

Abstract
Small cell lung cancer (SCLC) is an aggressive disease that accounts for approximately 14% of all lung cancers. In the United States, approximately 31,000 patients are diagnosed annually with SCLC. Despite numerous clinical trials, including at least 40 phase 3 trials since the 1970s, systemic treatment for patients with SCLC has not changed significantly in the past several decades. Consequently, the 5-year survival rate remains low at <7% overall, and most patients survive for only 1 year or less after diagnosis. Unlike nonsmall cell lung cancer (NSCLC), in which major advances have been made using targeted therapies, there are still no approved targeted drugs for SCLC. Significant barriers to progress in SCLC include 1) a lack of early detection modalities, 2) limited tumor tissue for translational research (eg, molecular profiling of DNA, RNA, and/or protein alterations) because of small diagnostic biopsies and the rare use of surgical resection in standard treatment, and 3) rapid disease progression with poor understanding of the mechanisms contributing to therapeutic resistance. In this report, the authors review the current state of SCLC treatment, recent advances in current understanding of the underlying disease biology, and opportunities to advance translational research and therapeutic approaches for patients with SCLC.
Keywords: small cell lung cancer, genomics, proteomics, translational research, novel therapies
INTRODUCTION
Where Are We Now?
Small cell lung cancer (SCLC) is the most aggressive form of lung cancer. In the United States, SCLC makes up 14% of lung cancers overall, with approximately 31,000 patients diagnosed annually.1 In the vast majority of patients, the development of SCLC is associated with tobacco exposure.2,3 This, combined with frequent TP53 mutation (>75%–90% of SCLCs),4–6 results in an aggressive, highly complex disease at the molecular level with a large number of mutations present in each tumor.7,8
Lack of early detection methods
Compared with non—small cell lung cancers (NSCLC), SCLC is characterized by a rapid doubling time and early, widespread metastases. Consequently, most patients (60%–70%) will have extensive-stage (ES) disease at the time of diagnosis (defined as cancer that has spread beyond the ipsilateral lung and regional lymph nodes and cannot be included in a single radiation field). Recently, results from the National Lung Screening Trial highlighted the aggressive nature of SCLC and its propensity for characteristic early hematogenous spread.9,10 That study proved the value of screening high-risk patients with low-dose computed tomography (CT) imaging for the early detection of lung cancers overall. However, for patients who had SCLC identified (in contrast to those who had NSCLC), early detection did not reduce the number of patients who were diagnosed with ES disease, nor did it have an evident impact on survival for patients with SCLC. Specifically, 86% of the 125 patients who had SCLC identified through this early detection program had advanced disease at diagnosis. On the basis of these results, there are currently no methods with proven efficacy for the early detection of SCLC.
Current treatment standards for limited-stage SCLC
The vigorous response of SCLC to frontline chemotherapy (roughly 60%–70% response rates11) and to radiation is starkly contrasted by its subsequent resistance to second-line and subsequent therapies after disease recurrence. In general, limited-stage (LS) SCLC (ie, cancer confined to the thorax in a single radiation field) is treated with concurrent chemoradiation, whereas ES-SCLC is treated with chemotherapy alone. The current state-of-the-art treatment for LS-SCLC has recently been reviewed in Cancer by Amini et al12 and typically includes cisplatin-etoposide chemotherapy in combination with radiation therapy (RT). For LS-SCLC, clinical trials have established the superiority of hyperfractionated RT and the importance of beginning RT as early in the treatment course as possible (preferably during cycle 1 of chemotherapy).13–19 With these multimodality treatments, up to 20% of patients will have long-term disease control. However, a majority will recur despite definitive chemoradiation.20,21 Additional progress in the treatment of SCLC has included the use of prophylactic cranial irradiation in patients with ES-SCLC and LS-SCLC who have a response to their initial platinum-based chemotherapy.22,23
Current treatment standards for ES-SCLC
For patients with ES-SCLC, front-line treatment is platinum-based chemotherapy. Most patients in the United States receive platinum-etoposide (EP) chemotherapy (with either carboplatin or cisplatin), and some patients receive platinum-irinotecan as an alternative, especially outside the United States.24 After relapse, topotecan is the only second-line drug approved by the US Food and Drug Administration (FDA). However, despite its indication in this setting, topotecan has produced disappointing response rates. As with other second-line therapies, responses are typically higher in patients who experiences longer disease control after frontline, platinum-based therapy. For example, response rates may be as high as 25% in patients who relapse >3 months after the completion of EP, but the rates are only 3% to 6% if patients relapse <3 months after EP.25 Other options after front-line therapy include taxanes, irinotecan, vinorelbine, and gemcitabine. A more recent arrival to the second-line setting is temozolomide (TMZ), which is attractive for its oral dosing and activity in central nervous system lesions (a 38% response rate was observed in patients who had brain metastasis in a phase 2 study).26 In the third-line setting, responses to chemotherapy are rare, and there is no consensus on treatment beyond first-line and second-line therapy.25
Clinical trials for SCLC
Given the high rates of recurrence, rapid development of treatment-resistant disease, and limited activity of existing therapies after relapse, current National Comprehensive Cancer Network (NCCN) guidelines support the use of clinical trials in the second-line and later settings after disease progression or recurrence.24 However, despite intensive efforts by clinical investigators, the list of unsuccessful drugs for SCLC is long. These include more than 40 failed phase 3 studies since the 1970s, including 3 studies that attempted to replicate results from the Japanese Cooperative Oncology Group which had suggested the superior activity of platinum-irinotecan27–30 and a platinum-pemetrexed study that was terminated early for inferiority relative to platinum-etoposide.31 Many more drugs did not make it far beyond the starting gates because of early negative data in phase 1 or 2 trials or limitations because of poor enrollment or early toxicity. These include imatinib, oblimersen, and bevacizumab (targeting c-Kit, B-cell leukemia 2 [Bcl2], and vascular endothelial growth factor [VEGF], respectively).32–34 To address these challenges and bring forward more effective drugs, the medical and scientific community will need to address existing barriers to SCLC research and leverage opportunities for progress in the field.
Few advances in therapeutic options
In the absence of effective approaches for early detection or prevention, effective treatments for patients diagnosed with SCLC become even more critical. However, as described above, therapeutic options have remained largely unchanged for over 3 decades.35 This finding, combined with the relative resistance of recurrent SCLC to salvage chemotherapy, has resulted in persistently dismal patient outcomes. Less than 7% of patients diagnosed with SCLC are alive 5 years after diagnosis (all stages), and <5% of patients with ES disease survive for >2 years.36
The lack of major therapeutic breakthroughs in SCLC is in stark contrast to nonsmall cell lung cancer (NSCLC), in which a growing number of mutations or gene fusions guide treatment selection for specific patient subsets. These include epidermal growth factor receptor (EGFR) and B-Raf proto-oncogene serine/threonine kinase (BRAF) mutations as well as anaplastic lymphoma kinase (ALK), v-ros avian UR2 sarcoma virus oncogene homolog 1 (ROS1), and ret proto-oncogene (RET) fusions.37 Because of the success of targeted therapies in NSCLC, molecular profiling (primarily in nonsquamous cancers) has become standard in clinical practice. For example, the current NCCN guidelines for the treatment of metastatic NSCLC recommend that oncologists have adequate tissue for molecular testing to establish histologic subtype, with repeat biopsy if necessary that can then be used for standard-of-care mutation testing (eg, for EGFR and ALK) performed during multiplexed/next-generation sequencing.37
By using Clinical Laboratory Improvement Amendments (CLIA)-certified, multiplexed platforms that test for DNA alterations in multiple genes simultaneously, like those in use at our institutions or others that are commercially available, it is possible to test for validated targets (eg, EGFR mutation, ALK fusion) that are critical for initial treatment selection while also screening for the presence of additional potentially drug-gable targets. Although most of these additional targets are not currently used to select front-line therapy, this information can subsequently be used to match patients to clinical trials or other therapies at the time of relapse based on the best available clinical data. Online resources like www.mycancergenome.org allow medical professionals to explore the most recent data around specific gene alterations, including existing clinical data for a given target and ongoing clinical trials for drugs directed against the target (or associated pathway).38
In addition to the genomic profiling that has been adopted into clinical practice, several research initiatives to catalog DNA, RNA, and protein profiles among lung squamous carcinomas and adenocarcinomas have accelerated the pace of discovery, such as The Cancer Genome Atlas (TCGA) (which includes >493 squamous carcinomas and 512 adenocarcinomas with molecular profiling to date).39–41 Unfortunately, similar efforts have not yet been possible in SCLC because of a lack of adequate tumor tissue (most patients are diagnosed with SCLC by fine-needle aspiration, and surgically resected specimens are relatively rare).
Overview
To improve outcomes for patients with SCLC, therapies are needed that 1) improve the durability of responses to front-line therapy (including an increased number of patients with LS-SCLC who have long-term survival) and 2) have activity after disease relapse. To achieve this, we need to apply an approach similar to what has been done in NSCLC and other cancers, including in-depth characterization of potential drug targets and activated pathways present in SCLC, followed by translation of the most promising drug targets into the clinic. Furthermore, beyond profiling of treatment-naive tumors, there is an urgent need to better understand what drives therapeutic resistance in recurrent SCLC—because chemotherapy resistance to second-line and later treatments remains a key factor in poor patient outcomes.
To make progress in these areas, there are several barriers specific to SCLC that must be addressed. These include, first and foremost, a lack of adequate tissue for molecular profiling. This is the result of rare surgical resections in SCLC, small diagnostic biopsies, and the absence of a validated biomarker (for treatment selection) that would necessitate more substantial diagnostic biopsies (and rebiopsies at the time of progression). Furthermore, given the rapid pace of disease progression, translation of new targets into the clinic will require multidisciplinary teams that can expedite biopsies and profiling to minimize the time to treatment initiation. In this review, we discuss the current state of SCLC treatment, established hallmarks of SCLC biology, existing challenges in translational research, and opportunities for progress.
Recent Advances in SCLC Translational Research
Several recent advances in SCLC research have contributed to the understanding of SCLC biology, including the development of new animal models of SCLC to help facilitate translational studies.42–44 Some of these findings have led to the identification of new potential drug targets that are now being investigated in clinical trials. For example, among 127 interventional studies currently enrolling SCLC patients, novel drug targets under investigation include poly(ADP-ripose) polymerase 1 (PARP1), immune checkpoints, stem cell targets, and fibroblast growth factor receptor (FGFR).45 In addition, novel bioinformatics approaches have opened new avenues for exploring existing data, including the identification of existing drugs that potentially could be repurposed for SCLC treatment (eg, tricyclic antidepressants).46,47 Other investigators have recently demonstrated the potential of “liquid biopsies”—obtaining SCLC cells from a patient’s circulation that can be propagated in mouse models for subsequent molecular profiling and drug sensitivity screening.48
Molecular hallmarks of SCLC—and their clinical implications
The current classification system for lung cancers from the World Health Organization (WHO 2004) recognizes 3 high-grade lung cancers of neuroendocrine (NE) origin.49 These include SCLC, combined SCLC (which contain areas of NSCLC), and large cell NE cancer (LCNEC) (a subset of NSCLC). It is noteworthy that previous studies have demonstrated similarities at the protein and messenger RNA (mRNA) levels between SCLC and LCNEC, suggesting a related underlying biology.50–52 SCLC and other high-grade NE lung cancers are distinct in their biology and clinical behavior from intermediate-grade and low-grade NE cancers like atypical and typical carcinoid.
SCLC is characterized by a nearly universal loss of tumor protein 53 (TP53) (75%–90% of patients)4–6 and retinoblastoma 1 (RB1) (approaching 100%)53,54 and by frequent 3p deletion.55 Beyond these shared molecular features, increased expression of cKit,56,57 amplification v-myc avian myelocytomatosis viral oncogene homolog (MYC) family members (MYC, MYC lung carcinoma-derived homolog 1 [MYCL1], and MYC neuroblastoma-derived homolog [MYCN]),58 and loss of phosphatase and tensin homolog (PTEN) have also been described in subsets of SCLC. These molecular hallmarks of SCLC (or SCLC subsets) were confirmed in 2 recent comprehensive genomic profiling studies of SCLC.7,8 Consistent with prior studies, loss of the tumor suppressors TP53 and RB1 were the 2 most common events identified by both groups. However, those studies also identified novel mutations (eg, those in epigenetic regulators) and driver mutations with well established roles in multiple cancer types (eg, MYC family genes, BCL2, PTEN, CREB binding protein [CREBBP], and FGFR1) (Fig. 1).
Figure 1.
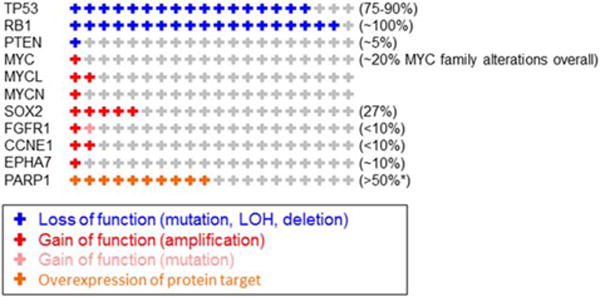
The frequency of genomic alterations is illustrated according to percentage in small cell lung cancer (SCLC). This is a representative list of the more common and/or potentially targetable alterations. An asterisk indicates the percentage that was positive for poly(ADP-ripose) polymerase 1 (PARP1) protein expression based on the number of SCLC tumors that had an immunohistochemical staining score of 31in 100% of tumor cells. TP53 indicates tumor protein 53; RB1, retinoblastoma 1; PTEN, phosphatase and tensin homolog; MYC, v-myc avian myelocytomatosis viral oncogene homolog; MYCL, v-myc avian myelocytomatosis viral oncogene lung carcinoma-derived homolog; MYCN, v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog; SOX2, sex-determining region Y box 2; FGFR1, fibroblast growth factor receptor 1; CCNE1, cyclin E1; EPHA7, ephrin receptor A7.
Myc alterations and targeting
Alterations in MYC family members were observed in approximately 20% of SCLC patient tumors. Unlike TP53 and RB1, which are tumor suppressors, MYC family members represent frequent oncogenes in SCLC and other cancer types. Although Myc has been recognized as a potentially important target for many years, designing a drug to directly inhibit Myc activity has been challenging. However, MYC amplified or driven tumors may be more sensitive to certain emerging targeted drugs, such as Aurora kinase or bromodomain inhibitors.25,59,60
In 1 recent example, a phase 2 clinical trial of single-agent alisertib (a highly selective Aurora kinase A inhibitor) produced response rates of 21% in 47 patients with recurrent or progressive SCLC.61 Among these, the highest response rates were observed in patients with platinum-refractory SCLC (defined as disease recurrence within 3 months of completing front-line platinum-based therapy), typically the most drug-resistant population. Correlative studies from SCLC patients treated on this trial are pending; however, data from preclinical models of SCLC, as well as other cancer types, suggest that tumors with Myc alterations may be especially sensitive to Aurora kinase inhibitors.62,63 The mechanism through which MYC-amplified or Myc-overexpressing tumors are sensitized to inhibition of Aurora kinase is still incompletely understood. However, previous studies suggest that Myc plays an important role in transcriptional regulation of Aurora kinases A and B, which, in turn, may provide a growth advantage in the absence of p53.63–66 Because of the key role of Aurora kinase A in mitotic spindle assembly, these drugs may be especially active in combination with taxane chemotherapies.
If the activity of Aurora kinase inhibitors is validated in subsequent SCLC clinical trials (such as a new study of paclitaxel with or without alisertib in second-line SCLC; National Clinical Trial no. NCT02038647), then Myc-amplified SCLC could be the first genomically defined subgroup of SCLC with a specific targeted agent. In addition to the MYC amplifications mentioned above, others biomarkers and/or targets that may predict response in specific subsets of SCLC patients include those with mutations or amplification in FGFR159 or sex-determining region Y box 2 (SOX2)8 and alterations affecting epigenetic changes, such as O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation (associated with response to TMZ).26
Overexpression of PARP1 protein and potential role of DNA repair inhibitors
Beyond mutations and changes in DNA copy number, overexpression of certain proteins in SCLC tumors or changes at the epigenetic level may represent new and important therapeutic targets. For example, proteomic profiling of a large panel of SCLC cell lines led to the identification of several potentially novel therapeutic targets.52 These include the DNA repair proteins PARP1 and checkpoint kinase 1 (Chk1) as well as enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) (a chromatin modulator). Increased expression of these proteins appears to be independent of alterations at the DNA level (mutation, amplification) in their corresponding genes (PARP1, CHEK1, and EZH2), emphasizing the value of comprehensive molecular profiling—ie, examining not just tumor DNA but alterations that occur at the protein or pathway level.
On the basis of this observation, several PARP inhibitors (eg, olaparib, rucaparib, BMN-673) were then investigated in preclinical models of SCLC and exhibited single-agent activity in cell lines and/or animal models.52,67 Those results led to the investigation of PARP inhibitors (eg, BMN-673 and veliparib) in patients with SCLC in several clinical trials (eg, NCT01286987, NCT01642251, and NCT01638546). The first results of PARP inhibitors in SCLC patients were recently presented at the 2014 American Society of Clinical Oncology annual meeting and, among other findings, exhibited single-agent activity of the PARP inhibitor BMN-673 in a subset of SCLC patients.68 Other ongoing studies are testing the combination of PARP inhibition with chemotherapy and include a TMZ/veliparib combination for second-line or third-line treatment (NCT01638546) and a first-line trial of veliparib with platinum-etoposide (Eastern Cooperative Oncology Group trial ECOG-E2511). Further studies are ongoing to identify biomarkers that may predict response to these drugs and include levels of PARP1 or other DNA repair proteins.67
Immunotherapy and other promising targets
Several immunotherapy approaches have been investigated or are under current investigation in SCLC, including vaccine studies, interferon-a, and several clinical trials of immune checkpoint inhibitors (for review, see Spigel and Socinski69). Immune checkpoint blockade, either alone or in combination with chemotherapy, represents a particularly promising approach to the treatment of this disease. For example, a randomized phase 2 study investigated the combination of ipilimumab (a monoclonal antibody targeting cytotoxic T-lymphocyte–associated antigen 4 [CTLA-4]) in combination with paclitaxel and carboplatin. One hundred thirty patients with treatment-naive were randomized to 1 of 3 treatment arms: 1) concurrent ipilimumab (ipilimumab plus chemotherapy for 4 cycles followed by 2 cycles of chemotherapy plus placebo), 2) phased ipilimumab (chemotherapy plus placebo for 2 cycles followed by chemotherapy plus ipilimumab for 4 cycles), or 3) chemotherapy plus placebo. In the patients who received phased ipilimumab, an improvement was observed in progression-free survival based on immunerelated response criteria (hazard ratio, 0.64; P 5 .03), with a trend for improved overall survival (12.5 months vs 9.1 months).70 Several trials of immunotherapy in the first-line and relapsed settings are now ongoing, such as the investigation of a programmed cell death protein 1 (PD-1) inhibitor (nivolumab) combined with ipilimumab (NCT01928394).
Another drug target with potential activity in SCLC is FGFR. Alterations (eg, amplification or mutation) in FGFR family members have been described in a small subset of SCLC tumors; and some, but not all, SCLC models with FGFR1 amplification have demonstrated sensitivity to FGFR inhibitors.59 Drugs targeting FGFR family members that are currently in clinical investigation for SCLC include JNJ-42756493 (a pan-FGFR inhibitor) and BIBF1120 (a multitargeted drug that inhibits FGFR, vascular endothelial growth factor receptor [VEGFR], and platelet-derived growth factor receptor [PDGFR]; NCT01703481 and NCT01441297). However, the extent to which SCLC with mutations and/or amplifications in FGFR family member genes are dependent on the FGFR pathway is not yet known.
Current Barriers and Challenges in Translational SCLC Research
There are several barriers that have made translational research in SCLC particularly difficult. These include 1) limited tissue available for study, 2) the molecular complexity of SCLC, 3) a poor understanding of the mechanisms of chemotherapy resistance in recurrent disease (including unique molecular alterations that may be acquired after initial treatment), and 4) the rapid pace of disease progression. In addition, investment in SCLC research has been low in recent years (possibly because of some of the limitations noted above in contrast to NSCLC, in which research tools, including tissue and model systems, are more plentiful). For example, in fiscal Year 2012, the National Cancer Institute (NCI) research portfolio contained 745 projects that included lung cancer research, but only 17 (approximately 2%) of those had a focus on SCLC.71
Contribution of SCLC pathobiology to scarce tissue resources for research
SCLC cells are readily recognized by light microscopy because of their characteristic appearance as small blue cells, often with “crush” artifact (an effect of cell fixing). Thus, a diagnosis can be made based on the presence of as few as 3 to 10 cancer cells (Fig. 2). The ability to establish a diagnosis of SCLC from a small number of malignant cells, combined with infrequent surgical resection and a lack of validated predictive biomarkers that would necessitate larger tissue biopsies, has significantly limited the availability of SCLC tissue for molecular profiling and other scientific studies. This is especially true for profiling technologies which currently work best on frozen tissue in adequate quantities, including in-depth whole genome sequencing of DNA and comprehensive expression analysis for RNA and protein.
Figure 2.
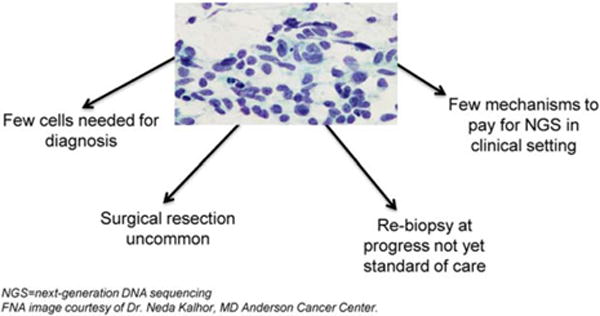
Tissue limitations in the study of small cell lung cancer are illustrated. NGS indicates next-generation DNA sequencing.
To date, whole-exome sequencing data (ie, DNA sequencing of protein-coding regions of the genome) has only been published on 82 SCLC tumors.7,42 This is in stark contrast to NSCLC, for which data from more than 1000 NSCLC tumors have been published.9,35,39,41 This small number of profiled tumors is especially problematic in a disease like SCLC, which carries one of the highest rates of mutations per tumor42 because of long-term tobacco exposure and near universal loss of p53 function.4 On average, SCLC tumors have greater than 4 times the number of mutations observed in breast cancer and almost 10 times the number in prostate cancer.42 The vast majority of mutations observed in SCLC patient tumors are passengers (those that do not meaningfully contribute to disease behavior or progression). This makes it more difficult to definitively identify those mutations that are drivers of cancer growth and invasion (including those that may be druggable).
The challenge of discriminating driver mutations from passengers is even more critical for cases in which a driver may only be present in a subset of SCLC patients. We know that driver genes—even those identified in only a minority of patients–still may have incredible clinical importance. For instance, although ALK fusions are identified in only 7% of lung adenocarcinomas, testing for ALK fusions has become a standard of care in the metastatic setting, because we have highly effective, FDA-approved drugs targeting ALK (crizotinib and ceritinib) that provide substantial clinical benefit for a majority of ALK-positive patients.72–74 However, the genomic studies of SCLC to date have been insufficiently powered to reliably identify recurrent mutations present in <10% of patients.
This point was illustrated by 2 studies that reported the frequency of amplification events in SCLC patient tumors. Whereas one reported FGFR1 amplifications in 6% of SCLC tumors,8 the other study did not identify any instances of FGFR1 amplification.7 This example illustrates the finding that we are likely missing other potentially druggable driver genes in our existing analyses that have potential therapeutic relevance.
Opportunities and Tools for Advancing Progress Improved research tools for the study of SCLC
Recent progress in modeling SCLC has included the development of a cohort of patient-derived xenografts (PDXs) and several genetically engineered mouse models (GEMMs). These animal models can be extremely helpful in dissecting the biology of disease and providing a system in which to test new drugs. Furthermore, a new, potentially transformative tool for animal models (and for expanding SCLC patient tissue for profiling) was recently reported by Hodgkinson et al, who demonstrated the feasibility of using circulating tumor cells (CTCs) to establish animal models (CTC-derived xenografts; [CDXs]) to propagate patient-derived SCLC cells collected from blood.48 This approach has the potential to allow for longitudinal sampling of SCLC cells from patient blood at the time of initial diagnosis and at relapse.
In addition, there are several established resources supported by the NCI and other National Institutes of Health programs that can be leveraged for SCLC research. These include the large drug screening effort in SCLC described below as well as a collection of core facilities, consortia, and networks to support a broad range of research activities, including molecular profiling, animal studies, and early detection initiatives (available at: http://resresources.nci.nih.gov; accessed September 1, 2014).
Clinical strategies to promote progress in the treatment of SCLC
Ultimately, our goal is to improve patient survival and quality of life. To do this, translating research findings into clinical trials and, subsequently, into new standard-of-care therapies for SCLC must be achieved. Successful drug development will require a multidisciplinary, translational approach that incorporates discovery and prioritization of candidate drugs and/or targets in preclinical models, as well as well-designed correlative studies as part of each clinical trial to better define those patients most likely to benefit from specific therapeutic approaches.
Toward this goal, a major initiative to characterize drug sensitivity is ongoing at the NCI through the Developmental Therapeutics Program, which is investigating more than 400 targeted drugs and 100 FDA-approved oncology therapies in a panel of >60 SCLC cell lines. Results from this drug screen and other ongoing preclinical efforts—combined with an integrated analysis of the molecular profiles of these SCLC cell lines75 (eg, mutations, amplifications, deletions, or alterations at the protein or pathway levels that correspond to drug sensitivity)—will provide important leads that can be validated in the laboratory and taken forward into clinical trials. As part of this, bioinformatics tools will allow us to mine these growing data sets for the new targets or biomarkers to maximize the value and use of a growing molecular resource/database in SCLC.47
Remarkably, only approximately 100 interventional clinical trials in SCLC have been registered at www.clinicaltrials.gov since December 2007. This number is clearly inadequate to meet the needs/demand that exists for SCLC patients—particularly with the goal of making a meaningful advance in patient outcomes in the short term. Critical clinical components of a successful SCLC translational research framework must also include 1) biopsy and rebiopsy programs to obtain adequate issue for molecular profiling, 2) mechanisms to support the logistics and funding of patient tumor profiling to identify potential drug targets (such as cooperative group programs or other laboratory protocols), and 3) clinical trial options for all stages of disease that meet the needs of our patients and respond to emerging discoveries/targets identified by ongoing profiling efforts or other research (eg, clinical trial consortiums, basket studies, SCLC master protocols). Figure 3 is an illustration of the key elements to a successful translational approach for the advancement of SCLC treatment, in which CLIA-approved biomarker testing to guide therapeutic selection is occurring in parallel to potentially transformative exploratory analyses.
Figure 3.
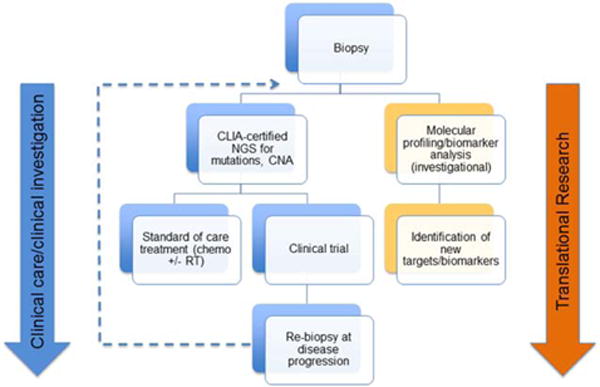
The framework for clinical-translational research in small cell lung cancer (SCLC) is illustrated. Tissue biopsies (fine-needle aspiration and/or core-needle) are required that provide ample material for integrated molecular analysis 1) to guide clinical decision making (eg, marker-selected clinical trial) and 2) for translational/exploratory scientific analysis (eg, the identification of new targets and/or biomarkers). Patient rebiopsies at the time of progression are necessary to assess for potential new therapeutic targets and to characterize molecular features/alterations acquired in resistant disease. CLIA indicates Clinical Laboratory Improvement Amendments; NGS, next-generation DNA sequencing; CNA, copy number alterations; RT, radiotherapy.
Furthermore, beyond SCLC of the lung, there is a similarly underserved group of patients with extrapulmonary small cell carcinoma. Partnerships of pulmonary and extrapulmonary small cell efforts also potentially could advance our understanding of SCLC biology and opportunities for clinical trials of shared targets. Although the aggressiveness of SCLC introduces challenges to clinical research (such as the need for efficient decision making, biopsy completion, and initiation of therapy), previous studies, such as the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination 1 (BATTLE1) trial and, more recently, the BATTLE2 trial (both for metastatic, relapsed NSCLC), have demonstrated the feasibility of obtaining fresh biopsies in a population of patients with advanced, chemotherapy-resistant disease.76
Summary
SCLC is an aggressive disease affecting >30,000 individuals per year in the United States. Most patients present with advanced disease and rapidly develop treatment resistance despite a high rate of responses to initial chemotherapy and radiation. Regardless of numerous clinical trials, treatment has not changed significantly for more than 30 years.
Progress in the treatment of SCLC has been limited by several factors, including limited tissue availability for translational research. Consequently, SCLC lags significantly behind NSCLC and other cancers in molecular profiling and the development of targeted therapies. Despite these challenges, there have been some notable recent discoveries that have led to clinical trials and have the potential to advance the field.
By using a multidisciplinary, collaborative, cross-institutional approach, we have the opportunity to meaningfully impact outcomes in the next 5 to 10 years. Important steps toward this goal will include a coordinated effort to collect adequate SCLC tumor tissue for research from treatment-naive and treatment-refractory patients, support for a scientific community of SCLC investigators, and a framework for translating the most promising discoveries into clinical trials and, ultimately, clinical practice.
Acknowledgments
FUNDING SUPPORT
L.B. is supported, in part, by the R. Lee Clark Fellow Award (supported by the Jeane F. Shelby Scholarship Fund), the MDACC Physician Scientist Award, and the NCI Cancer Clinical Investigator Team Leadership Award.
We thank Dr. Neda Kalhor for providing the cytology image of small cell lung cancer for Figure 2.
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Dr. Byers reports consulting fees from BioMarin and AbbVie outside the submitted work. Dr. Rudin reports consulting fees from AbbVie, Aveo/Biodesik, Boehringer Ingelheim, GlaxoSmithKline, and Merck outside the submitted work.
References
- 1.American Cancer Society. Cancer Facts & Figures 2014. Atlanta, GA: American Cancer Society; 2014. [Google Scholar]
- 2.Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol. 2014;9:892–896. doi: 10.1097/JTO.0000000000000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ettinger DS, Aisner J. Changing face of small-cell lung cancer: real and artifact. J Clin Oncol. 2006;24:4526–4527. doi: 10.1200/JCO.2006.07.3841. [DOI] [PubMed] [Google Scholar]
- 4.Miller CW, Simon K, Aslo A, et al. p53 mutations in human lung tumors. Cancer Res. 1992;52:1695–1698. [PubMed] [Google Scholar]
- 5.Takahashi T, Suzuki H, Hida T, Sekido Y, Ariyoshi Y, Ueda R. The p53 gene is very frequently mutated in small-cell lung cancer with a distinct nucleotide substitution pattern. Oncogene. 1991;6:1775–1778. [PubMed] [Google Scholar]
- 6.D’Amico D, Carbone D, Mitsudomi T, et al. High frequency of somatically acquired p53 mutations in small-cell lung cancer cell lines and tumors. Oncogene. 1992;7:339–346. [PubMed] [Google Scholar]
- 7.Peifer M, Fernandez-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aberle DR, DeMello S, Berg CD, et al. Results of the 2 incidence screenings in the National Lung Screening Trial. N Engl J Med. 2013;369:920–931. doi: 10.1056/NEJMoa1208962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon M, Argiris A, Murren JR. Progress in the therapy of small cell lung cancer. Crit Rev Oncol Hematol. 2004;49:119–133. doi: 10.1016/S1040-8428(03)00118-5. [DOI] [PubMed] [Google Scholar]
- 12.Amini A, Byers LA, Welsh JW, Komaki RU. Progress in the management of limited-stage small cell lung cancer. Cancer. 2014;120:790–798. doi: 10.1002/cncr.28505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiro SG, James LE, Rudd RM, et al. Early compared with late radiotherapy in combined modality treatment for limited disease small-cell lung cancer: a London Lung Cancer Group multicenter randomized clinical trial and meta-analysis. J Clin Oncol. 2006;24:3823–3830. doi: 10.1200/JCO.2005.05.3181. [DOI] [PubMed] [Google Scholar]
- 14.Fried DB, Morris DE, Poole C, et al. Systematic review evaluating the timing of thoracic radiation therapy in combined modality therapy for limited-stage small-cell lung cancer. J Clin Oncol. 2004;22:4837–4845. doi: 10.1200/JCO.2004.01.178. [DOI] [PubMed] [Google Scholar]
- 15.De Ruysscher D, Pijls-Johannesma M, Bentzen SM, et al. Time between the first day of chemotherapy and the last day of chest radiation is the most important predictor of survival in limited-disease small-cell lung cancer. J Clin Oncol. 2006;24:1057–1063. doi: 10.1200/JCO.2005.02.9793. [DOI] [PubMed] [Google Scholar]
- 16.Pijls-Johannesma MC, De Ruysscher D, Lambin P, Rutten I, Vansteenkiste JF. Early versus late chest radiotherapy for limited stage small cell lung cancer [serial online] Cochrane Database Syst Rev. 2005;(1):CD004700. doi: 10.1002/14651858.CD004700.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Ruysscher D, Pijls-Johannesma M, Vansteenkiste J, Kester A, Rutten I, Lambin P. Systematic review and meta-analysis of randomised, controlled trials of the timing of chest radiotherapy in patients with limited-stage, small-cell lung cancer. Ann Oncol. 2006;17:543–552. doi: 10.1093/annonc/mdj094. [DOI] [PubMed] [Google Scholar]
- 18.Huncharek M, McGarry R. A meta-analysis of the timing of chest irradiation in the combined modality treatment of limited-stage small cell lung cancer. Oncologist. 2004;9:665–672. doi: 10.1634/theoncologist.9-6-665. [DOI] [PubMed] [Google Scholar]
- 19.Turrisi AT, 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med. 1999;340:265–271. doi: 10.1056/NEJM199901283400403. [DOI] [PubMed] [Google Scholar]
- 20.Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur Respir J. 2010;35:202–215. doi: 10.1183/09031936.00105009. [DOI] [PubMed] [Google Scholar]
- 21.Johnson BE, Janne PA. Basic treatment considerations using chemotherapy for patients with small cell lung cancer. Hematol Oncol Clin North Am. 2004;18:309–322. doi: 10.1016/j.hoc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Auperin A, Arriagada R, Pignon JP, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med. 1999;341:476–484. doi: 10.1056/NEJM199908123410703. [DOI] [PubMed] [Google Scholar]
- 23.Slotman B, Faivre-Finn C, Kramer G, et al. Prophylactic cranial irradiation in extensive small-cell lung cancer. N Engl J Med. 2007;357:664–672. doi: 10.1056/NEJMoa071780. [DOI] [PubMed] [Google Scholar]
- 24.National Comprehensive Cancer Network. Small cell lung cancer. Available at: http://www.nccn.org/professionals/physician_gls/pdf/sclc.pdf Accessed July 18, 2014.
- 25.Simos D, Sajjady G, Sergi M, et al. Third-line chemotherapy in small-cell lung cancer: an international analysis. Clin Lung Cancer. 2014;15:110–118. doi: 10.1016/j.cllc.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide in patients with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res. 2012;18:1138–1145. doi: 10.1158/1078-0432.CCR-11-2059. [DOI] [PubMed] [Google Scholar]
- 27.Lara PN, Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–2535. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noda K, Nishiwaki Y, Kawahara M, et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med. 2002;346:85–91. doi: 10.1056/NEJMoa003034. [DOI] [PubMed] [Google Scholar]
- 29.Zatloukal P, Cardenal F, Szczesna A, et al. A multicenter international randomized phase III study comparing cisplatin in combination with irinotecan or etoposide in previously untreated small-cell lung cancer patients with extensive disease. Ann Oncol. 2010;21:1810–1816. doi: 10.1093/annonc/mdq036. [DOI] [PubMed] [Google Scholar]
- 30.Hanna N, Bunn PA, Jr, Langer C, et al. Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol. 2006;24:2038–2043. doi: 10.1200/JCO.2005.04.8595. [DOI] [PubMed] [Google Scholar]
- 31.Socinski MA, Smit EF, Lorigan P, et al. Phase III study of pemetrexed plus carboplatin compared with etoposide plus carboplatin in chemotherapy-naive patients with extensive-stage small-cell lung cancer. J Clin Oncol. 2009;27:4787–4792. doi: 10.1200/JCO.2009.23.1548. [DOI] [PubMed] [Google Scholar]
- 32.Rudin CM, Salgia R, Wang X, et al. Randomized phase II study of carboplatin and etoposide with or without the bcl-2 antisense oligonucleotide oblimersen for extensive-stage small-cell lung cancer: CALGB 30103. J Clin Oncol. 2008;26:870–876. doi: 10.1200/JCO.2007.14.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dy GK, Miller AA, Mandrekar SJ, et al. A phase II trial of imatinib (ST1571) in patients with c-kit expressing relapsed small-cell lung cancer: a CALGB and NCCTG study. Ann Oncol. 2005;16:1811–1186. doi: 10.1093/annonc/mdi365. [DOI] [PubMed] [Google Scholar]
- 34.Krug LM, Crapanzano JP, Azzoli CG, et al. Imatinib mesylate lacks activity in small cell lung carcinoma expressing c-kit protein: a phase II clinical trial. Cancer. 2005;103:2128–2131. doi: 10.1002/cncr.21000. [DOI] [PubMed] [Google Scholar]
- 35.Oze I, Hotta K, Kiura K, et al. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results [serial online] PLoS One. 2009;4:e7835. doi: 10.1371/journal.pone.0007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chute JP, Chen T, Feigal E, Simon R, Johnson BE. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol. 1999;17:1794–1801. doi: 10.1200/JCO.1999.17.6.1794. [DOI] [PubMed] [Google Scholar]
- 37.National Comprehensive Cancer Network. NCCN guidelines: non-small cell lung cancer. Available at: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#nscl Accessed July 18, 2014.
- 38.My Cancer Genome. Available at: http://www.mycancergenome.org/. Accessed July 18, 2014.
- 39.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National Cancer Institute, National Human Genome Research Institute. The Cancer Genome Atlas (TCGA) Research Network. Available at: http://cancergenome.nih.gov/ Accessed July 18, 2014.
- 41.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daniel VC, Marchionni L, Hierman JS, et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69:3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 44.Schaffer BE, Park KS, Yiu G, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010;70:3877–3883. doi: 10.1158/0008-5472.CAN-09-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Institutes of Health. ClinicalTrials.gov. Available at: www.clinicaltrials.gov. Accessed July 18, 2014.
- 46.Jahchan NS, Dudley JT, Mazur PK, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013;3:1364–1377. doi: 10.1158/2159-8290.CD-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Byers LA. Teaching an old dog new tricks: drug repositioning in small cell lung cancer. Cancer Discov. 2013;3:1333–1335. doi: 10.1158/2159-8290.CD-13-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hodgkinson CL, Morrow CJ, Li Y, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20:897–903. doi: 10.1038/nm.3600. [DOI] [PubMed] [Google Scholar]
- 49.Hirsch FR, Matthews MJ, Aisner S, et al. Histopathologic classification of small cell lung cancer. Changing concepts and terminology. Cancer. 1988;62:973–977. doi: 10.1002/1097-0142(19880901)62:5<973::aid-cncr2820620521>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 50.Jones MH, Virtanen C, Honjoh D, et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet. 2004;363:775–781. doi: 10.1016/S0140-6736(04)15693-6. [DOI] [PubMed] [Google Scholar]
- 51.Peng WX, Shibata T, Katoh H, et al. Array-based comparative genomic hybridization analysis of high-grade neuroendocrine tumors of the lung. Cancer Sci. 2005;96:661–667. doi: 10.1111/j.1349-7006.2005.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byers LA, Wang J, Nilsson MB, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helin K, Holm K, Niebuhr A, et al. Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc Natl Acad Sci U S A. 1997;94:6933–6938. doi: 10.1073/pnas.94.13.6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaye FJ. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene. 2002;21:6908–6914. doi: 10.1038/sj.onc.1205834. [DOI] [PubMed] [Google Scholar]
- 55.Wistuba II, Behrens C, Virmani AK, et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and 3 regions of frequent breakpoints. Cancer Res. 2000;60:1949–1960. [PubMed] [Google Scholar]
- 56.Rohr UP, Rehfeld N, Pflugfelder L, et al. Expression of the tyrosine kinase c-kit is an independent prognostic factor in patients with small cell lung cancer. Int J Cancer. 2004;111:259–263. doi: 10.1002/ijc.20252. [DOI] [PubMed] [Google Scholar]
- 57.Tamborini E, Bonadiman L, Negri T, et al. Detection of overex-pressed and phosphorylated wild-type kit receptor in surgical specimens of small cell lung cancer. Clinical Cancer Res. 2004;10:8214–8219. doi: 10.1158/1078-0432.CCR-04-1013. [DOI] [PubMed] [Google Scholar]
- 58.Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28(2 suppl):3–13. [PubMed] [Google Scholar]
- 59.Sos ML, Dietlein F, Peifer M, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A. 2012;109:17034–17039. doi: 10.1073/pnas.1207310109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melichar B, Adenis A, Havel L, et al. Phase (Ph) I/II study of investigational Aurora A kinase (AAK) inhibitor MLN8237 (alisertib): updated ph II results in patients (pts) with small cell lung cancer (SCLC), non-SCLC (NSCLC), breast cancer (BrC), head and neck squamous cell carcinoma (HNSCC), and gastroesophageal cancer (GE) [abstract] J Clin Oncol. 2014;31(suppl) Abstract 605. [Google Scholar]
- 62.Hook KE, Garza SJ, Lira ME, et al. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther. 2012;11:710–719. doi: 10.1158/1535-7163.MCT-11-0184. [DOI] [PubMed] [Google Scholar]
- 63.Brockmann M, Poon E, Berry T, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell. 2013;24:75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.den Hollander J, Rimpi S, Doherty JR, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood. 2010;116:1498–1505. doi: 10.1182/blood-2009-11-251074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu LY, Wood JL, Ye L, et al. Aurora A is essential for early embryonic development and tumor suppression. J Biol Chem. 2008;283:31785–31790. doi: 10.1074/jbc.M805880200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 67.Cardnell RJ, Feng Y, Diao L, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19:6322–6328. doi: 10.1158/1078-0432.CCR-13-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wainberg Z, Rafii S, Ramanathan R, et al. Safety and antitumor activity of the PARP inhibitor BMN673 in a phase 1 trial recruiting metastatic small-cell lung cancer (SCLC) and germline BRCA-mutation carrier cancer patients [abstract] J Clin Oncol. 2014;32(5s) Abstract 7522. [Google Scholar]
- 69.Spigel DR, Socinski MA. Rationale for chemotherapy, immunotherapy, and checkpoint blockade in SCLC: beyond traditional treatment approaches. J Thorac Oncol. 2013;8:587–598. doi: 10.1097/JTO.0b013e318286cf88. [DOI] [PubMed] [Google Scholar]
- 70.Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 71.National Cancer Institute. Group Scientific Framework for Small Cell Lung Cancer (SCLC) Bethesda, MD: National Cancer Institute, Small Cell Lung Cancer Working Group; 2014. [Google Scholar]
- 72.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 74.Mok T, Kim DW, Wu YL, et al. First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients (pts) with advanced ALK-positive non-squamous non-small cell lung cancer (NSCLC): results of a phase III study (PROFILE 1014) [abstract] J Clin Oncol. 2014;32(5s) Abstract 8002. [Google Scholar]
- 75.Teicher BA. Perspective: opportunities in recalcitrant, rare and neglected tumors. Oncol Rep. 2013;30:1030–1034. doi: 10.3892/or.2013.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Dis. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]