing
1. Introduction
The chemical synthesis of complex modified biomolecules allows for the development of tools for the study of fundamental questions regarding the evolution and function of living systems. Thus, it can be seen as an important part of the discipline of synthetic biology, an interdisciplinary field that brings together chemistry, biology, and biotechnology to design and engineer biological systems.[1] As a consequence of their fundamental significance in all living organism, nucleic acids and their modifications have become the focus of many research groups. In addition to the naturally occurring noncanonical nucleobases,[2] many examples of non-natural bases and base pairs have been described and reviewed,[3–6] including nucleobase shape mimics,[4] hydrophobic pairs,[5] metal-mediated base pairs,[6] and base pairs with altered hydrogen-bonding motifs.[7]
A special role in this context is taken by the concept of size-expanded nucleobases and their incorporation into novel genetic sets (for earlier reviews see Refs. [3,8]). New genetic sets are able to test the basic chemical questions of life: is the Watson—Crick DNA architecture[9] the only viable one, or can other structures function as well? Working outside the constraints of purine—pyrimidine geometry gives more flexibility in molecular designs to address these questions. As will be discussed in detail below, the increased dimensions of size-expanded nucleobases confer useful properties, such as high binding affinity, high helix stability, and fluorescence. These properties are interesting for directly addressing bioanalytical questions and they are also potentially useful for the development of new biotechnological tools. Several research groups have addressed this topic and worked out different concepts for size-expanded genetic sets containing nucleobases and pairs larger than the natural ones. These studies are outlined and discussed in this Minireview.
2. Synthesis, Properties, and Applications of Size-Expanded Nucleobases in DNA
2.1. Benzo-Homologation: xDNA and yDNA
A prominent strategy for expanding the size of a nucleobase is benzo-homologation. By addition of a benzene ring, a bicyclic purine is converted into a three-ring analogue, and a monocyclic pyrimidine into a bicyclic structure. This concept was first introduced by Leonard et al. nearly four decades ago when they prepared a “stretched out” analogue of adenine as well as the corresponding ribonucleoside.[10] The latter was used to probe ATP-dependent enzymes,[11] but was not studied in DNA (indeed, Leonard et al. considered it too large for DNA).
Inspired by this work, our research group has designed and studied a complete set of benzo-homologated (“xDNA”) deoxyribonucleosides.[12] This benzo-homologation leads to a 2.4 Å expansion of the nucleobases. Figure 1A shows the structures of the size-expanded nucleosides dxA, dxT, dxG, and dxC (1–4) in comparison to the natural nucleosides dA, dT, dG, and dC. Figure 1B shows space-filling models of the size-expanded nucleobases with calculated electrostatic potentials mapped on the surfaces. It is important that the four structures have analogous lengths and angular shifts in the hydrogen-bonding faces of the bases, thus enabling the formation of a regular helical conformation when paired. Furthermore, given their similar electrostatic charges, they are expected to retain the native hydrogen-bonding potential and thus pair normally with complementary nucleobases (see below).[12]
Figure 1.
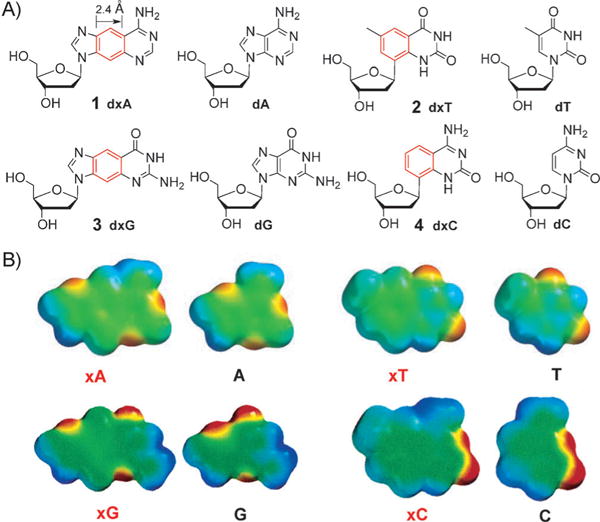
A) The size-expanded xDNA nucleosides 1–4 in comparison to the natural nucleosides. B) Space-filling models of size-expanded nucleobases with calculated electrostatic potentials mapped on the surface (red denotes negative potential and blue, positive). Adapted from Ref. [12] with permission. TBDPS =tert-butyldimethylsilyl.
Synthetic procedures for the preparation of 1, 2, 3, and 4 as well as for their corresponding phosphoramidite derivatives for automated synthesis have been developed and will only be described in brief here. For the synthesis of dxA (1), modified nucleobase 5 is coupled to chlorosugar 6, followed by conversion into nucleoside 1, which is converted into the corresponding phosphoramidite (1-PA) ready for solid-phase oligonucleotide synthesis (Scheme 1 A). For the synthesis of C-glycoside dxT (2), iodinated heterocycle 7 is coupled through Heck reactions[13] to compound 8, followed by deprotection to 2 and synthesis of 2-PA. Furthermore, 4-PA can be synthesized from 2 (Scheme 1B). The preparation of dxG (3) and 3-PA is accomplished with size-expanded nucleobase 9[10] (synthesized in 5 steps), which is coupled to ribose derivative 10 through Vorbrüggen glycosylation[14] (followed by reductive removal of the 2-OH group; Scheme 1C).[12]
Scheme 1.
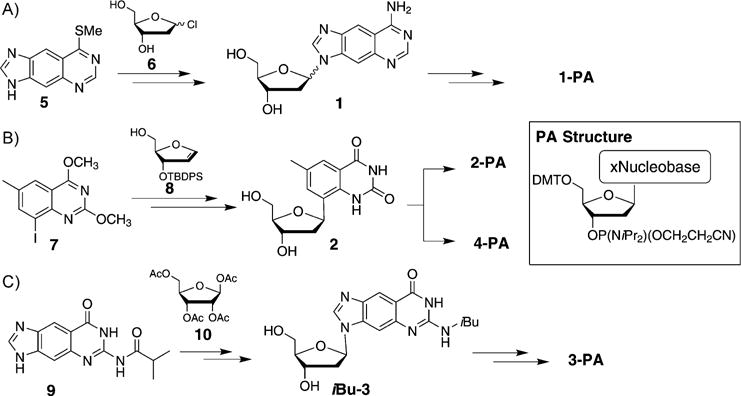
Syntheses of the size-expanded nucleosides and phosphoramidites for solid-phase DNA synthesis.[12]
The xDNA helix design involves pairing of a benzo-homologated base with a complementary normally sized base in every pair, thereby yielding a fully expanded duplex composed of eight genetic letters rather than the standard four. To study this arrangement, the xDNA phosphoramidites have been incorporated into a wide variety of different oligonucleotides in lengths up to 20 mer and containing either expanded monomers alone or both xDNA and DNA monomers (Figure 2A). A wide variety of thermodynamic and photophysical studies have been carried out with these oligonucleotides. The four xDNA bases are brightly fluorescent, with emission maxima at 380–410 nm. Interestingly, when grouped together their emission can change dramatically depending on the oligomer length, hybridization, and composition.[15] The experiments suggested multiple ways in which the expanded DNA bases (alone and in oligomers) could serve as useful tools in biophysical analysis and in biotechnological applications.[12]
Figure 2.
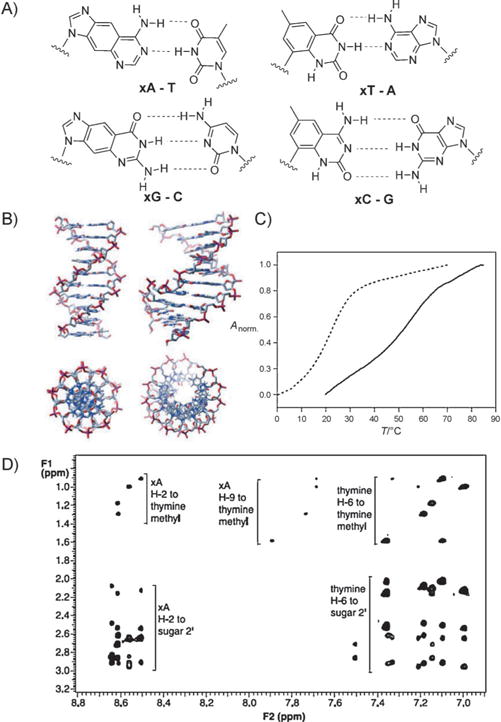
A) Structure of base pairs of size-expanded nucleobases with natural nucleobases. B) Structure of size-expanded DNA (xDNA) in comparison to natural DNA. C) Thermal denaturation plot of a size-expanded helix, with the self-complementary sequence d(xATxAxAT-xATTxAT) (control d(ATAATATTAT)), showing enhanced stability in comparison with analogue natural DNA duplex. D) 2D NMR spectrum confirming the structure (IUPAC standard numeration). Adapted from Ref. [12] with permission.
Computational studies have explored the origins of the optical absorption spectra of the xDNA bases[16] and further details about their hydrogen-bonding and stacking abilities.[17] Time-dependent density functional theory calculations revealed the changes to the frontier orbitals induced by the additional aromatic ring. The hypochromicity, resulting from stacking interactions, was calculated to be more pronounced in stacked xG-C and xA-T pairs than in natural G-C and A-T pairs. Calculations of the electronic structure in double-stranded xDNA have also been carried out and showed that xDNA has a smaller π → π* gap than natural B-DNA, which suggests that xDNA could be a candidate for efficient transport of positive and negative charge as well as nanowire applications.[18] The interaction between size-expanded guanine (xG) and different gold nanoclusters was recently studied using density functional theory.[19] The analysis of the local aromaticity has revealed relevant differences in the aromatic character of the xDNA bases.[20]
A number of experimental studies have been carried out on single xDNA bases to evaluate the steric and electronic effects of the added size.[21] For example, base-stacking studies of the xDNA bases revealed that the benzo-homologated bases stack much more strongly than do their natural counterparts, most likely because of the increased surface area of contact as well as the enhanced polarizability and hydrophobicity. Single substitutions of the xDNA bases in double-stranded natural DNAs are sterically destabilizing, which is relieved when multiple adjacent substitutions are used. However, Watson—Crick base pairs are still apparently formed despite local backbone strain at the DNA-xDNA junctions. The energetic cost of the 2.4 A stretching of an isolated base pair was estimated to be 1–2 kcalmol−1.
The uniform pairing of expanded bases with canonical bases in helices results in a stable, double helical, antiparallel xDNA structure. A wide range of investigations have explored the helix-forming properties and structure of xDNA (Figure 2B–D).[22] These include NMR and UV spectroscopy, stoichiometry, CD spectra, ionic-strength dependence, thermodynamic measurements, and molecular dynamics simulations.[23] xDNA is in nearly every case much more stable than natural DNA of an analogous sequence, because of the enhanced stacking of the enlarged base pairs. The structure is similar to B-DNA, with analogous pairing and sugar conformations. Interestingly, the larger diameter requires about 12 base pairs per 360° turn, compared with 10.5 pairs for natural DNA. Thermodynamic studies confirmed that xDNA is sensitive to single-base mismatches, thereby showing the same selectivity as natural DNA.[24] Thus, as a self-assembling system, xDNA performs as well or better than native DNA, and establishes that xDNA can function as an artificial genetic system from a chemical and physical point of view.
Next the focus turned to biochemical and biological questions: can the information stored in xDNA sequences be copied faithfully? Notably, xDNA is composed of eight letters, thus offering an extraordinary density of information storage. Studies on the processing of single and multiple xDNA pairs by DNA polymerase I Klenow fragment (Kf, an A-family sterically rigid enzyme) and by Sulfolobus solfataricus polymerase Dpo4 (a flexible Y-family polymerase) were performed to examine the potential of benzo-expanded DNA to encode and transfer biochemical information.[25,26] In most cases, the polymerases selected the correctly paired partner for each xDNA base, but with lower efficiency than the natural bases. Kinetic studies revealed that the flexible Dpo4 enzyme was much more efficient than Kf at extending the enlarged pairs, and further studies showed that Dpo4 could correctly synthesize up to four successive xDNA pairs on a template. Figure 3 shows the results for nucleoside insertion by Dpo4 (A), nucleotide insertion by Kf exo (B), primer/template base-pair extension by Kf exo (C), and base-pair extension by Dpo4 (D). The data suggest that for successful replication of xDNA and the design of other large base pairs, existing polymerases are suitable although limited, but that the use of multiple enzymes in combination or the identification and evolution of new polymerases will be helpful.
Figure 3.
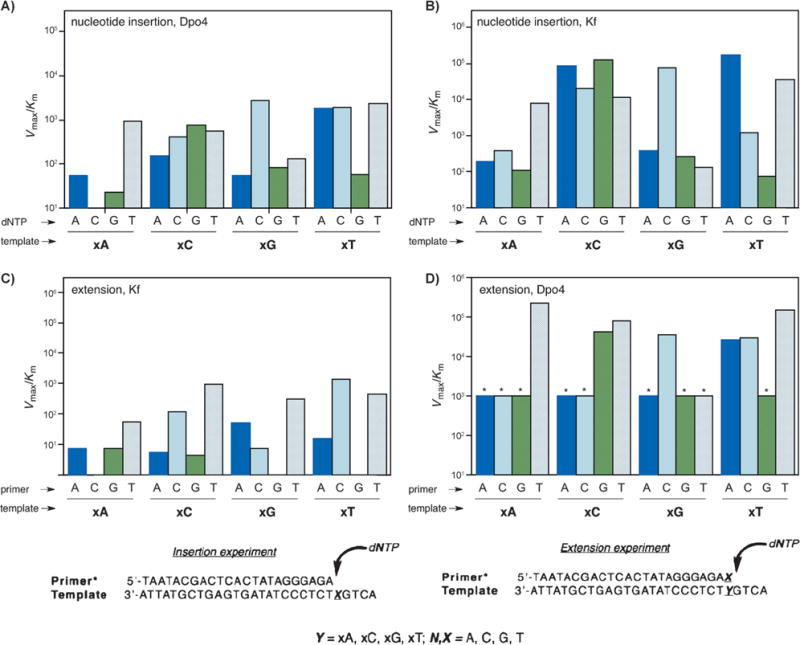
A–D) Comparison of the kinetic efficiencies and selectivities for the synthesis and extension of the xDNA pair for Dpo4 and Kf exo enzymes (see text for explanation). Bottom: primer:templates used in single nucleotide insertion and extension experiments. (Reproduced from Ref. [25] with permission from The Royal Society of Chemistry.)
Next the polymerase copying of xDNAwas tested in living bacteria. An approximately 7000 nt phage genome constructed to contain single xDNA bases was replicated in E. coli, which contains five different DNA polymerases, some of which are flexible Y-family enzymes. The results showed efficient bypass of some of the xDNA bases.[27] Tests were then carried out to see whether xDNA could be used to encode some of the amino acids of green fluorescent protein. Plasmid DNAs containing up to eight base pairs of xDNA were correctly read by cellular polymerases, thereby resulting in green fluorescent bacteria.[28] This was the first example of the use of an artificial genetic set to encode phenotype in a living organism, and is a significant step from the standpoint of synthetic biology.
Syntheses and a few replication studies have also been developed for a related structural design, called “wide DNA” (yDNA), which was developed to have a different vector of benzo-expansion for the three modified nucleosides dyA, dyT, and dyC (Figure 4A).[29] Highly stable double-stranded helices were formed in several sequence contexts. This result suggests the possibility of an eight-base genetic system based on the yDNA geometry, analogous to what has been developed for xDNA. Analysis of the fluorescence properties as well as binding selectivity and affinity studies of yDNA strands have been carried out.
Figure 4.
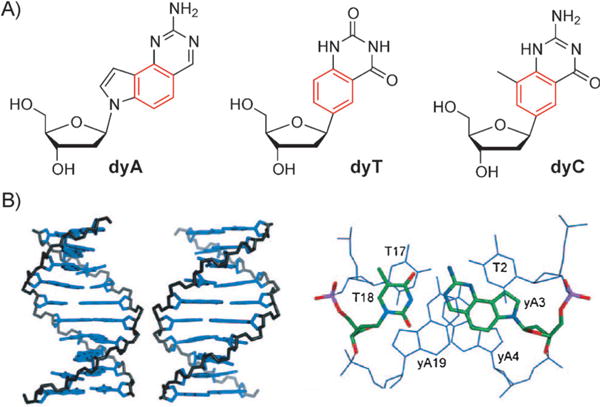
A) Structure of the three synthesized yDNA nucleosides. B) Modeled structure of yDNA helix and base pairs (see text for explanation). Adapted from Ref. [29] with permission.
Figure 4B shows views of the yDNA structure from the major groove and minor groove (left), as well as structures of three consecutive base pairs in the yDNA structure, which demonstrates pairing geometry and base-base overlap (right).
It was found in experiments addressing polymerase amplification, cloning, and gene expression of yDNA that polymerase reading of yDNA did occur with low efficiency; however, a strong potential for mismatching was found, mostly as a result of T-yT and T-yC wobble-type mispairing. This is different than xDNA, and likely arises from the distinct yDNA geometry, where wobble-type pairs can be formed more readily.[30]
After the above-mentioned successes in benzo-homolo-gation, we explored whether further size expansion might be possible. Thus, initial experiments exploring naphtho-homo-logation were undertaken.[31] The doubly widened naphtho-pyrimidines yyT and yyC were synthesized through Heck coupling reactions (Figure 5A) and incorporated into oligonucleotides through solid-phase chemistry. The deoxyribosides were fluorescent and had emission maxima of 446 nm and 433 nm, respectively. Sequences containing multiple substitutions of yyT and yyC paired opposite adenine and guanine were subsequently mixed and investigated. Data from UV mixing experiments, FRET experiments, as well as quenching and on-bead hybridization studies suggested that complementary “double-wide DNA” (yyDNA) strands are formed by self-assembly into helical complexes with 1:1 stoichiometry. Molecular models were built to compare the natural B-form DNA dodecamer with a yyDNA composed of yyT-A and yyC-G pairs (Figure 5B, AMBER force field, continuum water). The hypothesized base-pairing schemes are analogous to Watson—Crick pairing, but with glycosidic C1′-C1′distances widened by over 45 % to about 15.2 Å. The yyDNA hybridizations were tested on PEG-polystyrene beads under an epifluorescence microscope. In this study, two sets of beads were prepared with different DNA oligonucleotides covalently conjugated, one complementary to the yyDNA 5-CCTTCTCC sequence, and the other a scrambled control sequence. The yyDNA oligomer was incubated with beads, followed by washing. The digital fluorescence images (Figure 5 C) showed a significant increase in the fluorescence for the sequence-complementary beads (d,f), relative to background (b) and to the scrambled-sequence beads (c,e). These findings confirm the formation of a sequence-selective noncovalent complex between the yyDNA oligonucleotides and the complementary DNA sequence on the polymer beads.
Figure 5.
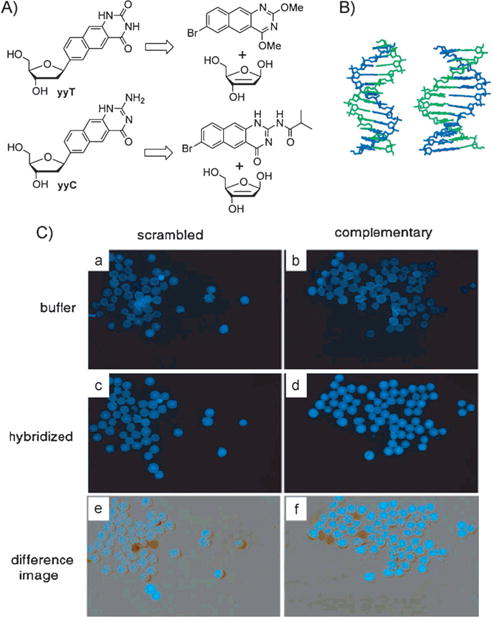
A) Retrosynthetic scheme for the synthesis of naphtho-homo-logated deoxyribonucleosides yyT and yyC. B) Structure of yyDNA (right) compared to natural DNA. C) Hybridization studies on 60 μm PEG-polystyrene beads. See the text for an explanation. Reproduced from Ref. [31] with permission.
The yyDNA studies have established a new, larger limit for the size of information encoding nucleic acid based systems. A detailed computational analysis of the different effects resulting from the exceptional structure of yDNA and yyDNA has recently been described by Sharma et al.[32] Another comprehensive theoretical study addressed the important topic of tautomerism.[33] Naphtho-homologated yyG and its five possible tautomers were investigated in terms of their electronic transitions, and the results were compared to that of yDNA bases showing interesting effects. For example, the absorption and emission maxima for yyG were blue-shifted after solvation in methanol, while the maxima of the tautomeric bases were red-shifted. In general, the visible fluorescence properties of yyDNA bases suggest their use as reporters in a wide range of analytical applications.
2.2. Purine—Purine Base Pairs
Another prominent example of size-expanded nucleobase pairing involves purine—purine base pairs. Such pairs, if oriented in an anti-anti arrangement, can possibly take on a dimension wider than Watson—Crick pyrimidine—purine pairs. Although the nucleobases by themselves are not necessarily “artificial”, purine–purine (Pu-Pu) pairing plays a special role in the expanded base-pair context because of its relevance to the origins of life. The investigation of these noncanonical base pairs is of high interest[34] because they raise the question of why the genetic information on Earth is stored in purine—pyrimidine base-pairing systems. It is well-established that purine—purine base pairs do occur in natural RNAs,[35] and Benner and co-workers have reported studies with isolated Pu-Pu pairs in otherwise natural DNA duplexes.[36] Jovin and co-workers described and characterized a parallel-stranded duplex DNA which contains segments of trans-purine—purine and purine—pyrimidine base pairs.[37]
Fully Pu-Pu-substituted DNAs have been studied by multiple research groups. A prominent example is the guanine—isoguanine and diaminopurine-7-deazaxanthine base pairs described by Heuberger and Switzer[38] (Figure 6A). The studies demonstrated that these purine—purine DNA duplexes have thermal stabilities and free energies that are very similar to the analogous purine—pyrimidine DNA duplexes. A number of oligonucleotides were synthesized containing these purine bases, and UV melting studies revealed that complementary strands associate, but that the single strands do not self-associate in the absence of their complement (Figure 6B). The interaction of these base pairs as well as of the adenine—inosine base pair with a wide range of DNA intercalators was also studied by Hud and co-workers.[39]
Figure 6.
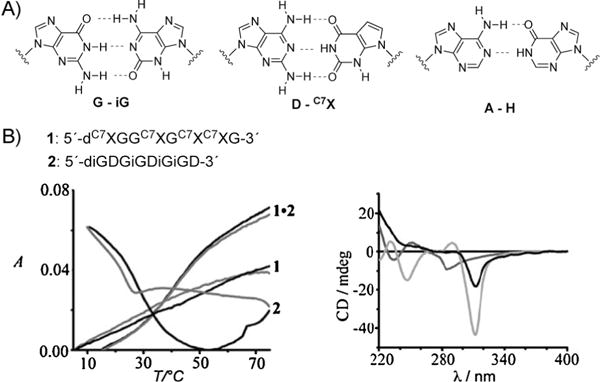
A) Structure of the purine—purine base pairs G-iG, D-C7X, and A-H (see text for explanation). B) Two single oligonucleotides containing complementary purine—purine base pairs as well as a denaturation profile and a CD spectrum of these oligonucleotides (see text). Reproduced from Ref. [38] with permission.
Battersby et al. performed thermal melting and structural studies with oligonucleotides containing adenine—hypoxanthine and guanine—isoguanine base pairs.[40] The results confirmed that nucleic acids containing these purine nucleobases can form stable duplexes.
Further studies with multiple purine—purine pairs have been described by Seela et al. and by Eschenmoser and co-workers, who have also addressed the helix formation issues of purine oligonucleotides paired with other strands that contained purines.[41] To date, no full structures have been determined by 2D NMR or crystallographic methods. Also, no tests of the ability of polymerase enzymes to copy homopurine DNA in the presence of complementary purine nucleotides have been reported, thus the templating power of Pu-Pu pairing remains to be determined.
These purine—purine base-pairing studies have a high significance for understanding the formation of nucleic acid structures and evolution of living systems. They should be considered as an alternative concept for biological information storage, and detailed comparisons with the purine—pyrimidine base pairs give us enlightening insights in to the origin of the genetic information storage systems on our planet.
2.3. Base Pairs Containing Naphthyridine and Imidazopyridopyrimidine
Minakawa, Matsuda et al. have been working on the development of size-expanded base-pairing motifs containing four hydrogen bonds. For example, they described the synthesis and properties of oligonucleotides containing imidazopyrimidine (Im) nucleosides (combinations diamino, dioxo, amino-oxo, oxo-amino; Figure 7A, top).[42] These tricyclic structures were synthesized by a Stille coupling reaction, followed by an intramolecular cyclization (Figure 7A, bottom). Incorporation of the large tricyclic nucleosides opposite one another in a complementary DNA helix resulted in duplex destabilization as a result of the size mismatch with adjacent Watson—Crick pairs, while incorporation of three adjacent tricyclic nucleosides per strand resulted in thermal and thermodynamic stabilization of the duplex. Further comparisons of the melting temperatures confirmed the 4-hydrogen-bond motif. In further studies, 1,8-naphthyridine (Na) C-nucleosides and their pairing properties in oligonucleotides were described,[43] as well as the synthesis and selective recognition of naphthyridine:imidazopyridopyrimidine base pairs (ImON:NaNO and Im-NO:NaON) by the Klenow fragment DNA polymerase (Figure 7B).[44] The Im bases can be considered as ring-expanded purine analogues toward the minor groove direction and the Na bases as ring-expanded analogues of pyrimidine toward the major groove direction. It was found that the DNA duplexes containing these artificial nucleobases are highly thermally stabilized (+ 8–9°C) as a result of the four non-canonical hydrogen bonds, the strong base stacking, and the shape compatibility of the Im:Na pair.
Figure 7.
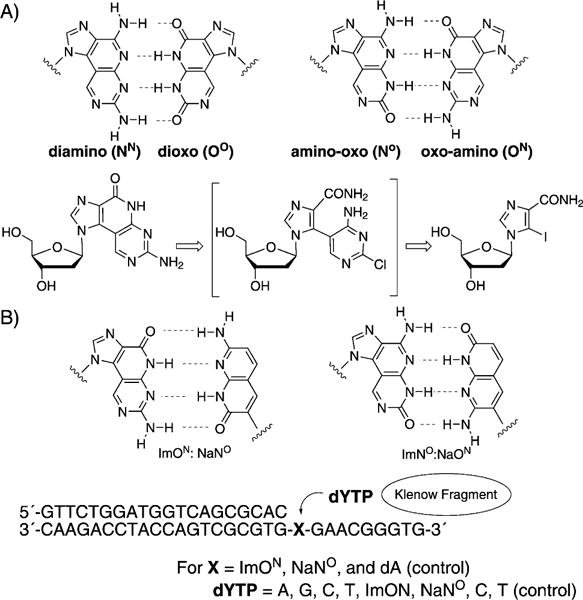
A) Imidazopyridopyrimidine base pairs and a retrosynthetic approach to these modified nucleotides. B) Imidazopyridopyrimidine: naphthyridine base pairs and an experimental design for testing the single nucleostide insertion by Klenow fragment. Adapted from Refs. [42–44] with permission.
In this study, the single nucleotide insertion by KF (selectivity towards natural dNTPs and selectivity towards noncanonical dNTPs) was tested in a series of experiments. A primer—template duplex was used with a DNA polymerase, as shown in Figure 7. As one example, the different dYTP nucleosides were incorporated against ImNO in the template. The triphosphates dATP and ImONTP were selectively incorporated against NaNO in the template by this enzyme, a result which is consistent with their electrostatic and shape complementarity. Im-Na-type pairs were selectively incorporated relative to the enlarged Im-Im-type pairs, which is consistent with the fact that Im:Na pairs are expected to be similar in size to natural purine :pyrimidine base pairs. Thus, it is not yet clear whether the novel Im-Im size-expanded system will be replicable with naturally occurring enzymes, or whether engineered polymerases may be required.
2.4. Alkynyl-Substituted Base Pairs
Another design for DNA-like systems with expanded diameters has been developed by Inouye and co-workers.[45] In this creative study, a set of four nucleosides is described wherein the non-natural bases are attached to the deoxyribose moiety through an acetylene linkage in the β configuration (Figure 8A). These nucleosides, stretched in size by about 3 Å, were incorporated into oligonucleotides through the corresponding phosphoramidites, and the resulting DNA was shown to form right-handed duplexes and triplexes with complementary DNA. A series of absorption experiments showed that the resulting duplexes have thermal stabilities very close to those of natural duplexes (Figure 8B). A spontaneous and sequence-selective hybridization occurred, thus making this artificial genetic system suitable for information storage and thus offering possible applications in synthetic biology if it can be replicated. Structural studies will be of great interest in this system to see how the helix and backbone geometry adapt to the presence of the alkyne linkages.
Figure 8.
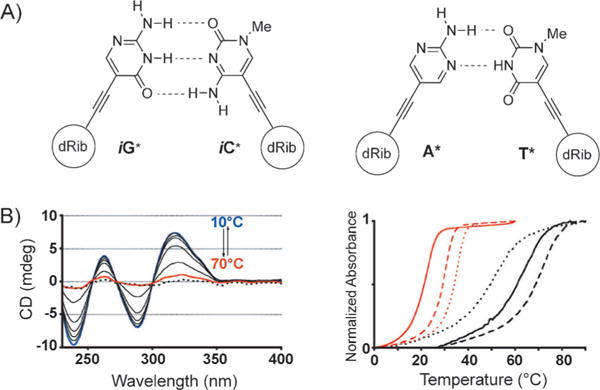
A) Artificial alkynyl base pairs iG*:iC* and A*:T*; B) Left: CD spectrum of duplex 5′-d(iG*)8/3′-d(iC*)8 at different temperatures from 10 (blue) to 70°C (red); the dotted line is the sum of each single-strand spectrum of d(iG*)8 and d(iC*)8; right: UV melting curves of duplex 5′-d(iG*)8/3′-d(iC*)8 (black) and further sequences with different d(iG*)/d(iC*) containing sequences. Adapted from Ref. [45] with permission.
2.5. Size-Expanded Base Pairs Driven by Forces Other Than Hydrogen Bonding
It has been demonstrated that hydrophobic packing and base-stacking interactions between nucleobases can be harnessed in the absence of hydrogen bonding to stabilize the formation of base pairs.[4,5,46] In this context, Romesberg and co-workers have designed nucleobases larger than pyrimidines that can be paired with themselves or other large partners, for example, 7-propynylisocarbostyril:7-propyliso-carbostyril (PICS:PICS),[47] azaindole:azaindole (7AI:7AI), and 3-methylnaphthalene:3-methylnaphthalene (3MN:3MN; Figure 9).[48] PICS:PICS has been shown to be more stable than natural base pairs and can be efficiently incorporated into DNA by the Klenow fragment of E. coli DNA polymerase I (KF). However, the natural DNA context in which these have been studied constricts the ability of such pairs to take on an expanded dimension; structural studies have shown that the large bases intercalate with each other, adapting to the native helix size.[4,5]
Figure 9.
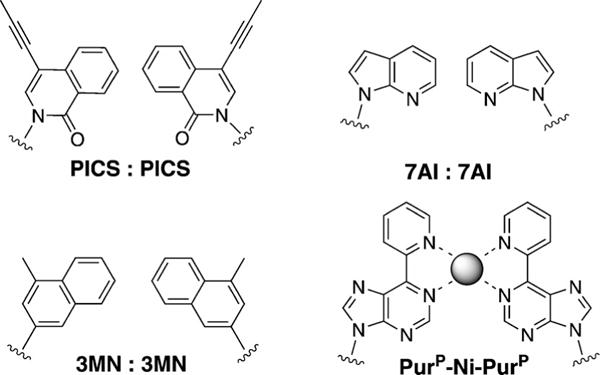
Non-hydrogen-bonded base pairs PICS:PICS, 7AI:7AI, 3MN:3MN, and PuP-Ni-PurP, which involve enlarged bases.[47–49]
Another novel strategy for the design of a stable base pair is metal-mediated pairing. If the bases are larger than pyrimidines, this can result in pairs of expanded size. A good example is the pyridylpurine system PurP-Ni2+-PurP developed by Switzer et al.[49] This has also been shown to be more stable than natural base pairs, and it appeared to have the potential to serve as the functional component of an ion-activated switch. There are additional examples of such non-hydrogen-bonded base pairs whose description fall beyond the scope of this Minireview; they can be found in the corresponding literature.[50]
3. Size-Expanded RNA (xRNA) Nucleosides
3.1. Benzo-Homologation of RNA
In the natural genetic system, RNA displays different biophysical and structural properties from DNA, and plays distinct biological roles as well. This knowledge led us to ask whether a size-expanded analogue of RNA might also be developed. Thus, we have undertaken the synthesis and study of xRNA in our laboratory.[51] The photophysical properties of these molecules, which are efficient fluorophores, as well as the enhanced stacking properties make these molecules highly interesting for a wide range of biochemical applications (Figure 10 A), and they are already being employed as biophysical and biological tools.
Figure 10.
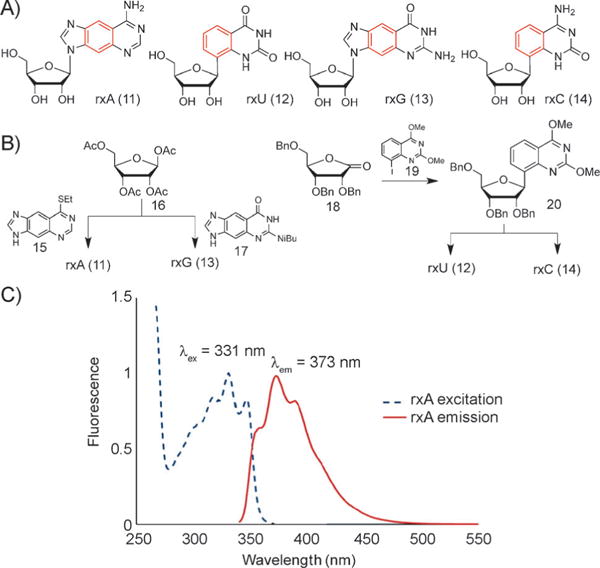
A) xRNA nucleosides rxA, rxU, rxG, and rxC. B) Synthesis schemes for these nucleosides. C) Absorption and emission spectrum of rxA. Reproduced from Ref. [51] with permission. Bn = benzyl.
The additional OH group of ribose compared to the deoxysugar of DNA adds distinct challenges to the syntheses of these compounds (Figure 10 B). In addition to this, a main issue is the glycosylation step. For the size-expanded purine derivatives rxA (11)[10] and rxG (13), this step is accomplished by coupling either nucleobase 15 (which is synthesized in 4 steps) or, respectively, nucleobase 17 (prepared in 5 steps) to tetraacetylribose 16 under Vorbrtiggen conditions,[14] followed by deprotection steps. For the size-expanded pyridine nucleosides rxU (12) and rxC (14), iodinated compound 19 (synthesized in 4 steps) is coupled to ribonolactone 18. The subsequent reactions are performed with different amounts of Lewis acid BBr3 to arrive at either rxU (12) or to rxC (14; for details see Ref. [51]). Photophysical data have been compiled for these compounds. For example, Figure 10C shows an absorption and emission spectrum for rxA (11).[51]
These xRNA nucleosides also have to be converted into the corresponding phosphoramidites for solid-phase synthesis of oligonucleotides (Figure 11 A). As a consequence of the additional OH group, the preparation of the RNA phosphoramidites is more challenging than that for the modified xDNA nucleotides. Recently the phosphoramidites rxA-PA (11-PA) and rxU-PA (12-PA) have been reported by our research group,[52] while the syntheses of the remaining two (rxC-PA and rxG-PA) are underway.
Figure 11.
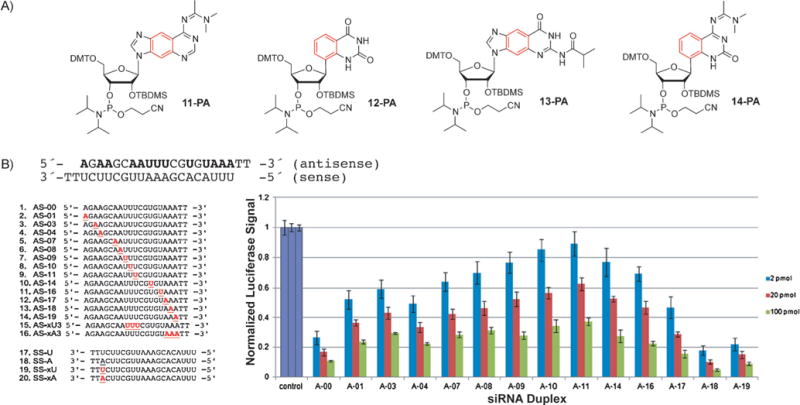
A) Structure of the xRNA phosphoramidites rxA (11-PA), rxU (12-PA), rxG (13-PA), and rxC (14-PA). B) Double-stranded RNA probes containing xRNA bases for RNA interference experiments and results of the RNAi luciferase activity assay with siRNAs containing the xRNA bases. Reproduced from Ref. [52] with permission. DMT = dimethoxytrityl.
Oligonucleotides containing these size-expanded RNA nucleobases were used as steric probes in RNA interference experiments. This cellular pathway is recognized as an important strategy for downregulation of specific genes in cells.[53] To test the effects of the steric size of siRNA on biological activity, a number of probes were synthesized with xA or xU in the guide strand (Figure 11B) and the resulting modified RNAs used to downregulate the luciferase gene in a dual-reporter luciferase assay in HeLa cells.[52] Although xRNA nucleobases in the guide strand reduced the activity at some central positions near the seed region (apparently because of adverse steric effects), they were well tolerated at the end. Most importantly, substitutions near the 3′-end increased the activity over that of wild-type siRNAs (Figure 11B, right). Further studies demonstrated that xRNA substitutions protected the siRNA against nuclease degradation, and also thermally stabilized the helices. Biologically active siRNAs containing up to three consecutive xRNA base pairs were described.
3.2. Further Examples of Size-Expanded Ribonucleosides
Seley-Radtke et al. described the design, synthesis, and preliminary biological activities of some heterocycle-expanded purine nucleosides (21 and 22, Figure 12A).[54] These thieno-expanded purine nucleosides were used for investigations of nucleic acid structure and function, and it was demonstrated that the Rd-RNA polymerase readily recognized the corresponding non-natural triphosphates. The enzyme exhibited remarkable differences in recognition. Furthermore, it has been shown that the incorporation of non-natural bases such as a thienylimidazole-substituted size-expanded nucleobase in aptamers can yield aptamers with augmented affinities for target proteins, for example, cell growth factors.[55]
Figure 12.
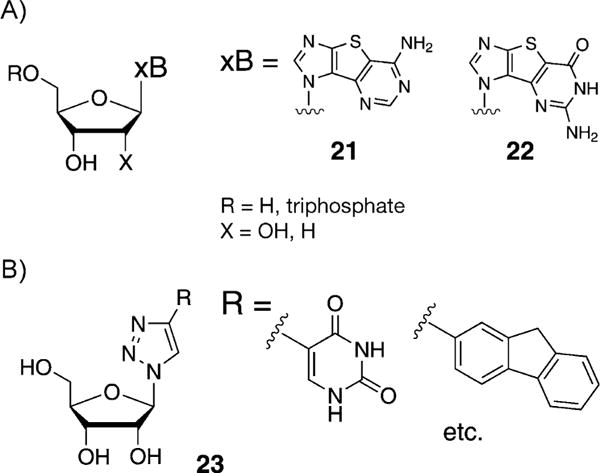
A) Thieno-expanded purine nucleosides described by Seley-Radtke et al.[54] B) Click fleximers described by Hudson and co-workers.[56]
Hudson and co-workers described the synthesis of nucleoside analogues incorporating 4-(5-pyrimidinyl)-1,2,3-triazoles as expanded nucleobase mimics (23, Figure 12B),[56,57] which was accomplished through azide-alkyne Huisgen cycloaddition[58] between the corresponding ribosyl azide and a variety of 5-alkynylpyrimidines. It was shown that the heterocycles of the unfused nucleobase prefer coplanar arrangements and that the anti-glycosidic conformer was favored. Neither of these expanded designs has yet been reported in an oligomeric form.
4. Conclusions and Outlook
Artificial genetic sets containing size-expanded base pairs have shown the ability to function in many, if not all, of the ways that the natural genetic system can. It can be concluded that a genetic base-paired molecular framework—from the chemical point of view—may not necessarily be limited to the size of the natural DNA helix that has evolved for living systems on Earth.
Despite this progress, there remain many interesting challenges ahead to be investigated with size-expanded genetic sets. The basic science of chemical evolution still invokes many questions; for example, the role of pKa values and tautomerization of the nucleobases on base pairing with respect to expanded base pairs and the effect on mismatch discrimination is of continuing interest.[59] Since expanded pairs can form stable helices, the implications for the selection of a purine—pyrimidine paradigm in nature will have to be discussed in detail.
Structural and functional questions also remain. For example, most of the expanded pairing systems mentioned above have not yet been studied structurally in the full helical context, and it will be interesting to see how the helical backbone adapts to the geometry of the noncanonical pairs. Furthermore, it is not yet known what the size limits are in expanded pairing systems; since naphtho-homologous pairing seems to self-assemble, it raises the question of whether even larger systems might be possible. Going beyond structure, function remains critically important as well. In this light, the development of efficient replicating enzymes for the size-expanded systems will be of high priority, as it will enable researchers to assemble longer sequences than can be made with a DNA synthesizer. Having longer expanded DNAs raises the interesting possibility of using their stability and rigidity (and perhaps charge conductivity) in self-assembling nanostructures. Moreover, the ability to amplify such systems raises the possibility of selection and evolution of such genetic sets. Finally, many applications in biology remain to be explored; in particular, we are interested in pursuing biochemical and biological studies of expanded RNAs to see whether they function analogously to RNAs, which not only carry genetic information but also act as ligands, switches, and enzymes in the cell.
The chemical expansion of the natural nucleoside architecture provides tools for the investigation of fundamental biochemical processes on Earth, thereby allowing us to learn about living processes and augment our admiration for the simplicity of structure and complexity of function that has evolved in nature. Furthermore, the concept of new genetic sets is relevant to the development of modified living systems for synthetic biology, and to the investigation of the viability of alternative life forms, which will be especially interesting if extraterrestrial life is eventually discovered.
Acknowledgments
M.W. thanks the German Academic Exchange Service (DAAD) for a postdoctoral fellowship. E.T.K. thanks the U.S. National Institutes of Health (GM063587, GM072705.
Biographies

Eric T. Kool studied at Miami University in Oxford, Ohio, and received his PhD at Columbia University in 1988 as an NSF Predoctoral Fellow. After postdoctoral research at Caltech, he began his career at the University of Rochester in 1990, and was promoted to Professor in 1997. In 1999 he joined the faculty at Stanford University, where he is the George and Hilda Daubert Professor of Chemistry.

Malte Winnacker received his diploma in chemistry at the University of Würzburg in 2006 and his PhD at the Ludwig-Maximi-lians-University of Munich in 2010 under the direction of Prof. Dr. T. Carell. He was then a postdoctoral DAAD fellow at Stanford University with Prof. Eric T. Kool. Currently, he is a scientific co-worker at the WACKER-Chair of Macromolecular Chemistry at the Technical University of Munich in the group of Prof. Dr. B. Rieger.
References
- 1.Benner SA. Nature. 2003;421:118. doi: 10.1038/421118a. [DOI] [PubMed] [Google Scholar]; Kool ET, Waters ML. Nat Chem Biol. 2007;3:70–73. doi: 10.1038/nchembio0207-70. [DOI] [PubMed] [Google Scholar]; Khalil AS, Collins JJ. Nat Rev Genet. 2010;11:367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For a good review, see; Carell T, Brandmayr C, Hienzsch A, Müller M, Pearson D, Reiter V, Thoma I, Thumbs P, Wagner M. Angew Chem. 2012;124:7220–7242. doi: 10.1002/anie.201201193. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:7110–7131. doi: 10.1002/anie.201201193. [DOI] [PubMed] [Google Scholar]
- 3.Chiba J, Inouye M. Chem Biodiversity. 2010;7:259–282. doi: 10.1002/cbdv.200900282. [DOI] [PubMed] [Google Scholar]; Krueger AT, Kool ET. Curr Opin Chem Biol. 2007;11:588–594. doi: 10.1016/j.cbpa.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kool ET, Morales JC, Guckian KM. Angew Chem. 2000;112:1046–1068. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Hirao I. Curr Opin Chem Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]; Henry AA, Romesberg FE. Curr Opin Chem Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Clever GH, Kaul C, Carell T. Angew Chem. 2007;119:6340–6350. doi: 10.1002/anie.200701185. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:6226–6236. doi: 10.1002/anie.200701185. [DOI] [PubMed] [Google Scholar]; Tanaka K, Clever GH, Takezawa Y, Yamada Y, Kaul C, Shionoya M, Carell T. Nat Nanotechnol. 2006;1:190–195. doi: 10.1038/nnano.2006.141. [DOI] [PubMed] [Google Scholar]; Meggers E, Holland PL, Tolman WB, Romesberg FE, Schultz PG. J Am Chem Soc. 2000;122:10714–10715. [Google Scholar]; Weizman H, Tor Y. J Am Chem Soc. 2001;123:3375–3376. doi: 10.1021/ja005785n. [DOI] [PubMed] [Google Scholar]
- 7.Benner SA. Acc Chem Res. 2004;37:784–797. doi: 10.1021/ar040004z. for nucleobase pairing studies in expanded Watson—Crick-like genetic information systems, see for example. [DOI] [PubMed] [Google Scholar]; Geyer CR, Battersby TR, Benner SA. Structure. 2003;11:1485–1498. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Krueger AT, Kool ET. Chem Biol. 2009;16:242–248. doi: 10.1016/j.chembiol.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson JD, Crick FHC. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 10.Leonard NJ, Kaźmierczak F, Rykowski A. J Org Chem. 1987;52:2933–2935. [Google Scholar]; Leonard NJ, Keyser GE. Proc Natl Acad Sci USA. 1979;76:4262–4264. doi: 10.1073/pnas.76.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]; Scopes DIC, Barrio JR, Leonard NJ. Science. 1977;195:296–298. doi: 10.1126/science.188137. [DOI] [PubMed] [Google Scholar]; Leonard NJ, Morrice AG, Sprecker MA. J Org Chem. 1975;40:356–363. doi: 10.1021/jo00891a021. [DOI] [PubMed] [Google Scholar]
- 11.Lessor RA, Gibson KJ, Leonard NJ. Biochemistry. 1984;23:3868–3873. doi: 10.1021/bi00312a012. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Gao J, Kool ET. J Org Chem. 2005;70:639–647. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]; Liu H, Gao J, Maynard L, Saito YD, Kool ET. J Am Chem Soc. 2004;126:1102–1109. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]; Liu H, Gao J, Lynch SR, Saito YD, Maynard L, Kool ET. Science. 2003;302:868–871. doi: 10.1126/science.1088334. for another synthesis approach to pyridine-streched 2′-deoxynucleosides, but without base pairs, see also. [DOI] [PubMed] [Google Scholar]; Clayton R, Davis ML, Fraser W, Li W, Ramsden CA. Synlett. 2002;9:1483–1486. [Google Scholar]
- 13.Heck RF. J Am Chem Soc. 1968;90:5531–5534. [Google Scholar]
- 14.Vorbrüggen H, Höfle G. Chem Ber. 1981;114:1256–1268. [Google Scholar]
- 15.Krueger AT, Kool ET. J Am Chem Soc. 2008;130:3989–3999. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]; Wilson JN, Kool ET. Org Biomol Chem. 2006;4:4265–4274. doi: 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]
- 16.Varsano D, Garbesi A, Di Felice R. J Phys Chem B. 2007;111:14012–14021. doi: 10.1021/jp075711z. [DOI] [PubMed] [Google Scholar]
- 17.McConnell TL, Wetmore SD. J Phys Chem B. 2007;111:2999–3009. doi: 10.1021/jp0670079. [DOI] [PubMed] [Google Scholar]
- 18.Fuentes-Cabrera M, Zhao X, Kent PRC, Sumpter BG. J Phys Chem B. 2007;111:9057–9061. doi: 10.1021/jp0729056. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Ren T, Yang X, Zhou L, Li X. Int J Quantum Chem. 2013 doi: 10.1002/qua.24436. [DOI] [Google Scholar]
- 20.Huertas O, Poater J, Fuentes-Cabrera M, Orozco M, Solà M, Luque FJ. J Phys Chem A. 2006;110:12249–12258. doi: 10.1021/jp063790t. [DOI] [PubMed] [Google Scholar]
- 21.Gao J, Liu H, Kool ET. J Am Chem Soc. 2004;126:11826–11831. doi: 10.1021/ja048499a. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Gao J, Kool ET. J Am Chem Soc. 2005;127:1396–1402. doi: 10.1021/ja046305l. [DOI] [PubMed] [Google Scholar]; Lynch SR, Liu H, Gao J, Kool ET. J Am Chem Soc. 2006;128:14704–14711. doi: 10.1021/ja065606n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blas JR, Huertas O, Tabares C, Sumpter BG, Fuentes-Cabrera M, Orozco M, Ordejon P, Luque FJ. J Phys Chem A. 2011;115:11344–11354. doi: 10.1021/jp205122c. [DOI] [PubMed] [Google Scholar]
- 24.Lu H, He K, Kool ET. Angew Chem. 2004;116:5958–5960. [Google Scholar]; Angew Chem Int Ed. 2004;43:5834–5836. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]
- 25.Lu H, Krueger AT, Gao J, Liu H, Kool ET. Org Biomol Chem. 2010;8:2704–2710. doi: 10.1039/c002766a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krueger AT, Lu H, Hojland T, Liu H, Gao J, Kool ET. Nucl Acids Res Symp Ser. 2008;52:233–234. [Google Scholar]
- 27.Delaney JC, Gao J, Liu H, Shrivastav N, Essigmann JM, Kool ET. Angew Chem. 2009;121:4594–4597. doi: 10.1002/anie.200805683. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:4524–4527. doi: 10.1002/anie.200805683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krueger AT, Peterson LW, Chelliserry J, Kleinbaum DJ, Kool ET. J Am Chem Soc. 2011;133:18447–18451. doi: 10.1021/ja208025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, Lynch SR, Lee AHF, Kool ET. ChemBioChem. 2009;10:2530–2538. doi: 10.1002/cbic.200900434. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lu H, He K, Kool ET. Angew Chem. 2004;116:5958–5960. [Google Scholar]; Angew Chem Int Ed. 2004;43:5834–5836. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]
- 30.Chelliserrykattil J, Lu H, Lee AHF, Kool ET. ChemBioChem. 2008;9:2976–2980. doi: 10.1002/cbic.200800339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee AHF, Kool ET. J Am Chem Soc. 2006;128:9219–9230. doi: 10.1021/ja0619004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma P, Lait LA, Wetmore SD. Phys Chem Chem Phys. 2013;15:2435–2448. doi: 10.1039/c2cp43910g. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Ren T. Int J Quantum Chem. 2013;113:1225–1233. [Google Scholar]
- 34.Wächtershäuser G. Proc Natl Acad Sci USA. 1988;85:1134–1135. doi: 10.1073/pnas.85.4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Stombaugh J, Zirbel CL, Westhof E, Leontis NB. Nucleic Acid Res. 2009;37:2294–2312. doi: 10.1093/nar/gkp011. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nagaswamy U, Voss N, Zhang Z, Fox GE. Nucleic Acid Res. 2000;28:375–376. doi: 10.1093/nar/28.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geyer CR, Battersby TR, Benner SA. Structure. 2003;11:1485–1498. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 37.Evertsz EM, Rippe K, Jovin TM. Nucleic Acid Res. 1994;22:3293–3303. doi: 10.1093/nar/22.16.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heuberger BD, Switzer C. ChemBioChem. 2008;9:2779–2783. doi: 10.1002/cbic.200800450. [DOI] [PubMed] [Google Scholar]
- 39.Buckley R, Enekwa CD, Williams LD, Hud NV. ChemBioChem. 2011;12:2155–2158. doi: 10.1002/cbic.201100375. [DOI] [PubMed] [Google Scholar]
- 40.Battersby TR, Albalos M, Friesenhahn MJ. Chem Biol. 2007;14:525–531. doi: 10.1016/j.chembiol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 41.a) Seela F, Melenewski A, Wei C. Bioorg Med Chem Lett. 1997;7:2173–2176. [Google Scholar]; b) Groebke K, Hunziker J, Fraser W, Peng L, Diederichsen U, Zimmermann K, Holzner A, Leumann C, Eschenmoser A. Helv Chim Acta. 1998;81:375–474. [Google Scholar]; c) Seela F, Wei C, Malenewski A, Feiling E. Nucleosides Nucleotides. 1998;17:2045–2052. [Google Scholar]
- 42.Minakawa N, Kojima N, Hikishima S, Sasaki T, Kiyosue A, Atsumi N, Ueno Y, Matsuda A. J Am Chem Soc. 2003;125:9970–9982. doi: 10.1021/ja0347686. [DOI] [PubMed] [Google Scholar]
- 43.Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew Chem. 2005;117:602–604. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:596–598. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]
- 44.Minakawa N, Ogata S, Takahashi M, Matsuda A. J Am Chem Soc. 2009;131:1644–1645. doi: 10.1021/ja807391g. [DOI] [PubMed] [Google Scholar]
- 45.Doi Y, Chiba J, Morikawa T, Inouye M. J Am Chem Soc. 2008;130:8762–8768. doi: 10.1021/ja801058h. [DOI] [PubMed] [Google Scholar]
- 46.Kool ET. Curr Opin Chem Biol. 2000;4:602–608. doi: 10.1016/s1367-5931(00)00141-1. [DOI] [PubMed] [Google Scholar]; Guckian KM, Kool ET. Angew Chem. 1997;109:2942–2945. [Google Scholar]; Angew Chem Int Ed Engl. 1997;36:2825–2828. [Google Scholar]
- 47.McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]
- 48.Ogawa AK, Wu Y, McMinn DL, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:3274–3287. [Google Scholar]
- 49.Switzer C, Sinha S, Kim PH, Heuberger BD. Angew Chem. 2005;117:1553–1556. doi: 10.1002/anie.200462047. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:1529–1532. doi: 10.1002/anie.200462047. [DOI] [PubMed] [Google Scholar]
- 50.Lavergne T, Degardin M, Malyshev DA, Quach HT, Dhami K, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2013;135:5408–5419. doi: 10.1021/ja312148q. [DOI] [PMC free article] [PubMed] [Google Scholar]; Walsh JM, Beuning PJ. J Nucl Acids. 2012 doi: 10.1155/2012/530963. ID 530963. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang L, Schultz PG. Chem Commun. 2002:1–11. doi: 10.1039/b108185n. [DOI] [PubMed] [Google Scholar]
- 51.Hernández AR, Kool ET. Org Lett. 2011;13:676–679. doi: 10.1021/ol102915f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernández AR, Peterson LW, Kool ET. ACS Chem Biol. 2012;7:1454–1461. doi: 10.1021/cb300174c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meister G, Tuschl T. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]; Dykxhoorn DM, Novina CD, Sharp PA. Nat Rev Mol Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 54.Seley-Radtke KL, Zhang Z, Wauchope OR, Zimmermann SC, Ivanov A, Korba B. Nucleic Acids Symp Ser. 2008;52:635–636. doi: 10.1093/nass/nrn321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimoto M, Yamashige R, Matsunaga K-i, Yokoyama S, Hirao I. Nat Biotechnol. 2013;31:453–458. doi: 10.1038/nbt.2556. [DOI] [PubMed] [Google Scholar]
- 56.St Amant AH, Bean LA, Guthrie JP, Hudson RHE. Org Biomol Chem. 2012;10:6521–6525. doi: 10.1039/c2ob25678a. [DOI] [PubMed] [Google Scholar]
- 57.For the first description of these kind of “fleximers”, see; Seley KL, Zhang L, Hagos A. Org Lett. 2001;3:3209–3210. doi: 10.1021/ol0165443. [DOI] [PubMed] [Google Scholar]; Seley KL, Zhang L, Hagos A, Quirk S. J Org Chem. 2002;67:3365–3373. doi: 10.1021/jo0255476. [DOI] [PubMed] [Google Scholar]
- 58.Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Wiley; New York: 1984. pp. 1–176. [Google Scholar]; Huisgen R. Proc Chem Soc. 1961:357–396. [Google Scholar]
- 59.Krishnamurthy R. Acc Chem Res. 2012;45:2035–2044. doi: 10.1021/ar200262x. [DOI] [PMC free article] [PubMed] [Google Scholar]