

Abstract
OFD1, now recognized as a ciliopathy, is characterized by malformations of the face, oral cavity and digits, and is transmitted as an X-linked condition with lethality in males. Mutations in OFD1 also cause X-linked Joubert syndrome (JBTS10) and Simpson–Golabi–Behmel syndrome type 2 (SGBS2). We have studied 55 sporadic and six familial cases of suspected OFD1. Comprehensive mutation analysis in OFD1 revealed mutations in 37 female patients from 30 families; 22 mutations have not been previously described including two heterozygous deletions spanning OFD1 and neighbouring genes. Analysis of clinical findings in patients with mutations revealed that oral features are the most reliable diagnostic criteria. A first, detailed evaluation of brain MRIs from seven patients with cognitive defects illustrated extensive variability with the complete brain phenotype consisting of complete agenesis of the corpus callosum, large single or multiple interhemispheric cysts, striking cortical infolding of gyri, ventriculomegaly, mild molar tooth malformation and moderate to severe cerebellar vermis hypoplasia. Although the OFD1 gene apparently escapes X-inactivation, skewed inactivation was observed in seven of 14 patients. The direction of skewing did not correlate with disease severity, reinforcing the hypothesis that additional factors contribute to the extensive intrafamilial variability.
Keywords: OFD1 mutation, orofaciodigital syndrome, ciliopathy
Introduction
Thirteen forms of orofaciodigital syndrome (OFDS) have been described, with extensive overlap in the clinical presentation between the various forms. The main clinical findings in OFD type I (OFD1; MIM# 311200) have been known since Papillon-Léage and Psaume summarized features in three reports from the literature together with their own patients and noted that only females were affected (Papillon-Léage andPsaumesyndrome[Papillon-Léage and Psaume, 1954]. Additional single patientswere described until it was finally recognized as a new syndrome with the most common features being summarized by Gorlin and Psaume in 1962 [Gorlin and Psaume, 1962]. Predominant clinical features are: malformations of the face (frontal bossing, hypertelorism, prominent root of the nose with a large funnel, pseudo-cleft in the upper lip, variable degree of alopecia), oral cavity (hyperplastic frenulae, cleft or high-arched palate, supernumerary or malpositioned teeth, and a multilobulated tongue), and digits (brachydactyly, syndactyly, clinodactyly, camptodactyly, polydactyly, and hypoplastic thumbs). Central nervous system involvement occurs in as many as 50% of patients [Connacher et al., 1987; Gorlin and Psaume, 1962; Towfighi et al., 1985] and polycystic kidney disease is characteristic of OFD1 [Gurrieri et al., 2007; Thauvin-Robinet et al., 2006; Toriello, 2009]. OFD1 patients often develop renal failure, requiring dialysis, and renal transplantation in late childhood or adulthood [Odent et al., 1998; Stapleton et al., 1982]. OFD 1 is transmitted as an X-linked trait with prenatal lethality in males [Toriello, 1993] and the mapping of the locus toXp22.2 to 22.3 [Feather et al., 1997] was followed by detection ofmutations in theCXORF5 gene, subsequently named OFD1 (MIM# 300170) [Ferrante et al., 2001]. OFD1 is the second of only two known genes for the OFDSs.
OFD1 has been recognized as a ciliopathy [Toriello, 2009] as the OFD1 protein is localized to the centrosome and basal body of primary cilia [Romio et al., 2003, 2004] and has been shown to be required for primary cilia formation and left–right symmetry [Ferrante et al., 2006]. The phenotype of patients with OFD1 is in keeping with other ciliopathies in which midline defects and cystic kidney disease are recurrent features. The phenotypic spectrum associated with OFD1 mutations has recently been extended to include macrocephaly, severe intellectual disability, and ciliary dyskinesia in male patients with Type 2 Simpson– Golabi–Behmel syndrome [SGBS2; MIM# 300209, Budny et al., 2006] and symptoms of classical Joubert syndrome including the molar tooth sign on brain imaging in males with X-linked Joubert syndrome (JBTS10; MIM# 300804 [Coene et al., 2009, Field et al., 2012]). In both GBS2 and JBTS10, females are not affected. Mutations in the TMEM216 gene (MIM# 613277), which is associated with the ciliopathies Joubert (JBTS2; MIM# 608091), and Meckel (MKS2; MIM# 603194) syndromes, have recently been identified in two patients with Varadi–Papp syndrome, also known as OFD type VI [OFD6;MIM# 277170, Valente et al., 2010].
The OFD1 protein has five predicted coil–coil domains and a LisH domain and Northern blot analysis showed widespread expression in pancreas, kidney, skeletal muscle, liver, lung, placenta, brain, and heart [de Conciliis et al., 1998]. Animal models of OFD1 have been generated in mouse and zebrafish, and the mouse phenotype reiterates the main features seen in human patients, albeit with increased severity which was postulated to result from lack of X inactivation in the mouse [Ferrante et al., 2003; Macca and Franco, 2009]. In 9- and 14-week-old mouse embryos OFD1 was expressed in metanephric mesenchyme, oral mucosa, lung, heart, nasal and cranial cartilage, and brain [Romio et al., 2003]. Ofd1 inactivation in zebrafish resulted in a typical ciliary phenotype with bent body, laterality defects, and edema [Ferrante et al., 2009]. Conditional limb mesoderm-specific inactivation of Ofd1 in mouse resulted in polydactyly and shortened long bones [Bimonte et al., 2011]. Sequencing of the OFD1 coding and intronic flanking regions detects mutations in aproximately 80% of patients with a clear clinical diagnosis of OFD1 [Thauvin-Robinet et al., 2009]. To date, 99 different mutations have been identified in OFD1 patients [Macca and Franco, 2009]. The majority (58%) are small insertions or deletions resulting in a frameshift, with missense and nonsense mutations accounting for only 23% [Macca and Franco, 2009]. The location of mutations causing OFD1 extends only to exon 17 out of 23 coding exons [Macca and Franco 2009; Prattichizzo et al., 2008], with mutations 3′ of exon 17 having been found to cause JBTS10 in males [Coene et al., 2009]. The first large intragenic deletions in OFD1 (seven in total), detected via quantative PCR and FAM-6 assays were found in 23% of patients in whom sequencing had not detected a mutation, thus increasing the detection rate to 85% [Thauvin-Robinet et al., 2009]. There have been many attempts at delineating a genotype–phenotype correlation for OFD1 [Gurrieri et al., 2007; Thauvin-Robinet et al., 2006].Mutations in exons 3, 8, 9, 13, and 16 have been associated with intellectual disability [Ferrante et al., 2001], and renal cysts withmutations in exon 12 [Prattichizzo et al., 2008].
The inter- and intrafamilial clinical variability in OFD1 is extensive and the reasons for this are still not clear. There is evidence that OFD1 escapes X inactivation in humans [Carrel et al., 1999; Carrel and Willard, 2005; de Conciliis et al., 1998]. However, it has been shown that genes that escape inactivation are expressed at a lower level on the inactive X (Xi) than on the active chromosome and that the escape from inactivation therefore may not be complete [Carrel and Willard, 2005]. Skewed inactivation has been previously shown in 30% of families with OFD1 [Thauvin-Robinet et al., 2006] and it has been repeatedly suggested that X-inactivation may in fact play a role in the extensive clinical variability [Franco and Ballabio, 2006; Morleo and Franco, 2008].
The aims of this study were manifold: first, to further explore the inter- and intrafamilial phenotypic variability in OFD1 in a large cohort of patients, and in particular to investigate and compare the brain malformations in patients with OFD1 mutations; second, to further investigate the extent of skewing of X-chromosome inactivation in patients from families with more than one affected individual; and lastly, to establish the frequency of deletions in OFD1, using a different method of analysis from what has previously been published, namely, quantitative PCR(qPCR) of the entire coding region.
Materials and Methods
Mutation Nomenclature
Amino acid substitutions were named with the first methionine encoded by the ATG start codon that is designated as amino acid number “1” following the journal guidelines (www.hgvs.org/mutnomen).
Patients
Sixty-one unrelated patients were referred to us for OFD1 mutation analysis. Informed consent was obtained from each patient or the patient’s parents for the molecular genetic testing. Consent for the publication of the clinical and molecular genetic data and the use of photographs of the patients was obtained for patients in whom mutations were found. Clinicians completed a clinical checklist to confirm the clinical diagnosis and to distinguish between OFD1 and other forms of OFDS. Brain magnetic resonance images (MRI) were obtained when available.
PCR and DNA Sequencing
Genomic DNA was extracted from peripheral blood using standard techniques (DNA extraction kit for mammalian blood, GE health Sciences, Amersham, UK) or obtained directly from referring doctors. PCR and sequencing of the 23 coding OFD1 exons and at least 50 bp of flanking intronic sequences was performed using primers designed in our laboratory (primers available upon request). Purified PCR products were directly sequenced using the ABI Prism BigDye Terminator Cycle Sequencing Kit (version 1.1, Applied Biosystems, Foster City, CA) and capillary electrophoresis on an ABI Prism 3100 Genetic Analyzer.Mutation analysis was performed using the SeqPilot program (JSI, Kippenheim, Germany). The missense mutation c.422T>G in Exon 6 was tested by PCR and restriction enzyme digestion with HpyCH4V (New England Biolabs, Frankfurt, Germany).
Quantitative PCR
In patients in whom no mutation was detected by sequence analysis, qPCR was performed to search for intragenic deletions or duplications. Initially, primers were designed to amplify fragments of 100–300 bp within or flanking each of the 23 OFD1 coding exons. Subsequently, for routine deletion/duplication screening, we designed a set of 10 primer pairs in exons 1, 4/5, 6, 8, 9, 11, 15, 19, 21, and 23with a resolution of maximum5.3 kB. Additional primers in the ATXN3L, EGFL6, TCEANC, RAB9A, GPM6B, and GEMIN8 genes flanking OFD1 were used to determine the size of the deletions in patients 29 and 30. All primers used for qPCR are provided in Supp. Table S1. qPCR was performed on an ABI 7900HT Fast Real-Time PCR System (PE Applied Biosystems, Norwalk, CT) using SYBR-Green according to the method of Borozdin et al. (2004). The reaction was performed in white 384-well qPCR plates (ABgene, Hamburg, Germany) using 10 ng of DNA per well. Reactions contained 0.25 mM of each primer and 5 μl of QuantiTect SYBRH Green PCR Master Mix (Qiagen, Hilden, Germany) in a total of 10 μl. Assays included DNA standards in a final concentration of 5.0, 2.5, 1.25, or 0.625 ng/μl, a no-template control, or 2.5 ng/μl of patient DNA in replicates (n = 6). Cycling conditions were 50°C for 2 min, 95°C for 15 min, and 40 cycles of 94°C (15 sec), 58°C (15 sec), and 72°C (one min). The same conditions were applied to all amplicons. Each amplicon was run at least in duplicate with the average values used in calculation.To normalize the values generated for each amplicon, a subtelomeric amplicon (3p1) and an amplicon from an established gene (SALL4ex3) were used as standards [Borozdin et al., 2004].
To avoid the generation of nonspecific products, melting curve analysis of products was routinely undertaken following the amplification. Quantitative data were normalized against a normal female diploid reference genome by calculating the ratio relative to the average amount of reference amplicons, for each test amplicon. In this manner, ratio values of 1.0 indicated the presence of a diploid situation in females, and values of 0.5 or 1.5 indicate partial haploidy or partial triploidy, respectively.
RNA Extraction and cDNA Analysis
RNA was extracted from transformed lymphocyte cell lines using standard methods (RNAeasy kit, Qiagen, Hilden, Germany) and cDNA was produced by reverse transcription of RNA using Superscript 3 (Invitrogen Carlsbad, CA). cDNA was analyzed in family 1 by PCR and DNA sequencing using primers within exons 1 (Primer OFD1 Ex1F-RT: ACTAAACTCGGGCCGCGG and 3 (Primer OFD1 Ex3R-RT: GCAGTTCTCCACTCAATACAGGGTG). cDNA transcripts were either sequenced directly or extracted from agarose gel after electrophoresis and cloned into pGEM-T easy Vector (Promega Corporation, Madison, WI). Individual clones were sequenced using T7 and SP6 primers.
X-Inactivation Assay and Microsatellite Marker Analysis X-inactivation was studied in lymphocytes in the females from five families using a PCR-based assay with fluorescently labelled primers for differential methylation of Hha1 sites in the first exon of the androgen receptor (AR) gene [Allen et al., 1992].We quantified the degree of skewing by analyzing 6-Fam-labelled PCR products on an ABI 3100 sequencer. Analysis was performed using GeneScan software (Applied Biosystems, Foster city, CA). DNA from a healthy male was always included in the analysis as a control for HhaI digestion. Five microsatellite markers: DSX8022, DXS8014, DXS6810, DXS988, and DXS1194 located between OFD1 (Xp22.1) and AR (Xq12) were similarly genotyped in each of the females tested, to identify the OFD1 mutation allele in the patients and carriers.
Bioinformatic Resources
The OFD1 reference sequence NM_003611.1 was used for the comparison of patient sequences (http://www.ncbi.nlm.nih.gov/genbank/). Nucleotide changes were verified on the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway) and at the NCBI single nucleotide polymorphism (SNP) database (www.ncbi.nlm.nih.gov/projects/SNP). Mutations predicted to affecting splicing were checked for their pathogenicity using splice prediction software (www.fruitfly.org/seq_tools/splice.html) and Human Splicing Finder (http://www.umd.be/HSF/) and all other mutations were assessed for pathogenicity using Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation Taster (http://neurocore.charite.de/MutationTaster/).
Results
Clinical Findings
Mutation analysis was initially performed in single patients from 61 different families who were referred to us with suspected OFD1. Six of the patients were males with features of OFD1 and in whom mosaicism or the possibility of a mild mutation was suspected. No mutations were detected inDNA extracted from blood lymphocytes in these male patients. Mutations were found in 30 of the remaining 55 female index patients (54.5%). Completed clinical checklists were returned for 28 patients including mothers and sisters in whom mutations had been confirmed, and limited clinical findings were available from an additional three patients in whom OFD1 mutations had been found. The results of these findings are summarized in Table 1, together with the summarized findings of those from previous studies for comparison [Macca and Franco, 2009].
Table 1.
Comparison of the Clinical Findings in the Patients with OFD1 Mutations in This Report with Previously Published Cohorts
| Presence of clinical features in % (n of patients in which symptom was observed/assessed) | |||
|---|---|---|---|
| Clinical Features | Gorlin et al. 2001 | Macca and Franco 2009 | This study |
| Craniofacial | 82.8 (24/29) | ||
| Brittle or sparse hair/alopecia | 15 to 65 | 21.5 (110/126) | 40.7 (11/27) |
| Facial dysmorphism | 25 to 75 | 69.1 (87/126) | 80.8 (21/26) |
| Facial milia | 10 to 35 | 29.4 (37/126) | 60.0 (3/5) |
| Frontal bossing | 40.0 (8/20) | ||
| Downslanting palpebral fissures | 51.8 (14/27) | ||
| Hypertelorism/telecanthus | 50.0 (13/26) | ||
| Epicanthus | 26.9 (7/26) | ||
| Broad nasal ridge | 48.1 (13/27) | ||
| Microretrognathia/retrognathia | 50.0 (13/26) | ||
| Cleft lip/pseudo-cleft of the upper lip | 35 to 45 | 32.6 (44/135) | 57.7 (15/26) |
| Abnormal ears | 30.7 (8/26) | ||
| Oral | 96.8% (122/126) | 100 (31/31) | |
| Teeth abnormalities | 20 to 50 | 43.3 (58/134) | 87.0 (20/23) |
| Missing/supernumary teeth | 87.0 (20/23) | ||
| Enamel hypoplasia | 20.0 (3/15) | ||
| Oral frenula | 75 to 80 | 63.7 (86/135) | 78.6 (22/28) |
| Tongue anomalies | 75 | 84.1 (106/126) | 78.6 (22/28) |
| Bifid tongue | 64.3 (18/28) | ||
| Tongue hamartomas | 67.8 (19/28) | ||
| Cleft/high-arched palate | 35 to 80 | 49.6 (67/135) | 71.4 (20/28) |
| Bifid uvula | 8.3 (2/24) | ||
| Skeletal | 88.1 (111/126) | 80.0 (24/30) | |
| Syndactyly | 55.5 (15/27) | ||
| Clinodactyly | 47.4 (64/135) | 50.0 (13/26) | |
| Metacarpal shortening/brachydactyly | 63.7 (86/135) | 52.0 (3/25) | |
| Polydactlyly | 23.0 (31/135) | 12.0 (3/25) | |
| Tibia dysplasia/shortening | 8.6 (2/23) | ||
| Hip dysplasia | 28.6 (2/7) | ||
| Kidneys | |||
| Cystic kidney disease | 15 to 50 | 37.3 (50/134) | 26.1 (6/23) |
| Under 18 years | 0 | ||
| Over 18 years | 100.0 (6/6) | ||
| Neurological | |||
| Central nervous system (CNS) involvement | 50 | 48.4 (61/126) | 64.5 (20/31) |
| Mental retardation | 28.9 (39/135) | 28.0 (7/25) | |
| Psychomoter retardation | 19.2 (5/26) | ||
| MRI available | 22.5 (7/31) | ||
| Hydrocephalus/porencephaly | 61.5 (8/13) | ||
| Cortical dysgenesis | 66.6 (4/6) | ||
| Corpus callosum dysgenesis/agenesis | 81.2 (13/16) | ||
| Cysts | 53.8 (7/13) | ||
| Epilepsy | 16.0 (4/25) | ||
Age at first diagnosis of the index patients ranged from birth to 30 years. Not surprisingly, the most common clinical features affected the mouth and face, with all patients having at least one oral anomaly and 82.8% of patients having a facial feature. Facial dysmorphism was most common (80.8% of patients), and included midface hypoplasia, frontal bossing, hypertelorism/telecanthus, a broad nasal bridge, downslanting palpebral fissures, and microretrognathia. Facial milia were found in three of five patients in whom milia were investigated. Cleft lip or a pseudo-cleft of the upper lip occurred frequently (57.7%), as did brittle or sparse hair (40.7%). Abnormal ears were reported in a third of patients and where specified were described as small and low-set.
The most common oral abnormality was missing or supernumerary teeth (87.0%), with most patients also having tongue anomalies (78.6%) including tongue hamartomas (67.8%), and/or a bifid tongue (64.3%). Oral frenulae (78.6%) and a cleft or high-arched palate (71.4%) were frequent findings. Skeletal abnormalities were slightly less frequent than the oral and facial features and were present in 80.0% of patients. Syndactyly was the most common symptom (55.5%), followed by clinodactyly (50.0%) and brachydactyly (52.0%). Interestingly, when the skeletal finding was unilateral, the left hands and feet were apparently more often affected than the right: of the seven cases of unilateral syndactyly for which details were provided, the left hand or foot only was affected in six patients. Two of the three patients reported with polydactyly had an extra hallux on the left foot, the third patient had bilateral polydactyly. Other interesting findings included: type II atrial-septal defects in both mother and daughter of family 27, a ventricular septum defect in the index patient of family 15, and myxomatous changes of the mitral and tricuspid valves in patient 30. Patient 26 had severe hyperopia at +6 diopters. No hepatic or pancreatic cysts were reported.
Clinical datawere available from the affected mothers and daughters in six families and the most pertinent features in four mother–daughter pairs are shown in Figure 1(A–R). In each of the families, the mother was very mildly affected and was only assessed for signs of OFD1 after confirmation of the diagnosis of OFD1 in her daughter. Phenotypes of the affected individuals in the four families shown in Figure 1 and are described in detail in the online Supporting Information.
Figure 1.
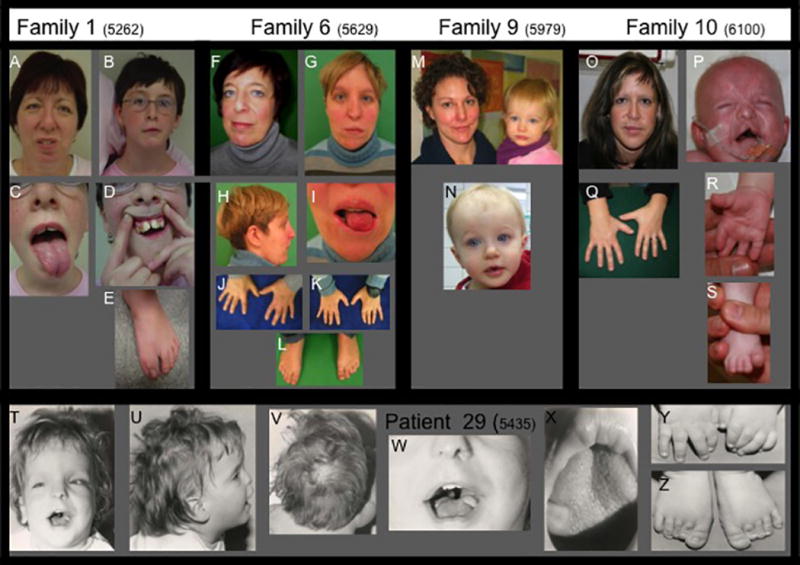
Mother–daughter pairs showing some of the typical clinical findings in OFD1. The pedigree symbols for the family members refer to the pedigrees shown in Fig. 3. A–E Family 1: the mother (II-2, A) and her youngest, more severely affected daughter (III-2, B–E); F–L Family 6: mother (I-1, F, J) and her daughter (II-1, G–I, K–L); M, N Family 9: mother and daughter, O–S Family 10: mother (I-1, O and Q) and daughter (II-2, P–S). T–Z: The only affected individual in family 29, in whom the entire OFD1 gene is deleted on one allele. Typical facial features include frontal bossing (G, M, N, T), a broad nasal ridge (G, N, T), downslanting palpebral fissures (A, F, N, T), facial asymmetry (A, B, F, G, O), and midface hypoplasia (B, G, T). Typical oral features shown include cleft lip and bifid tongue (C, I, P), tongue hamartomas (C, I, X). Skeletal features include polydactyly (surgically corrected in E), brachydactyly (J, K, R, Y), clinodactyly (J, K, Q), and syndactyly (J, K). Alopecia is shown in patient 29 in U and V.
Neurological Findings and Brain MRI
Intellectual disability was reported in 12 of 26 patients (46.1%) in whom it could be assessed, and varied from learning disability (two patients), speech developmental delay (one patient), psychomotor retardation (two infants), and intellectual disability (seven patients). The most consistent structural brain anomaly reported on the clinical checklist was agenesis or dysgenesis of the corpus callosum (81.2%). Brain MRI was available from seven patients and representative images are shown in Figure 2. All available imaging data sets were retrospectively studied and evaluated according to the criteria in Supp. Table S2. There was substantial variation in the findings, with the complete OFD1 brain phenotype consisting of complete agenesis of the corpus callosum with large single or multiple interhemispheric cysts, striking cortical infolding of gyri on both mesial and lateral surfaces, additional periventricular nodular heterotopia that appear unconnected to areas of infolded cortex, ventriculomegaly that was often asymmetric, mild molar tooth malformation of the midbrain, and moderate to severe cerebellar vermis hypoplasia. In less severely affected individuals, the callosal defect was less severe with partial agenesis, and the interhemispheric cysts were much smaller or absent. The vermis hypoplasia could be less severe but was always present. Only one patient (patient 27) appeared to have a molar tooth malformation (Fig. 2V–X), although another (patient 10) had an enlarged inferior tectum and mildly thick but downslanting superior cerebellar peduncles (Fig. 2H). Other variable features included hippocampal hypoplasia (Fig. 2I,Q), reduced volume of white matter (Fig. 2A, I, Q), thick anterior commissure, and mild brainstem hypoplasia or compression. Of the patients shown in Figure 2, patient 1 III-1 was not intellectually impaired, but had a learning disability and the intellectual status of patients 10 and 14 could not be assessed at ages 2 and 4 months, respectively. The remaining four patients all had intellectual disability.
Figure 2.
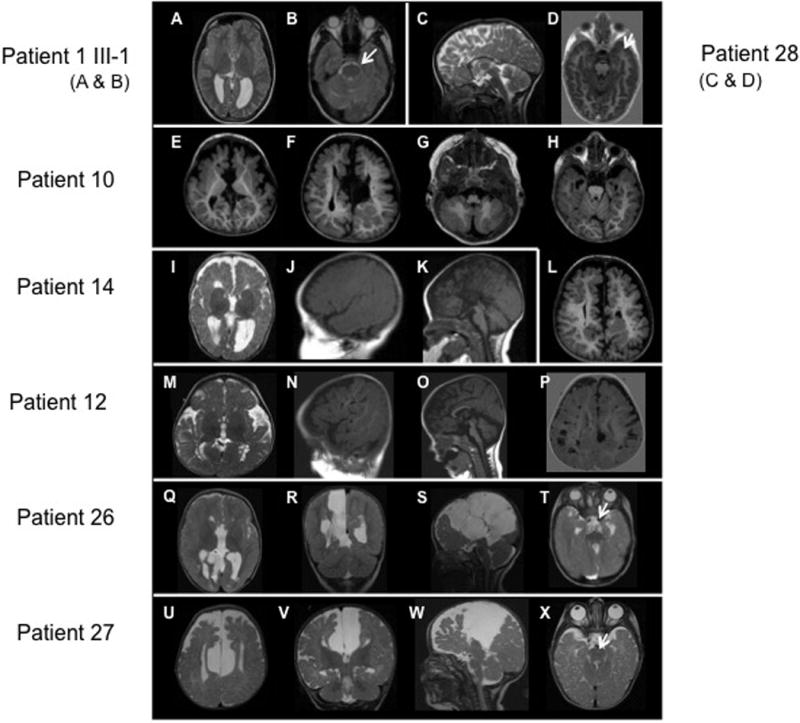
Brain MRIs of patients with OFD1 mutations. A, B: Patient II-1 in family 1, showing mild asymmetry with the right frontal lobe smaller than the left (A, B), reduced white matter volume, and mildly enlarged lateral ventricle (A). C, D: Patient 28 showing a short, thin, and dysmorphic corpus callosum. E–H and L: Patient II-2 in family 10, showing a large interhemispheric cyst (F), I–K: Patient 14, showing reduced white matter volume (I), mildly enlarged and widely separated lateral ventricles (I), severe partial agenesis of the corpus callosum (K), and possibly subtle cerebellar hypoplasia (K). M–P: Patient 12, showing a prominent bossed forehead (N, O), numerous large perivascular (Virchow–Robin) spaces in the white matter (mostly posteriorly) (N and P), a short, thin corpus callosum, and mild cerebellar vermis hypoplasia (O). Q–T: Patient 26, showing multiple, large interhemispheric cysts displacing the right hemisphere laterally (Q–S), reduced white matter volume (Q, R), a markedly enlarged third ventricle with an uncertain connection to the interhemispheric cysts (Q, R), and complete agenesis of the corpus callosum (Q, S). U–X: Patient II-1 in family 27, showing a large interhemispheric cyst displacing both hemispheres laterally (U–W), reduced white matter volume, particularly frontally (U, V), a markedly enlarged third ventricle that appears to connect to the interhemispheric cysts (V, W), complete agenesis of the corpus callosum (W), mildly small cerebellum with abnormally small anterior vermis, and mildly small posterior fossa (W).
OFD1 Intragenic Mutations
The mutations found in this study are listed in Table 2 and have been submitted to the LOVD database (www.lovd.nl/OFD1). Twenty-two of the mutations are novel and previously unreported. All of the intragenic mutations found were located in the first 16 exons of OFD1 (out of 23 total exons). Within these first 16 exons, mutations were found in all coding exons except exons 4 and 10, and were relatively evenly distributed amongst the exons, although exons 2 and 7 were affected most often (5 mutations each), followed by exon 12 (4 mutations) and exon 16 (three mutations, or the splicing thereof). The mothers of 20 patients were tested for the intragenic mutation found in their daughters. Seven mothers (35.0%) were found to be heterozygous carriers of the mutation; in two families themutation was also found in an affected sister of the index patient. In two families, the maternal grandmothers of the index patients were also tested and found not to carry the mutation. In both cases haplotype analysis revealed that the mutation had probably arisen on the grandpaternal allele.
Table 2.
OFD1 Mutations Found in the Patients in This Study
| Family | Exon/intron | DNA change | Amino acid change | Mutation | Familial? | Reference |
|---|---|---|---|---|---|---|
| 1 | Intron 1 | c.13-10T>A het. | – | Splice | Yes 2 sisters and mother affected | This report |
| 2 | Exon 2 | c.63insT | p.Lys21Aspfs∗8 | Frameshift | No | This report |
| 3 | Exon 2 | c.52G>T | p.Glu18∗ | Nonsense | Not tested | This report |
| 4 | Intron 2 | c.111+2T>C | – | Splice | Not tested | [Prattichizzo et al. 2008] |
| 5 | Intron 2 | c.111+3A>G | – | Splice | Not tested | This report |
| 6 | Exon 3 | c.148insG | p.His50Alafs∗26 | Frameshift | Yes mother affected | This report |
| 7 | Exon 3 | c.275_276delCT | p.Ser92Cysfs∗24 | Frameshift | No | This report |
| 8 | Exon 5 | c.400_403delGAAA | p.Glu134Ilefs∗9 | Frameshift | No | [Prattichizzo et al. 2008] |
| 9 | Exon 6 | c.422T>G | p.Met141Arg | Missense | Yes mother affected | This report |
| 10 | Exon 6 | c.508_509delGA | p.Asp170Phefs∗4 | Frameshift | Yes mother affected | This report |
| 11 | Intron 6 | c. 518-1G>A | – | Splice | No | This report |
| 12 | Exon 7 | c.541dupG | p.Asp181Glyfs∗22 | Frameshift | Not tested | This report |
| 13 | Exon 7 | c.607_610delTATA | p.Tyr203Argfs∗4 | Frameshift | Not tested | This report |
| 14 | Exon 7 | c.616_617delGA | p.Glu206Asnfs∗16 | Frameshift | No | [Prattichizzo et al. 2008] |
| 15 | Exon 7 | c.614_617delGAGA | p.Arg205Lysfs∗1 | Frameshift | No | This report |
| 16 | Exon 8 | c.710dupA | p.Tyr238Valfs∗1 | Frameshift | No | [Prattichizzo et al. 2008] |
| 17 | Exon 9 | c.877_878delAT | p.Met293Glyfs∗14 | Frameshift | No | [Prattichizzo et al. 2008] |
| 18 | Exon 11 | c.1099C>T | p.Arg367∗ | Nonsense | No | [Prattichizzo et al. 2008] |
| 19 | Intron 11 | c.1130-20_1130- 16delTTGGT | – | Splice | Yes (de novo in mother (mat. grandparents tested) | This report |
| 20 | Intron 11 | c.1130-1G>A | – | Splice | No | This report |
| 21 | Exon 12 | c.1190dupA | p.Asn397Lysfs∗11 | Frameshift | Not tested | This report |
| 22 | Exon 12 | c.1193_1196delAAT C | p.Gln398Leufs∗1 | Frameshift | Not tested | [Prattichizzo et al. 2008] |
| 23 | Exon 13 | c.1363_1366delAAA C | p.Lys455Asnfs∗13 | Frameshift | No | This report |
| 24 | Exon 14 | c.1468G>T | p.Glu490∗ | Nonsense | Not tested | This report |
| 25 | Exon 15 | c.1612C>T | p.Gln538∗ | Nonsense | Yes mother affected | This report |
| 26 | Exon 16 | c.1859_1860delCCin sG | p.Ser620Cysfs∗8 | Frameshift | Not tested | This report |
| 27 | Exon 16 | c.1979_1980delCT | p.Ser660Cysfs∗39 | Frameshift | Yes mother and sister affected | [Prattichizzo et al. 2008] |
| 28 | Exon | 16 c.1990dupC | p.Leu665Thrfs∗35 | Frameshift | No | This report |
| 29 | Deletion of whole OFD1 gene | Large deletion | No | This report | ||
| 30 | Deletion of whole OFD1 gene | Large deletion | Not tested | This report | ||
All splice mutations were tested using two splice prediction programs and were predicted to affect the splicing of the OFD1 transcript. The only missense mutation found, c.422T>G (p.Met141Arg), was not detected in 140 sequenced X chromosomes from patients with suspected OFD1, nor in 108 X chromosomes from healthy controls. Although the methionine at position 141 is not conserved in non-mammalian vertebrates, the programs PolyPhen and Mutation Taster both predicted a structural change to the protein. This change has not been documented in either the 1000 Genomes Database (http://browser.1000genomes.org/index.html) or the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/) (May 2012).
Detection of an Alternative Transcript with an Additional Exon
In Family 1, the mutation c.13-10T>A was predicted to create a new splice site in intron 1, although both thewild type and predicted new splice sites are weak. cDNA reverse transcribed from RNA extracted from transformed lymphocytes from the carrier mother, healthy father and both affected daughters was sequenced using forward and reverse primers located in exons 1 and 3 respectively, to test the effect of the splice mutation. Several cDNA fragments were detected (Supp. Fig. S1A), with two prominent transcripts of approximately 400 bp detected predominantly in the more severely affected daughter, III-2. Sequencing of the transcripts before gel separation revealed that intron 1 had been retained in the mutated allele. Cloning of the transcripts from patient III-2, her mother and healthy father, to sequence individual cDNA fragments, confirmed this result in the affected daughter, III-2 (Supp. Fig. S1C). The analysis of different transcripts from the healthy father revealed a transcript with the inclusion of an additional exon between exons 2 and 3, and which we have named exon 2b (Supp. Fig. S1D and E). This exon is 195 bp, is predicted to encode an additional 65 amino acids, and would not disrupt the open reading frame when included between exons 2 and 3. The splice acceptor consensus sequence for exons 2 and 2b, ag/TC, is identical. The additional exon 2b is documented in the Ensembl and UCSC databases, but to the best of our knowledge has not been included in OFD1 mutation analysis. We sequenced this exon in 32 patients with suspected OFD1 and in whom we had not previously detected a mutation, but did not detect any additional mutations.
Complete Deletion of OFD1 and Neighbouring Genes in Two Patients
Quantitative PCR of the entire OFD1 coding region was performed in 30 patients in whom no mutation had been found via DNA sequencing. Deletions spanning the entire OFD1 gene as well as neighbouring genes were identified in two patients (Supp. Fig. S2). Neither deletion was present in the patients’ parents. In Patient 29, the deletion was found to be a minimum of 143.242 kB and maximum of 145.437 kB and distally encompassed exons 11 and 12 of the EGFL6 gene, and the complete TCEANC, RAB9A, and TRAPPC2 genes 5 of OFD1. The proximal (3′) breakpoint appeared to be between OFD1 and the GPM6B gene because an amplicon in the last exon of GPM6B (exon 7) showed 100% amplification. The deletion in patient 30, which spanned a minimum of 94.185 kB and maximum of 248.074 kB began in intron 1 of RAB9A and spanned the entire TRAPPC2 gene 5′ of OFD1 and ended in the 152.698 kB large intron 1 of the GPM6B gene proximal to OFD1. The clinical details of patients 29 and 30 are provided in the online Supporting Information.
Genotype–Phenotype Correlation
Since various genotype–phenotype correlations have been previously reported [Prattichizzo et al., 2008; Thauvin-Robinet et al., 2006] we tested for correlations in our patient sample. A correlation between missense or splice mutations and cleft or high-arched palate has previously been reported. Of the 19 of our patients reported to have a cleft or high-arched palate, 5 had splice mutations (26.3%), and 1 single detected missense mutation. Of the remaining 13 patients, two had the deletion of the entire OFD1 gene (10.5%), nine (52.9%) had frameshift mutations and two had nonsense mutations (11.8%). These findings do not deviate significantly from the expected values for the different mutation types in the complete group of patients. Intellectual disability has previously been reported to be prevalent in patients with mutations in exons 3, 8, 9, 13, and 16. In our seven patients in whom intellectual disability was reported, two mutations affected the splicing of exons 2 and 3 and the remaining were frameshift mutations starting in exons 7 (one) and 16 (three patients). Five of the seven patients shown in Figure 2 with structural brain phenotypes had frameshift mutations starting in either exons 7 or 16. Four of these patients were at an age at which intellectual disability could be diagnosed. Cystic kidneys have been reported to occur more frequently in patients with mutations in exons 9 and 12; however, none of our three patients with mutations in exons 9 and 12 had cystic kidneys, although they were all under 18 years of age. Of the six patients who did have cystic kidneys, three had mutations in exon 3.
X-Inactivation
An analysis of X-inactivation was performed in families 1, 6, 10, 19, and 27, and in two patients with the deletion of OFD1 (Fig. 3). Haplotyping using five polymorphic microsatellite markers between the Androgen receptor gene, AR, and OFD1, was performed in the families to track the allele carrying the OFD1 mutation. Skewed inactivation was apparent in seven of the 14 patients tested; however, no consistent pattern of X-inactivation was observed, with the only correlation with degree of severity of the disorder being apparent in Family 1. Although a crossover was revealed on the mutation allele, it appeared that the mutation allele was preferentially inactivated in the mother (Figs. 3 II-2 and 1A) and lesser affected daughter (Fig. 3 III-1). Both alleles appeared to be active in the more severely affected daughter (Figs. 3 III-2 and 1B). On the other hand, in family 10, both the mother and daughter showed skewed inactivation with the mutation allele preferentially active. The mother (Figs. 3 I-1 and 1O) is very mildly affected whereas her daughter (Figs. 3 II-2 and 1P) is severely affected. The two patients with the deletion of OFD1 showed different patterns, with both alleles apparently active in patient 29 and skewed inactivation in patient 30 (Fig. 3F).
Figure 3.
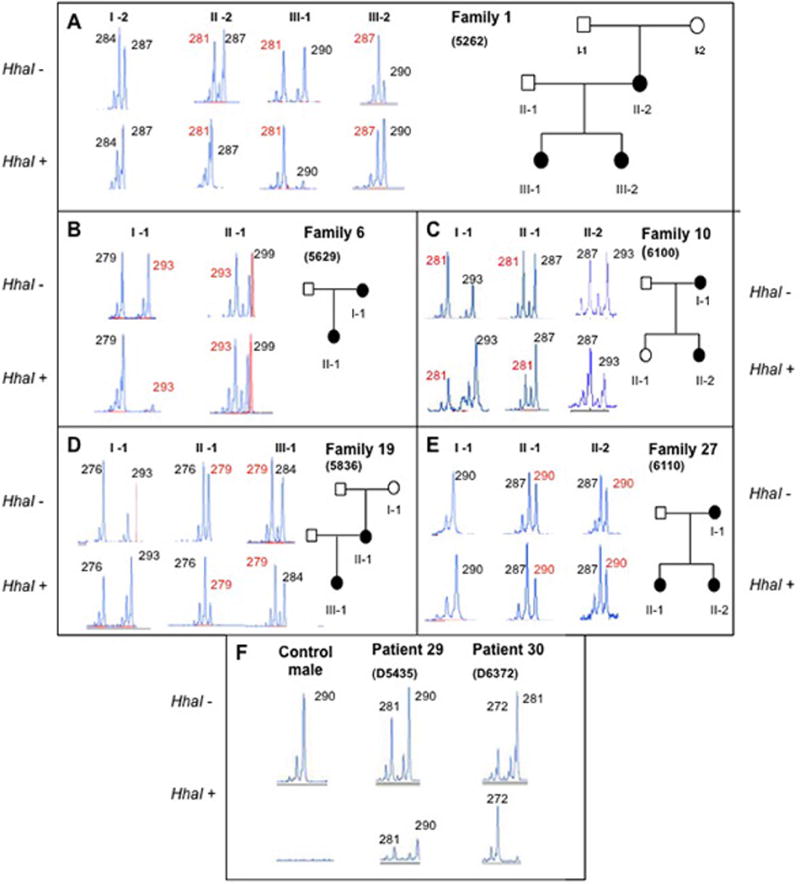
The analysis of possible X-inactivation in familial cases of OFD1 and two patients with deletion of the entire gene. A: Family 1; B: Family 6; C: Family 10; D: Family 19; and E: Family 27. F: Patients 29 and 30 with deletion of OFD1, and the results of the assay in DNA from a healthy male used as a control. Alleles of the CAG repeat in the androgen receptor gene, AR, on Xq22 are shown before (HhaI-) and after (HhaI+) restriction enzyme digestion, and the affection status of the individuals tested is show on the pedigrees. The sizes of the alleles are shown in bp, and the allele carrying the OFD1 mutation is labelled red, bold, and underlined, when it was possible, via haplotyping, to track the mutant allele in the family. The segregation of the AR alleles in family 24 was not informative, so that the mutation allele could not be identified.
Discussion
Diseases caused by defects in primary cilia are usually associated with an astoundingly broad spectrum of pathology, probably related to the near-ubiquitous presence of cilia in the mammalian body plan. Commonly observed phenotypes include polydactyly, craniofacial abnormalities, structural brain malformations, situs inversus, obesity, diabetes, and polycystic kidney disease, as exemplified by Bardet–Biedl syndrome (BBS) [Nigg and Raff, 2009]. OFD1, which has recently been recognized as a ciliopathy [Toriello, 2009] is no exception to the rule. The OFD1 gene product is localized to the centrosome and the basal body of primary cilia [Coene et al., 2009; Ferrante et al., 2006; Romio et al., 2004], and a knockout mouse model of OFD1 showed the main clinical features of OFD1 in an exaggerated form: laterality defects, craniofacial (severe cleft palate) and limb abnormalities, as well as cystic kidneys [Ferrante et al., 2006].
Clinical variability between sporadic cases is high [Macca and Franco, 2009; Morleo and Franco, 2008; Prattichizzo et al., 2008], and intrafamilial variability is possibly the rule rather than the exception [Donnai et al., 1987; Gorlin and Psaume, 1962; Salinas et al., 1991; Scolari et al., 1997; Thauvin-Robinet et al., 2006]. Morleo and Franco (2008) estimated that 30% of familial cases with mutations in OFD1 show a high degree of phenotypic variability in the clinical manifestations of the syndrome. On the basis of the present study, in which intrafamilial phenotypic variability was apparent in the six families for whom we had detailed clinical data, this percentage may be much higher. In all cases, as in previous reports, mothers of patients presented with a milder phenotype than that of their daughters. This can probably be ascribed to ascertainment bias and the observation of features in the mothers of patients only after the presentation of severe symptoms in at least one daughter. Thus many cases of mild OFD1 may well be underdiagnosed.
The summary of our clinical findings showed that all patients had at least one oral diagnostic feature, whereas craniofacial and digital features were not present in all. A comparison of the prevalence of the features noted with those previously reported revealed that many features, including brittle or sparse hair, a clefted or pseudo-clefted upper lip, a clefted or high-arched palate, and tooth abnormalities were more frequently reported in our patients than in the literature. This possibly underscores the importance of a thorough oral investigation of patients in making the diagnosis of OFD type I. OFD1 appears to associated with a large number of additional, variable symptoms that have been intermittently reported: A bifid uvula has been previously reported [Gorlin and Psaume, 1962; Salinas et al., 1991], we found it in two patients, and the uvula was completely missing in our patient 29 with the deletion of the entire OFD1 gene. Decreased hearing acuity has been reported in a few cases [Doege et al., 1964; Prattichizzo et al., 2008]; one-third of our patients were reported to have abnormal ears; however, hearing loss was not reported. Patient III-2 in family 1 was reported to have been diagnosed with Kawasaki syndrome as a child due to unexplained recurrent fever. Interestingly, recurrent fevers were also reported in a patient with X-linked JBTS and a mutation in OFD1 [Coene et al., 2009]. Upper airway congestion, recurrent infections, and congenital heart disorders are usually associated with disorders of motile cilia; however, upper airway congestion and recurrent ear infections have been reported in patients with OFD1 in the literature [Salinas et al., 1991] and were reported in our patient 23. Congenital heart disease was reported in 16% of our patients (4/25).
Patients with polycystic kidney disease often have less obvious additional anomalies in the liver, spleen, heart, and brain [Chang and Ong, 2008]. Fibrocystic liver and pancreas disease were recently reported in two adult females diagnosed with OFD1 [Chetty-John et al., 2010], and it was suggested that thesemay be under-recognized features of OFD1. Neither liver nor pancreatic disease was reported for any of the 28 patients in this study for whom the clinical checklist was completed, indicating that fibrocystic liver and pancreas disease are probably rare findings. Brain malformations were, however, more common and diverse. Although CNS malformations in patients with OFD1 have often been described [Connacher et al., 1987; Odent et al., 1998; Salinas et al., 1991; Towfighi et al., 1985], to date there has not been a comprehensive study of brain MRIs from patients with OFD1 mutations. Although corpus callosum dysgenesis is the most common finding (82% of patients in this study), perhaps the most characteristic findings of the brain phenotype are large single or multiple interhemispheric cysts and striking cortical infolding. Renal cysts in polycystic kidney disease are associated with increased cell proliferation and often also with a loss of cell polarity [Jonassen et al., 2008]; however, the cause of the intracerebral brain cysts is still unclear. Hydrocephalus has often been associated with motile cilia and ciliary dyskinesia and aplasia [De Santi et al., 1990; Greenstone et al., 1984; Jabourian et al., 1986]. Although in many patients a structural brain malformation was found, only 16.7% of patients had epilepsy. Sixty-eight percent of patients showed signs of intellectual disability.
Many receptors and ion channels are expressed on the membrane of primary cilia and modulate various signalling pathways, including those of sonic hedgehog (Shh) [Wechsler-Reya and Scott, 1999], Wnt [Simons et al., 2005], and platelet-derived growth factor [PDGFα, Schneider et al., 2005], that are involved in diverse processes important in development and homeostasis [Lee and Gleeson, 2010]. One could postulate that the disruption of these pathways known to be important for cell proliferation, fate determination, migration, and neural tube patterning [Huangfu et al., 2003; Schneider et al., 2005; Simons et al., 2005; Wallingford and Habas, 2005; Wechsler-Reya and Scott, 1999] plays a role in the brain phenotype. Patients with Meckel–Gruber or Bardet–Biedl syndrome can display occipital encephalocoele, macrocephaly, holoprosencephaly, and other structural brain defects [Ahdab-Barmada and Claassen, 1990; Rooryck et al., 2007]. Patients with Joubert syndrome (JBTS) sometimes have polymicrogyria [Dixon-Salazar et al., 2004; Giordano et al., 2009; Senocak et al., 2010]. Thus, ciliary function is likely required for some aspects of neuronal development and organization within the cerebral cortex, although the mechanisms remain to be determined. Cerebellar hypoplasia is a striking feature of JBTS and many other ciliopathies [Lee and Gleeson, 2010]. Interestingly, the molar tooth sign (MTS), which is characteristic of patients with Joubert syndrome [Quisling et al., 1999] and Joubert Syndrome related disorders (JSRD) [Gleeson et al., 2004], and which was found in the patients with X-linked Joubert syndrome and mutations in OFD1 [Coene et al., 2009], and in patients with OFD6 with and without mutations in TMEM216 [Poretti et al., 2012; Valente et al., 2010], was found in only one of our patients.
The OFD1 gene is one of the almost 20% of X-chromosomal genes that escape inactivation [Carrel et al., 1999; Carrel and Willard, 2005]. Carrel and Willard (2005) have shown that there are substantial differences in the level of expression from the inactive (Xi) alleles of genes escaping inactivation in different cell lines, thereby indicating that females are considerably heterogeneous for the levels of X-linked gene expression. Although it has been shown that there is still some expression from some of the genes escaping inactivation, no expression of the OFD1 gene was shown in the somatic cell lines tested [Carrel et al., 1999; de Conciliis et al., 1998]. In fact, the more severe OFD1 phenotype in a mouse model has been attributed to the fact that Ofd1 does not escape inactivation in the mouse [Prattichizzo et al., 2008]. Thauvin-Robinet et al. (2006) investigated X-inactivation in 23 females with OFD1 mutations, including eight sporadic and 15 patients belonging to five families and found incomplete, but nonrandom inactivation in 30% of patients. Complete, skewed X-inactivation was shown in different tissues from a severely affected fetus, leading the authors to conclude that the severe cerebral and renal phenotype could be explained by X-inactivation [Thauvin-Robinet et al., 2011]. In an attempt to shed more light on the question of X-inactivation and the OFD1 gene, we investigated the patterns of inactivation in five families with at least two affected females, using haplotype analysis to track the mutation allele. Although substantial skewing was apparent in more than half of the patients, there was no consistent correlation between inactivation and the mutant OFD1 allele. Therefore, although it appears that X-inactivation may in fact be playing some role in the expression of the disorder, the situation is likely much more complex with other factors predominating.
Most OFD1mutations identified to date lead to a premature truncation of the protein [Ferrante et al., 2001; Prattichizzo et al., 2008; Rakkolainen et al., 2002; Romio et al., 2003; Thauvin-Robinet et al., 2006], presumably resulting in a loss-of-function. In a large study of 81 patients with mutations in OFD1 [Prattichizzo et al., 2008], most of the mutations (53.7%) resulted in a frameshift and presumably premature protein truncation. Missense, splice and nonsense mutations (13.4%, 16.4%, and 16.4%, respectively) were in theminority, screening for deletions or duplications was not performed. Of the 30 different mutations that we identified, 17 (56.7%) resulted in a frameshift, 6 (20.0%) were predicted to affect splicing, four (13.3%) were nonsense mutations, one (3.3%) a missense mutation, and two (6.7%) were large deletions spanning the entire OFD1 gene. The combination of results of Thauvin-Robinet (2006) with those of previously reported cases showed that the majority of mutations (65.5%) were located in exons 3, 8, 9, 13, and 16 [Prattichizzo et al., 2008]. Our results differ in that the 26 intragenic mutations in our cohort were fairly evenly distributed between exons two to 16, with most mutations affecting exons 2, 7, 12, and 16 and no mutations in exons 4 and 10. The mutation identified in patients with SGBS2 occurred in exon 16 and those in patients with JBTS10 in exon 21. Using in vitro assays, Coene et al. (2009) demonstrated that the mutations in SGBS2 and JBTS10 had a less deleterious effect both on the binding of OFD1 to Lebercilin and on the cellular localization of OFD1 protein, compared to mutations causing OFD1, providing one possible explanation for the nonlethal phenotype in males with SGBS2 and JBTS. However, more recently a deletion in OFD1 exon 8 was found in a male patient with JBTS10 [Field et al., 2012], indicating that the regulation of phenotypic severity is more complex. Coene et al. (2009) also demonstrated a transcript lacking exon 10, which may imply that mutations in exon 10 do not lead to OFD1. The same may well apply to the additional exon 2b, which is alternatively included in transcripts and in which we found no mutations in female patients with OFD1.
We found the heterozygous de novo deletion of the whole OFD1 gene in two patients. One deletion of approximately 144 kB (Patient 29) spans the TCEANC, RAB9A, andTRAPPC2 genes and interrupts the EGFL6 gene. The other deletion (Patient 30) disrupts RAB9A, spans TRAPPC2 and OFD1 and ends in the large first intron of GPM6B. Neither of the two patients with deletions spanning other genes in addition toOFD1showed any extra features not attributable to the deletion of OFD1.
The cilium is an extremely complicated organelle, and its assembly can be disrupted in many different ways. Different mutations in a single “cilia gene” causing different disorders, such as that observed for the OFD1 gene, is a common theme in ciliopathies [Delous et al., 2007; Garcia-Gonzalez et al., 2007]. Moreover, there is increasing evidence that single patients may carry deleterious or potentially deleterious genetic alterations in more than one “cilia gene” [Tory et al., 2007]. Initial studies to compile ciliary and centrosome proteomes and analyze their interaction networks [Inglis et al., 2006], suggest that complex networks of ciliary proteins function to mediate partially overlapping and non-redundant functions. [Zaghloul and Katsanis, 2010] have argued that mutational load and the complex interactions between causal and modifying alleles may contribute to phenotypic variability. In addition, recent studies have shown that OFD1 also localizes to the nucleus and interacts with proteins involved in the chromatin-remodeling complex [Giorgio et al., 2007], suggesting that some of the clinical features observed in OFD1 may be due to the nuclear function of the protein [Macca and Franco, 2009]. The identification of additional genes causing other forms of OFDS will no doubt provide further clues to other pathways in which OFD1 is involved.
Supplementary Material
Acknowledgments
We are grateful to the patients and their families for their willing cooperation in this study. We thank Elke Jantz-Schuble, Elke Troppmann, Christine Hodler, and Angela Steiert for excellent technical assistance.
Footnotes
Disclosure statement: The authors declare no financial conflict of interest.
References
- Ahdab-Barmada M, Claassen D. A distinctive triad of malformations of the central nervous system in the Meckel-Gruber syndrome. J Neuropathol Exp Neurol. 1990;49(6):610–20. doi: 10.1097/00005072-199011000-00007. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Brown CJ. Polymorphic X-chromosome inactivation of the human TIMP1 gene. Am J Hum Genet. 1999;65(3):699–708. doi: 10.1086/302556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimonte S, De Angelis A, Quagliata L, Giusti F, Tammaro R, Dallai R, Ascenzi MG, Diez-Roux G, Franco B. Ofd1 is required in limb bud patterning and endochondral bone development. Dev Biol. 349(2):179–91. doi: 10.1016/j.ydbio.2010.09.020. [DOI] [PubMed] [Google Scholar]
- Borozdin W, Boehm D, Leipoldt M, Wilhelm C, Reardon W, Clayton-Smith J, Becker K, Muhlendyck H, Winter R, Giray O, others SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J Med Genet. 2004;41(9):e113. doi: 10.1136/jmg.2004.019901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura M, Lenzner S, Latos-Bielenska A, Ropers HH. A novel X-linked recessive mental retardation syndrome comprisingmacrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171–178. doi: 10.1007/s00439-006-0210-5. [DOI] [PubMed] [Google Scholar]
- Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci U S A. 1999;96(25):14440–4. doi: 10.1073/pnas.96.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–4. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Chang MY, Ong AC. Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and treatment. Nephron Physiol. 2008;108(1):p1–7. doi: 10.1159/000112495. [DOI] [PubMed] [Google Scholar]
- Chetty-John S, Piwnica-Worms K, Bryant J, Bernardini I, Fischer RE, Heller T, Gahl WA, Gunay-Aygun M. Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD I) Am J Med Genet A. 152A(10):2640–5. doi: 10.1002/ajmg.a.33666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B, van Wijk E, Gorden NT, others OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85(4):465–81. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connacher AA, Forsyth CC, Stewart WK. Orofaciodigital syndrome type I associated with polycystic kidneys and agenesis of the corpus callosum. J Med Genet. 1987;24(2):116–8. doi: 10.1136/jmg.24.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Conciliis L, Marchitiello A, Wapenaar MC, Borsani G, Giglio S, Mariani M, Consalez GG, Zuffardi O, Franco B, Ballabio A, others Characterization of Cxorf5 (71–7A), a novel human cDNA mapping to Xp22 and encoding a protein containing coiled-coil alpha-helical domains. Genomics. 1998;51(2):243–50. doi: 10.1006/geno.1998.5348. [DOI] [PubMed] [Google Scholar]
- De Santi MM, Magni A, Valletta EA, Gardi C, Lungarella G. Hydrocephalus, bronchiectasis, and ciliary aplasia. Arch Dis Child. 1990;65(5):543–4. doi: 10.1136/adc.65.5.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, others The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39(7):875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, others Mutations in the AHI1 gene, encoding Jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75(6):979–87. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege TC, Thuline HC, Priest JH, Norby DE, Bryant JS. Studies of a Family with the Oral-Facial-Digital Syndrome. N Engl J Med. 1964;271:1073–8. doi: 10.1056/NEJM196411192712101. [DOI] [PubMed] [Google Scholar]
- Donnai D, Kerzin-Storrar L, Harris R. Familial orofaciodigital syndrome type I presenting as adult polycystic kidney disease. J Med Genet. 1987;24(2):84–7. doi: 10.1136/jmg.24.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feather SA, Woolf AS, Donnai D, Malcolm S, Winter RM. The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3. Hum Mol Genet. 1997;6(7):1163–7. doi: 10.1093/hmg/6.7.1163. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Barra A, Truong JP, Banfi S, Disteche CM, Franco B. Characterization of the OFD1/Ofd1 genes on the human and mouse sex chromosomes and exclusion of Ofd1 for the Xpl mouse mutant. Genomics. 2003;81(6):560–9. doi: 10.1016/s0888-7543(03)00091-0. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, others Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–76. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, Wilson SW. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet. 2009;18(2):289–303. doi: 10.1093/hmg/ddn356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–7. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, Gardner A, Glubb G, Hobson L, Corbett M, Friend K, Willis-Owen S, Gecz J. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20:806–809. doi: 10.1038/ejhg.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev. 2006;16(3):254–9. doi: 10.1016/j.gde.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalez MA, Menezes LF, Piontek KB, Kaimori J, Huso DL, Watnick T, Onuchic LF, Guay-Woodford LM, Germino GG. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. 2007;16(16):1940–50. doi: 10.1093/hmg/ddm141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano L, Vignoli A, Pinelli L, Brancati F, Accorsi P, Faravelli F, Gasparotti R, Granata T, Giaccone G, Inverardi F, others Joubert syndrome with bilateral polymicrogyria: clinical and neuropathological findings in two brothers. Am J Med Genet A. 2009;149A(7):1511–5. doi: 10.1002/ajmg.a.32936. [DOI] [PubMed] [Google Scholar]
- Giorgio G, Alfieri M, Prattichizzo C, Zullo A, Cairo S, Franco B. Functional characterization of the OFD1 protein reveals a nuclear localization and physical interaction with subunits of a chromatin remodeling complex. Mol Biol Cell. 2007;18(11):4397–404. doi: 10.1091/mbc.E07-03-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, Graham JM, Jr, Maria BL, Barkovich AJ, Dobyns WB. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A(2):125–34. doi: 10.1002/ajmg.a.20437. discussion 117. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Psaume J. Orodigitofacial dysostosis–a new syndrome. A study of 22 cases. J Pediatr. 1962;61:520–30. doi: 10.1016/s0022-3476(62)80143-7. [DOI] [PubMed] [Google Scholar]
- Greenstone MA, Jones RW, Dewar A, Neville BG, Cole PJ. Hydrocephalus and primary ciliary dyskinesia. Arch Dis Child. 1984;59(5):481–2. doi: 10.1136/adc.59.5.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: review and diagnostic guidelines. Am J Med Genet A. 2007;143A(24):3314–23. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Inglis PN, Boroevich KA, Leroux MR. Piecing together a ciliome. Trends Genet. 2006;22(9):491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Jabourian Z, Lublin FD, Adler A, Gonzales C, Northrup B, Zwillenberg D. Hydrocephalus in Kartagener’s syndrome. Ear Nose Throat J. 1986;65(10):468–72. [PubMed] [Google Scholar]
- Lee JH, Gleeson JG. The role of primary cilia in neuronal function. Neurobiol Dis. 38(2):167–72. doi: 10.1016/j.nbd.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151C(4):318–25. doi: 10.1002/ajmg.c.30224. [DOI] [PubMed] [Google Scholar]
- Morleo M, Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet. 2008;45(7):401–8. doi: 10.1136/jmg.2008.058305. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139(4):663–78. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Odent S, Le Marec B, Toutain A, David A, Vigneron J, Treguier C, Jouan H, Milon J, Fryns JP, Verloes A. Central nervous system malformations and early end-stage renal disease in oro-facio-digital syndrome type I: a review. Am J Med Genet. 1998;75(4):389–94. [PubMed] [Google Scholar]
- Papillon Léage Mme, Psaume J. Hereditary abnormality of the buccal mucosa: abnormal bands and frenula. Revue Stomatol. 1954;55(4):209–27. [PubMed] [Google Scholar]
- Poretti A, Vitiello G, Hennekam RC, Arrigoni F, Bertini E, Borgatti R, Brancati F, D’Arrigo S, Faravelli F, Giordano L, others Delineation and Diagnostic Criteria of Oral-Facial-Digital Syndrome Type VI. Orphanet J Rare Dis. 7(1):4. doi: 10.1186/1750-1172-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008;29(10):1237–46. doi: 10.1002/humu.20792. [DOI] [PubMed] [Google Scholar]
- Quisling RG, Barkovich AJ, Maria BL. Magnetic resonance imaging features and classification of central nervous system malformations in Joubert syndrome. J Child Neurol. 1999;14(10):628–35. doi: 10.1177/088307389901401002. discussion 669–72. [DOI] [PubMed] [Google Scholar]
- Rakkolainen A, Ala-Mello S, Kristo P, Orpana A, Jarvela I. Four novel mutations in the OFD1 (Cxorf5) gene in Finnish patients with oral-facial-digital syndrome 1. J Med Genet. 2002;39(4):292–6. doi: 10.1136/jmg.39.4.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15(10):2556–68. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- Romio L, Wright V, Price K, Winyard PJ, Donnai D, Porteous ME, Franco B, Giorgio G, Malcolm S, Woolf AS, others OFD1, the gene mutated in oral-facial-digital syndrome type 1, is expressed in the metanephros and in human embryonic renal mesenchymal cells. J Am Soc Nephrol. 2003;14(3):680–9. doi: 10.1097/01.asn.0000054497.48394.d2. [DOI] [PubMed] [Google Scholar]
- Rooryck C, Pelras S, Chateil JF, Cances C, Arveiler B, Verloes A, Lacombe D, Goizet C. Bardet-biedl syndrome and brain abnormalities. Neuropediatrics. 2007;38(1):5–9. doi: 10.1055/s-2007-981466. [DOI] [PubMed] [Google Scholar]
- Salinas CF, Pai GS, Vera CL, Milutinovic J, Hagerty R, Cooper JD, Cagna DR. Variability of expression of the orofaciodigital syndrome type I in black females: six cases. Am J Med Genet. 1991;38(4):574–82. doi: 10.1002/ajmg.1320380416. [DOI] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861–6. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scolari F, Valzorio B, Carli O, Vizzardi V, Costantino E, Grazioli L, Bondioni MP, Savoldi S, Maiorca R. Oral-facial-digital syndrome type I: an unusual cause of hereditary cystic kidney disease. Nephrol Dial Transplant. 1997;12(6):1247–50. doi: 10.1093/ndt/12.6.1247. [DOI] [PubMed] [Google Scholar]
- Senocak EU, Oguz KK, Haliloglu G, Topcu M, Cila A. Structural abnormalities of the brain other than molar tooth sign in Joubert syndrome-related disorders. Diagn Interv Radiol. 16(1):3–6. doi: 10.4261/1305-3825.DIR.2673-09.1. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, others Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37(5):537–43. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton FB, Bernstein J, Koh G, Roy S, 3rd, Wilroy RS. Cystic kidneys in a patient with oral-facial-digital syndrome type I. Am J Kidney Dis. 1982;1(5):288–93. doi: 10.1016/s0272-6386(82)80027-9. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, others Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43(1):54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Franco B, Saugier-Veber P, Aral B, Gigot N, Donzel A, Van Maldergem L, Bieth E, Layet V, Mathieu M, others Genomic deletions of OFD1 account for 23% of oral-facial-digital type 1 syndrome after negative DNA sequencing. Hum Mutat. 2009;30(2):E320–9. doi: 10.1002/humu.20888. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Lesca G, Aral B, Gigot N, Lambert S, Gueneau L, Macca M, Franco B, Huet F, Zabot MT, others Cerebral dysgenesis does not exclude OFD I syndrome. Am J Med Genet A. 155(2):455–7. doi: 10.1002/ajmg.a.33812. [DOI] [PubMed] [Google Scholar]
- Toriello HV. Oral-facial-digital syndromes, 1992. Clin Dysmorphol. 1993;2(2):95–105. [PubMed] [Google Scholar]
- Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A(5):1089–95. doi: 10.1002/ajmg.a.32799. [DOI] [PubMed] [Google Scholar]
- Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, others High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18(5):1566–75. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]
- Towfighi J, Berlin CM, Jr, Ladda RL, Frauenhoffer EE, Lehman RA. Neuropathology of oral-facial-digital syndromes. Arch Pathol Lab Med. 1985;109(7):642–6. [PubMed] [Google Scholar]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, others Mutations in TMEM 216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 42(7):619–25. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB, Habas R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132(20):4421–36. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22(1):103–14. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Zaghloul NA, Katsanis N. Functional modules, mutational load and human genetic disease. Trends Genet. 26(4):168–76. doi: 10.1016/j.tig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.