

Abstract
The 2013–2015 outbreak of Ebola virus disease in Guinea, Liberia, and Sierra Leone was unprecedented in the number of documented cases, but there have been few published reports on immune responses in clinical cases and their relationships with the course of illness and severity of Ebola virus disease. Symptoms of Ebola virus disease can include severe headache, myalgia, asthenia, fever, fatigue, diarrhea, vomiting, abdominal pain, and hemorrhage. Although experimental treatments are in development, there are no current U.S. Food and Drug Administration–approved vaccines or therapies. We report a detailed study of host gene expression as measured by microarray in daily peripheral blood samples collected from a patient with severe Ebola virus disease. This individual was provided with supportive care without experimental therapies at the National Institutes of Health Clinical Center from before onset of critical illness to recovery. Pearson analysis of daily gene expression signatures revealed marked gene expression changes in peripheral blood leukocytes that correlated with changes in serum and peripheral blood leukocytes, viral load, antibody responses, coagulopathy, multiple organ dysfunction, and then recovery. This study revealed marked shifts in immune and antiviral responses that preceded changes in medical condition, indicating that clearance of replicating Ebola virus from peripheral blood leukocytes is likely important for systemic viral clearance.
Introduction
During the 2013–2015 Ebola virus disease (EVD) outbreak in Guinea, Liberia, and Sierra Leone, the World Health Organization reported 28, 616 cases with 11, 310 deaths (1, 2). This outbreak, caused by a Makona variant of Ebola virus (EBOV), had an estimated 40% case fatality rate (3, 4).
EBOVs infect numerous cell types, including monocytes, macrophages, dendritic cells (DCs), endothelia, epithelia, and hepatocytes (3). Infection leads to suppression of antiviral responses, alterations in proinflammatory responses, and immune cell dysregulation (5, 6). Severe EVD in humans occurs after EBOV exposure to non-intact skin or mucosa. Clinical illness onset follows an average incubation of 6 days and is characterized by neuromuscular weakness, fever, and asthenia (7–9). High-level viremia and broad tissue tropism result in widespread infection (10). Complex pathogenesis due to direct viral-mediated cellular injury and indirect host-mediated injury contributes to multiple organ dysfunction (MOD) (11). Hypovolemic shock, attributable to gastrointestinal fluid losses from recurrent episodes of emesis and large-volume watery diarrhea, contributes to tissue ischemia and mortality (8). Although much has been learned about EVD in humans from this recent large outbreak, detailed molecular studies of EVD in humans are critical because significant gaps remain in our knowledge of the molecular aspects of disease and associated clinical correlates.
Here, daily global gene expression responses were measured in peripheral blood from a 34-year-old male health care worker exposed to EBOV in Sierra Leone and evacuated to National Institutes of Health (NIH) on day 7 (d7) of clinical illness through onset of critical illness and recovery (12). This patient received supportive care without experimental therapies and, after recovery and viral clearance from blood, was released from the hospital on d33. Kinetics of gene expression were characterized using Pearson correlation and principal components analyses (PCAs) to identify key transition points. Gene ontology and pathway mapping of these major transition points, and the overall gene expression response, were performed to characterize host responses that correlated with EVD, including replication of virus in peripheral blood leukocytes (PBLs), serum viral load, coagulopathy, critical illness, and recovery.
Results
PBL global gene expression response shows distinct phases correlating with viral replication and different stages of illness
To determine the kinetics of viral replication in PBLs and its effect on gene expression, we performed strand-specific quantitative reverse transcription polymerase chain reaction (qRT-PCR) to measure both EBOV nucleoprotein (NP) mRNA as a marker of active viral replication and NP vRNA as a marker of presence of virions. EBOV NP mRNA was detected from d7 to d11, peaked at d9 [cycle threshold (Ct) = 31], and then declined to below detection by d12 (Fig. 1A). EBOV NP vRNA was detected from d7 to d14, peaked at d8 to d9 (Ct = 29), and then decreased to below detection by d15. These data showed that PBL EBOV replication, as indicated by the presence of NP mRNA, ceased by d12, and the presence of EBOV genomic RNA, as measured by NP vRNA, was cleared from PBLs by d15. The peak of PBL-associated EBOV NP mRNA and vRNA on d8 to d9 approximated the peak of serum EBOV glycoprotein (GP) RNA on d7 to d8, reported previously (12). EBOV RNA in PBLs peaked just before the patient was downgraded to critical condition on d10. The level of PBL-associated NP mRNA showed a sudden rise on d7 to d9, suggesting that during the start of infection, rapid replication in a small population of cells, likely myeloid cells, drove serum EBOV RNA to peak on d9.
Fig. 1. Overall gene expression profile in peripheral blood shows distinct phases during EVD and recovery.

(A) Kinetics of EBOV NP mRNA and vRNA in PBLs as measured by strand-specific qRT-PCR during course of illness. Serum EBOV GP RNA qRT-PCR data previously published in (12) are shown for comparison with PBL replication. (B) Heatmap showing expression levels of genes (n = 8660 sequences) that showed ≥2-fold change in expression level in at least 30% of time points relative to a pool of 29 healthy adult volunteers. For all figures, each heatmap column represents gene expression data from a microarray experiment comparing patient peripheral blood at individual time points relative to pooled RNA isolated from peripheral blood from healthy volunteers (n = 29). Genes shown in red were increased, genes shown in blue were decreased, and genes in black indicate no change in expression in EVD patient relative to healthy volunteers. (C) Pairwise Pearson correlation coefficient similarity matrix of expression values of these 8660 sequences. *Previously reported in (12).
Microarrays were used to measure gene expression responses in PBLs on 21 of the 26 days of inpatient care (d7 to d22, d24, d25, d28, d31, and d33) and 4 outpatient visits (d59, d103, d180, and d270). Expression values from the patient's PBL at each time point were compared to expression in a pool of PBL RNA from 29 healthy adult donors. This analysis showed that 8660 sequences were differentially expressed with at least a twofold change in concentration in at least 30% of time points (Fig. 1B). Pearson analysis was performed to understand the relationship between gene expression changes and patient's course of EVD.
A two-dimensional similarity matrix (Fig. 1C), composed of pair-wise Pearson correlation coefficients of gene expression levels, showed highest similarity between samples taken on d7 to d13 (phase 1), d14 to d21 (phase 2), d22 to d24 (phase 3), and d25 to d28 (phase 4) and during outpatient visits between d59 and d270 (phase 5). This analysis showed that gene expression responses in PBLs underwent several phases from onset of critical illness through recovery, generally preceding changes in clinical classification.
Gene expression differences correlated with changes in serum EBOV RNA levels
Correlation analysis was next performed using clinical data, including EBOV GP RNA in serum (systemic viral load), EBOV NP mRNA in PBLs (viral replication in PBLs), D-dimer and platelet counts (coagulopathy), and serum creatinine as a surrogate marker for critical illness. Pearson analysis was performed to identify host transcripts whose expression correlated with kinetics of serum EBOV GP RNA, with a Pearson correlation coefficient of ≥0.6. This value was chosen to maximize our ability to identify transcripts whose expression levels changed in accordance with changes in serum EBOV GP RNA levels and to identify subtle transition points in host responses during the course of inpatient illness. Similar to PBL gene expression in Fig. 1C, the Pearson similarity matrix (Fig. 2A) revealed four phases with three major transition points in gene expression responses that were positively correlated (n = 2690) and negatively correlated (n = 1071) with serum EBOV GP RNA Ct values (Fig. 2B). These marked transitions in gene expression profiles occurred on d13 to d14 (phase 1 to 2), d20 to d21 (phase 2 to 3), and d25 to d28 (phase 3 to 4).
Fig. 2. Gene expression correlating with kinetics of serum EBOV load.

(A) Pairwise Pearson correlation coefficient similarity matrix of expression values of transcripts positively correlated (≥0.6) with EBOV GP RNA levels. (B) Heatmap showing overall expression level of genes with positive (n = 2690 sequences) or negative (n = 1071 sequences) correlation with serum Ebola GP RNA (12). (C) Scatterplot showing relative expression of genes at d13/d14. Red points indicate sequences with higher expression on d13 (n = 2350), and blue points indicate sequences with higher expression on d14 (n = 655). (D) Scatterplot showing relative expression of genes at d20 and d21. Red points indicate sequences with higher expression on d20 (n = 550), and blue points indicate sequences with higher expression on d21 (n = 431). (E) Scatterplot showing relative expression of genes at d25/d28. Red points indicate sequences with higher expression on d25 (n = 132), and blue points indicate sequences with higher expression on d28 (n = 1688).
Gene ontology mapping of positively correlated transcripts showed activation of antiviral, inflammatory, oxidative stress, DNA damage, cell death, and wound healing responses (table S1). Antiviral responses included type I interferon (IFN) signaling and pattern recognition receptor signaling, whereas immune and inflammatory responses included innate immune response markers, antigen processing, and presentation of peptide antigen via major histocompatibility complex (MHC) class I, response to IFN-γ, leukocyte activation, cellular response to cytokine stimulus, regulation of interleukin-6 production, and response to stress. A marked increase in expression of genes involved in response to oxidative stress and DNA damage was also observed, including signal transduction by p53, G1 DNA damage checkpoint, response to DNA damage, and activation of cell death responses, including apoptotic signaling, tumor necrosis factor (TNF)– mediated signaling, and regulation of programmed cell death, along with expression of genes involved in coagulation, wounding, and repair responses.
Gene ontology analysis of transcripts negatively correlated with serum EBOV GP RNA Ct values was dominated by repair-related pathways, including regulation of ion transport, cell-cell signaling, anatomical structure morphogenesis, anatomical structure development, cell differentiation, system development, and multicellular organism development. Intriguingly, 49% of sequences (n = 529) negatively correlated with serum EBOV GP RNA Ct values were unmapped using gene ontology terms (table S2).
To identify host responses underlying these transitions, we directly compared gene expression profiles of d13 and d14, d20 and d21, and d25 and d28 (Fig. 2, C to E). Analysis of individual transition points, phase 1 to 2 (d13 to d14), phase 2 to 3 (d20 to d21), and phase 3 to 4 (d25 to d28), revealed that different pathways dominated different transition points.
Gene expression correlated with host response transition on d13 to d14 of illness
To determine pathways differentiating d13 to d14, we directly compared the expression of transcripts positively correlated with serum EBOV GP RNA Ct values on these days. This analysis identified 1695 transcripts ≥1.5-fold more highly expressed on d13 relative to d14 and 655 transcripts ≥1.5-fold more highly expressed at d14 relative to d13 (Fig. 2C). Ontology analysis of transcripts with higher d13 expression showed enrichment of antiviral, innate immune, inflammatory, reactive oxygen species (ROS), and cell death responses (table S3). These included type I IFN signaling, response to IFN-γ, regulation of cytokine secretion, respiratory burst by NADH (reduced form of nicotinamide adenine dinucleotide)–dependent system producing hydrogen peroxide, super-oxide anions and hydroxyl radicals, positive regulation of nitric oxide synthase biosynthesis, regulation of TNF superfamily production, and regulation of endopeptidase activity involved in apoptotic signaling.
Gene ontology analysis of transcripts with higher d14 expression was dominated by cell proliferation and adenosine 5′-triphosphate (ATP) biosynthesis, ubiquitin-mediated proteolysis, and DNA damage (including mitochondrial DNA damage), and DNA damage–associated cell cycle arrest, nuclear factor κB (NF-κB)–inducing kinase (NIK)/ NF-κB signaling, and chromatin assembly (table S4). The d13 to d14 transition was also associated with induction of MHC class II genes in peripheral blood (fig. S1), and serum immunoglobulin analysis showed that Ebola- and GP-specific immunoglobulin Gs(IgGs) were detected starting on d13 to d14 (fig. S2).
Gene expression correlated with host response transition on d20 to d21 of illness
An identical approach was used to identify transcripts with ≥1.5-fold higher expression on d20 versus d21. This analysis identified 550 transcripts with higher expression on d20 and 431 transcripts with elevated expression on d21 (Fig. 2D). Gene ontology analysis showed that transcripts with higher d20 expression (n = 550) were dominated by innate immune response markers, cytokine-mediated signaling, defense response to virus, mitotic cell cycle process, and regulation of protein metabolic process (table S5). In contrast, gene ontology analysis of transcripts with higher d21 expression showed repair, oxidative phosphorylation, and metabolic responses, including respiratory electron transport chain, nucleoside triphosphate metabolism, cellular respiration, mitochondrial ATP synthesis–coupled electron transport, oxidative phosphorylation, and glycosyl compound metabolism (table S6). The d20 to d21 transition was also associated with appearance of neutralizing antibodies (fig. S3), clearance of EBOV GP RNA from serum, a decrease in total PBL counts, end of renal and respiratory failure, and upgrading from critical to serious condition.
Gene expression correlated with host response transition on d25 to d28 of illness
The d25 to d28 transition was similarly assessed and identified 132 transcripts with ≥1.5-fold higher expression on d25 relative to d28 (Fig. 2E). The lack of enriched biological processes was likely due to the small number of sequences (table S7). However, several type I IFN-stimulated genes (ISGs), including IFITM1, IFITM2, IFITM3, IFIT1, IFI6, and SOCS3, were identified. Several neutrophil function–related genes (MPO and CD177) and innate inflammatory response–related genes (IL1R2, LY96, MMP9, and TNFAIP6) were also present. Apoptosis-related genes (including DEDD2 and OLFM4) were also identified with higher expression on d25. Several genes (SOCS3, IL1R2, TNFAIP6, and OLFM4) are negative regulators of cytokine-related responses, suggesting a role in down-regulating inflammatory and apoptosis-related responses.
Gene ontology of sequences with ≥1.5-fold higher d28 expression (n = 1688) showed activation of antigen presentation–related responses and a slight reactivation of antiviral and inflammatory responses (table S8), although with much lower expression than was observed on d7 to d13 (fig. S4). These included response to type I IFN signaling, positive regulation of leukocyte-mediated immunity, IFN-γ–mediated signaling, antigen processing and presentation of exogenous peptide antigen via MHC class I, and Fc-γ receptor signaling and phagocytosis.
Gene expression correlated with PBL EBOV NP mRNA levels
To characterize the effect of EBOV replication and clearance from PBLs on host gene expression in PBLs, we performed correlation analysis (Pearson coefficient ≥ 0.6) using PBL EBOV NP mRNA Ct values. This analysis identified transcripts (n = 444) with expression levels that correlated with PBL EBOV replication (Fig. 3A). Ontology analysis revealed significant enrichment for antiviral, inflammatory, and cell death–related responses (table S9), including type I IFN signaling, IFN-γ–mediated signaling, cytokine-mediated signaling, and positive regulation of programmed cell death. Cell death– and immune response– correlated genes included CASP1, CASP4, RIPK3, CXCL1, IL1B, IL2RG, LY96, and high levels of C2 (>20-fold increase at peak between d8 and13). This population was dominated by expression of type I ISGs that markedly decreased between d13 and d14 (Fig. 3B). These genes included canonical ISGs, such as ADAR, IRF7, IRF9, OAS1, OAS2, OAS3, IFIT1, IFIT3, MX1, IFI35, IFI44, ISG15, and IFI6. Multiplex qRT-PCR was also performed on a subset of type I IFNs and ISGs and showed that three type I IFN genes (IFNB1, IFNA6, and IFNA21), cytosolic viral RNA sensor (IFIH1), and ISGs (IFI27, IFI44, IFI6, IFIT1, IFIT2, IFIT3, ISG15, MX1, and OAS1) were more highly expressed on d13. (Fig. 3C; additional qRT-PCR data in fig. S5). Transcripts with higher d14 expression were related to activation of lymphocytes, including IFNG and IRF4, and may represent a shift from an innate immune cell–dominated response on d13 to a lymphocyte-predominant response on d14. Direct comparison of cytokine signaling and cell death–related pathways on d13/d14 showed that these were similarly more highly expressed at d13 than on d14 (Fig. 3, D and E, respectively). Thus, the transition between phases 1 and 2 was driven predominantly by decreased antiviral response–related gene expression and an increase in adaptive immune response markers.
Fig. 3. Gene expression associated with Ebola viral replication in PBLs.

(A) Quantification of EBOV NP mRNA (qPCR Ct) in PBLs and heatmap showing expression of genes with positive correlation (≥0.6) with PBL EBOV NP mRNA. (B) Heatmap showing expression levels of key ISGs. (C) Graph showing relative expression of selected ISGs from (B) as determined by qRT-PCR. (D) Scatterplot showing relative expression of cytokine signaling–related genes (n = 51) at d13/d14. (E) Scatterplot showing relative expression of cell death–related genes (n = 29) at d13 and d14. Red points indicate sequences with higher expression on d13, and blue points indicate sequences with higher expression on d14.
Gene expression associated with distinct phases of innate and adaptive immune responses
Expression profiles of inflammatory mediators (n = 3518) were examined to identify immune response markers critical for controlling viral replication and mediating viral clearance. Of these, 1029 transcripts showed at least a twofold difference in expression relative to normal controls in at least four time points. A correlation matrix, composed of pairwise Pearson correlation coefficients of transcript expression values, demonstrated high similarity between samples taken on d7 to d13 (phase 1), d14 to d21 (phase 2), d22 to d24 (phase 3), and d25 to d270 (phase 4) (Fig. 4A). These inflammation-related gene expression–driven phases were comparable with the phases of gene expression correlating with EBOV GP RNA in serum (Fig. 2A).
Fig. 4. Distinct phases of innate antiviral and adaptive immune response markers during EVD.

Analysis of inflammation-related gene expression (n = 1029 transcripts) with ≥2-fold change in expression level in at least four individual time points. (A) Pairwise Pearson correlation coefficient similarity matrix of expression values of these transcripts. (B) Heatmap showing expression of innate antiviral and adaptive immune response markers. (C) Total white blood cell (WBC) count and differential analysis of immune cell populations.
K-means analysis of these transcripts further identified groups of potentially co-regulated genes based on expression level and pattern (Fig. 4B) that generally correlated with viral kinetics in either PBLs or serum. Transcripts in groups I (n = 183) and II (n = 34) were most highly expressed from d7 to d13 (Fig. 4B). During this period, serum EBOV GP RNA levels peaked (d8) followed by decline (Fig. 1A). After d13, expression levels of these transcripts decreased but remained elevated until d25 before decreasing to near-baseline levels. During this period, serum EBOV GP RNA levels continued to decline until they become undetectable at d24. Groups I and II represent some of the most highly expressed transcripts at any time point and include classic antiviral response–related genes (IFIT1-3, ISG15, IFI27, IFI35, and IFI44) and type I and II IFN signaling (OAS2, OAS3, Mx1, IRF7, and IRAK3) that are likely important for attenuating viral replication. Upstream analysis identified key antiviral response–related transcription regulators (STAT1, STAT2, STAT3, and IRF1/IRF7) and multiple IFNs (IFNL1 and IFNα/β/γ) as upstream regulators (table S10). That expression of these transcripts remained high despite a concomitant decrease in serum EBOV RNA levels suggests a role in control of viral replication.
Expression of transcripts in groups III (n = 61), IV (n = 375), and V (n = 229) were generally consistent from d7 to d24, after which levels declined (table S10). Transcripts in these groups were dominated by genes involved in T and B lymphocyte responses and coincided with both clearance of EBOV RNA from peripheral blood and an increase in Ebola-specific IgG titers (figs. S2 and S3). Sequences in group III were the most highly expressed and remained elevated even at d270. Gene ontology analysis showed enrichment for lymphocyte-related functions, including April-mediated signaling, B lymphocyte activating factor signaling, interleukin-6 (IL-6) signaling, and granzyme A signaling. Further, there were genes associated with lymphocyte homeostasis/ proliferation (CD70, TNFRSF13B, TNFRSF17, IRF5, SOCS3, HAVCR2, and MAPK14) and protein folding/trafficking (FKBP1A and HSPA5). Predicted upstream regulators of these sequences included cytokines (CD40LG, IFN-γ, TNF, IFNα, IL-2, and IL-3) and the transcription factor signal transducer and activator of transcription 1 (STAT1) that function in both innate and adaptive immune responses (table S10). Transcripts in group IV were also enriched for adaptive immune response functions, death receptor signaling, including the FAS pathway (CASP8, CASP9, CFLAR, and FADD). Consistent with death receptor signaling, there was enrichment of genes associated with epithelial cell death and lymphocyte development/activation/proliferation, possibly reflective of cell-mediated immune responses targeting infected cells. Predicted upstream regulators of this group were consistent with T and B lymphocyte responses [including CD40LG, CD3, T cell receptor (TCR), IL-2, IL4, BCR, and IFN-γ] (table S10). Transcripts in group V also represented processes required for clearance of infected cells, including genes that regulate T lymphocyte development/differentiation. In addition, neutrophil recruitment/activation (CCL3, CCL4, CD14, and ITGAM), ROS, and macrophage/antigen-presenting cell–related genes were enriched, possibly reflecting repair processes after cell-mediated tissue damage (table S10). Collectively, immune response–related gene expression encompassing d7 to d24 was dominated by T and B lymphocyte–mediated activity, coinciding with the steady decline and subsequent clearance of EBOV RNA from serum, likely reflective of responses critical for clearance of infected cells and development of protective immunity.
Groups VI (n = 95) and VII (n = 52) included transcripts with expression levels that increased after viral clearance from serum (Fig. 4B). Sequences in group VI showed slightly elevated levels until d24, at which time they increased further (table S10). Sequences in group VII showed decreased levels relative to controls until d24 when they increased. The expression level of transcripts in both groups remained elevated throughout the remaining time points, coinciding with patient recovery. Some genes are involved in hematopoiesis/immune cell function, including development of pro-T lymphocytes (BB3, CD3D, IL2RB, CD27, Bax, and ZFPM1), recruitment of memory T lymphocytes (CCL5 and CCR10), differentiation of DC (CSFIR), and Nurr77 signaling (BCL2, MEF2D, and RXRA). These processes may reflect renewal of homeostatic immune cell populations after viral clearance.
Total PBL numbers were elevated between d8 and d17 and again on d21 and d24, after which they returned to normal range (4230 to 9070 cells/μl) (Fig. 4C). At no point during the illness were they below normal range, although it is possible that leukopenia and lymphopenia (commonly described during EVD) (13, 14) occurred in this patient before d7. The monocyte population steadily increased beginning at d7 and was above normal range (300 to 820 cells/μl) between d10 and d24, peaking at d15 and then declining (Fig. 4C). The neutrophil population was elevated above normal range (1780 to 5380 cells/μl) from d8 to d25 (Fig. 4C). Lymphocytes were within normal range (1320 to 3570 cells/μl) throughout all but d14 to d15 when they were elevated, coinciding with clearance of NP vRNA from PBL. The multiple phases of increased lymphocytes in PBL possibly reflected alternating replenishment from lymphopoiesis and extravasation into tissues to target infected cells. Notably, return of all immune cell subsets to normal range also coincided with clearance of EBOV RNA from serum.
EVD is associated with moderate induction of key inflammatory chemokines and cytokines that generally peak before critical illness phase of EVD
Dysregulated cytokine production has been linked to lethal EBOV infection (13–17). The critical phase of illness (d10 to d19) was not associated with the highest levels of inflammatory mediators at either the mRNA or protein level in this patient (figs. S6 to S9). Increased mRNA levels of only limited inflammatory mediators were observed throughout the time course, and levels generally peaked before d10. The expression profiles of chemokines/receptors and cytokines/receptors that showed at least a twofold difference in expression level at one time point are represented in Fig. 5 (A and B, respectively). Of classical inflammatory mediators, only IL1-b mRNA was elevated relative to healthy controls consistently from d7 to d12. IL-15 mRNA, which is secreted by monocytes in response to viral infection and induces proliferation of natural killer (NK) cells (18) and memory CD8 T cell survival (19), was increased until d12, and its subsequent decrease was possibly related to clearance of EBOV NP mRNA in PBLs (Fig. 1A). The mRNAs encoding several other cytokines, including IL-21, IL-26, IL-32, IL-11, IL-27, and IL-17F, were transiently increased on isolated days. Expression levels of other cytokines that showed differential regulation were decreased, suggesting either suppression or absence of cells that express these mediators.
Fig. 5. Strongest induction of inflammatory mediators and receptors generally occurred during early EVD.

Heatmaps depicting expression levels of mRNAs encoding chemokines/receptors (A) and cytokines/receptors (B) during EVD and recovery. Gray indicates that the sequence was not detected.
Chemokine mRNAs, particularly the CC class, were generally more highly and consistently elevated than cytokines, although many also had levels lower than controls. The levels of CCL3L3, CCL3, CXCL2, and CXCL1 mRNAs were generally higher at earlier time points, although CCL3 continued to be elevated until later time points. CCL4 mRNA levels were highest on d7 to d8 and d16 to d24, remaining elevated until well after viral clearance from serum. CCL23 mRNA levels, highly chemotactic for resting T lymphocytes and monocytes (20), were highest on d11 to d13, preceding an increase in peripheral lymphocyte and monocyte populations (Fig. 4C). CCL25 mRNA levels, thought to play a role in recruiting progenitor T lymphocytes to the thymus, were highest between d13 and d22, roughly coinciding with critical illness, but remained elevated until d28. Levels peaked at d15 that corresponded to the highest PBL counts (Fig. 4C). Although not as highly induced as CCL25, CXCL16 mRNA peak levels also corresponded generally to the critical phase of illness (Fig. 5A). Transcript levels of many other chemokines (CCL15, CCL24, CCL21, CCL27, CXCL5, and CXCL14) were lower than normal controls (Fig. 5B), particularly during the presence of EBOV in serum.
Selected cytokines and chemokines present in peripheral blood were also measured by enzyme-linked immunosorbent assay (ELISA) (figs. S6 to S9) and generally supported mRNA expression data in that many classical inflammatory mediators were not induced. Those that were elevated typically peaked before onset of critical illness, when tissue injury preceding organ failure was highest. Of cytokines measured, TNFα, IL-12p70, IL-2, IL-4, and IL-5 (fig. S6) were not detectable above background at any time point. IL-1β was only detected on d7, whereas other mediators were detectable on d7 to d8/d9 (IL-13, IL-17a, and IFN-γ) (fig. S7). IL-8 was highest at d7, after which it decreased but remained detectable until d22, just before clearance of virus from serum. Similarly, IL-10 decreased significantly from d7 and became undetectable by d14. IL-6 levels were detectable from d7 followed by an increase on d9, peaking on d11 and then decreasing rapidly to low levels and becoming undetectable by d17. IL-7 was intermittently detectable (d7, d11, d12, d20, and d21) (fig. S8). Granulocyte colony-stimulating factor (G-CSF), which stimulates production of granulocytes (21), was highest on d7 and then decreased significantly by d8, just before an increase in neutrophils between d9 and 10 (Fig. 4C and fig. S9). It remained detectable at low levels until d22. Granulocyte-macrophage colony-stimulating factor (GM-CSF), which stimulates production of granulocytes and monocytes (22), showed a similar expression pattern, with the highest levels occurring on d7 to d8 and then decreasing until it was undetectable by d12 (fig. S9). This was just before a gradual increase in monocytes that occurred between d9 and d15 (Fig. 4C). The chemokines CCL2 (MCP-1) and CCL4 [macrophage inflammatory protein-1 β (MIP-1β)] were both elevated to varying degrees (fig. S9). The levels of CCL2 were also highest on d7, decreasing on d8 but remaining relatively constant until becoming undetectable on d14. Levels of CCL4 were also highest on d7 but remained relatively high for all time points analyzed (up to d31), consistent with increased mRNA expression until d103 (Fig. 5A). This observation, along with inflammatory gene expression, suggests that some aspects of the inflammatory response remained stimulated well after viral clearance from serum in this patient, possibly resulting from maintenance of virus in immune-privileged sites such as the eye and testes (23–26).
mRNAs encoding receptors for cytokines and chemokines generally showed higher and more sustained expression than the ligands, particularly the cytokine and CXC class of chemokine receptors (Fig. 5, A and B). Whereas some cytokine receptors (IL11RA, IL9R, IL7, IL17RE, IL15RA, CXCR5, and CXCR7) showed decreased expression, many were highly expressed over multiple days, including components of the IL-1 receptor (IL1R2, IL1RN, and IL1R1), IL-18 receptor (IL18R1 and IL18RAP), and others (IL17RA, IL15RA, IL10RB, IL4RB, IL6RA, IL21R, IL2RB, and IL2RA). Transcripts of many chemokine receptors, including CCR1, CCR2, CCRL2, CCR5, CXCR1, CXCR3, and CXCR6, were also highly expressed over multiple time points, whereas others showed decreased expression (CCR3, CCR7, CXCR5, and CXCR7). Because chemokine/cytokine receptors are commonly expressed on immune cells, the general lack of high levels of CXC chemokine/cytokine mRNAs may be more related to dysregulated immune cell function than the absence of specific immune cell subsets.
Gene expression underlying clinical markers of coagulopathy suggest viral-mediated death of macrophages, neutrophil activation, and increased phagocytosis of immune cells during active viral replication
Perturbations in coagulation have been observed during EVD (27–29). Abnormal platelet levels, D-dimer (fibrin degradation product), and aPTT (activated partial thromboplastin time) occurred between d7 and d15, consistent with coagulopathy previously described in this patient (12). Specifically, D-dimer levels were elevated (>20 μg/μl) from at least d7, returning to normal levels (5 μg/μl) around d16 (Fig. 6A). Platelet counts remained below normal levels (lower limit of normal ∼150,000/μl) until d16, increased to almost 500, 000 at d20, and then declined but remained within normal range through d33 (Fig. 6A).
Fig. 6. Gene expression correlating with clinical parameters of coagulopathy during EVD.
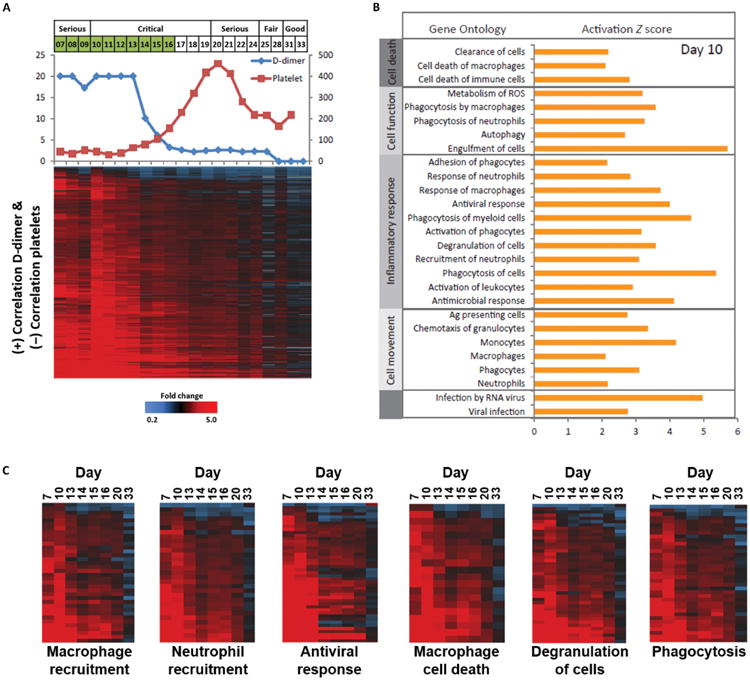
(A) Serum D-dimer (μg/ml, left axis) and platelet levels (K/μl, right axis) and heatmap showing expression profiles of transcripts (n = 1016) with expression levels that are correlated positively with serum D-dimer levels (≥.6) and negatively with platelet levels (≤-0.6). (B) Gene ontology analysis of transcripts, with expression levels correlating with serum D-dimer and platelet values (activation Z score is based on d10 expression values). (C) Expression profiles of transcripts involved in macrophage recruitment, neutrophil recruitment, antiviral response, macrophage cell death, degranulation, and phagocytosis.
D-dimer and platelet levels throughout the illness were used as reference profiles to identify transcripts whose expression level correlated with clinical parameters of coagulopathy to better understand underlying pathological processes. Correlation analysis (≥0.6) identified 112 and 1516 transcripts that negatively and positively correlated with D-dimer levels, respectively, between d7 and d33 and were also twofold differentially expressed in at least four time points (fig. S10 and tables S11 and S12). Similarly, 1060 and 56 transcripts were identified that negatively and positively, respectively, correlated with platelet levels between d7 and d33 (fig. S11 and tables S13 and S14). Not surprisingly, there was significant overlap in transcripts that showed positive correlation with D-dimer and negative correlation with platelet levels (n = 1060) (table S15). Expression profiles of these transcripts, along with D-dimer and platelet levels, are shown in Fig. 6A. Most showed elevated expression levels that decreased parallel to D-dimer levels decreasing and platelet levels increasing. These likely represent cellular processes that were being attenuated after resolution of coagulopathy, although they generally remained elevated to some degree beyond clinical resolution of coagulopathy at d15.
Gene ontology analysis of these 1060 transcripts revealed enrichment for genes associated with innate immune responses including macrophage/monocyte- and neutrophil-related activities (Fig. 6, B and C). Specifically, expression of classic antiviral response genes (including IFIT1, IFIT2, IFIT3, IFITM1, IFITM2, OAS1, OAS2, STAT1, and MX1) and neutrophil recruitment/degranulation (CXCR1, CXCR2, and others) was increased coincident with elevated D-dimer and depleted platelet levels (Fig. 6C). Genes involved in macrophage recruitment (including IL1b, CCR1, and ITBG2), death (including inflammasome-related NLRC4 and AIM2), and phagocytosis/engulfment of cells also correlated with clinical parameters of coagulopathy (Fig. 6C). One possible explanation for these gene expression changes is that coagulopathy results from viral replication and associated cytopathic effect in macrophages and DCs. Subsequent engulfment of infected cells by macrophages and neutrophils may then induce fibrin clot formation, possibly via activated neutrophils mediating thromboinflammatory injury. The decrease in D-dimer levels between d13 and d14 coincided with both clearance of EBOV NP mRNA from PBL by d12 (Fig. 1A) and significant attenuation of antiviral response gene expression, with resolution of coagulopathy occurring at d15.
Critical phase of illness characterized by decreasing serum EBOV RNA levels and transitions in lymphocyte, cell death, and organ repair–related gene expression
The patient's clinical condition deteriorated from serious to critical on d10 to d19, after which it was upgraded back to serious (12). This period reflected MOD associated with EVD. These transition points correlated closely with serum creatinine levels, which began rising at d10, peaked at d15, and returned to normal levels by about d20 (Fig. 7A). Therefore, creatinine levels were used as a surrogate marker of transitions to and from critical illness. Transcripts with expression kinetics correlating with serum creatinine levels were identified to gain insight into possible pathological processes underling the critical phase of illness.
Fig. 7. Gene expression correlating with the critical phase of EVD.

(A) Serum creatinine values (U/liter, left axis) and EBOV GP levels (qPCR Ct, right axis), with heatmap showing expression profiles of transcripts with positive (≥0.6, n = 576) or negative (≥−0.6, n = 420) correlation with serum creatinine levels. (B) Gene ontology analysis of transcripts, with expression levels correlating with serum creatinine values (activation Z score is based on d15 expression values).
Expression profiles of 420 transcripts (table S16) showed a negative correlation with creatinine levels and also a twofold difference in expression level compared to healthy controls across at least three sample times (Fig. 7A). Most showed similar or slightly decreased levels relative to healthy controls from d7 to d10, followed by a general decrease until d14/d15, after which they returned to near baseline. A subset of these transcripts showed increased expression relative to healthy controls on d7 to d10, followed by decreased expression between d11 and d20, and subsequent return to increased levels relative to healthy controls (Fig. 7A).
Gene ontology analysis of these transcripts showed enrichment for lymphocyte-related gene expression including recruitment, development, differentiation, proliferation, and activation of lymphocytes (Fig. 7B). Expression levels of certain lymphocyte activation markers (FasLG, granzyme K, and ICOS), NK markers (KLRB1, KLRC3, and KLRD1), and T lymphocyte markers (CD3D, CD83, CD8A, CD8B, and IL2RB) generally decreased from d9 to d14/d15 and then began to increase until d18/d19 (fig. S12). In contrast, total lymphocyte counts began increasing at d13 and peaked at d15 before decreasing again (Fig. 4C), suggesting that a subset of lymphocytes was leaving the periphery through either apoptosis, as suggested by positive activation Z score for lymphocyte apoptosis (Fig. 7B), or extravasation into tissues. Serum EBOV RNA level was continually dropping during this phase (Fig. 1A), possibly as a result of cell-mediated immune responses targeting infected tissue. EBOV is known to have a broad tissue tropism (3, 11), resulting in direct virally mediated cytopathology and MOD. Organ injury leading to MOD was reached by d9/d10, manifested by rising serum creatinine, down-trending but elevated hepatic enzymes, and onset of respiratory failure (12). A transition to organ recovery was observed by d14/d15, manifested by decreasing creatinine and normalization of hepatic enzymes and serum creatinine phosphokinase levels. Cytotoxic T lymphocyte responses during this period of critical illness likely contributed to viral clearance from tissues tipping the balance toward tissue repair and recovery. This hypothesis is supported by the observation that inflammation-related gene expression during this phase was dominated by adaptive immune response markers and decreasing EBOV RNA levels (Fig. 4B).
Positively correlated transcripts (n = 576) (table S17) generally showed slightly elevated levels relative to healthy controls and began increasing at d10 parallel to serum creatinine levels, peaking at d15 followed by gradual decrease, although they still generally remained slightly elevated. Gene ontology analysis of these transcripts shows increased activity (based on positive activation Z score) of numerous processes associated with tissue repair/recovery, including cell survival, development/proliferation of endothelial cells and angiogenesis (BMP6, ANG, GAS6, NCOA3, EDF1, PDCL3, PLOD3, SCARB1, and CLIC4), and epithelial tissue development (Fig. 7B). Processes predicted to be decreased based on expression level at d15 include organ death, apopto-sis, and necrosis (Fig. 7B).
The immune-related genes were dominated by antiviral responses and adhesion/activation (CD36, ITGB1, CCR2, and MIF) of antigen-presenting cells including macrophage/monocytes (Fig. 7B). There was also enrichment of genes related with proliferation and adhesion of immune cells (including IL27RA, CCR2, and CXCR3) and phagocytosis/ engulfment of cells (ClQA, CD36, Syk, Tusc2, and GAPVD1), possibly due to clearance of apoptotic bodies resulting from lymphocyte targeting of infected cells. In support of this, enriched pathways included the unfolded protein response (including ATF6, DNAJC3, HSP90B1, and HSPA5) and NRF2-mediated oxidative stress response (such as DNAJC3, DNAJC10, GSTP1, GSK3M, and TXNRD1) (fig. S13). Genes associated with oxidative phosphorylation, the predominant mechanism used by lymphocytes to fuel their effector functions (30), were also overrepresented and included ATP5G1, COX6B1, and numerous genes encoding NADH–ubiquinone oxidoreductase subunits (NDUFA3, NDUFB7, NDUFB11, NDUFS1, NDUFS7, NDUFS8, and NDUFV3) (fig. S13).
Collectively, these data suggest that the critical phase of illness was associated with declining serum EBOV RNA levels, possibly related to cell-mediated immune responses targeting infected tissues, and increasing activation of organ repair processes. Further, peak expression of these genes at d15 suggests that it represented a key transition point in patient illness when underlying cellular processes shifted from cellular/ tissue damage to organ/endothelium repair. d15 also corresponded to both peak serum creatinine levels and peripheral lymphocyte counts and clearance of EBOV NP vRNA from PBL (Fig. 1A). Subsequent decreasing levels of tissue repair–related gene expression likely reflected attenuation of these responses, as viral/immune response–mediated tissue damage was reduced after viral clearance along with reparative processes in tissues and organs.
To understand the complexity of EVD in this patient, a plot integrating PBL gene expression, antibody responses, and clinical symptoms (12) is shown in Fig. 8. Daily global gene expression responses were spatially arranged into a two-dimensional coordinate system using values from Eigen rows 1 and 2 derived from PCA of the gene expression data from d7 through recovery and outpatient sampling. The timing of major clinical symptoms, including diarrhea, fever, coagulopathy, and MOD, is overlaid. Diarrhea occurred during peak serum and PBL EBOV RNA, as well as type IIFN responses. Virus-specific IgMs were already present at low levels at d7 (fig. S14) and the appearance of GP-specific and whole-virus IgG around d14; neutralizing antibodies were not detected until after d21. During recovery, “flare-ups” of PBL gene expression responses on d59 and d103 were observed; however, these later samples were PCR-negative for GP RNA (fig. S15).
Fig. 8. Model of patient host responses and clinical disease.

PCA-derived model of patient PBL gene expression responses during illness course and recovery. Day of illness is indicated by numbered circles colored by patient medical grade and spatially arranged using Eigen row coordinates. (A) Host defense and immune response markers correlated with peak viral load and clearance of replicating virus from PBL on d7 to d13. (B) Activation of T lymphocytes and switch toward adaptive immune responses associated with appearance of Ebola GP–specific IgG and declining serum EBOV RNA levels on d14 to d15. (C) Continuation of adaptive immune responses and appearance of virus-specific IgGs correlated with decreasing serum EBOV RNA and increased mitogenic and metabolic responses on d16 to d20. (D) Adaptive immune and repair responses associated with appearance of neutralizing antibodies and clearance of serum EBOV RNA on d21 to d24. (E) Similarity of PBL gene expression before patient discharge on d25 to d33 and continuing in outpatient samples on d59 to d270. Timing of clinical symptoms (co-agulopathy, diarrhea, fever, and MOD) is shown as reported in (12).
Discussion
Molecular correlates of disease severity, symptoms, and outcome during human EBOV infection are just beginning to be elucidated. A large-scale study of T lymphocyte responses in EVD cases during the 2013-2015 epidemic demonstrated differences in T lymphocyte activation between survivors and fatalities (17). Specifically, increased expression of inhibitory molecules CTLA-4 and PD-1 by CD8+ and CD4+ T cells correlated with greater inflammatory responses and fatal infections. Kinetics and amplitude of viral replication have also been shown to be an important factor in fatal versus nonfatal EVD (31–33). A report demonstrated that patients with a qRT-PCR Ct of <24 had a 22% survival rate compared with 87% survival in patients with Ct of ≥24 (34). Similarly, a study of seven patients with EVD provided supportive care and different therapeutic interventions showed that more severe disease was associated with increased viremia and higher expression of proinflammatory- and coagulopathy-related biomarkers and decreased type I IFN responses (29).
Whereas expression of many proinflammatory cytokines and chemokines in this patient did not peak until later in infection, expression of CCL3 (MIP-1α) peaked on or before d7, suggesting the critical role of macrophage infection during early phases of EVD noted previously (35, 36). The highest expression of several ISGs and the antiviral gene RSAD2 (viperin) occurred on the first day of PBL RNA sampling (d7) and then decreased, whereas PBL EBOV NP mRNA increased through d9. EBOV NP mRNA and vRNA levels peaked in PBLs at about d9 and then were no longer detected on d12 and d15, respectively, and correlated with the marked decrease in type I IFN responses starting d14. This suggests that expression of these genes was responsible, at least in part, for clearance of virus from PBLs, along with virus-specific IgM already present at low levels at d7 and the appearance of GP-specific and whole-virus IgG around d14; neutralizing antibodies were not detected until after d21. Activation of ROS, DNA damage, and macrophage cell death responses were also highest during d7 to d13, but were not associated with concomitant loss of monocytes, but rather an increase in monocytes peaking on d15. It is unclear whether clearance of EBOV from PBLs was related to death of infected macro-phages, previously described in non-human primates (37), or if macrophages were able to halt viral replication without undergoing death. d13 to d15 also marked the transition from an innate immune-dominated to an adaptive immune-dominated response. Although liver dysfunction first appeared in this patient before d7, and transition from tissue injury to repair occurred by d14/d15, features of MOD in this patient persisted through d21 (12). The clinical grade of this patient was still serious, but d21 marks a further transition in the equilibrium between damage and repair responses that coincided with detection of neutralizing antibodies. Recovery in this patient was marked by an overall decrease in infection-correlated gene expression responses that persisted from discharge on d33 through d270, with observed flare-ups of PBL gene expression responses on d59 and d103; however, these later samples were PCR-negative for GP RNA. These data are compatible with recent studies showing persistent EBOV infection in immune-privileged sites and prolonged recovery from EVD (23, 38–41). McElroy et al. (29) also found evidence of robust T lymphocyte responses during EBOV infection, with sustained activation of CD8 T lymphocytes long after clearance of virus from serum, suggesting continued antigen stimulation (11). However, these flare-ups could also indicate exposure to other community-acquired viruses during outpatient sampling.
Although this patient had critical care support and recovered from EVD, severity of MOD suggests a fatal outcome if care was provided in a more resource-limited setting. The peak serum creatinine levels in this patient were consistent with those observed during fatal EVD (27). Moreover, plasma levels of some cytokines/chemokines were generally more consistent with levels reported for fatal cases (27). A patient evacuated from Sierra Leone to Germany for treatment showed a similar disease course, where clinical condition deteriorated after peak viral load and just before a significant drop in serum viral titers. This also correlated with a rise in PBL and creatinine levels (42).
Excessive cytokines, lymphocyte apoptosis leading to immuno-suppression, and absence of antiviral IFN response have all been suggested to play a role in fatal EVD (13, 14, 16, 17). Ruibal et al. (17) reported that plasma levels of selected cytokines (including TNF, IL8, MIP-1α, and MIP-1β) were significantly higher in fatal EVD than in patients who survived. A study of 42 nonsurvivors and 14 survivors from five separate EVD outbreaks found that fatal outcomes were associated with hypersecretion of numerous proinflammatory cytokines (14). Similarly, analysis of samples from the 2000 outbreak of Sudan virus in Uganda found that levels of IL-6, IL-8, IL-10, and MIP-1b, but not IL-1B, IP10, or RANTES, were higher in patients with fatal infections (15). Surprisingly, but similar to the current patient, levels of TNF and IFN-γ were not generally increased compared to control patients (15). Although differences in cytokine levels were considered statistically significant in some cases, there was extensive variation in the levels among patients and sampling time with respect to clinical course was generally not reported. Serum cytokine/chemokine levels measured in the patient here were more consistent with levels reported for fatal cases but peaked several days before onset of critical illness. Furthermore, TNF, a key proinflammatory cytokine, was undetected in the serum, whereas IL-1β was only detected on d7. Because physiological effects of cytokines are rapid and short-lived, with transcriptional responses typically peaking within hours (43, 44), it is unclear what direct role, if any, they play in pathological events underlying critical illness.
Leukopenia has been reported in some cases of EVD (13, 16, 45, 46) and has been correlated with severity of infection (13). Despite the severity of the infection in the current patient, from d8, PBL populations were either within normal range or slightly elevated. Lymphocytes were at lower range of normal at d7 and then increased, suggesting early depletion and then recovery of this subset, with highest levels corresponding to critical phase of illness. Gene ontology analysis of transcripts with expression kinetics corresponding with serum creatinine levels, a surrogate marker for critical illness, predicted a decrease in lymphocyte activity and increase in lymphocyte apoptosis based on decreased expression levels of specific lymphocyte-related gene expression.
Lymphocyte apoptosis has been proposed as a cause of leukopenia during EVD (13, 14). Elevated CD95 expression in T lymphocytes was reported to be responsible for massive lymphocyte apoptosis, although no direct measurement of apoptosis was performed (14). However, CD95 exerts both pro- and anti-apoptotic signaling and modulates TCR/CD3-driven signal initiation in a dose-dependent manner, with a high dose of CD95 agonist (including CD95L) silencing T cells through blocking early TCR-driven induction, whereas a lower dose of agonist actually augments lymphocyte activation/proliferation (47). Elevated CD95 levels on lymphocytes may therefore not always be indicative of apoptosis. Sanchez et al. (13) reported that fatal cases of EVD have reduced numbers of peripheral activated CD8+ T lymphocytes compared to survivors. However, these patients also had much higher levels of serum EBOV RNA, and decreased circulating T lymphocyte populations could result from these cells infiltrating infected tissues. Extensive lymphocyte apoptosis as a cause of the decreased expression of T lymphocyte–specific genes cannotberuled outinthe current patient becauseno direct measurementofapoptosis was conducted. However, changes in peripheral lymphocyte numbers and decreasing serum viral levels suggest a subset of lymphocytes leaving the PBL pool and targeting infected tissue. This scenario would also result in decreased abundance of lymphocyte-specific gene expression in peripheral blood. The question of apoptosis versus tissue infiltration is difficult to answer without analyses of infected tissues.
This study was limited to a detailed characterization of daily changes in PBL gene expression responses in a single patient from early symptomatic disease to critical illness and through recovery. While adding to our understanding of this patient's disease, it remains to be shown how it applies in the larger context of human EVD, including both survivors and fatalities. Nonetheless, it highlights the benefits of supportive medical care, as well as the need to perform larger host response studies. Although we observed similarities between this patient and studies in nonhuman primates (48–52), we also identified several important differences such as a lack of a pronounced “cytokine storm” and absence of pronounced lymphopenia during peak illness in this patient. Improving our ability to reduce severity and high mortality rates of EVD will increasingly rely on studies that integrate critical care medicine and detailed, longitudinal host response studies in both humans and nonhuman primates.
Materials and Methods
Study design
Daily blood samples were collected during inpatient care of an individual infected with EBOV beginning before critical illness and continuing through recovery. Global expression of RNA isolated from PBLs in these samples was measured by expression microarray. Each RNA sample was labeled and hybridized in duplicate on individual arrays (n = 2). All samples (except for d25 and d180) were analyzed by microarray twice (n = 4). Expression data were correlated with other clinical information, including viral RNA concentration by qRT-PCR, EBOV-specific antibodies by ELISA and PRNT80 assays, and D-dimer, platelet, and serum creatinine measured using standard clinical microbiology laboratory methods. The study was not blinded.
Patient information
Blood samples from the EBOV-infected patient (clinicaltrials.gov identifier NCT02363322) and healthy volunteers (clinicaltrials.gov identifier NCT0138664) were obtained under clinical protocols approved by the National Institute of Allergy and Infectious Diseases (NIAID) Institutional Review Board and were conducted in accordance with provisions of the Declaration of Helsinki and good clinical practice guidelines. The clinical case of this patient was previously described (12).
RNA isolation and expression microarray analysis
Total RNA was isolated from whole blood using PAXgene Blood RNA Kit IVD (Qiagen) and resuspended in TRIzol LS before removal from BSL-4 under supervision of the Biosurety Program of the NIH and in accordance with Division of Select Agents and Toxins at the Centers for Disease Control and Prevention. RNA quality was assessed using Bioanalyzer (Agilent Technologies). Gene expression profiling was performed using Agilent Human Whole Genome 44K microarrays. Fluorescent probes were prepared using Agilent QuickAmp Labeling Kit. Each RNA sample was labeled and hybridized in duplicate on individual arrays (n = 2) except for d25 and d180. Spot quantitation was performed using Agilent's Feature Extractor software, and data were entered into the custom database SLIMarray (http://slimarray.systemsbiology.net) and uploaded into Genedata Analyst 9.0 (Genedata). Complete MIAME-compliant (53) microarray data set has been deposited in the National Center for Biotechnology Information (NCBI)'s Gene Expression Omnibus (GEO) (54) and is accessible at GEO GSE93861. TIBCO Spotfire Analyst 6.0 was used for analysis and heatmap creation.
WBC counts
WBC differential counts were performed at NIH Clinical Center by the Department of Laboratory Medicine using standard methods.
Quantitative EBOV and EBOV GP IgG ELISA
Serum samples and EBOV-specific IgG reference standard were serially diluted and quantified using horseradish peroxidase-labeled anti-human IgG secondary antibody (KPL). Detailed methods are in the Supplementary Materials.
EBOV IgM end titer ELISA
Serum samples were added to ELISA plates coated with irradiated whole EBOV antigen, as described in (55). Detailed methods are in the Supplementary Materials.
Plaque reduction neutralization titration assay
Serially diluted serum was mixed and incubated with 100 plaque-forming units of EBOV, and percent neutralization was determined relative to wells treated with media alone, as described in (55). Detailed methods are in the Supplementary Materials.
Quantitative cytokine analysis by Bio-Plex assay
Multiplex cytokine analysis was completed for serum samples using a magnetic bead–based Bio-Plex Pro Human Cytokine 17-plex Assay kit (Bio-Rad) and FLEXMAP 3D instrument (Luminex Corporation). Concentration of each sample analyte was determined using standard curves and reported as picograms per milliliter. Normal human serum (Sigma-Aldrich; catalog #H4522) was used as a negative control.
Quantitative reverse transcription polymerase chain reaction
Reverse transcription of equivalent amounts of total RNA was performed using SuperScript III First-Strand Synthesis System (Life Technologies Inc.). Primers for Ebola NP vRNA were as follows: forward, 5′-TCATGCGTACCAAGGAGATTAC-3′; reverse, 5′-GCTTCACTCCATCACACTTCT-3′; probe, 5′-TCAAGTATTTGGAAGGGCACGGGT-3′. Primers for Ebola NP mRNA were as follows: forward, 5′-GCGTTAAGCCACAGTTATAGCC-3′; reverse, 5′-GGGCAGGCTAGTAGGTAAGTTAT-3′; probe, 5′-TGGTAACTCAATATCTTAGCCAGCGA-3′. qRT-PCR was performed in duplicate using an ABI QuantStudio 6 Flex Real-Time PCR system.
Statistics
Data normalization was performed in Genedata Analyst using central tendency followed by relative normalization using pooled RNA isolated from blood of healthy volunteers (n = 29) as a reference. Statistical analysis was performed using individual replicates. Pearson and PCA were performed using Genedata Analyst 9.0. For Pearson correlation analysis, r ≥ 0.6 was used. For visualization, values from replicates were combined using arithmetic mean. Ingenuity Pathway Analysis, Panther (56, 57), and Entrez Gene (www.ncbi.nlm.nih.gov/gene) were used for gene ontology and pathway analysis using Bonferroni correction.
Supplementary Material
Acknowledgments
We thank P. Jahrling, L. Hensley, and the staff of the NIAID Integrated Research Facility for sample storage and initial processing. Complete MIAME-compliant microarray data set has been deposited in NCBI GEO GSE93861.
Funding: Division of Intramural Research, NIAID, and NIH. K.S. and D.B. were supported by Defense Threat Reduction Agency (DTRA) (HDTRA1-13-C-0055). United States Army Medical Research Institute of Infectious Diseases and J.M.D. acknowledge DTRA (CB10166) for support.
Footnotes
Author contributions: J.C.K., K.-A.W., and J.K.T. conceived and designed experiments, analyzed and interpreted data, drafted the article, and revised the article. J.K. conceived and designed experiments, acquired data, and revised the article. K.B.J., R.D.A., D.B. and K.S. acquired data. A.S.H., S.W.S., R.M.J., and J.M.D. acquired and interpreted data. M.J.M. and R.T.D. provided clinical samples. D.S.C. conceived and designed experiments, provided clinical samples, and revised the article. All authors approved the final version.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.World Health Organization. Ebola Situation Report—30 March 2016. World Health Organization; 2016. [Google Scholar]
- 2.Spengler JR, Ervin ED, Towner JS, Rollin PE, Nichol ST. Perspectives on West Africa Ebola virus disease outbreak, 2013-2016. Emerg Infect Dis. 2016;22:956–963. doi: 10.3201/eid2206.160021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rousgeron V, Feldmann H, Grard G, Becker S, Leroy EM. Ebola and Marburg haemorrhagic fever. J Clin Virol. 2015;64:111–119. doi: 10.1016/j.jcv.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de La Vega MA, Stein D, Kobinger GP. Ebolavirus evolution: Past and present. PLOS Pathog. 2015;11:e1005221. doi: 10.1371/journal.ppat.1005221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen AL. Host factors in Ebola infection. Annu Rev Genomics Hum Genet. 2016;17:333–351. doi: 10.1146/annurev-genom-083115-022446. [DOI] [PubMed] [Google Scholar]
- 6.Baseler L, Chertow DS, Johnson KM, Feldmann H, Morens DM. The pathogenesis of Ebola virus disease. Annu Rev Pathol. 2017;12:387–418. doi: 10.1146/annurev-pathol-052016-100506. [DOI] [PubMed] [Google Scholar]
- 7.Singh G, Kumar A, Singh K, Kaur J. Ebola virus: An introduction and its pathology. Rev Med Virol. 2016;26:49–56. doi: 10.1002/rmv.1863. [DOI] [PubMed] [Google Scholar]
- 8.Chertow DS, Kleine C, Edwards JK, Scaini R, Giuliani R, Sprecher A. Ebola virus disease in West Africa—Clinical manifestations and management. N Engl J Med. 2014;371:2054–2057. doi: 10.1056/NEJMp1413084. [DOI] [PubMed] [Google Scholar]
- 9.Bente D, Gren J, Strong JE, Feldmann H. Disease modeling for Ebola and Marburg viruses. Dis Model Mech. 2009;2:12–17. doi: 10.1242/dmm.000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet. 2011;377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martines RB, Ng DL, Greer PW, Rollin PE, Zaki SR. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J Pathol. 2015;235:153–174. doi: 10.1002/path.4456. [DOI] [PubMed] [Google Scholar]
- 12.Chertow DS, Nath A, Suffredini AF, Danner RL, Reich DS, Bishop RJ, Childs RW, Arai AE, Palmore TN, Lane HC, Fauci AS, Davey RT. Severe meningoencephalitis in a case of Ebola virus disease: A case report. Ann Intern Med. 2016;165:301–304. doi: 10.7326/M15-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez A, Lukwiya M, Bausch D, Mahanty S, Sanchez AJ, Wagoner KD, Rollin PE. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: Cellular responses, virus load, and nitric oxide levels. J Virol. 2004;78:10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLOS Negl Trop Dis. 2010;4:e837. doi: 10.1371/journal.pntd.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutchinson KL, Rollin PE. Cytokine and chemokine expression in humans infected with Sudan Ebola virus. J Infect Dis. 2007;196(suppl 2):S357–S363. doi: 10.1086/520611. [DOI] [PubMed] [Google Scholar]
- 16.Agrati C, Castilletti C, Casetti R, Sacchi A, Falasca L, Turchi F, Tumino N, Bordoni V, Cimini E, Viola D, Lalle E, Bordi L, Lanini S, Martini F, Nicastri E, Petrosillo N, Puro V, Piacentini M, Di Caro A, Kobinger GP, Zumla A, Ippolito G, Capobianchi MR. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. 2016;7:e2164. doi: 10.1038/cddis.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruibal P, Oestereich L, Lüdtke A, Becker-Ziaja B, Wozniak DM, Kerber R, Korva M, Cabeza-Cabrerizo M, Bore JA, Koundouno FR, Duraffour S, Weller R, Thorenz A, Cimini E, Viola D, Agrati C, Repits J, Afrough B, Cowley LA, Ngabo D, Hinzmann J, Mertens M, Vitoriano I, Logue CH, Boettcher JP, Pallasch E, Sachse A, Bah A, Nitzsche K, Kuisma E, Michel J, Holm T, Zekeng EG, Garcia-Dorival I, Wolfel R, Stoecker K, Fleischmann E, Strecker T, Di Caro A, Avsic-Zupanc T, Kurth A, Meschi S, Mely S, Newman E, Bocquin A, Kis Z, Kelterbaum A, Molkenthin P, Carletti F, Portmann J, Wolff S, Castilletti C, Schudt G, Fizet A, Ottowell LJ, Herker E, Jacobs T, Kretschmer B, Severi E, Ouedraogo N, Lago M, Negredo A, Franco L, Anda P, Schmiedel S, Kreuels B, Wichmann D, Addo MM, Lohse AW, De Clerck H, Nanclares C, Jonckheere S, Van Herp M, Sprecher A, Xiaojiang G, Carrington M, Miranda O, Castro CM, Gabriel M, Drury P, Formenty P, Diallo B, Koivogui L, Magassouba N, Carroll MW, Gunther S, Munoz-Fontela C. Unique human immune signature of Ebola virus disease in Guinea. Nature. 2016;533:100–104. doi: 10.1038/nature17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbist KC, Klonowski KD. Functions of IL-15 in anti-viral immunity: Multiplicity and variety. Cytokine. 2012;59:467–478. doi: 10.1016/j.cyto.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. 2003;170:5018–5026. doi: 10.4049/jimmunol.170.10.5018. [DOI] [PubMed] [Google Scholar]
- 20.Novak H, Müller A, Harrer N, Günther C, Carballido JM, Woisetschlager M. CCL23 expression is induced by IL-4 in a STAT6-dependent fashion. J Immunol. 2007;178:4335–4341. doi: 10.4049/jimmunol.178.7.4335. [DOI] [PubMed] [Google Scholar]
- 21.Roberts AW. G-CSF: A key regulator of neutrophil production, but that's not all! Growth Factors. 2005;23:33–41. doi: 10.1080/08977190500055836. [DOI] [PubMed] [Google Scholar]
- 22.Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukocyte Biol. 2016;100:481–489. doi: 10.1189/jlb.3RU0316-144R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brainard J, Pond K, Hooper L, Edmunds K, Hunter P. Presence and persistence of Ebola or Marburg virus in patients and survivors: A rapid systematic review. PLOS Negl Trop Dis. 2016;10:e0004475. doi: 10.1371/journal.pntd.0004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deen GF, Knust B, Broutet N, Sesay FR, Formenty P, Ross C, Thorson AE, Massaquoi TA, Marrinan JE, Ervin E, Jambai A, McDonald SL, Bernstein K, Wurie AH, Dumbuya MS, Abad N, Idriss B, Wi T, Bennett SD, Davies T, Ebrahim FK, Meites E, Naidoo D, Smith S, Banerjee A, Erickson BR, Brault A, Durski KN, Winter J, Sealy T, Nichol ST, Lamunu M, Stroher U, Morgan O, Sahr F. Ebola RNA persistence in semen of Ebola virus disease survivors—Preliminary report. N Engl J Med. 2015 doi: 10.1056/NEJMoa1511410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez LL, De Roo A, Guimard Y, Trappier SG, Sanchez A, Bressler D, Williams AJ, Rowe AK, Bertolli J, Khan AS, Ksiazek TG, Peters CJ, Nichol ST. Persistence and genetic stability of Ebola virus during the outbreak in Kikwit, Democratic Republic ofthe Congo, 1995. J Infect Dis. 1999;179(suppl. 1):S170–S176. doi: 10.1086/514291. [DOI] [PubMed] [Google Scholar]
- 26.Varkey JB, Shantha JG, Crozier I, Kraft CS, Lyon GM, Mehta AK, Kumar G, Smith JR, Kainulainen MH, Whitmer S, Stroher U, Uyeki TM, Ribner BS, Yeh S. Persistence of Ebola virus in ocular fluid during convalescence. N Engl J Med. 2015;372:2423–2427. doi: 10.1056/NEJMoa1500306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schieffelin JS, Shaffer JG, Goba A, Gbakie M, Gire SK, Colubri A, Sealfon RS, Kanneh L, Moigboi A, Momoh M, Fullah M, Moses LM, Brown BL, Andersen KG, Winnicki S, Schaffner SF, Park DJ, Yozwiak NL, Jiang PP, Kargbo D, Jalloh S, Fonnie M, Sinnah V, French I, Kovoma A, Kamara FK, Tucker V, Konuwa E, Sellu J, Mustapha I, Foday M, Yillah M, Kanneh F, Saffa S, Massally JL, Boisen ML, Branco LM, Vandi MA, Grant DS, Happi C, Gevao SM, Fletcher TE, Fowler RA, Bausch DG, Sabeti PC, Khan SH, Garry RF K. G. H. L. F. Program; Viral Hemorrhagic Fever Consortium; WHO Clinical Response Team. Clinical illness and outcomes inpatients with Ebola in Sierra Leone. N Engl J Med. 2014;371:2092–2100. doi: 10.1056/NEJMoa1411680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson AJ, Martin DS, Maddox V, Rattenbury S, Bland D, Bhagani S, Cropley I, Hopkins S, Mepham S, Rodger A, Warren S, Chowdary P, Jacobs M. Thromboelastography in the management of coagulopathy associated With Ebola virus disease. Clin Infect Dis. 2016;62:610–612. doi: 10.1093/cid/civ977. [DOI] [PubMed] [Google Scholar]
- 29.McElroy AK, Harmon JR, Flietstra TD, Campbell S, Mehta AK, Kraft CS, Lyon MG, Varkey JB, Ribner BS, Kratochvil CJ, Iwen PC, Smith PW, Ahmed R, Nichol ST, Spiropoulou CF. Kinetic analysis of biomarkers in a cohort of US patients with Ebola virus disease. Clin Infect Dis. 2016;63:460–467. doi: 10.1093/cid/ciw334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol. 2010;184:4062–4068. doi: 10.4049/jimmunol.0903002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanini S, Portella G, Vairo F, Kobinger GP, Pesenti A, Langer M, Kabia S, Brogiato G, Amone J, Castilletti C, Miccio R, Zumla A, Capobianchi MR, Di Caro A, Strada G, Ippolito G INMI-EMERGENCY EBOV Sierra Leone Study Group. Blood kinetics of Ebolavirus in survivors and nonsurvivors. J Clin Invest. 2015;125:4692–4698. doi: 10.1172/JCI83111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de La Vega MA, Caleo G, Audet J, Qiu X, Kozak RA, Brooks JI, Kern S, Wolz A, Sprecher A, Greig J, Lokuge K, Kargbo DK, Kargbo B, Di Caro A, Grolla A, Kobasa D, Strong JE, Ippolito G, Van Herp M, Kobinger GP. Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J Clin Invest. 2015;125:4421–4428. doi: 10.1172/JCI83162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Duan HJ, Chen HY, Ji YJ, Zhang X, Rong YH, Xu Z, Sun LJ, Zhang JY, Liu LM, Jin B, Zhang J, Du N, Su HB, Teng GJ, Yuan Y, Qin EQ, Jia HJ, Wang S, Guo TS, Wang Y, Mu JS, Yan T, Li ZW, Dong Z, Nie WM, Jiang TJ, Li C, Gao XD, Ji D, Zhuang YJ, Li L, Wang LF, Li WG, Duan XZ, Lu YY, Sun ZQ, Kanu AB, Koroma SM, Zhao M, Ji JS, Wang FS. Age and Ebola viral load correlate with mortality and survival time in 288 Ebola virus disease patients. Int J Infect Dis. 2016;42:34–39. doi: 10.1016/j.ijid.2015.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowe SJ, Maenner MJ, Kuah S, Erickson BR, Coffee M, Knust B, Klena J, Foday J, Hertz D, Hermans V, Achar J, Caleo GM, Van Herp M, Albarino CG, Amman B, Basile AJ, Bearden S, Belser JA, Bergeron E, Blau D, Brault AC, Campbell S, Flint M, Gibbons A, Goodman C, McMullan L, Paddock C, Russell B, Salzer JS, Sanchez A, Sealy T, Wang D, Saffa G, Turay A, Nichol ST, Towner JS. Prognostic indicators for Ebola patient survival. Emerg Infect Dis. 2016;22:217–223. doi: 10.3201/eid2202.151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnittler HJ, Feldmann H. Marburg and Ebola hemorrhagic fevers: Does the primary course of infection depend on the accessibility of organ-specific macrophages? Clin Infect Dis. 1998;27:404–406. doi: 10.1086/517704. [DOI] [PubMed] [Google Scholar]
- 36.Bray M, Geisbert TW. Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol. 2005;37:1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 37.Geisbert TW, Hensley LE, Gibb TR, Steele KE, Jaax NK, Jahrling PB. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab Invest. 2000;80:171–186. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 38.Uyeki TM, Erickson BR, Brown S, Mc Elroy AK, Cannon D, Gibbons A, Sealy T, Kainulainen MH, Schuh AJ, Kraft CS, Mehta AK, Lyon GM, III, Varkey JB, Ribner BS, Ellison RT, III, Carmody E, Nau GJ, Spiropoulou C, Nichol ST, Ströher U. Ebola virus persistence in semen of male survivors. Clin Infect Dis. 2016;62:1552–1555. doi: 10.1093/cid/ciw202. [DOI] [PubMed] [Google Scholar]
- 39.Sow MS, Etard JF, Baize S, Magassouba N, Faye O, Msellati P, Toure AI, Savane I, Barry M, Delaporte E Postebogui Study Group. New evidence of long-lasting persistence of Ebola virus genetic material in semen of survivors. J Infect Dis. 2016;214:1475–1476. doi: 10.1093/infdis/jiw078. [DOI] [PubMed] [Google Scholar]
- 40.Fischer RJ, Judson S, Miazgowicz K, Bushmaker T, Munster VJ. Ebola virus persistence in semen ex vivo. Emerg Infect Dis. 2016;22:289–291. doi: 10.3201/eid2202.151278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chughtai AA, Barnes M, Macintyre CR. Persistence of Ebola virus in various body fluids during convalescence: Evidence and implications for disease transmission and control. Epidemiol Infect. 2016;144:1652–1660. doi: 10.1017/S0950268816000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf T, Kann G, Becker S, Stephan C, Brodt HR, de Leuw P, Grünewald T, Vogl T, Kempf VA, Keppler OT, Zacharowski K. Severe Ebola virus disease with vascular leakage and multiorgan failure: Treatment of a patient in intensive care. Lancet. 2015;385:1428–1435. doi: 10.1016/S0140-6736(14)62384-9. [DOI] [PubMed] [Google Scholar]
- 43.Lanford RE, Guerra B, Lee H, Chavez D, Brasky KM, Bigger CB. Genomic response to interferon-α in chimpanzees: Implications of rapid downregulation for hepatitis C kinetics. Hepatology. 2006;43:961–972. doi: 10.1002/hep.21167. [DOI] [PubMed] [Google Scholar]
- 44.Chiewchengchol D, Wright HL, Thomas HB, Lam CW, Roberts KJ, Hirankarn N, Beresford MW, Moots RJ, Edwards SW. Differential changes in gene expression in human neutrophils following TNF-α stimulation: Up-regulation of anti-apoptotic proteins and down-regulation of proteins involved in death receptor signaling. Immun Inflamm Dis. 2016;4:35–44. doi: 10.1002/iid3.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wauquier N, Padilla C, Becquart P, Leroy E, Vieillard V. Association of KIR2DS1 and KIR2DS3 with fatal outcome in Ebola virus infection. Immunogenetics. 2010;62:767–771. doi: 10.1007/s00251-010-0480-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liddell AM, Davey RT, Jr, Mehta AK, Varkey JB, Kraft CS, Tseggay GK, Badidi O, Faust AC, Brown KV, Suffredini AF, Barrett K, Wolcott MJ, Marconi VC, Lyon GM, III, Weinstein GL, Weinmeister K, Sutton S, Hazbun M, Albarino CG, Reed Z, Cannon D, Stroher U, Feldman M, Ribner BS, Lane HC, Fauci AS, Uyeki TM. Characteristics and clinical management of a cluster of 3 patients with Ebola virus disease, including the first domestically acquired cases in the United States. Ann Intern Med. 2015;163:81–90. doi: 10.7326/M15-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paulsen M, Valentin S, Mathew B, Adam-Klages S, Bertsch U, Lavrik I, Krammer PH, Kabelitz D, Janssen O. Modulation of CD4+ T-cell activation by CD95 co-stimulation. Cell Death Differ. 2011;18:619–631. doi: 10.1038/cdd.2010.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geisbert TW, Strong JE, Feldmann H. Considerations in the use of nonhuman primate models of Ebola virus and Marburg virus infection. J Infect Dis. 2015;212(suppl 2):S91–S97. doi: 10.1093/infdis/jiv284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong G, Richardson JS, Pillet S, Patel A, Qiu X, Alimonti J, Hogan J, Zhang Y, Takada A, Feldmann H, Kobinger GP. Immune parameters correlate with protection against ebola virus infection in rodents and nonhuman primates. Sci Transl Med. 2012;4:158ra146. doi: 10.1126/scitranslmed.3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E, Hensley LE. Pathogenesis of Ebola hemorrhagic fever in primate models: Evidence that hemorrhageis not a direct effect of virus-induced cytolysis of endothelial cells. Am J Pathol. 2003;163:2371–2382. doi: 10.1016/S0002-9440(10)63592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: Possible involvement of the tumor necrosisfactor receptor superfamily. Immunol Lett. 2002;80:169–179. doi: 10.1016/s0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 52.Rubins KH, Hensley LE, Wahl-Jensen V, Daddario Di Caprio KM, Young HA, Reed DS, Jahrling PB, Brown PO, Relman DA, Geisbert TW. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 2007;8:R174. doi: 10.1186/gb-2007-8-8-r174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M. Minimum information about a microarray experiment (MIAME)—Toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 54.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression andhybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howell KA, Qiu X, Brannan JM, Bryan C, Davidson E, Holtsberg FW, Wec AZ, Shulenin S, Biggins JE, Douglas R, Enterlein SG, Turner HL, Pallesen J, Murin CD, He S, Kroeker A, Vu H, Herbert AS, Fusco ML, Nyakatura EK, Lai JR, Keck ZY, Foung SK, Saphire EO, Zeitlin L, Ward AB, Chandran K, Doranz BJ, Kobinger GP, Dye JM, Aman MJ. Antibody treatment of Ebola and Sudan virus infection via a uniquely exposed epitope within the glycoprotein receptor-binding site. Cell Rep. 2016;15:1514–1526. doi: 10.1016/j.celrep.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.