

Abstract
Sarcomas are a rare subgroup of pediatric cancers comprised of a variety of bone and soft-tissue tumors. While significant advances have been made in improving outcomes of patients with localized pediatric sarcomas since the addition of systemic chemotherapy to local control many decades ago, outcomes for patients with metastatic and relapsed sarcoma remain poor with few novel therapeutics identified to date. With the advent of new technologies to study cancer genomes, transcriptomes and epigenomes, our understanding of sarcoma biology has improved tremendously in a relatively short period of time. However, much remains to be accomplished in this arena especially with regard to translating all of this new knowledge to the bedside. To this end, a meeting was convened in Philadelphia, PA on April 18, 2015 sponsored by the QuadW foundation, Children’s Oncology Group and CureSearch for Children’s Cancer that brought together sarcoma clinicians and scientists from North America to review the current state of pediatric sarcoma biology and ongoing/planned genomics based clinical trials in an effort to identify and bridge knowledge gaps that continue to exist at the current time. At the conclusion of the workshop, three key objectives that would significantly further our understanding of sarcoma were identified and a proposal was put forward to develop an all-encompassing pediatric sarcoma biology protocol that would address these specific needs. This review summarizes the proceedings of the workshop.
Keywords: pediatric sarcoma, genomics, precision medicine, molecular profiling, patient derived xenografts, sarcoma biology
Introduction
Sarcomas are rare mesenchymal tumors arising from the bone or soft tissue that affect all ages but are relatively more common in the pediatric age group. Despite their rarity, they constitute significant mortality burden of about 13% of cancer related deaths in patients 0–19 years of age[1–3]. Each of the sarcoma sub-types has a distinct genetic profile and phenotype with some such as osteosarcoma characterized by a highly unstable and complex genome while others such as Ewing sarcoma characterized by a translocation between EWSR1 gene and a variety of ETS partners as the single major oncogenic driver[4]. Similarly, rhabdomyosarcoma can be molecularly sub-classified into fusion positive (PAX-FOXO1 translocation) and fusion negative RMS[5]. Despite the fact that the biology of individual sarcoma sub-types is vastly different, their treatment has historically been very similar including a combination of conventional chemotherapeutics, surgery and radiation. In addition, limited non-clinical prognostic markers exist for adequate risk stratification of patients to allow for therapy modifications (intensification of therapy for poor-risk groups or reduction of therapy for good-risk groups) for the majority of pediatric sarcomas. With the exception of recurrent RMS and a few rare soft tissue sarcomas, molecularly targeted therapies have not been incorporated into standard treatment at either diagnosis or recurrence and consequently therapy relies almost exclusively on cytotoxic chemotherapy.
While the current therapeutic approaches have been successful in improving outcomes of patients with localized sarcomas to 5-year event-free survival rates, of 60–70%, survival for patients with metastatic disease remains below 20–30% and those with relapsed disease is below 10–20%[2, 3, 6, 7]. In addition, pediatric patients who are cured of their disease are at a very high risk of long term morbidity and mortality due to the adverse effects of the toxic therapies to which they are exposed to at a young age. Therefore, an urgent unmet need remains to identify novel therapeutic strategies for pediatric sarcomas which can only be fulfilled by continued efforts to develop a deeper understanding of the biology of each individual subtype both in the context of the tumor itself as well as the host environment. As each of these individual sarcomas are extremely rare and will continue to be further molecularly sub-classified as novel genetic aberrations are discovered, it is imperative that research efforts are undertaken in a collaborative fashion to make the most impact in patient care.
A workshop, supported by the QuadW foundation, Children’s Oncology Group (COG) and the CureSearch foundation, was held on April 18, 2015 bringing together basic and translational scientists as well as clinical researchers from North America to review the current state of the art of pediatric sarcoma biology as well as genomic profiling technologies with the overarching objective of identifying areas of further research opportunities that would be complementary to ongoing efforts by individual institutions and national cooperative groups such as the COG. At the end of the workshop, three potential high-impact areas of research were identified that need to be addressed at the current time. These include: 1) establishment of better measures to identify patient prognosis and response to therapy by analysis of circulating tumor DNA; 2) analysis of a variety of germline genetic alterations such as in the gene TP53 that may impact development and progression of sarcomas; and 3) development of novel techniques of freezing tissue samples that would allow for creation of patient derived xenografts (PDXs) in the context of standard therapy or clinical trials. Herein, we provide a brief overview of the proceedings of this workshop
Genomic Analyses of Pediatric Sarcomas
The spectrum of technologies to identify genetic make-up of tumors has evolved over time from conventional cytogenetics to next generation sequencing[8, 9]. Several large efforts to characterize the molecular landscape of sarcomas using next generation sequencing technology platforms are ongoing. While these technologies, and the techniques to interpret vast amounts of data, are being continuously refined, the meeting highlighted comprehensive genome sequencing efforts across the more common pediatric sarcomas including osteosarcoma, Ewing sarcoma and rhabdomyosarcoma. In addition to detailing the tumor biology of these sarcomas, the contribution of germline aberrations towards an increased predisposition to develop these tumors was also discussed.
Osteosarcoma- Tumor genomics
Several investigators have previously published sequencing (either targeted mutation, whole genome or whole exome) results from sample sets of osteosarcoma tumors[10, 11]. A common theme that emerges from most of these studies is that the osteosarcoma genome is complex with a large number of structural variations (SVs) noted with a relatively small number of recurring exonic mutations. The most commonly mutated gene is TP53 often through an inactivating translocation in intron 1[10]. Other recurrent somatic alterations that have been identified with some frequency in osteosarcoma include RB1, ATRX, PIK3CA, AKT1 and DLG2 genes. The largest dataset for osteosarcoma somatic genome sequencing that exists is from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) which is an NCI led initiative including COG, Texas Children’s Hospital, Mount Sinai, and international collaborators including the Hospital for Sick Children in Toronto as well as collaborators form Brazil and Japan. Dr. Paul Meltzer is the lead investigator for the osteosarcoma TARGET sequencing effort. The TARGET data are consistent with previously published results in showing numerous somatic SVs which disrupt genes causing copy number alterations (CNAs). SVs can exhibit a pattern of localized hypermutation called kataegis in some tumors as well as chromothripsis depending on the degree of SVs. Extensive loss of heterozygosity is seen in exonic regions of osteosarcoma tumors. As previously published, notable high frequency of disruptions are seen in the TP53 gene as well as in alternative lengthening of telomere pathway[12, 13]. Beyond those, relatively few non-synonymous mutations are seen in osteosarcoma on the order of less than 30 missense mutations per genome. In this “regularly irregular” genome, using integrated mutation analysis as well as pathway analysis, the three most frequently affected pathways that are identified are the loss of the p53 pathway, gain of function of the alternative telomere-lengthening (ALT) pathway and the PI3 kinase pathways. Alterations in the PI3 kinase pathway had previously been identified in other studies with varying methodologies[11, 14, 15]. In summary, the challenges that remain with the work completed so far include lack of potentially targetable mutations in any significant frequency to validate in a clinical trial, potential clonal heterogeneity within tumors, lack of understanding of epigenetic events, and lack of genomic data in relapsed or refractory tumors as well as metastatic tumors in osteosarcoma.
Osteosarcoma- Role of the germline
Osteosarcoma incidence peaks in adolescence during the pubertal growth spurt and has been associated with bone growth, tall stature, high birth-weight, and therapeutic radiation[16–18]. There have been many candidate gene and pathway based studies to try to understand the germline genetics of osteosarcoma[18]. In 2013, the first genome-wide association study (GWAS) of osteosarcoma was completed[19]. Two novel loci associated with risk of osteosarcoma achieved genome-wide significance: rs1906953 at 6p21.3, which maps to the GRM4 gene and rs7591996 in a gene desert on 2p25. This GWAS showed that common SNPs are associated with risk of osteosarcoma. Using the patient data from this GWAS[19] and from four independent studies, a multi-stage genome-wide association study of osteosarcoma metastasis at diagnosis was conducted in to determine whether germline genetic variation contributes to risk of metastasis[20]. This study identified a genome-wide significant SNP, rs7034162, in the NFIB gene at 9p24.1 strongly associated with metastasis in European osteosarcoma patients, as well as in patients of African and Brazilian ancestry. The rs7034162 risk allele was also significantly associated with worse overall survival. Osteosarcoma cell line analyses further suggested that NFIB is an osteosarcoma metastasis susceptibility gene[20]. Importantly, this study suggests a connection between germline genetics and osteosarcoma metastasis at diagnosis, the leading cause of death in osteosarcoma patients.
Osteosarcoma is a hallmark of certain inherited cancer syndromes such as Li-Fraumeni syndrome (LFS), which is caused by autosomal dominant, germline TP53 mutations[21, 22]. However, most osteosarcoma cases are sporadic without a known genetic cause. A large study was conducted to determine the prevalence of germline TP53 mutations in unselected osteosarcoma cases[23]. They observed a high frequency of young osteosarcoma cases (≤29 years of age) carrying a known LFS or likely LFS-associated mutation (3.4%) or rare exonic variant (5.7%), compared with none observed in cases 30 years of age or older. This suggests that genetic susceptibility to young onset osteosarcoma is distinct from adult onset osteosarcoma; and, that genetic counseling and TP53-mutation testing in young patients with osteosarcoma should be considered. Additional genome-wide association analysis and exome sequencing studies are underway to further evaluate the role of common and rare germline genetic variation in osteosarcoma risk and outcome.
Ewing Sarcoma- Tumor Genomics
Unlike the genomic complexity of osteosarcoma, the Ewing sarcoma genome is primarily characterized by the fusion between EWS and ETS partners with 80–90% being EWSR1-FLI1 and 5–10% being EWS-ERG. Several other novel translocations have been described in patients with Ewing like tumors such as CIC-DUX4, ETV6-NTRK3, BCOR-CCNB3 and EWSR1-NFATC2[24–29]. While these tumors histologically appear similar to Ewing sarcoma their gene expression patterns cluster distinctly from EWSR1-ETS driven tumors. These findings pose the question of whether translocation testing should be performed for every patient diagnosed histologically as Ewing sarcoma and whether the disease should be defined molecularly as opposed to histologically analogous to efforts in RMS. Three groups have published the genomic landscape of Ewing sarcoma and have found consistent results[30–32]. Beyond the characteristic translocation, variants that are relatively common include mutations in the tumor suppressors STAG2 and TP53 as well as CDKN2A deletions (Figure 1). While alterations in TP53 and CDKN2A had previously been known to be present in Ewing sarcoma, they have not thus far proven to be prognostic when evaluated in a subset of patients treated with multi-agent interval compressed chemotherapy[33, 34]. On the other hand, STAG2 loss may be associated with advanced disease and decreased overall survival[30, 31].
Figure 1.
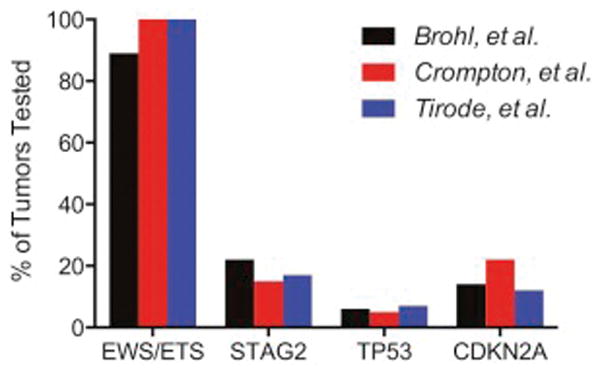
Percent of Ewing sarcoma with EWS/ETS translocations, STAG2 loss, TP53 abnormalities, and CDKN2A deletions. Adapted from Brohl, et al, PLoS Genet. 2014, Crompton, Stewart, et al, Cancer Discov. 2014, and Tirode, et al, Cancer Discov. 2014[30–32].
Although Ewing sarcoma is relatively homogenous at diagnosis with low mutational burden, preliminary evidence suggests an increase in mutational burden and clonal heterogeneity is present at relapse. Whether these mutations may be therapeutically targetable, remains to be determined. Once again, one of the limitations of the genomic data available so far is the relative lack of analysis of relapsed and metastatic samples. Ewing sarcoma provides a unique opportunity for serial disease monitoring through circulating tumor cells or cell free circulating tumor DNA due to the presence of a known translocation that is tumor specific. An optimally sensitive and specific test could aid in risk stratification, could be helpful prognostically, could allow for genomic information without a biopsy and aid in the detection of relapse. Pilot studies to test the feasibility of detecting the EWSR1-FLI1 translocation in patient plasma are underway.
Ewing Sarcoma- Role of the Germline
Ewing sarcoma is more common in Caucasian population as compared to Asian or African populations. The genetic basis for this predisposition in patients of European descent has not yet been established but indicates an underlying predisposition to disease risk. Some of this risk may be related to the GGAA microsatellite polymorphisms in the NROB1 gene[35] or other GGAA microsatellite regions across the genome[36]. Recently, a susceptibility variant in the enhancer region of EGR2 has been identified representing the first such germline determinant that predisposes to development of Ewing sarcoma [37], and this also may be related to its impact on GGAA microsatellite length[37, 38]. Other clinical associations such as an increased incidence of Ewing sarcoma noted in patients with umbilical and inguinal hernia raises the question of whether there is a germline (inherited) basis for such an association[39]. There are also a handful of familial cohorts reported in the literature. The Utah Population Database (UPDB) has been investigated to find kindred of Ewing sarcoma patients or family histories that had greatly increased incidence of Ewing sarcoma over multiple generations compared to what would be expected in the general population and five such pedigrees were identified in this database (unpublished). In addition, a review of 114 Ewing sarcoma cases with familial and genealogy data available for at least three generations showed an increased risk for developing other cancers in first degree relatives such as colon, female genital, prostate and lung cancer[40], and similar findings of different types of cancer in families with Ewing sarcoma in the UPDB also have been described[41]. These preliminary observations have led to a large international study, Project GENESIS (Genetics of Ewing sarcoma International Study, COG AEPI10N5), to study the genetic epidemiology of Ewing sarcoma. This study is ongoing but the data gathered so far supports the findings from the UPDB with there being an increased risk of cancer in families with a patient with Ewing sarcoma, including several patients with familial Ewing sarcoma (Figure 2).
Figure 2.
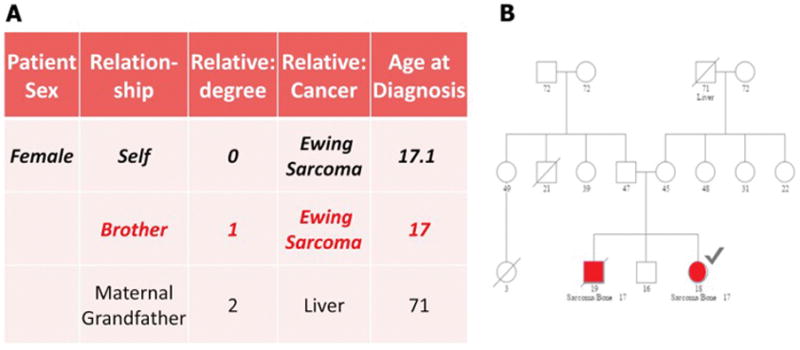
Representative example of familial Ewing sarcoma in a female patient (index case marked with a checkmark) and her brother shown in A. Tabular form and B. As a pedigree chart.
Rhabdomyosarcoma- Tumor genomics
Amongst soft-tissue sarcomas, rhabdomyosarcoma is the only sarcoma whose genomic landscape has been studied in detail. Two studies have been published which used Next Generation Sequencing to characterize these tumors [42, 43]. The major observations are that there are two distinct genomic subtypes of the disease, those characterized by a PAX gene rearrangement and those without. While both subtypes have a relatively low mutation rate, the presence of a PAX gene rearrangement seems to be associated with a particularly low rate of mutation within the coding region (Figure 3). Fusion negative tumors frequently acquire mutations along the receptor tyrosine kinase/RAS/PI3K pathway, including in NRAS, HRAS, KRAS, PIK3CA, NF1 and FGFR4, which are potentially amenable to targeted therapy[42, 43]. Additional observations include recurrent mutations in BCOR and FBXW7 but their clinical significance remains to be determined. A subset of rhabdomyosarcoma tumors harbor point mutations in the muscle development transcription factor MYOD1[44, 45]. Interestingly, these mutations appear to lead to a muscle development block and are associated with activating mutations in PIK3CA, which confers an aggressive phenotype of embryonal rhabdomyosarcoma. Recently, dystrophin has been identified as a tumor suppressor gene and an intragenic deletion in this gene leads to progression of myogenic tumors to aggressive sarcomas. Dystrophin inactivation has been found in 100% of embryonal rhabdomyosarcoma tumors[46]. Finally, an initial analysis of the clonal and evolutionary history of rhabdomyosarcoma has recently been completed [47]. This work identified clonal populations and mutations within primary tumor biopsies. The authors identified loss of heterozygosity as an early event within the development of fusion negative tumors. In addition, they identify mutation of PKN1, which confers a developmental block, as an early event in a small subset of tumors.
Figure 3.
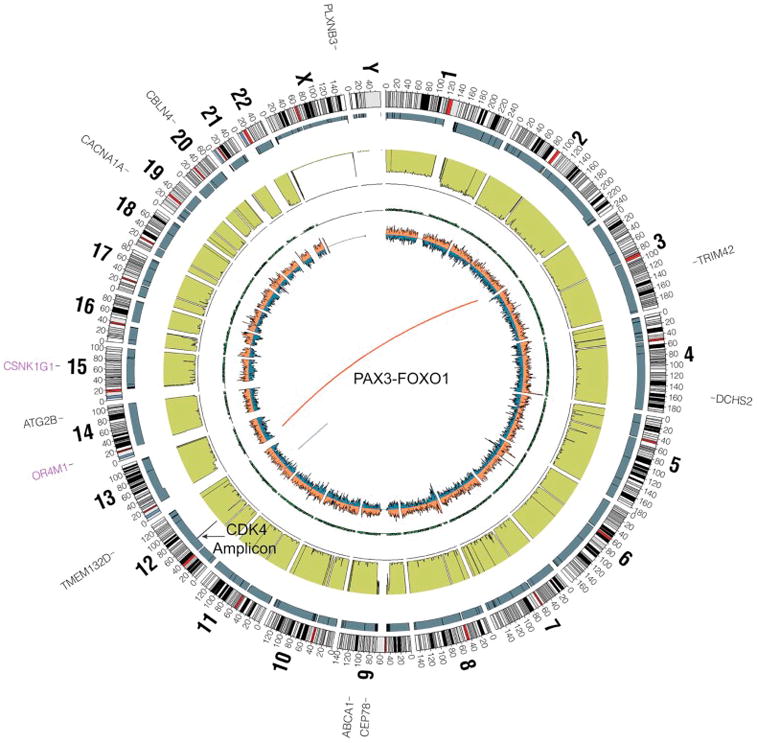
A representative circos plot of whole genome sequencing from a 13 year old patient with alveolar rhabdomyosarcoma demonstrating a translocation between chromosome 2 to 13 (PAX3-FOXO1), amplification of a region on chromosome 12 including the gene CDK4, and relatively few point mutations. The represented tracks from outside to inside include genes with point mutations (black) and stop gains (purple), copy number (gray), lesser allele frequency (green), the density of heterozygous snps (orange) and homozygous snps (blue).
Rhabdomyosarcoma- Role of the germline
Although less is known about genetic susceptibility of rhabdomyosarcoma, there is evidence defining germline mutations in a subset of patients. Additionally, there appear to be differences in terms of genetic risk for embryonal rhabdomyosarcoma and alveolar rhabdomyosarcoma. This is evident in the age-incidence patterns of these two phenotypes, as embryonal rhabdomyosarcoma incidence peaks around the age of 5 years compared to alveolar rhabdomyosarcoma, which has a relatively sustained incidence throughout childhood. An important indicator of genetic susceptibility to rhabdomyosarcoma is that about 5% of rhabdomyosarcoma cases are associated with an underlying cancer predisposition syndrome such as neurofibromatosis type 1, Li Fraumeni, DICER1, Costello sydrome, or Noonan syndrome. Interestingly, these syndromes are more common among those with embryonal rhabdomyosarcoma[48]. Data from COG and the UPDB revealed a higher odds ratio for having a family history of cancer specifically in patients with embryonal rhabdomyosarcoma[49]. About 13% of patients with rhabdomyosarcoma meet the criteria for diagnosis of Li Fraumeni syndrome with embryonal rhabdomyosarcoma having more positive cases than alveolar rhabdomyosarcoma. Furthermore increased incidence of associated birth defects, such as clubfoot, limb defects, genital defects, cleft palate and tracheoesophageal fistula has been seen in patients with rhabdomyosarcoma[50]. To determine the genetic basis of RMS epidemiology, studies are underway to evaluate susceptibility using multiple methods including genome-wide association analyses and whole-exome sequencing.
Beyond Genomics
While continued characterization of both germline and somatic variations is vital to improving our understanding of the fundamental biology of sarcomas, there is also a clear understanding that this genomic analysis will not provide a complete explanation of pathogenesis for sarcomas nor will genomics reveal all possible avenues for new therapies. Other tumor-related factors such as the mechanisms affecting the transcription of DNA (epigenetics), mechanisms affecting drug sensitivity and resistance as well as the impact of the host environment including the host immune system are equally important part of this intricate web of oncogenesis. Improved model systems that recapitulate the disease process in humans are vital. These models may include developing low passage primary cell cultures from patient tumors or patient derived xenografts (PDX) in mice that can be used to perform comprehensive genomic and epigenetic analysis of tumors as well as perform functional drug screens to determine drug sensitivities and mechanisms of drug resistance. These additional studies may aid in the treatment decision-making in future genomic clinical trials.
Epigenetics
Epigenetic modifications have been shown to be the driving force in several cancers especially pediatric cancers that have hallmark translocations leading to fusion proteins. Many sarcomas such as Ewing sarcoma, rhabdomyosarcoma and synovial sarcoma also have characteristic translocations and the fusion proteins generated from these translocations typically act as transcription factors. The detailed mechanism of how this leads to cancer remains unknown in many cases. One area of active research is the complexity of chromatin remodeling complexes. Data from sequencing studies show that more than 20% of human tumors have mutations in mSWI/SNF (BAF complexes)[51]. In synovial sarcoma, a tumor characterized by SS18-SSX fusion protein, the fusion protein competes for binding with the wild type SS18 leading to an altered complex formation which lacks the tumor suppressor gene BAF47[52]. This leads to activation of the SOX gene by reversing the polycomb-mediated repression, which in turn leads to cell proliferation. This cascade of events is triggered by a 2-amino acid region in the SSX tail leading to the altered BAF complex subunit composition. While, transcription factors are historically difficult to target with small molecules, the data on BAF complexes in synovial sarcoma suggests that the area of the SSX tail altered due a 2 amino acid substitution may potentially be druggable in the future. In addition to acting on individual proteins in the complex, targeting the ATP dependent steps may also prove to be a path towards therapy in the future. As epigenetically targeted therapies continue to be studied in clinical trials, it is anticipated that this overall approach may lead to improvements in therapy for patients with translocation-associated sarcomas.
Preclinical Model Systems
Several investigators are building resources of patient derived xenograft PDX models that closely mimic human disease with the overarching objective of using them towards in vivo modeling for precision oncology. The specific goals of these efforts include utilizing PDXs for target discovery, testing of novel approaches, and biomarker development with identification of predictive signatures for therapy resistance. Figure 4A and B delineates the process of developing PDXs and the timeline for their use for clinical drug testing respectively. For example, The Jackson Laboratory has developed over 450 PDXs across 20 tumor types with more than 25 clinical collaborators across the country. These include 17 pediatric cancer models spanning sarcoma, CNS malignancies and other pediatric solid tumors. With rare exceptions PDXs closely resemble patient tumors in terms of tumor heterogeneity, histology, and genomics. PDXs are a powerful platform for translational cancer research but as with any model system there are limitations. For example, as tumors are passaged, human stroma is replaced with that of the mouse. In addition, because profoundly immunodeficient mice are typically used to generate PDX models the system is not well-suited for modeling the role of the human immune system in cancer therapy. To overcome this limitation new “humanized” mouse models are currently under development[53]. There has been progress in utilizing PDXs as “avatars” to evaluate therapies for individual patients and initial data suggests that responses in PDX are similar to responses in patients. However, these findings remain to be validated in larger cohorts of PDXs and corresponding patients.
Figure 4.
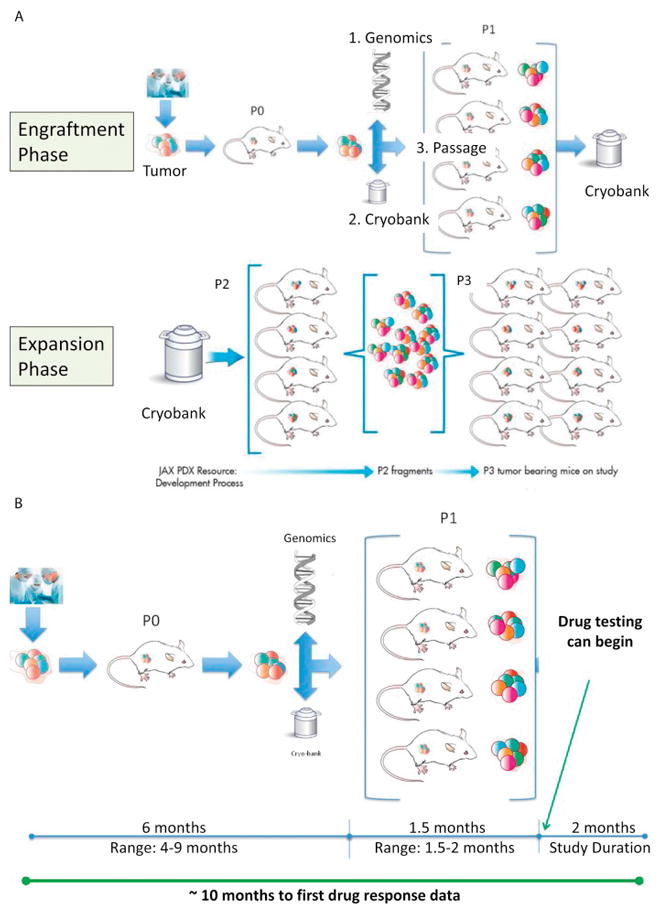
A. Schematic for the process of generation of mouse models of patient-derived xenografts (PDX) consisting of 2 phases- 1) Engraftment phase whereby fresh tumor from a patient is implanted in the flank of mice; once engrafted, the tumor undergoes detailed analysis to confirm that it is similar to the original implanted tumor and 2) Expansion phase whereby small aliquots of tumor are re-implanted in a large group of mice to enable clinical drug testing. In each of the phases, a portion of the tumor is frozen for future testing. B. Depicts the timeline for generation and expansion of PDX in mice to initiate clinical testing.
The Host
The immune system has long been thought to play a role in cancer pathogenesis and a variety of strategies, such as tumor vaccines, monoclonal antibodies, adoptive T cell therapy, donor leukocyte infusions etc., have been employed to harness the host immune system to fight and eradicate cancer cells. In parallel to rapid advances in unraveling the genomic landscape of cancer, improved understanding of a variety of immune checkpoint pathways that contribute to cancer pathogenesis has led to major breakthroughs in developing effective pan-immune therapy strategies against adult cancers. The explosion of knowledge in both these fields has occurred simultaneously but in parallel without major overlap between the two. However, a unique opportunity exists to leverage the knowledge gained from genomic discoveries to identify novel immunotherapy cell surface targets against which immune therapies can be developed. Towards this goal, a multi-institutional consortium, Pediatric Cancer Dream Team, funded by Stand Up to Cancer has been established to conduct in-depth analysis of existing genomic databases such as Oncogenomics and TARGET using powerful bioinformatics platforms like CBIO portal in the context of comparing tumor with normal tissue. The results from this integrative analysis are then sorted through a novel framework called IMmunotarget Prioritization And Capture Tool (IMPACT), which helps prioritize and track the discovered targets (Figure 5). The high- priority targets identified through these genomic analyses are then being validated at the protein level by immunohistochemistry using normal and tumor tissue banks. This effort has identified potential targets of therapeutic interest that are currently being validated. Efforts such as these are possible because of COG’s tumor banking efforts, with data analysis and statistical support provided in part by the QuadW foundation, that date back over a decade. These studies have collected available tumor tissue, often mandated for therapeutic trial entry along with associated clinical information providing a critical resource to answer key clinical questions and validate or refute observations in smaller cohorts[54, 55].
Figure 5.
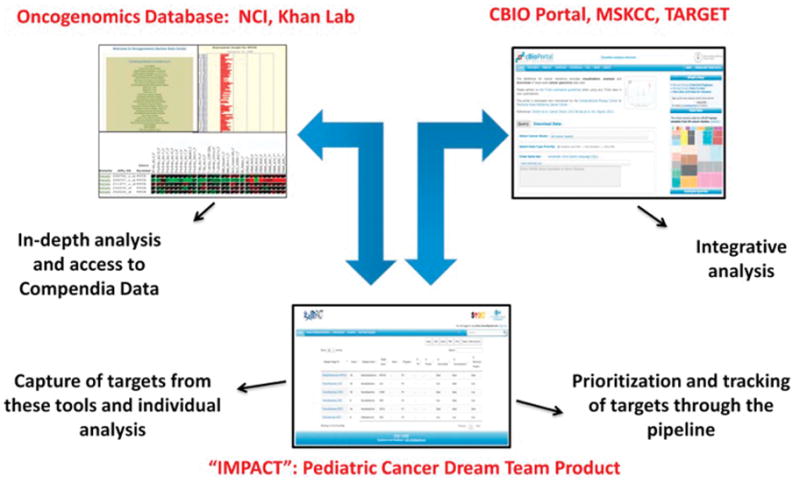
Schematic representing the workflow of the Pediatric Cancer Dream Team Project; Abbreviations: NCI, National Cancer Institute; MSKCC, Memorial Sloan Kettering Cancer Center; TARGET, Therapeutically Applicable Research to Generate Effective Treatments; IMPACT, IMmunotarget Prioritization And Capture Tool.
One of the limitations of the COG tumor banking efforts has been a paucity of banking tumors from metastatic sites as well as at the time of relapse. In addition, every disease committee in COG has had individual protocols for tumor banking that have varied in collection criteria for both tissues as well as clinical data. To overcome these challenges a new all-encompassing tumor banking protocol called Project Every Child is going to replace all of the individual disease-based banking efforts with a goal of standardizing and streamlining processes for tumor and data collection.
Bench to bedside
The ultimate goal of all cancer research including genomic, proteomic and epigenetic analysis is to be able to use this information for clinical decision making to improve patient outcomes. Over time, the molecular techniques and the depth of genetic analysis have evolved from using fixed number of common cancer related gene panels to whole genome and transcriptome analysis using next generation sequencing technologies. Newer and faster technologies keep evolving rapidly and the major challenge is the interpretation of the vast volumes of data being generated by these assays. Several major cancer centers such as Memorial Sloan Kettering Cancer Center (MSK) and Dana Farber Cancer Institute (DFCI) have led the development of institutional platforms to molecularly profile cancer patients and the majority of these platforms were initially focused in adult tumors.
The MSK-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) is a hybridization capture-based next-generation sequencing assay for targeted deep sequencing of all exons and selected introns of 410 key cancer genes including sarcoma genes such as EWSR1, in formalin-fixed, paraffin-embedded tumors. Data on 15 pediatric and young adult sarcoma patients accumulated from prospective clinical testing using MSK-IMPACT clinical next generation sequencing (NGS) assay[56, 57], showed examples of RAS-mutated embryonal rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, and MYOD1 L122R positive rhabdomyosarcoma[45]. In addition, the NGS data generated by MSK-IMPACT can pinpoint the genomic EWSR1-FLI1 or EWSR1-ERG rearrangement in a given patient with Ewing sarcoma which can then be used to design patient-specific PCR assays for circulating tumor DNA detection by digital PCR, enabling longitudinal disease monitoring in the context of an ongoing research protocol.
A joint venture between DFCI, Boston Children’s Hospital and Brigham and Women’s Hospital called Profile, is an effort which started as a mass spectrometry based platform for 471 predefined alleles across 41 genes (Sequenom) and subsequently incorporated next generation sequencing across 300 genes (Illumina)[58]. Lessons learned from rolling out this homogenous panel were that the technology could be relatively rapidly set up and validated (9 months), but that the clinical execution from consents, sample accessioning, analytics, data storage, and financially supporting the infrastructure remain to be major challenges. Pediatric patients represented about 5% of patients participating in this service. Profile has demonstrated good specificity and sensitivity for point mutations, amplifications and deletions across tumor types. Several diagnoses were changed after the sequencing results reveled a translocation or set of mutations that lead to better characterization of the tumor type with obvious therapeutic implications as well. Approximately 60% of adult patients who had their tumors profiled showed a mutation with potential clinical utility, which included both mutations that are necessary for cancer development or those that may be useful in the context of a particular tumor.
Completed/Ongoing Pediatric Cancer Precision Medicine Protocols
In line with the efforts to molecularly analyze adult tumors in order to identify actionable mutations, similar clinical genomics endeavors are ongoing in pediatric cancers. Most of these protocols have shared objectives of 1) establishing feasibility of prospectively enrolling pediatric patients on a clinical protocol to molecularly analyze their tumors (primarily solid tumors) either at diagnosis or relapse; 2) determining the frequency with which potentially actionable mutations are identified in pediatric tumors; 3) evaluating the frequency and spectrum of incidental germline findings that may have a clinical impact and 4) assessing the frequency with which a drug or an agent may be available for therapeutic intervention in pediatric patients with cancer. The protocols may vary in the platforms/technologies they use for molecular profiling and bioinformatic analysis of the sequenced genomic data. A few of these are described below.
iCAT1
iCAT (individualized cancer therapy) 1 was a multi-institutional protocol prospectively enrolling pediatric patients with relapsed/refractory non-CNS solid tumors with the primary objective of determining the feasibility of identifying genetic alterations in patient tumors to be able to make a clinical therapeutic recommendation. The platforms used for molecular analysis varied through the duration of the protocol based on the current available panels/technologies. The genetic alterations identified were mutations, translocations and copy number variants. The results of this study showed that it was feasible to run a multi-institutional molecular profiling protocol, accrual was faster than anticipated, 60% of enrolled patients had a bone or soft-tissue sarcoma diagnosis and an iCAT recommendation was made in approximately 31% of the patients[59].
GAIN consortium
GAIN (Genomic Assessment Informs Novel Therapy) is the successor study to iCAT1 with 12 collaborating institutions to assess the clinical impact of genomic analysis of tumors in recurrent/refractory pediatric tumors. This study will include a pilot hematologic malignancy cohort along with a large solid tumor cohort (825 patients over 3 years). The planned sequencing platform includes selected NGS panel on all patients along with whole exome sequencing in selected patients with the potential to incorporate CLIA RNA sequencing once available. The results of the molecular analysis will be discussed in a multi-disciplinary tumor board similar to iCAT1 and provided to patient’s treating physician for discussion with the patients. The study will follow the outcome/response of all patients enrolled whether they were treated based on the recommendation made by the molecular analysis report or not. In addition, the study includes linked pre-clinical evaluations such as creation of low passage cell cultures and patient derived xenografts for further genomic and drug response analysis.
BASIC3
Baylor College of Medicine Advancing Sequencing in Childhood Cancer Care (BASIC3) is a clinical protocol at Texas Children’s Hospital (TCH) prospectively enrolling unselected children with newly diagnosed solid and CNS tumors on a trial evaluating the diagnostic yield and impact of CLIA-certified exome sequencing on treating physicians and patients’ families. To date, the study has enrolled >240 patients, >360 parents and 20 treating oncologists at TCH. Approximately 83% of the families have shown enthusiasm to enroll at the time of diagnosis with the majority of the remaining denying enrollment due to being overwhelmed with the child’s care as opposed to specific concerns with regard to genomic testing (Figure 6)[60]. Separate reports for tumor and germline sequencing analysis are generated and returned to the primary oncologist and family with the aid of genetic counselors along with being placed in the medical record. The data from the first 150 patients enrolled on this study revealed approximately 25% of tumor reports to include somatic mutations classified as being of established or potential clinical utility, and about 8% of germline reports identifying pathogenic or potentially pathogenic germline mutations in cancer susceptibility genes[61].
Figure 6.
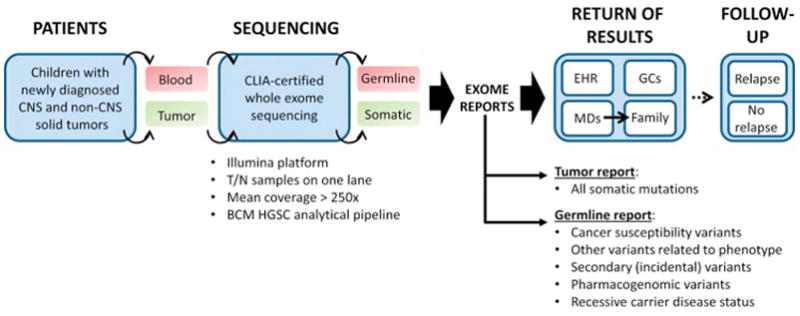
Clinical tumor whole exome sequencing and reporting performed on BASIC3 study. Abbreviations: CNS, central nervous system; T, tumor; N, normal; BCM HGSC, Baylor College of Medicine Human Genome Sequencing Center; EHR, electronic health record, GC, genetic counselor; diagram adapted from: Scollon S et al., Genome Med 2014 Sep17;6(9):69. doi: 10.1186/s13073-014-0069-3. eCollection 2014. PubMed PMID: 25317207[60].
PEDS-MIONCOSEQ
This is a pediatric precision medicine trial at University of Michigan ongoing since 2012. This study is enrolling patients with refractory solid and hematologic malignancies for analysis of tumor exome, normal exome and tumor RNA in a CLIA-certified environment with the objectives of determining feasibility of real-time sequencing analysis in pediatric refractory tumors, determining the proportion of patients with potentially actionable findings both within the tumor and the germline and the proportion of these findings that are actually acted upon along with determining major barriers that prevent action on these findings. 89% of families approached agreed for obtaining a biopsy, with the same percentage opting in to receive germline results. The majority of the biopsies were performed by interventional radiology without complications. To date more than 150 patients have been consented with adequate tissue was obtained in 87% of the patients. The time from consent to making recommendations back to the primary physician and patient has been 7 weeks on average and 46% of patients have had reported actionable findings, including 10% patients harboring pathogenic, clinically relevant germline findings[62].
In summary, all of the above clinical genomics studies have provided tremendous insight into the feasibility, operational infrastructure and key components of conducting such studies as well as the landscape of genetic mutations occurring in patients with newly diagnosed and relapsed pediatric tumors. The take home messages include:
it is feasible to perform genome scale tumor and germline analysis in clinically relevant timeframes
patients and families are extremely enthusiastic about participating in such studies;
potentially actionable tumor alterations can be identified in a substantial minority of pediatric patients;
strong bioinformatics and data curation experts are a key component of the team along with physicians, pathologists, surgeons, genetic counselors and genomicists;
an upfront plan to handle and disclose germline findings that may have an impact on patients’ and their families’ needs to be in place prior to undertaking these studies and this includes having genetic counselors as part of the team; and
there is an urgent need to conduct prospective trials of precision medicine in order to evaluate the utility of this approach for children with solid tumors.
The next generation of trials will have to incorporate actively evaluating the clinical impact of suggested treatment recommendations both by making such therapies available to pediatric patients as well as by assessing response to such therapies.
Planned Precision Medicine Initiatives
National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI-MATCH) initiative: This is a national adult precision medicine trial that recently opened to enrollment in August 2015. It is being coordinated by the ECOG-ACRIN research group and is available to all sites in the US participating in the National Clinical Trial Network. The primary objective of the trial is to ascertain response rates to molecularly targeted therapies selected based on genetic aberrations identified by DNA sequencing of patients’ relapsed solid tumors or lymphomas. The trial has pre-defined treatment arms for specific genetic abnormalities that are targetable by the agents available on the trial and has the flexibility to add/drop arms as it progresses.
Similar to the NCI-MATCH trial, the Pediatric MATCH (Molecular Analysis for Therapy Choice) study, a collaborative effort between the National Cancer Institute and the Children’s Oncology Group, is being planned as a COG-wide phase II precision oncology trial evaluating the use of genomically directed therapies for pediatric patients with refractory solid tumors and lymphomas. Study subjects will have a tumor biopsy at the time of disease recurrence in order to provide tissue for sequencing on a targeted cancer gene mutation panel. Pre-existing definitions will be used to assign subjects to study arms if an actionable mutation is identified, with approximately 25% of subjects screened anticipated to have such a mutation. The primary objective of this study will be to assess objective response rates in patients treated with a priori specified genomic alterations treated with pathway targeted agents. Secondary objectives will include additional non-clinical sequencing of study tumor samples to characterize the genomic landscape of relapsed/refractory solid tumors and comparison to FFPE samples obtained from the time of diagnosis. Germline sequencing using the same mutation panel is planned to be conducted in parallel to the tumor sequencing in order to clarify whether mutations identified in cancer susceptibility genes are somatic (tumor-specific) or germline in nature: a separate germline report will be returned after interpretation of these germline results by clinical geneticists. Sequencing and analysis pipelines will be adapted from those developed for the ongoing adult NCI-MATCH study, with modifications made as needed for pediatric cancer-specific targets and study agents.
Opportunities to bridge knowledge gaps
With the advent of novel genomic technologies such as next generation sequencing, rapid progress has been made in understanding the biology and genomic landscape of pediatric sarcomas. However, much remains to be learnt especially in the context of translating this knowledge into the clinic to improve risk stratification, disease monitoring, predicting therapy response, patient outcomes with respect to improving survival and decreasing therapy related morbidity and last but certainly not the least, cancer prevention. With those goals in mind, there was unanimous consensus amongst the pediatric sarcoma clinical and laboratory researchers that continued prospective biology studies are needed to achieve these objectives. Three key areas of knowledge gaps were identified which included:
paucity of available tumor biology data on relapsed, metastatic and refractory tumors as most of the efforts to date have focused on analysis of primary tumors, which are believed to be genomically different;
lack of prognostic biomarkers to better risk stratify patients at original diagnosis as well as lack of biomarkers to predict early relapse and
lack of model systems that accurately depict a specific patient’s tumor and therefore impede our ability to characterize each individual patient’s tumor and predict response to specific therapies.
To bridge these knowledge gaps in parallel and complementary to the current ongoing efforts of individual institutions as well as cooperative groups such as COG, a proposal was put forward to develop an all-encompassing national pediatric sarcoma biology protocol with three primary objectives:
In the context of therapeutic trials or standard therapy for patients with sarcomas, assessing the prognostic value of the presence or absence of circulating tumor DNA and a determination of its disappearance as predictors of chemotherapy response, disease status and survival.
To determine the status of a number of germline genetic alterations such as the TP53 gene in patients diagnosed with sarcomas in the context of therapeutic trials or standard therapy for patients with sarcomas, as predictors of chemotherapy response, disease status and survival.
To implant fresh tumor samples obtained from patients with metastatic and recurrent sarcomas into immunocompromised mice to establish patient derived xenograft models for use testing relevant therapeutic agents.
The specific aims of this proposed biology protocol are currently under development with individual lead investigators identified in each of the three domains but the details of the best and fastest methods to accomplish these aims remain to be determined. Each of them may be incorporated individually into currently open or upcoming therapeutic trials within COG so that adequate collection of biological samples required to successfully complete these aims can be ensured. These research objectives will be complementary to COG’s ongoing efforts to improve sample and data collection through Project Every Child initiative and to those collected on patients enrolled on the Pediatric-MATCH trial. Ultimately improved longitudinal tracking of patient outcomes coupled to biology studies would optimize translational opportunities going forward including serial biologic testing from initial diagnoses to relapse and metastasis.
Highlights.
A summary of proceedings from translational research workshop on pediatric sarcomas
Discusses biology advancements in pediatric sarcomas
Discusses ongoing/planned precision medicine trials in pediatric oncology
Outlines key areas of future research in pediatric sarcomas
These include- evaluate circulating tumor DNA, germline variants and develop PDXs
Acknowledgments
We would like to thank the QuadW foundation, Children’s Oncology Group and the CureSearch foundation for organizing this workshop
Footnotes
Disclosures: The authors have no conflicts of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. National Cancer Institute, SEER Program. Bethesda, MD: NIH; 1999. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995. [Google Scholar]
- 2.Gorlick R, Janeway K, Lessnick S, Randall RL, Marina N. Children’s Oncology Group’s 2013 blueprint for research: Bone tumors. Pediatric blood & cancer. 2013;60:1009–15. doi: 10.1002/pbc.24429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawkins DS, Spunt SL, Skapek SX. Children’s Oncology Group’s 2013 blueprint for research: Soft tissue sarcomas. Pediatric blood & cancer. 2013;60:1001–8. doi: 10.1002/pbc.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I, Volk C, Thiery JP, Olschwang S, Philip I, Berger MP, et al. Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12) Cancer Genet Cytogenet. 1988;32:229–38. doi: 10.1016/0165-4608(88)90285-3. [DOI] [PubMed] [Google Scholar]
- 5.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–46. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 6.Weiss A, Gill J, Goldberg J, Lagmay J, Spraker-Perlman H, Venkatramani R, Reed D. Advances in therapy for pediatric sarcomas. Curr Oncol Rep. 2014;16:395. doi: 10.1007/s11912-014-0395-z. [DOI] [PubMed] [Google Scholar]
- 7.Amankwah EK, Conley AP, Reed DR. Epidemiology and therapies for metastatic sarcoma. Clin Epidemiol. 2013;5:147–62. doi: 10.2147/CLEP.S28390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morozova O, Marra MA. From cytogenetics to next-generation sequencing technologies: advances in the detection of genome rearrangements in tumors. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2008;86:81–91. doi: 10.1139/O08-003. [DOI] [PubMed] [Google Scholar]
- 9.Hastings R, de Wert G, Fowler B, Krawczak M, Vermeulen E, Bakker E, Borry P, Dondorp W, Nijsingh N, Barton D, Schmidtke J, van El CG, Vermeesch J, Stol Y, Carmen Howard H, Cornel MC. The changing landscape of genetic testing and its impact on clinical and laboratory services and research in Europe. Eur J Hum Genet. 2012;20:911–6. doi: 10.1038/ejhg.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, Parker M, Rusch M, Nagahawatte P, Wu J, Mao S, Boggs K, Mulder H, Yergeau D, Lu C, Ding L, Edmonson M, Qu C, Wang J, Li Y, Navid F, Daw NC, Mardis ER, Wilson RK, Downing JR, Zhang J, Dyer MA. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choy E, Hornicek F, MacConaill L, Harmon D, Tariq Z, Garraway L, Duan Z. High-throughput genotyping in osteosarcoma identifies multiple mutations in phosphoinositide-3-kinase and other oncogenes. Cancer. 2012;118:2905–14. doi: 10.1002/cncr.26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Cai L, Wei RX, Hu H, Jin W, Zhu XB. Different expression of alternative lengthening of telomere (ALT)-associated proteins/mRNAs in osteosarcoma cell lines. Oncol Lett. 2011;2:1327–32. doi: 10.3892/ol.2011.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suva ML, Benes CH, Haber DA, Boussin FD, Zou L. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science. 2015;347:273–7. doi: 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupte A, Baker EK, Wan SS, Stewart E, Loh A, Shelat AA, Gould CM, Chalk AM, Taylor S, Lackovic K, Karlstrom A, Mutsaers AJ, Desai J, Madhamshettiwar PB, Zannettino AC, Burns C, Huang DC, Dyer MA, Simpson KJ, Walkley CR. Systematic Screening Identifies Dual PI3K and mTOR Inhibition as a Conserved Therapeutic Vulnerability in Osteosarcoma. Clin Cancer Res. 2015;21:3216–29. doi: 10.1158/1078-0432.CCR-14-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, Cowley GS, Bhatt AS, Rheinbay E, Pedamallu CS, Helman E, Taylor-Weiner A, McKenna A, DeLuca DS, Lawrence MS, Ambrogio L, Sougnez C, Sivachenko A, Walensky LD, Wagle N, Mora J, de Torres C, Lavarino C, Dos Santos Aguiar S, Yunes JA, Brandalise SR, Mercado-Celis GE, Melendez-Zajgla J, Cardenas-Cardos R, Velasco-Hidalgo L, Roberts CW, Garraway LA, Rodriguez-Galindo C, Gabriel SB, Lander ES, Golub TR, Orkin SH, Getz G, Janeway KA. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014;111:E5564–73. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirabello L, Pfeiffer R, Murphy G, Daw NC, Patino-Garcia A, Troisi RJ, Hoover RN, Douglass C, Schuz J, Craft AW, Savage SA. Height at diagnosis and birth-weight as risk factors for osteosarcoma. Cancer Causes Control. 2011;22:899–908. doi: 10.1007/s10552-011-9763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115:1531–43. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:548151. doi: 10.1155/2011/548151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, Flanagan AM, Tirabosco R, Andrulis IL, Wunder JS, Gokgoz N, Patino-Garcia A, Sierrasesumaga L, Lecanda F, Kurucu N, Ilhan IE, Sari N, Serra M, Hattinger C, Picci P, Spector LG, Barkauskas DA, Marina N, de Toledo SR, Petrilli AS, Amary MF, Halai D, Thomas DM, Douglass C, Meltzer PS, Jacobs K, Chung CC, Berndt SI, Purdue MP, Caporaso NE, Tucker M, Rothman N, Landi MT, Silverman DT, Kraft P, Hunter DJ, Malats N, Kogevinas M, Wacholder S, Troisi R, Helman L, Fraumeni JF, Jr, Yeager M, Hoover RN, Chanock SJ. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet. 2013;45:799–803. doi: 10.1038/ng.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirabello L, Koster R, Moriarity BS, Spector LG, Meltzer PS, Gary J, Machiela MJ, Pankratz N, Panagiotou OA, Largaespada D, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, Caminada de Toledo SR, Petrilli AS, Patino-Garcia A, Sierrasesumaga L, Lecanda F, Andrulis IL, Wunder JS, Gokgoz N, Serra M, Hattinger C, Picci P, Scotlandi K, Flanagan AM, Tirabosco R, Fernanda Amary M, Halai D, Ballinger ML, Thomas DM, Davis S, Barkauskas DA, Marina N, Helman L, Otto GM, Becklin KL, Wolf NK, Weg MT, Tucker M, Wacholder S, Fraumeni JF, Jr, Caporaso NE, Boland JF, Hicks BD, Vogt A, Burdett L, Yeager M, Hoover RN, Chanock SJ, Savage SA. A genome-wide scan identifies variants in NFIB associated with metastasis in patients with osteosarcoma. Cancer Discov. 2015 doi: 10.1158/2159-8290.CD-15-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, Schiffman JD. At-risk populations for osteosarcoma: the syndromes and beyond. Sarcoma. 2012;2012:152382. doi: 10.1155/2012/152382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, Thomas DM, Mitchell G. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. 2014;11:260–71. doi: 10.1038/nrclinonc.2014.41. [DOI] [PubMed] [Google Scholar]
- 23.Mirabello L, Yeager M, Mai PL, Gastier-Foster JM, Gorlick R, Khanna C, Patino-Garcia A, Sierrasesumaga L, Lecanda F, Andrulis IL, Wunder JS, Gokgoz N, Barkauskas DA, Zhang X, Vogt A, Jones K, Boland JF, Chanock SJ, Savage SA. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6:146–51. doi: 10.1038/ng0294-146. [DOI] [PubMed] [Google Scholar]
- 25.Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, Takizawa T, Nakamura T. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125–37. doi: 10.1093/hmg/ddl136. [DOI] [PubMed] [Google Scholar]
- 26.Jeon IS, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, Shapiro DN. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene. 1995;10:1229–34. [PubMed] [Google Scholar]
- 27.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–5. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 28.Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PC. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15:2259–68. doi: 10.1158/1078-0432.CCR-08-2184. [DOI] [PubMed] [Google Scholar]
- 29.Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, Perrin V, Coindre JM, Delattre O. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–6. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 30.Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, Reynaud S, Rio-Frio T, Hedlund E, Wu G, Chen X, Pierron G, Oberlin O, Zaidi S, Lemmon G, Gupta P, Vadodaria B, Easton J, Gut M, Ding L, Mardis ER, Wilson RK, Shurtleff S, Laurence V, Michon J, Marec-Berard P, Gut I, Downing J, Dyer M, Zhang J, Delattre O. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–53. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, Thorner AR, de Torres C, Lavarino C, Sunol M, McKenna A, Sivachenko A, Cibulskis K, Lawrence MS, Stojanov P, Rosenberg M, Ambrogio L, Auclair D, Seepo S, Blumenstiel B, DeFelice M, Imaz-Rosshandler I, Schwarz-Cruz YCA, Rivera MN, Rodriguez-Galindo C, Fleming MD, Golub TR, Getz G, Mora J, Stegmaier K. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4:1326–41. doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- 32.Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, Liao H, Wen X, Gerard J, Kim JS, Lopez Guerrero JA, Machado I, Wai DH, Picci P, Triche T, Horvai AE, Miettinen M, Wei JS, Catchpool D, Llombart-Bosch A, Waldman T, Khan J. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10:e1004475. doi: 10.1371/journal.pgen.1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP, Weiss AR. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:4148–54. doi: 10.1200/JCO.2011.41.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerman DM, Monument MJ, McIlvaine E, Liu XQ, Huang D, Monovich L, Beeler N, Gorlick RG, Marina NM, Womer RB, Bridge JA, Krailo MD, Randall RL, Lessnick SL. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62:759–65. doi: 10.1002/pbc.25340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monument MJ, Johnson KM, McIlvaine E, Abegglen L, Watkins WS, Jorde LB, Womer RB, Beeler N, Monovich L, Lawlor ER, Bridge JA, Schiffman JD, Krailo MD, Randall RL, Lessnick SL. Clinical and biochemical function of polymorphic NR0B1 GGAA-microsatellites in Ewing sarcoma: a report from the Children’s Oncology Group. PloS one. 2014;9:e104378. doi: 10.1371/journal.pone.0104378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, Formey A, Patel A, Gymrek M, Thapar V, Deshpande V, Ting DT, Hornicek FJ, Nielsen GP, Stamenkovic I, Aryee MJ, Bernstein BE, Rivera MN. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26:668–81. doi: 10.1016/j.ccell.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grunewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MM, Mirabeau O, Cidre-Aranaz F, Tirode F, Zaidi S, Perot G, Jonker AH, Lucchesi C, Le Deley MC, Oberlin O, Marec-Berard P, Veron AS, Reynaud S, Lapouble E, Boeva V, Rio Frio T, Alonso J, Bhatia S, Pierron G, Cancel-Tassin G, Cussenot O, Cox DG, Morton LM, Machiela MJ, Chanock SJ, Charnay P, Delattre O. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet. 2015;47:1073–8. doi: 10.1038/ng.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.EWSR1-FLI1 Induces EGR2 via a GGAA Microsatellite in Ewing Sarcoma. Cancer Discov. 2015;5:OF11. [Google Scholar]
- 39.Valery PC, Holly EA, Sleigh AC, Williams G, Kreiger N, Bain C. Hernias and Ewing’s sarcoma family of tumours: a pooled analysis and meta-analysis. Lancet Oncol. 2005;6:485–90. doi: 10.1016/S1470-2045(05)70242-4. [DOI] [PubMed] [Google Scholar]
- 40.Novakovic B, Goldstein AM, Wexler LH, Tucker MA. Increased risk of neuroectodermal tumors and stomach cancer in relatives of patients with Ewing’s sarcoma family of tumors. J Natl Cancer Inst. 1994;86:1702–6. doi: 10.1093/jnci/86.22.1702. [DOI] [PubMed] [Google Scholar]
- 41.Jones KB, Schiffman JD, Kohlmann W, Randall RL, Lessnick SL, Cannon-Albright LA. Complex genotype sarcomas display familial inheritance independent of known cancer predisposition syndromes. Cancer Epidemiol Biomarkers Prev. 2011;20:751–7. doi: 10.1158/1055-9965.EPI-10-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, Tolman C, Hurd L, Liao H, Zhang S, Bogen D, Brohl AS, Sindiri S, Catchpoole D, Badgett T, Getz G, Mora J, Anderson JR, Skapek SX, Barr FG, Meyerson M, Hawkins DS, Khan J. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, Wu G, Bradley C, McEvoy J, Pappo A, Spunt S, Valentine MB, Valentine V, Krafcik F, Lang WH, Wierdl M, Tsurkan L, Tolleman V, Federico SM, Morton C, Lu C, Ding L, Easton J, Rusch M, Nagahawatte P, Wang J, Parker M, Wei L, Hedlund E, Finkelstein D, Edmonson M, Shurtleff S, Boggs K, Mulder H, Yergeau D, Skapek S, Hawkins DS, Ramirez N, Potter PM, Sandoval JA, Davidoff AM, Mardis ER, Wilson RK, Zhang J, Downing JR, Dyer MA. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24:710–24. doi: 10.1016/j.ccr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232:300–7. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 45.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, Lim D, Marchetti A, Viale A, Pirun M, Socci ND, Qin LX, Sciot R, Bridge J, Singer S, Meyers P, Wexler LH, Barr FG, Dogan S, Fletcher JA, Reis-Filho JS, Ladanyi M. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Marino-Enriquez A, Bennett RR, Zhu M, Shen Y, Eilers G, Lee JC, Henze J, Fletcher BS, Gu Z, Fox EA, Antonescu CR, Fletcher CD, Guo X, Raut CP, Demetri GD, van de Rijn M, Ordog T, Kunkel LM, Fletcher JA. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet. 2014;46:601–6. doi: 10.1038/ng.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, Liao H, Catchpoole D, Skapek SX, Barr FG, Hawkins DS, Khan J. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet. 2015;11:e1005075. doi: 10.1371/journal.pgen.1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemminki K, Li X. A population-based study of familial soft tissue tumors. J Clin Epidemiol. 2001;54:411–6. doi: 10.1016/s0895-4356(00)00331-0. [DOI] [PubMed] [Google Scholar]
- 49.Lupo PJ, Danysh HE, Plon SE, Curtin K, Malkin D, Hettmer S, Hawkins DS, Skapek SX, Spector LG, Papworth K, Melin B, Erhardt EB, Grufferman S, Schiffman JD. Family history of cancer and childhood rhabdomyosarcoma: a report from the Children’s Oncology Group and the Utah Population Database. Cancer Med. 2015;4:781–90. doi: 10.1002/cam4.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang P, Grufferman S, Khoury MJ, Schwartz AG, Kowalski J, Ruymann FB, Maurer HM. Association of childhood rhabdomyosarcoma with neurofibromatosis type I and birth defects. Genetic epidemiology. 1995;12:467–74. doi: 10.1002/gepi.1370120504. [DOI] [PubMed] [Google Scholar]
- 51.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nature reviews Immunology. 2012;12:786–98. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borinstein SC, Beeler N, Block JJ, Gorlick R, Grohar P, Jedlicka P, Krailo M, Morris C, Phillips S, Siegal GP, Lawlor ER, Lessnick SL Committee COGESB. A Decade in Banking Ewing Sarcoma: A Report from the Children’s Oncology Group. Front Oncol. 2013;3:57. doi: 10.3389/fonc.2013.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glover J, Krailo M, Tello T, Marina N, Janeway K, Barkauskas D, Fan TM, Gorlick R, Khanna C Group COGOB. A summary of the osteosarcoma banking efforts: A report from the Children’s Oncology Group and the QuadW Foundation. Pediatr Blood Cancer. 2015;62:450–5. doi: 10.1002/pbc.25346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, Berger MF, Ladanyi M. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug discovery today. 2015 doi: 10.1016/j.drudis.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacConaill LE, Garcia E, Shivdasani P, Ducar M, Adusumilli R, Breneiser M, Byrne M, Chung L, Conneely J, Crosby L, Garraway LA, Gong X, Hahn WC, Hatton C, Kantoff PW, Kluk M, Kuo F, Jia Y, Joshi R, Longtine J, Manning A, Palescandolo E, Sharaf N, Sholl L, van Hummelen P, Wade J, Wollinson BM, Zepf D, Rollins BJ, Lindeman NI. Prospective enterprise-level molecular genotyping of a cohort of cancer patients. J Mol Diagn. 2014;16:660–72. doi: 10.1016/j.jmoldx.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janeway KJ, DuBois SG, Glade Bender JL, Kim A, Parker E, Church A, Crompton BD, Stegmaier K, Shusterman S, London WB, Lindeman NI, Diller L, Rodriguez-Galindo C, Harris MH. Multicenter study assessing tumor molecular profiles in advanced pediatric solid tumors. J Clin Oncol. 2014;32:5s. [Google Scholar]
- 60.Scollon S, Bergstrom K, Kerstein RA, Wang T, Hilsenbeck SG, Ramamurthy U, Gibbs RA, Eng CM, Chintagumpala MM, Berg SL, McCullough LB, McGuire AL, Plon SE, Parsons DW. Obtaining informed consent for clinical tumor and germline exome sequencing of newly diagnosed childhood cancer patients. Genome medicine. 2014;6:69. doi: 10.1186/s13073-014-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parsons DW, Chintagumpala MM, Berg SL, López-Terrada DH, Roy A, Kerstein RA, Scollon S, Hilsenbeck SG, Ramamurthy U, Eng CM, Yang Y, Gibbs YA, Wheeler DA, Street RL, McCullough LB, McGuire AL, Monzon FA, Plon SE. Implementation and evaluation of clinical exome sequencing in childhood cancer care: The BASIC3 study. J Clin Oncol. 2013;31 [Google Scholar]
- 62.Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, Dillman JR, Rabah RM, Kunju LP, Everett J, Raymond VM, Ning Y, Su F, Wang R, Stoffel EM, Innis JW, Roberts JS, Robertson PL, Yanik G, Chamdin A, Connelly JA, Choi S, Harris AC, Kitko C, Rao RJ, Levine JE, Castle VP, Hutchinson RJ, Talpaz M, Robinson DR, Chinnaiyan AM. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA. 2015;314:913–25. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]