

Abstract
Septins are cytoskeletal proteins involved in diverse biological processes including cytokinesis, cell morphogenesis, motility, and ciliogenesis. Septins form various filamentous structures in vitro and in vivo, but the higher-order architecture of septin structures in vivo remains poorly defined. The best understood system in this respect is the budding yeast Saccharomyces cerevisiae, where septins form a ring structure that undergoes multiple stages of remodeling during the cell cycle. In this chapter, we describe a method for visualizing supramolecular septin structures in yeast at high spatial resolution using platinum replica electron microscopy. This approach can be applied to further understand the regulation of assembly and remodeling of septin higher-order structures, as well as the relationship between septin architecture and function.
INTRODUCTION
Septins are a group of GTP-binding proteins that are conserved from fungi to mammals (Bridges & Gladfelter, 2014; Dolat, Hu, & Spiliotis, 2014; Hall, Russell, & Pringle, 2008; McMurray & Thorner, 2009; Oh & Bi, 2011). Septins play important roles in diverse cellular processes including cytokinesis (Hartwell, 1971; Kinoshita et al., 1997), cell migration (Dolat et al., 2014; Tooley et al., 2009), neuronal morphogenesis (Tada et al., 2007; Xie et al., 2007), and ciliogenesis (Dash et al., 2014; Hu et al., 2010; Kim et al., 2010; Zhai et al., 2014). Dysregulation of such functions has been implicated in cancer, neuropathy, and infertility (Dolat et al., 2014; Hall & Russell, 2004).
Septins form nonpolar, rod-shaped heterooligomers that serve as blocks of higher-order structures, such as filaments, bundles, rings, and gauzes (Bridges & Gladfelter, 2014; Caudron & Barral, 2009; McMurray & Thorner, 2009; Oh & Bi, 2011). In cells, septin structures are often associated with a discrete region of the plasma membrane and act as a scaffold or diffusion barrier between membrane compartments (Barral, Mermall, Mooseker, & Snyder, 2000; Takizawa, DeRisi, Wilhelm, & Vale, 2000). To understand how septins perform their diverse roles, one must understand the architecture, dynamics, and regulation of septin higher-order structures in vivo.
The organization and remodeling of septin higher-order assemblies is best understood in the budding yeast Saccharomyces cerevisiae where septins were first discovered (Byers & Goetsch, 1976a, 1976b; Haarer & Pringle, 1987; Hartwell, 1971; Sanders & Field, 1994). Decades of work spanning multiple disciplines and research groups have revealed the essential steps of septin filament organization, remodeling, and dynamics during the cell cycle in budding yeast. Septins were first visualized at the bud neck by thin section electron microscopy (EM), where they appeared as 10 nm striations perpendicular to the bud neck spaced out by 28 nm gaps (Byers & Goetsch, 1976a). These striations emerge early during bud formation and disappear just prior to cytokinesis. The presence of these striations is dependent upon four genes: CDC3, CDC10, CDC11, and CDC12 (Byers & Goetsch, 1976b; Frazier et al., 1998) that were earlier identified in a cell cycle genetic screen as being essential for cytokinesis. Immunofluorescence studies (Ford & Pringle, 1991; Haarer & Pringle, 1987; Kim, Haarer, & Pringle, 1991) suggested that the protein products of these genes [which came to be collectively referred to as “septins” (Sanders & Field, 1994)] might colocalize with the striations. When it was later discovered that purified preparations of these four septins form double filaments in vitro (Frazier et al., 1998), the idea arose that the septins formed a filamentous ring structure at the bud neck.
At the micron scale, fluorescence microscopy has unveiled the key remodeling stages of the septin cytoskeleton during the cell cycle (Cid, Adamikova, Sanchez, Molina, & Nombela, 2001; Haarer & Pringle, 1987; Kim et al., 1991; Lippincott, Shannon, Shou, Deshaies, & Li, 2001). When the bud site is first selected, septins accumulate in either a patch or ring (Fig. 1A). Dynamics studies (Caviston, Longtine, Pringle, & Bi, 2003; Dobbelaere, Gentry, Hallberg, & Barral, 2003) showed that subunits are continuously recruited into the nascent structure. Upon bud emergence, septins unambiguously form a ring structure at the bud neck referred to as the “hourglass” (Fig. 1B and C). Septin subunits are immobilized within the hourglass, suggesting that septin filaments form a very stable structure at this point. At cytokinesis, the hourglass splits into the “double-ring” structure (Fig. 1D). Subunits are initially mobile and then become immobilized within the double ring. When abscission completes, each cell, mother and daughter, inherits one of the rings from the double-ring structure (Fig. 1E), which is subsequently disassembled at the beginning of the next cell cycle.
FIGURE 1.

Septin architecture and remodeling during the cell cycle of Saccharomyces cerevisiae. (A–E) Schematic of septin ring remodeling during the cell cycle as observed at low spatial resolution. (F) An unroofed spheroplast containing a preserved cortical septin structure. (G–I) Septin structures visualized by platinum replica EM isolated from (G) the early hourglass stage, (H) the precytokinesis, transition stage, and (I) the double-ring stage. Left panels show overview images of full structures. Boxed regions in the left panels are enlarged in the middle panels. Right panels are schematics of filament organization with respect to the bud neck for each structure. Some double filaments are pseudocolored as green and single filaments as orange in the micrographs. All scale bars are 200 nm except for panel (f), as noted. (See color plate)
At the nanoscale, the organization of septin filaments within the hourglass structures had remained controversial. Given that in vitro double septin filaments are approximately 10 nm in diameter, the initial thin section EM studies suggested that the septin hourglass is composed of circumferential double filaments that wrap around the bud neck at a spacing of 28 nm (Byers & Goetsch, 1976a). At cytokinesis, no electron density patterns were observed at the bud neck in these studies, so the organization of filaments in the double-ring structure was completely unknown. Decades later, more advanced cryo-EM tomography studies would extend this model by detecting what resembled cross-linking filaments that intersect the 10 nm striations at irregular intervals of ~10–20 nm (Bertin et al., 2012).
This model of the hourglass was challenged by an alternative interpretation of the bud-neck striations. It was proposed that these striations reflect differences in heavy metal staining of different septin subunits rather than filament orientation (Field et al., 1996; Frazier et al., 1998). In line with this hypothesis, an alternative model arose that had double filaments in the hourglass oriented parallel to the mother–bud axis (Field et al., 1996). This model was later supported by polarized fluorescence microscopy experiments, which showed that the dipole of GFP-labeled septin subunits during the hourglass stage was ordered parallel to the mother–bud axis (DeMay et al., 2011; Vrabioiu & Mitchison, 2006). This dipole underwent a 90 degree rotation upon hourglass splitting, suggesting that the filaments are reorganized circumferentially in the double-ring structure (DeMay et al., 2011; Vrabioiu & Mitchison, 2006).
Recently, we sought to resolve the discrepancy between these two models. To do this, we used a method of visualizing septin structures in situ that was reported by David Drubin’s and John Hartwig’s labs (Rodal, Kozubowski, Goode, Drubin, & Hartwig, 2005). Avital Rodal, who spear-headed this project, was able to reveal endogenous septin structures in yeast while seeking a way to image actin patches associated with endocytosis by EM (Rodal et al., 2005). In brief, the method she developed involved mechanical rupture (unroofing) of yeast spheroplasts to clear the cytoplasm and expose cortical cytoskeleton structures (Fig. 1F). Rodal et al. described two distinct septin structures whose identity was confirmed by immunogold staining: (1) “gauzes” composed of ~300- to 400-nm-long double filaments arranged in parallel arrays and (2) rings formed of circumferential filaments that were up to multiple microns in length. These experiments, however, were performed with unsynchronized cells, so it was not possible to attribute different septin structures to a specific stage of the cell cycle.
To address this limitation, we used a modification of this approach to visualize septin organization in cells synchronized at three different stages of the cell cycle: (1) the small-budded, early hourglass stage (Fig. 1B), (2) the transition stage just prior to ring splitting (Fig. 1C), and (3) the double-ring stage during cytokinesis (Fig. 1D). We found that the morphology of septin structures changed dramatically depending on the stage of the cell cycle. Specifically, we found that septin structures at the early hourglass stage were composed of parallel arrays of double filaments (Fig. 1G) (Ong, Wloka, Okada, Svitkina, & Bi, 2014). The lengths of these filaments (300–400 nm) were too short to wrap around a bud neck. Rather, this organization is consistent with the model that filaments are ordered parallel to the mother–bud axis (Fig. 1G). At the double-ring stage, septin arrays contained circumferentially arranged filaments of varying lengths up to 4 μm long (Fig. 1I) (Ong et al., 2014), suggesting that septin filaments reoriented by 90 degrees during hourglass-to-double-ring transition. This observation is in agreement with the model based on polarized fluorescence microscopy studies (Fig. 1I) (DeMay et al., 2011; Vrabioiu & Mitchison, 2006). Our analysis of septin structures at a transition stage just prior to cytokinesis (Fig. 1C) suggested that the architectural switch occurs primarily by assembly of circumferential filaments and disassembly of longitudinal filaments rather than by physical rotation of preexisting filaments. Indeed, the transitional septin structures were predominantly composed of double filaments arranged in parallel sheets, as in the early hourglass, but with the addition of singular filaments that intersect the double filaments on the membrane-proximal side. These singular filaments are spaced at a regular interval of ~29 nm (the length of a septin octameric complex), their diameter is that of a single septin filament, and their formation is compromised in mutants lacking Shs1 (the fifth septin family member acting in cytokinesis). Therefore, we hypothesize that the intersecting filaments are Shs1-containing septin filaments (Ong et al., 2014). Together, our observations allowed us to settle a prominent debate in the field and propose a new model for septin filament organization and remodeling during the cell cycle in budding yeast (Fig. 1).
Our results also demonstrated that the unroofing-replica EM approach can be used to analyze in vivo septin ultrastructure (Ong et al., 2014) (Fig. 1). The high cytoplasmic density and budded morphology of S. cerevisiae have previously impeded the ability to study in vivo septin structures in detail. The sample preparation approach we describe in this chapter combines the unroofing procedure developed by Rodal et al. 2005 and our platinum replica EM protocol, which has been extensively used for analyses of cytoskeleton architecture (Svitkina, 2016; Svitkina & Borisy, 2006). This approach has provided unparalleled views of distinct septin architectures and revealed a course of their remodeling during the yeast cell cycle. It has also afforded the high spatial resolution required for quantitative dimensional analysis. We have successfully used this technique to analyze mutant and synchronized cell populations. This method is likely compatible with drug treatments or other types of experimental conditions to test a variety of hypotheses. Given these advantages, this method has the potential to provide unprecedented insights into the regulation of septin structure and the relationship between filament organization and septin functions in cytokinesis and beyond.
1. PREPARATION OF CELL CORTICES BY UNROOFING SPHEROPLASTS
While unroofed cell cortices are compatible with other whole-mount EM approaches, we utilize platinum replica EM, which provides the advantages of sample stability, three-dimensional views, and high spatial resolution. In metal replica (or rotary shadowing) EM, contrast is created by coating the sample in a vacuum with a metal. Platinum is commonly used because it has optimal combination of a low melting temperature and a small grain size. Because replica EM provides surface views of the sample, structures located inside the cell need to be exposed to shadowing by removal of the plasma membrane, which can be achieved by chemical (detergent extraction) or mechanical (unroofing) means. Since septins are typically associated with the plasma membrane, detergent extraction cannot fully preserve dynamic septin structures, making unroofing the most suitable method (see Fig. 2 for a schematic of the workflow).
FIGURE 2.
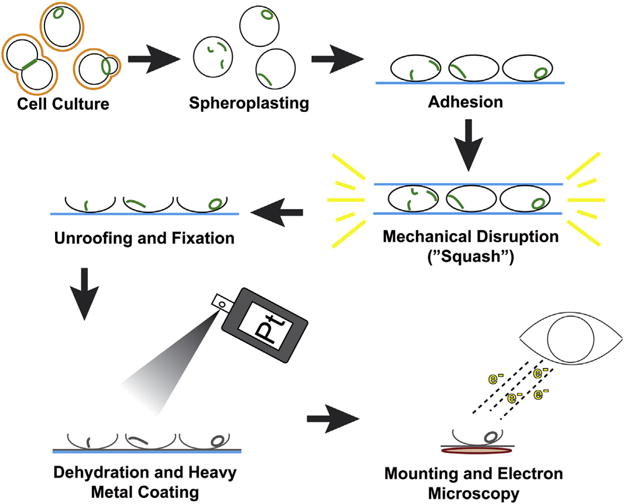
Schematic of overall workflow for preparing platinum replicas of unroofed, cell cortices.
In application to yeast cells that have a cell wall outside the plasma membrane, unroofing should be preceded by cell wall removal to generate wall-less cells termed spheroplasts. The production of high-quality spheroplasts is critical to the success of this protocol. Incomplete removal of the cell wall results in excess of debris in the final preparation, which can prevent clear visualization of cytoskeletal structures. After trying a number of variations, we have found that the following protocol produces highly stable spheroplasts with minimal cell wall contamination. Spheroplasts are then mechanically disrupted to produce unroofed cell cortices affixed to glass coverslips. This technique was first described in Rodal et al. (2005), which we adapted for our analysis of the septin cytoskeleton in budding yeast (Ong et al., 2014). Shortly after unroofing, the samples are fixed in EM-grade glutaraldehyde. Cortices can be immediately processed for EM after glutaraldehyde fixation or stored for up to 4 weeks at 4°C.
1.1 MATERIALS
Yeast culture grown to log phase, 10–50 mL
- TE buffer
- 100 mM Tris
- 100 mM EDTA
- pH 8.0
β-Mercaptoethanol
- Spheroplast buffer (freshly made prior to each experiment)
- 10 mM PIPES
- 1M Sorbitol
- pH 6.5
Zymolyase 100T, 2000 U/mL or 20 mg/mL in 50% glycerol in 10 mM PIPES, pH 6.5 (Amsbio, Cat #120493-1)
Trapezoid-shaped coverslips (can be made by cutting up 18 × 18 mm coverslips into four pieces with a diamond pencil)
High molecular weight poly-D-lysine, 1 mg/mL in water (Sigma Cat #6407)
Sterilized, MilliQ water
- 2X KHMgE buffer
- 140 mM KCl
- 40 mM HEPES
- 10 mM MgCl2
- 6 mM EGTA
- pH 7.5
Glutaraldehyde, 25% (EM grade, Polyscience Inc., #01909-10 or Sigma # 49626-25 mL)
1.2 EQUIPMENT
Temperature controlled centrifuge capable of holding 50 mL conical tubes
Dissection microscope
Temperature controlled water bath with agitator
Temperature controlled centrifuge capable of holding a 12-well plate
12-well plates
4 small (35 mm) petri dishes
1.3 METHODS
Grow a cell culture under desired conditions (Ong et al., 2014). For a culture synchronized to the early hourglass stage, we treated bar1Δ cells with alpha-factor pheromone to arrest them at the unbudded stage. The pheromone was then washed out to allow cells to reach the small-budded stage. For a culture synchronized to the double-ring stage, cdc15-2 cells were arrested prior to mitotic exit at the restrictive temperature. Cells were then shifted back to the permissive temperature to allow them to reach the double-ring stage.
Centrifuge 10–50 mL of yeast culture at 23°C or room temperature in a conical tube for 5 min at 4300 g.
Remove supernatant and weigh the cell pellet.
Resuspend the cells in 1.4 mL of TE buffer per gram of cells.
Adjust the volume to 3.5 mL per gram of cells with sterilized, deionized water.
Add 17.5 μL of β-mercaptoethanol per gram of cells. Mix by tapping the tube.
Incubate the cells in a 30°C water bath with moderate agitation for 15 min.
Centrifuge cells at 23°C or room temperature for 5 min at 4300 g.
Remove supernatant and add one or two volumes of spheroplast buffer to wash the cells. Resuspend the cells gently by tapping the tube.
Centrifuge cells again at 23°C or room temperature for 5 min at 4300 g.
Remove the supernatant and gently resuspend the cells in 4 mL of spheroplast buffer per gram of cells.
Add 50U or 25 μL of Zymolyase 100T per gram of cells.
Incubate the cells in a 30°C water bath with moderate agitation for 1 h.
During this incubation, place a trapezoidal cut coverslip into each of the wells in the 12-well plate. All the coverslips should be placed in the same orientation so that the side on which the cells are adhered to is unambiguous.
Place 10 μL of poly-D-lysine in a spot at the center of each coverslip. Incubate at 23°C or room temperature for 20 min.
Aspirate off excess poly-D-lysine. Place 10–20 μL of sterilized, MilliQ water onto the same spot to wash the coverslip. Shortly after, gently aspirate the water off. In our experience, excessively washing the coverslip or using a larger volume of water results in fewer cells adhering to the coverslip.
Let the coverslips dry for at least 30 min.
Centrifuge the cells at 4°C for 5 min at 4300 g. Remove the supernatant and gently resuspend in an excess volume of spheroplast buffer. For the rest of the protocol, keep cells on ice between steps.
Repeat this washing step two more times.
After three total washes, resuspend cells to an approximate concentration of one to two optical density measured at 600 nm. Higher cell concentrations work fine with this protocol.
Verify that you have efficiently spheroplasted your cells by mounting a small volume of the suspension on a glass slide and examining it with a dissection microscope. More than 99% of your cells should have lost their characteristic budding shape and now be spherical (Fig. 3A).
Add enough spheroplast suspension to each of the well of the plate such that you completely submerge the coverslips. Centrifuge the 12-well plate at 4°C for 5 min at 3000 rpm. The coverslips will have a cloudy appearance after this.
Fill one 35-mm petri dish with spheroplast buffer and three more dishes with 1X KHMgE buffer (dilute the 2X stock with sterilized, deionized water).
-
Fill another 12-well tissue culture plate with 1X KHMgE buffer containing 2% glutaraldehyde.
The following steps (steps 25–31) should be done quickly but gently to prevent your sample from drying out or being perturbed.
Pick up a coverslip with adherent cells with forceps and gently dip it into the petri dish filled with spheroplast buffer.
Dip into a petri dish filled with 1X KHMgE.
Carefully place the coverslip into the third dish filled with 1X KHMgE (Fig. 3B).
Repeat this process (steps 25–27) with another cell-coated coverslip, except this time place the coverslip cell side down on top of the previous coverslip (Fig. 3C).
Carefully press down on top of the two sandwiched coverslips with a blunt pair of tweezers (Fig. 3D). The coverslips should be securely pressed together and not sliding against one another. Sliding motions can result in the cells getting “smeared” instead of properly unroofed. Maintain light pressure for 5 s. Again, avoid letting the coverslips slide against one another as you lift your tweezers to relieve pressure. Typically, a successful “squash” will result in a loss of cloudiness in the region of overlap between the two coverslips.
Carefully separate the coverslips from one another. This is best done with two pairs of forceps: one fine and one blunt. Using your blunt forceps, pin down the exposed corner of the top coverslip, causing the opposite corner to separate from the bottom coverslip and pivot up. Then use the fine forceps to grab the exposed corner of the top coverslip and carefully lift it up (see Fig. 3E). Again, avoid letting the coverslips slide against one another during this process.
After separation, quickly dip the removed top coverslip into the final dish of 1X KHMgE buffer and then gently place it into a well of the 12-well plate filled with 1X KHMgE + 2% glutaraldehyde. Repeat this for the bottom coverslip.
After completing steps 25–31 for all of the coverslips, allow the samples to incubate in fixative for at least 20 min at 23°C or room temperature.
To verify that you have successfully produced unroofed cell cortices, mount a coverslip onto a glass slide and visualize with differential inference contrast (DIC) microscopy. You should observe faint, circular imprints of the cells (Fig. 3F). The presence of some intact spheroplasts in the preparation is normal, but if they make up the majority of the sample, the unroofing step of the protocol may have been unsuccessful. If the coverslip appears completely bare of cells or debris, there may have been a failure in adhering the cells to the coverslips. The coverslip that you image under DIC cannot be processed for EM and should be discarded.
You can seal the 12-well plate with parafilm and store your remaining coverslips at 4°C for up to 4 weeks before proceeding to platinum replica production.
FIGURE 3.

Unroofing of spheroplasted yeast cells. (A) Differential inference contrast (DIC) image of spheroplasted cells. (B) Placement of bottom coverslip (green (light gray in print versions)) into petri dish of KHMgE buffer. (C) Positioning of top coverslip (purple (dark gray in print versions)) such that there is a region of partial overlap (yellow (gray in print versions)). (D) Application of force with blunt forceps to mechanically disrupt the cells that are sandwiched between the two coverslips. (E) Technique for separating the coverslips from one another. (F) DIC image of fixed, unroofed cell cortices. Faint imprints of unroofed cells can be seen as debris. The appearance of some intact spheroplasts is normal.
2. PLATINUM REPLICA PRODUCTION
After exposing the intracellular structures and fixing the cortices, samples need to be dried in conditions preserving their structure and three-dimensionality. If simply dried in open air, biological samples are severely flattened and distorted by the surface tension of water. Two main approaches are employed to dry samples for replica EM: freeze-drying (or freeze-etching) and critical point drying (CPD). In the former procedure, ice from quickly frozen samples is fully (freeze-drying) or partly (freeze-etching) sublimated in vacuum. In the CPD procedure, the temperature and pressure of the liquid-immersed sample are raised above the critical point for the surrounding liquid. Under these conditions, liquid and gas coexist as a supercritical fluid and surface tension does not develop in the absence of a phase boundary. For most liquids, including water, the critical point is too extreme to be of practical use. Carbon dioxide that has a relatively low critical point (31.3°C and 1072 psi or 72.9 atm) is a fluid of choice for CPD. Therefore, the liquid in the sample needs to be completely exchanged out for liquid CO2 prior to drying. Because CO2 has limited solubility in water, ethanol is used as a transitional liquid, which is miscible with either water or CO2 in any proportion.
The unroofing approach reported by Rodal et al. (2005) for visualization of septin in yeast by EM was used in combination with rapid freezing and deep-etching. We instead use CPD, because of greater reliability, efficiency, and yield of high-quality samples with this drying technique. Since dehydration with ethanol can introduce some distortions, we use additional fixation steps with tannic acid and uranyl acetate that provide additional structural integrity to the sample (Svitkina, 2016; Svitkina & Borisy, 1998). Preparing platinum replicas from glutaraldehyde-fixed samples for EM analysis involves the following major phases: (1) two additional steps of chemical fixation, (2) ethanol dehydration, (3) CPD, (4) rotary shadowing with platinum followed by carbon coating on top of platinum layer to improve the physical stability of the replica, and (5) mounting replicas on EM grids (Fig. 2).
2.1 MATERIALS
2.1.1 Additional chemical fixation
Distilled water
Tannic acid (Fisher, # MK-1764-125), 0.1% in distilled water (made fresh prior to each experiment)
Uranyl acetate (SPI Supplies, # 02624-MB), 0.2% in distilled water. (Note: Some batches of uranyl acetate are poorly soluble; use a stirrer and enough time to dissolve; if necessary, centrifuge to remove undissolved residue.)
2.1.2 Ethanol dehydration
100% ethanol.
Graded ethanol solutions in distilled water: 10%, 20%, 40%, 60%, 80%, 100%. (Note: These solutions should be made up at least a day ahead of time as small air bubbles are generated when ethanol is mixed with water. These bubbles can damage the sample. Letting the ethanol dilutions sit overnight allows enough time for the bubbles to leave the solution.)
Dehydrated 100% ethanol: To remove traces of water that could be absorbed by ethanol, wash 4 Å molecular sieves (Fisher, #M514-500) free of dust with multiple changes of water and bake overnight at 160°C. After cooling, combine 50–100 g of molecular sieves with 500 ml of 100% ethanol and seal with parafilm for storage.
0.2% uranyl acetate in 100% ethanol. (Note: Use within a day, as uranyl acetate can precipitate from ethanol solution during storage.)
Microscope lens tissue with large mesh size (eg, Electron Microscopy Sciences, #71712-01).
2.1.3 Critical point drying
Liquid dehydrated carbon dioxide (bone dry grade) in a tank with siphon (also referred to as a tank with deep tube).
2.1.4 Platinum and carbon coating
0.76-mm tungsten wire (Ted Pella, # 27-3-20)
3-mm carbon rods (Ted Pella, # 61-15)
0.2-mm platinum wire (Electron Microscopy Sciences, # 73500)
Double-sided tape (Scotch)
Post-it notes
2.1.5 Replica mounting onto EM grids
~10% hydrofluoric acid solution (Fisher, #A622333-5000) (Caution: Hydrofluoric acid is highly volatile and toxic. Please use in a fume hood and wear gloves and eye protection while handling. Consult with your facilities’ work safety department for other safety measures required by your institution.)
Formvar-coated, copper EM grids with a low mesh size (we use 50 mesh size)
Gentle, colorless detergent (we use Ivory brand hand soap)
2.2 EQUIPMENT
2.2.1 Chemical fixation
12-well plates
Forceps
2.2.2 Ethanol dehydration
Stainless steel mesh sample holder (Fig. 4)
Two stainless steel mesh scaffolds (Fig. 4)
Stirring bars (or paperclips for low scaffolds) thoroughly cleaned with 100% ethanol
Stirrer plate
Two 50-mL beakers
12-well plates (for incubations)
FIGURE 4.

Equipment for ethanol exchange. (A) Disassembled components labeled. (B) Assembled ethanol exchange apparatus. See Section 2.3.2.
2.2.3 Critical point drying
Critical point dryer. We use the Samdri PVT-3D critical point dryer (Tousimis)
Water- and oil-absorbing filter attached to the CO2 tank (#8784A, Tousimis)
Air flow meter (#8770–45, Tousimis)
Desiccator (Note: We use an airtight container partially filled with Drierite. A Kimwipe is placed over the Drierite so that the sample holder can rest on top of it.)
2.2.4 Platinum and carbon coating
Vacuum evaporator with rotary stage. We use the Auto 306 coater (Boc Edwards).
2.2.5 Replica mounting onto EM grids
12-well plates
A 3- to 5-mm platinum wire loop with a handle
2.3 METHODS
2.3.1 Chemical fixation
Take the coverslips from glutaraldehyde solution (see Section 2.3.3) and transfer them into the aqueous 0.1% tannic acid; incubate for 20 min at 23°C or room temperature (all incubations will be at this temperature for the remainder of the protocol). (Note: For all washes and incubation treatments, do not exchange liquids by aspirating off solutions with a pipette. This can perturb the sample. Instead, transfer the coverslips between dishes of solution using fine forceps. Keep transfer time to a minimum to avoid air-drying of your samples.)
Briefly wash two times with distilled water and incubate in a third wash for 5 min.
Briefly wash an additional two times with distilled water and transfer coverslips into 0.2% uranyl acetate in distilled water; incubate for 20 min.
Transfer coverslips into distilled water.
Cut lens paper into rectangular pieces to fit the size of the sample holder or a little larger. Minor wrinkles formed when the paper pieces are placed into the holder promote looser packing of the coverslips and facilitate the liquid exchange during dehydration and CPD.
Place the sample holder into a wide container with distilled water. Insert a piece of lens paper into the bottom of the sample holder (Fig. 4). Transfer a coverslip onto the top of this lens paper, keeping the cell side up. All components should be completely under the water.
Repeat this process of sandwiching each coverslip with pieces of lens tissue. If you are processing coverslips of different sample types, be sure to keep track of the order in which you place the coverslips into the holder. In the end, you should have all of your samples loosely stacked in the sample holder, each separated by a piece of lens tissue. Place a final piece of lens tissue over the top most coverslip and put on the sample holder lid. Do not overload the sample holder, as this will prevent complete exchange of liquids during dehydration and CPD.
2.3.2 Ethanol dehydration
Put a stirring bar and a mesh scaffold into two different 50-mL beakers (Fig. 4B). A paperclip thoroughly cleaned with 100% ethanol works well in place of a stir bar.
Fill one beaker with 10% ethanol.
Transfer the sample holder into the beaker containing 10% ethanol. Be sure that you have enough ethanol in the beaker such that your samples are completely submerged. Place the beaker with the sample holder onto a stir plate and incubate for 5 min.
Fill the other beaker with 20% ethanol. Transfer the sample holder into this beaker and incubate with stirring again for 5 min.
Repeat this 5 min incubation step with the following sequence of ethanol concentrations: 40%, 60%, and 80%. After 80%, do two such incubations with 100% ethanol.
Fill a new beaker with 0.2% uranyl acetate dissolved in ethanol. Transfer the sample holder into this beaker and incubate for 20 min without agitation.
Transfer the sample into a beaker with a scaffold filled with 100% ethanol. Incubate for 5 min with stirring.
Repeat this incubation with 100% ethanol one more time.
Perform two more incubations with dehydrated ethanol dried over molecular sieve. Carefully take ethanol from the top of the bottle without perturbing beads to avoid picking up dust.
2.3.3 Critical point drying
If you are using a device other than the Samdri PVT-3D, follow the operation instructions for your dryer. (Note: Automatic procedures incorporated into some CPD devices do not give satisfactory results for replica EM purposes. It is advisable to switch such machines to a manual mode and follow the procedure described here, as close as possible.)
Turn on the critical point dryer (CPD). Check that all valves are closed before opening the CO2 tank.
Fill the CPD sample chamber with enough dehydrated ethanol to submerge the sample holder.
Transfer the sample holder quickly and carefully into the chamber. Close the chamber.
Cool the chamber down to 10–15°C. This temperature needs to be maintained until the heating phase of drying.
Open the inlet valve to allow CO2 to fill the chamber. Wait for 5 min.
Open the outlet valve until you see small bubbles in the chamber, indicating liquid exchange. Do not open the outlet valve anymore beyond this point or the level of liquid in the chamber may go below that of your sample holder. Keep the outlet valve open for 30 s, physically shake the critical point dryer to mix the contents of the chamber and assist with liquid exchange. (Note: If your CPD is equipped with a stirrer, you can turn on stirring instead of shaking the machine.)
Close the outlet valve after the 30 s. Wait for 5 min. During this time, check that the chamber is still at 10–15°C. If not, cool the chamber to return it to an appropriate temperature.
Repeat steps 7 and 8 for another nine liquid exchanges. This should completely exchange the ethanol in your sample for CO2. Check whether ethanol is still being released from the exhaust pipe by blotting it with a paper towel. If ethanol is still being released, do another one or two exchanges.
After the final exchange, wait until the chamber is completely filled with CO2 (no bubbles) before closing the inlet valve and the CO2 tank. The outlet valve should also have been closed by this point.
Turn on the heat. Wait until the temperature reaches 38–40°C. The pressure will also increase and reach approximately 1250 psi, at which point the safety valve may open temporarily to alleviate the pressure and keep it at a constant level. This is normal and requires no action.
Connect the air flow meter to the exhaust.
Open the bleed valve very slightly, such that the air flow meter reads ~2.5 SCFH (standard cubic feet per hour). Maintain this flow rate by adjusting the bleed valve accordingly until the pressure in the chamber reaches 0 psi. The release of pressure should take approximately 10 min.
When the pressure in the chamber has completely dissipated, open the chamber. Quickly transfer the sample holder into the desiccator. Exposure to water or humidity can damage your sample, so do not leave the sample holder out in open air for any longer than necessary.
Turn off the CPD.
2.3.4 Platinum and carbon coating
If you are using a device other than the Boc Edwards Auto 306 Coater, follow the operating instructions for your equipment. Wear powderless gloves while doing work inside the evaporator.
Cut a 6-cm piece of tungsten wire. To assure proper conductivity, clean off oxidation of the wire with sand paper followed by a rinse with 100% ethanol. Dry off with Kimwipes.
Cut a piece of platinum wire, clean with 100% ethanol and dry with Kimwipes. The length of platinum wire required to achieve proper thickness of coating depends on the distance between the platinum source and the sample stage. For the distance of ~10 cm, 16 mm of 0.2-mm-thick platinum wire will give the required ~2 nm thickness of coating. More precise measurements of the platinum layer thickness can be obtained by equipping the vacuum evaporator with a thickness monitor.
Bend the tungsten wire three times such that the middle of the wire has a wide “V” shape with straight ends that align with one another (Fig. 5A). The ends of the wire should be long enough so they can fit into the evaporator mount.
Tightly wrap the platinum wire around the bottom point of the “V” in the tungsten wire (Fig. 5B).
Load the platinum-tungsten assembly into the evaporator such that the bottom of the “V” with the platinum is oriented down, toward the rotary stage. Adjust the position of the platinum holder so that it is at a 45 degrees angle from the sample stage.
Sand one end of a carbon rod so that the end has a very flat surface (Fig. 5C).
With another carbon rod, use the rod sharpener to create a thin portion at one end of the rod 1 mm in diameter and 5–7 mm in length. Use sand paper to make a sharp point on this thin end of the rod (Fig. 5C).
Load the carbon rods into the evaporator such that the flat end of the first rod and the pointed end of the second rod make secure contact with one another. The meeting point of these two rods should be centered above the samples. One of the rods is spring-loaded, so that rods remain in contact during carbon evaporation.
To mount the samples on the rotary stage, apply a post-it note to each side of a piece of double-stick Scotch tape such that the sticky side of the notes face away from the tape. Cut off the nonsticky portions of the post-it notes. This will leave you with a thin sandwich having weak post-it note adhesive on each side. It will be used for securing the samples to the rotary stage while allowing you to safely remove samples from the stage after coating.
Place this sandwich onto the middle of the rotary stage, securing it lightly. You can test that it is secured properly by turning on the stage to the desired speed briefly.
Using forceps, open up the sample holder containing your coverslips. Place each coverslip onto the post-it note sandwich such that only a small portion of the coverslip actually touches the adhesive. This should be enough to secure the coverslip to the stage. Again, you can test that the samples are secure by turning on the stage briefly. Also, be sure to keep track of the sample order as you mount the coverslips if you are processing different sample types. You can write on the post-it note with pen to label each coverslip to aid in this. (Note: Sample mounting should be performed quickly to limit exposure of the sample to the humidity in the open air. It is highly recommended to run a powerful dehumidifier in the room to avoid damage of samples by ambient humidity during sample mounting.)
Close the evaporator assembly and pump down the chamber pressure to under 5 × 10−6 mbar.
Turn on the rotary stage. Coat the sample first with ~2 nm of platinum and then 3.5–5 nm of carbon.
Vent the evaporator chamber. Open the chamber and remove the post-it note with attached samples. The samples are very stable at this point and can be stored in a petri dish or similar container indefinitely.
FIGURE 5.

Setup of evaporation materials. (A) Tungsten wire for mounting platinum. (B) Platinum wire wrapped around the tip of the “V” in the tungsten wire. (C) Carbon rods. See Section 2.3.4.
2.3.5 Replica mounting onto EM grids
Mix a drop of hand soap with about 20 mL of deionized water. This stock solution of soap should be made fresh before use.
In a 12-well plate, fill a required number of wells (one per coverslips) with ~10% hydrofluoric acid using plastic pipets. To make replica handling easier, make sure the fill level is as flat as possible and fairly close to the top.
Fill the same number of wells with distilled water and add a trace amount of the soap stock solution to decrease the surface tension of water. For this purpose, dip a platinum loop into the stock soap solution, take it out (this will create a film on the loop), and dip the loop into a water-filled well. The final concentration of detergent is ~10−5%.
Fill additional two sets of wells with deionized water.
With a razor blade, lightly graze the surface of a coated coverslip (while it is still attached to the post-it note) in a grid pattern to divide up the replica into pieces sized approximately 2 × 2 mm. You may notice looking at the coverslip that there are marks with the appearance of water drops. This is a result of the poly-lysine coating on the coverslip and the regions of the replica with these markings tend to have a higher number of cell cortices given that these are the regions with the most poly-lysine. When going to mount the replica pieces onto grids, favor these regions.
With forceps, detach the coverslip from the post-it note. From a short distance, gently place the coverslip into the surface of the hydrofluoric acid in a well. Do not drop the coverslip in at an angle or else the replica may sink to the bottom. Instead, the coverslip should be completely parallel with the surface of the liquid to remain afloat.
After a few seconds of floating on the hydrofluoric acid solution, the coverslip will detach and sink, leaving the replica floating on the surface of the solution. The replica should break apart into small squares, according to the pattern you etched into it.
Using the platinum loop, transfer pieces of the replica (focusing on the “watermarked” portions) to the adjacent well filled with soapy water. To transfer, completely submerge the platinum loop into the well and position the loop under the piece you want to transfer. Lift the loop to pick up the piece. Make contact with the solution in the well you are transferring to, keeping the loop completely parallel with the solution. Gently submerge the loop, leaving the replica piece to float in the solution.
Allow each replica piece to incubate in the soap water for at least 5 s before transferring to the adjacent water-filled well.
Once all desired replica pieces have been treated with soap water and transferred into the third well filled with water, transfer a piece to the fourth well, also filled with water.
Pick up a grid with forceps, taking note of which side of the grid contains the formvar film. Press the grid into the water with the film facing down before submerging the grid. This motion allows the surface tension of the water to secure the film to the grid.
Once the grid is submerged, rotate it so the formvar film is facing upward. Position the grid underneath the replica piece and lift it up out of the water at a slight angle to capture the replica.
Allow the mounted replica to dry completely before storing or imaging.
3. IMAGING AND ANALYSIS
Imaging of replica samples can be done using any transmission electron microscope. We image our samples on a JEM 1011 transmission electron microscope operated at 100 kV (JEOL USA, Peabody, MA). Images are captured with an Orius 832.10 W charge-coupled device camera (Gatan, Warrendale, PA). Other imaging setups are also appropriate.
A goal of a typical experiment is to determine the distribution of structure types under a particular condition (eg, mutation, cell synchronization, etc.). To do this, scan your sample systematically and image structures as you go. Poorly preserved structures with only a few filaments can be disregarded. We classify our structures by the three major stages we have characterized: (1) the early hourglass composed of short (300–400 nm), double filaments arranged into parallel arrays, (2) the double ring composed of long, variably length filaments arranged circumferentially, and (3) the transition structure, composed of parallel arrays of double filaments intersected with singular linker filaments.
Note that cells lose the signature budded shape upon spheroplasting, leading to a change in membrane topology at the bud neck. This can result in fragmentation of septin rings which can be observed by EM as partial structures. Given the high-level organization of filaments in these partial structures, the reproducibility of observing this organization between preparations, and the fact that certain structure types are enriched in response to cell synchronization, we conclude that filament organization is not perturbed within fragmented structures. One can unambiguously classify these partial structures into the three major structure types based on filament length and orientation.
Hourglass structures are most prevalently observed in wild-type cells grown at log phase (Ong et al., 2014; Rodal et al., 2005). This is consistent with the fact that a majority of cells are in the small to mid-sized budded stage under these conditions. Double-ring structures are observed at a rate of about 25% (Ong et al., 2014). The transition structure is rarely observed under such conditions given that cells spend a very small fraction of time in this stage with respect to the cell cycle. To analyze this structure, we arrested cells prior to mitotic exit using a temperature-sensitive cdc15 mutant.
One may desire to analyze the effect of a mutation or treatment on a particular stage of septin remodeling. In this case, it would be best to synchronize cells to enrich structures of the desired stage in the preparation. We have used this approach to analyze the transition structure in shs1 and myo1 mutants (Ong et al., 2014). This method can be used with many types of experimental challenges, making it valuable for investigating the proteins that regulate septin filament organization and remodeling.
4. CORRELATIVE LIGHT AND ELECTRON MICROSCOPY
We used correlative light and electron microscopy (Svitkina, 2016; Svitkina & Borisy, 1998; Svitkina & Borisy, 2006) to confirm that isolated structures were septins. This involves preparing unroofed cortices on coverslips marked with a gold pattern using a finder grid as mask. Samples are then put through immunofluorescence staining for the protein of interest (in our case, Cdc11) and imaged using fluorescence microscopy prior to processing for EM. The grid position of objects found by fluorescence imaging can be recorded and correlated with structures visualized by EM. In our case, we were able to correlate the positions of rings visualized by fluorescence with ring structures in EM.
Note that with this technique, the lower resolution of light microscopy limits the ability to precisely determine the position of a protein within EM-visualized structures. Immunogold staining of the protein of interest is better suited for determining the nanoscale localization of individual proteins within cytoskeletal structures and is fully compatible with our replica EM protocol [for recent examples, see (Collins, Warrington, Taylor, & Svitkina, 2011; Jones, Korobova, & Svitkina, 2014; Shutova, Spessott, Giraudo, & Svitkina, 2014)].
4.1 MATERIALS
Glass coverslips marked with gold pattern: Shadow coverslips with gold through a finder grid, remove the grid and bake coverslips at 160°C overnight to prevent dislocation of gold grains
Petri dish compatible with your fluorescence microscopy setup
Sterile PBS buffer
Sodium borohydrate (NaBH4), 2 mg/mL and 5 mg/mL solutions in PBS
Glycine buffer, 1% w/v in PBS
Bovine serum albumin (BSA) block buffer, 1% w/v in PBS
Parafilm
Vacuum grease
Cdc11 antibody (y-415, Santa Cruz Biotechnology), 1:100 in BSA buffer
Large petri dish
Antirabbit secondary antibody of choice (Note: We use an Alexa Fluor 488 antirabbit at 1:500 in BSA buffer)
Aluminum foil
Glutaraldehyde, 2% in KHMgE buffer
4.2 EQUIPMENT
Inverted fluorescence microscope
Diamond pencil
4.3 METHODS
For all washes and treatments, whenever possible, exchange solutions by transferring coverslips between dishes of solutions instead of aspirating. When solutions need to be exchanged in the same well, aspirate and apply solutions very gently and slowly to avoid disturbing the sample. All incubations are performed at 23°C or room temperature.
Instead of using plain coverslips, perform Section 1.1 of the protocol using gold grid marked coverslips.
After producing unroofed, fixed cell cortices (ie, after finishing Section 1.1), wash your coverslips in sterile PBS by dipping the coverslip in two wells or dishes filled with PBS and then letting the coverslips sit in a third well for at least 5 min.
Transfer coverslips to dishes or wells with 2 mg/mL NaBH4 and incubate on a shaker for 10 min. Check on the coverslips during the course of the incubation to assure that the coverslips remain submerged in liquid and that the bubbles are being shaken off from the agitation of the shaker.
Repeat step 3 but with 5 mg/mL solution of NaBH4.
Incubate coverslips in 1% glycine solution for 10 min. Agitation is not needed.
Wash coverslips in PBS three times, as in step 3.
Block coverslips in BSA buffer for 30 min.
Spot ~10 μL of diluted Cdc11 antibody onto a small piece of parafilm for each of the coverslips you are staining. Place each coverslip cell-side down onto each drop of antibody solution. Incubate for 1.5 h in a large petri dish sealed with parafilm to retain the moisture.
Wash the coverslips as in step 3 except five times instead of three.
Incubate in desired secondary antibody for 1.5 h in the same manner as step 9. Cover the petri dish as the incubation is being performed in with aluminum foil to maintain the integrity of the fluorescence signal.
Wash the coverslips as in step 10.
Cut a hole into the bottom of a small petri dish for each coverslip you are processing. The hole should be larger than the finder grid but smaller than the size of the coverslip. Use a piece of sand paper to make sure that the bottom of the petri dish is completely flat. This will be important for creating a liquid impermeable seal when attaching the coverslip to the bottom of the dish.
Cover each coverslip with a small piece of parafilm such that the parafilm covers the finder grid but leaves the edges of the coverslip exposed. This will keep your sample moist during the attachment process.
Apply a thin layer of vacuum grease around the edges of the hole of the dish on the bottom surface.
Attach the coverslip to the bottom of the dish. Avoid letting the vacuum grease touch the gold finder grid on the coverslip. Use only gentle pressure, otherwise you risk breaking the coverslip.
Gently add PBS to the dish to assure that you have a leak-proof seal between the coverslip and the bottom of the dish. If there is a leak, try repositioning the coverslip carefully or detaching it completely and adding more vacuum grease to get a better seal. Once a good seal is achieved, very carefully remove the piece of parafilm covering the sample by lifting, avoiding sliding motions between the coverslip and parafilm.
Image the coverslip by fluorescence microscopy, recording positions on the grid where there are objects of interest. Keep exposure of fluorescent light to the sample at a minimum as extended exposure can cause nanoscale damage to the sample that is detectable by EM.
Exchange the PBS in the dish for 2% glutaraldehyde and incubate for at least 20 min to refix your cells.
When you are ready to process your coverslips for EM, exchange out the glutaraldehyde in the dish with two washes of water.
Place the dish into a large petri dish of water such that the dish and coverslip are completely submerged.
Using a diamond pencil, cut out a piece of the coverslip containing the grid. Be careful not to let the coverslip slide around too much. Avoid getting vacuum grease on your grid.
Detach the cut piece of coverslip from the dish. Keeping this piece submerged in water, trim off the edges that have any residual vacuum grease if needed.
Wash the coverslip once in a fresh petri dish of water. Proceed to processing for EM as described in Sections 2.3.1–2.3.4.
When it comes time to mount the replica onto an EM grid (Section 2.3.5), cut around the area of interest on the grid with a razor blade or similar tool. Then, cut straight lines radiating out from this region of interest. This will ensure the region dissociate from the rest of the replica once the coverslip is placed onto the hydrofluoric acid solution. This is best done under a dissection microscope so you can visualize the fiducial marks with respect to your cuts.
After the soap and water washes, transfer the replica piece with your region of interest onto an EM grid. Do so as described in Section 2.3.5, except taking special care of positioning the replica onto the grid such that the regions where you marked an object of interest do not overlap with the grid bars. The copper portions of the grid cannot be imaged by EM. Instead, you want the areas you want to image to lay over the transparent windows of the grid. This may take some patience and care, but one should be confident of the positioning of the replica prior to lifting the grid from the water to secure the replica onto the grid.
When imaging, find the recorded fiducial marks associated with the object of interest to correlate your fluorescence signals with structures visualized by EM.
Acknowledgments
We thank Avital Rodal, Satoshi Okada, Jonathan Chia, and Steve Jones for technical advices. K.O. is supported by T32HD083185-01and T32-GM007229. This work is supported by NIH grants GM095977 (to T.S.) and GM115420 (to E.B.).
References
- Barral Y, Mermall V, Mooseker MS, Snyder M. Compartmentalization of the cell cortex by septins is required for maintenance of cell polarity in yeast. Molecular Cell. 2000;5:841–851. doi: 10.1016/s1097-2765(00)80324-x. [DOI] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Pierson J, Thai L, McDonald KL, Zehr EA. Three-dimensional ultrastructure of the septin filament network in Saccharomyces cerevisiae. Molecular Biology of the Cell. 2012;23:423–432. doi: 10.1091/mbc.E11-10-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Gladfelter AS. Fungal pathogens are platforms for discovering novel and conserved septin properties. Current Opinion in Microbiology. 2014;20:42–48. doi: 10.1016/j.mib.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Goetsch L. A highly ordered ring of membrane-associated filaments in budding yeast. The Journal of Cell Biology. 1976a;69:717–721. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Loss of the filamentous ring in cytokinesis-defective mutants of budding yeast. The Journal of Cell Biology. 1976b;70:35a. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudron F, Barral Y. Septins and the lateral compartmentalization of eukaryotic membranes. Developmental Cell. 2009;16:493–506. doi: 10.1016/j.devcel.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Longtine M, Pringle JR, Bi E. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Molecular Biology of the Cell. 2003;14:4051–4066. doi: 10.1091/mbc.E03-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Adamikova L, Sanchez M, Molina M, Nombela C. Cell cycle control of septin ring dynamics in the budding yeast. Microbiology. 2001;147:1437–1450. doi: 10.1099/00221287-147-6-1437. [DOI] [PubMed] [Google Scholar]
- Collins A, Warrington A, Taylor KA, Svitkina T. Structural organization of the actin cytoskeleton at sites of clathrin-mediated endocytosis. Current Biology. 2011;21:1167–1175. doi: 10.1016/j.cub.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash SN, Lehtonen E, Wasik AA, Schepis A, Paavola J, Panula P. Sept7b is essential for pronephric function and development of left-right asymmetry in zebrafish embryogenesis. Journal of Cell Science. 2014;127:1476–1486. doi: 10.1242/jcs.138495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMay BS, Bai X, Howard L, Occhipinti P, Meseroll RA, Spiliotis ET. Septin filaments exhibit a dynamic, paired organization that is conserved from yeast to mammals. The Journal of Cell Biology. 2011;193:1065–1081. doi: 10.1083/jcb.201012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere J, Gentry MS, Hallberg RL, Barral Y. Phosphorylation-dependent regulation of septin dynamics during the cell cycle. Developmental Cell. 2003;4:345–357. doi: 10.1016/s1534-5807(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Dolat L, Hu Q, Spiliotis ET. Septin functions in organ system physiology and pathology. Biological Chemistry. 2014;395:123–141. doi: 10.1515/hsz-2013-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolat L, Hunyara JL, Bowen JR, Karasmanis EP, Elgawly M, Galkin VE. Septins promote stress fiber-mediated maturation of focal adhesions and renal epithelial motility. The Journal of Cell Biology. 2014;207:225–235. doi: 10.1083/jcb.201405050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, Al-Awar O, Rosenblatt J, Wong ML, Alberts B, Mitchison TJ. A purified Drosophila septin complex forms filaments and exhibits GTPase activity. The Journal of Cell Biology. 1996;133:605–616. doi: 10.1083/jcb.133.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Developmental Genetics. 1991;12:281–292. doi: 10.1002/dvg.1020120405. [DOI] [PubMed] [Google Scholar]
- Frazier JA, Wong ML, Longtine MS, Pringle JR, Mann M, Mitchison TJ. Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. The Journal of Cell Biology. 1998;143:737–749. doi: 10.1083/jcb.143.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Molecular and Cellular Biology. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall PA, Russell SE. The pathobiology of the septin gene family. Journal of Pathology. 2004;204:489–505. doi: 10.1002/path.1654. [DOI] [PubMed] [Google Scholar]
- Hall PA, Russell SEH, Pringle JR. The septins. John Wiley & Sons, Ltd; 2008. [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Experimental Cell Research. 1971;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329:436–439. doi: 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Korobova F, Svitkina T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. The Journal of Cell Biology. 2014;205:67–81. doi: 10.1083/jcb.201401045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. The Journal of Cell Biology. 1991;112:535–544. doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010;329:1337–1340. doi: 10.1126/science.1191184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Kumar S, Mizoguchi A, Ide C, Kinoshita A, Haraguchi T. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes & Development. 1997;11:1535–1547. doi: 10.1101/gad.11.12.1535. [DOI] [PubMed] [Google Scholar]
- Lippincott J, Shannon KB, Shou W, Deshaies RJ, Li R. The Tem1 small GTPase controls actomyosin and septin dynamics during cytokinesis. Journal of Cell Science. 2001;114:1379–1386. doi: 10.1242/jcs.114.7.1379. [DOI] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Septins: molecular partitioning and the generation of cellular asymmetry. Cell Division. 2009;4:18. doi: 10.1186/1747-1028-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Bi E. Septin structure and function in yeast and beyond. Trends in Cell Biology. 2011;21:141–148. doi: 10.1016/j.tcb.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nature Communications. 2014;5:5698. doi: 10.1038/ncomms6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Kozubowski L, Goode BL, Drubin DG, Hartwig JH. Actin and septin ultrastructures at the budding yeast cell cortex. Molecular Biology of the Cell. 2005;16:372–384. doi: 10.1091/mbc.E04-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Field CM. Cell division. Septins in common? Current Biology. 1994;4:907–910. doi: 10.1016/s0960-9822(00)00201-3. [DOI] [PubMed] [Google Scholar]
- Shutova MS, Spessott WA, Giraudo CG, Svitkina T. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Current Biology. 2014;24:1958–1968. doi: 10.1016/j.cub.2014.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods in Enzymology. 1998;298:570–592. doi: 10.1016/s0076-6879(98)98045-4. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy G. Correlative light and electron microscopy studies of cytoskeletal dynamics. 3rd. Vol. 3. Elsevier; 2006. [DOI] [PubMed] [Google Scholar]
- Svitkina T. Imaging cytoskeleton components by electron microscopy. Methods in Molecular Biology. 2016;1365:99–118. doi: 10.1007/978-1-4939-3124-8_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Simonetta A, Batterton M, Kinoshita M, Edbauer D, Sheng M. Role of septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Current Biololgy. 2007;17:1752–1758. doi: 10.1016/j.cub.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa PA, DeRisi JL, Wilhelm JE, Vale RD. Plasma membrane compartmentalization in yeast by messenger RNA transport and a septin diffusion barrier. Science. 2000;290:341–344. doi: 10.1126/science.290.5490.341. [DOI] [PubMed] [Google Scholar]
- Tooley AJ, Gilden J, Jacobelli J, Beemiller P, Trimble WS, Kinoshita M. Amoeboid T lymphocytes require the septin cytoskeleton for cortical integrity and persistent motility. Nature Cell Biology. 2009;11:17–26. doi: 10.1038/ncb1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrabioiu AM, Mitchison TJ. Structural insights into yeast septin organization from polarized fluorescence microscopy. Nature. 2006;443:466–469. doi: 10.1038/nature05109. [DOI] [PubMed] [Google Scholar]
- Xie Y, Vessey JP, Konecna A, Dahm R, Macchi P, Kiebler MA. The GTP-binding protein septin 7 is critical for dendrite branching and dendritic-spine morphology. Current Biology. 2007;17:1746–1751. doi: 10.1016/j.cub.2007.08.042. [DOI] [PubMed] [Google Scholar]
- Zhai G, Gu Q, He J, Lou Q, Chen X, Jin X. Sept6 is required for ciliogenesis in Kupffer’s Vesicle, the Pronephros, and the neural tube during early embryonic development. Molecular and Cellular Biology. 2014;34:1310–1321. doi: 10.1128/MCB.01409-13. [DOI] [PMC free article] [PubMed] [Google Scholar]