

Abstract
Mounting evidence suggests neuroinflammation is a key process in glaucoma, yet the precise roles are not known. Understanding these complex processes, which may also be a key in other common neurodegenerations such as Alzheimer’s disease, will lead to targeted therapeutics for a disease that affects as many as 80 million people worldwide. Here, we define neuroinflammation as any immune-relevant response by a variety of cell types including astrocytes, microglia, and peripherally derived cells occurring in the optic nerve head and/or retina. In this review article, we first discuss clinical evidence for neuroinflammation in glaucoma and define neuroinflammation in glaucoma. We then review the inflammatory pathways that have been associated with glaucoma. Finally, we set out key research directions that we believe will greatly advance our understanding of the role of neuroinflammation in glaucoma. This review arose from a discussion of neuroinflammation in glaucoma at the 2015 meeting of the The Lasker/IRRF Initiative for Innovation in Vision Science. This manuscript sets out to summarize one of these sessions; “Inflammation and Glaucomatous Neurodegeneration”, as well as to review the current state of the literature surrounding neuroinflammation in glaucoma.
1. Introduction
An increasing body of literature, especially from various animal models, suggests neuroinflammation (generally considered to be immune responses relevant to the central nervous system (CNS)) is a key process in glaucoma (Xu et al., 2009; Baltmr et al., 2010; Chen et al., 2013; Soto and Howell, 2014). However, the contribution neuroinflammation plays is not clear. To understand neuroinflammation in glaucoma, it is important to determine (a) what types of neuroinflammatory processes are involved, (b) which sites and which cell types are involved, (c) the timing of neuroinflammatory responses and (d) whether these processes are beneficial or damaging. It is important to note that, to date, much of the supporting data for a role of neuroinflammation in glaucoma has been generated in animal models. Therefore, moving forward, it is important to fully explore neuroinflammation in both human subjects and animal models relevant to glaucoma.
It is important to standardize the terminology when referring to neuroinflammation. In non-neural tissues, inflammation usually refers to an influx of leukocytes from the blood. In the CNS, ‘neuroinflammation’ is also used to refer to microglial, and in some cases, astrocyte, responses, without necessarily leukocyte influx from the blood. The difficulty is that almost all CNS pathology is accompanied by microglial responses, and so there is no longer a special term for classical inflammatory processes in the CNS, as occurs, for example, in infection or Multiple Sclerosis. Throughout this review, ‘neuroinflammation’ in glaucoma will be used to distinguish between inflammatory processes taking place in CNS-associated structures such as the retina and optic nerve head (ONH), from inflammation in other locations in the eye such as the anterior compartments that may be more relevant to intraocular pressure (IOP) elevation.
The main body of the article (2. Neuroinflammation in glaucoma) is divided into four parts that reflect the following main topics: clinical relevance (2.1), the major sites to consider (2.2), the cell types involved (2.3), and the neuroinflammatory processes that seem worthy of further investigation (2.4). We stress that this article is not an extensive review of the literature; it is more a summary of the discussions that took place during the meeting. For those interested in a more comprehensive review of the literature, we recommended a number of relevant review articles (e.g., References (Wax and Tezel, 2009; Ren and Danias, 2010; Nickells et al., 2012; Vohra et al., 2013; Kuehn, 2014; Soto and Howell, 2014) and see Figure 1). The article ends with suggestions on the most impactful and achievable 5-year goals moving forward (part 3).
Figure 1. Neuroinflammation in glaucoma.
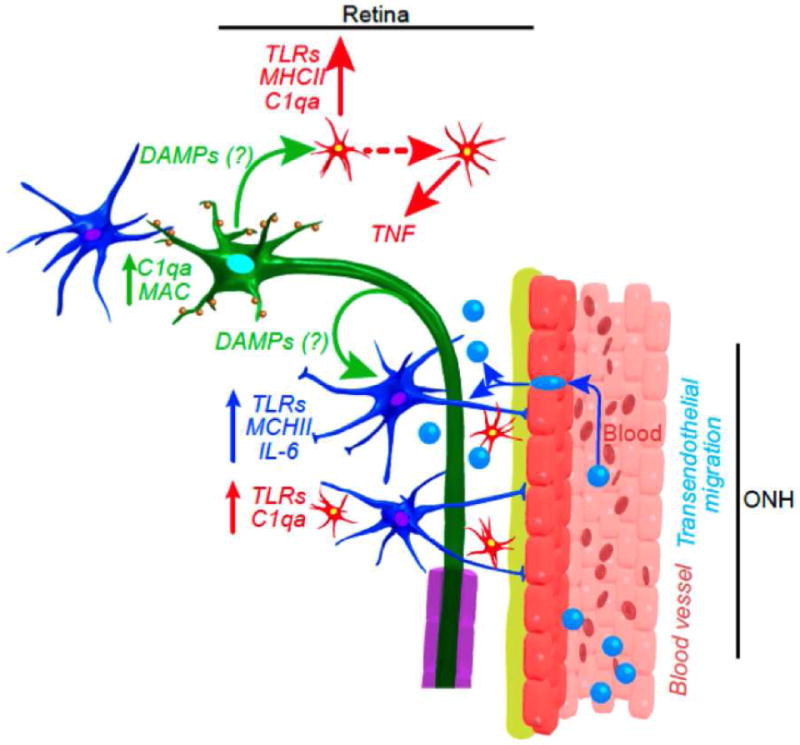
Many aspects of neuroinflammation in glaucoma are similar to what has been shown in other diseases. Although much is still to be determined, initiation of immune responses may occur after the release of DAMPs from RGCs (green), astrocytes (blue with processes) and/or microglia (red with processes). The TLRs expressed in glial cells activate the production and secretion of cytokines such as those of the IL-1 family. A secondary expression of cytokines, such as TNF-a in microglia and IL-6 in astrocytes, is induced, leading to an amplified inflammatory response, that may include infiltration of peripherally derived immune cells (blue, no processes). These neuroinflammatory responses are likely modulated by complement proteins, such as C1qa, in the ONH. In addition, intrinsic up-regulation of complement molecules in RGCs (such as C1qa and C3) occurs early and mediates synaptic dysfunction. From Soto and Howell 2015 (Soto and Howell, 2014).
2. Neuroinflammation in glaucoma
2.1. Clinical relevance of neuroinflammation in glaucoma
Paradoxically, both ocular inflammation (uveitis) and the treatment of inflammatory conditions with steroids can both lead to glaucoma. In the case of uveitis, a form of glaucoma called inflammatory, or uveitic, glaucoma can result from obstruction of drainage structures in the anterior segment as a secondary consequence of the ocular inflammation (Bodh et al., 2011; Siddique et al., 2013; Baneke et al., 2015). In the case of steroid-induced glaucoma, the steroids are generally thought to be deleterious, not by their action in reducing inflammation, but rather by acting directly to affect the drainage structures, among other things, by increasing extracellular matrix deposition by trabecular meshwork cells, which increases outflow resistance and consequently increased intraocular pressure (Clark et al., 1995; Tektas et al., 2011; Overby et al., 2014; Raghunathan et al., 2015; Taurone et al., 2015). This paradoxical relationship between inflammation and glaucoma (both inflammation and anti-inflammatory treatment can give you glaucoma), together with the absence of obvious large-scale lymphocyte infiltration into the retina (Yang et al., 2001; Tezel and Wax, 2004), have contributed to the generally held notion among clinicians that human glaucoma is not an inflammatory disease. And yet, many studies have reported microglial responses in glaucoma, which are considered neuroinflammation in other neurodegenerative disorders (Tezel and Wax, 2004; Johnson et al., 2007; Bosco et al., 2011; Howell et al., 2011; Johnson et al., 2011; Astafurov et al., 2014; Bosco et al., 2015).
Indeed, a significant fraction of glaucoma patients have small optic disk hemorrhages visible upon ophthalmic examinations, which are generally associated with disease progression (Airaksinen and Heijl, 1983; Drance, 1989; Gordon and Piltz-Seymour, 1997; Wang et al., 2006; Uhler and Piltz-Seymour, 2008; De Moraes et al., 2012). Given that only those bleeds that are seen during the very brief time that patients are undergoing ophthalmic examinations are considered (and only those in front rather than behind the lamina cribrosa would be visible), it is quite possible that small focal bleeds may occur in most and maybe all glaucoma patients (Law et al., 2001). Since hemorrhages necessarily mean that there is a breach of the blood-retina barrier, circulating immune cells certainly enter the retina, including in the nerve fiber layer where these hemorrhages are most common (Drance, 1989; Choi et al., 2007; Jeoung et al., 2008; Kurvinen et al., 2010; Hwang et al., 2014), which is the layer in the retina carrying the delicate axons which are damaged in glaucoma. So, are these hemorrhages a red herring, or are they evidence that a focal inflammatory event has occurred? Such lesions could certainly cause damage to axons. Even if they are not the primary event, they may contribute to the spread of a more focal insult, or, alternatively, they may just be reporting the occurrence of some other neuroinflammatory event. What would happen if optic disk hemorrhages where in some way prevented? Maybe nothing, maybe glaucoma damage would be more severe, or maybe glaucoma would be prevented.
Optic nerve vasculature is also special in terms of its permeability so that could provide another mechanism for contact of axons with immune cells or other immune system non-cellular components.
The infiltration of circulating monocytes, which can occur transiently and focally in many if not most glaucomas, is just one feature of neuroinflammation. There is extensive data that both the cellular hallmarks of neuroinflammation, astrogliosis and microgliosis, are also parts of the human disease. Both in the retina and, most importantly in the ONH, the principal microglial cells show signs of increased reactivity in glaucoma (Bosco et al., 2011; Howell et al., 2011; Qu and Jakobs, 2013; Seitz et al., 2013; Howell et al., 2014). Similarly, the microglia also become highly reactive in the human disease, extensively so in the ONH (Neufeld, 1999; Yuan and Neufeld, 2001). Furthermore, numerous pathways now known to be critical regulators of neuroinflammation have been shown to show gene-expression differences in glaucoma, or to confer genetic risk to developing glaucoma, as is discussed further below. Thus, not only is there likely infiltration of circulating monocytes, but the more chronic signs of neuroinflammation are also present in human glaucoma.
Thus, it may be time to abandon the notion that glaucoma does not have an inflammatory component. It is time to ask what kind of neuroinflammation occurs, where does it occur, is it relevant to disease progression, and, most importantly, whether modulation of the neuroinflammation can be used therapeutically. In light of animal studies that are beginning to show that modulation of neuroinflammation may be a promising therapeutic strategy (Bosco et al., 2008; Howell et al., 2011; Bosco et al., 2012; Howell et al., 2012; Howell et al., 2013; Howell et al., 2014), the group felt a renewed effort to study neuroinflammation in human glaucoma is warranted. There certainly are hurdles to incorporating clinical diagnostics of inflammation into routine glaucoma practice that cannot be ignored. Glaucoma is asynchronous in the two eyes, and different parts of the retina and optic nerve are going through different phases of the disease process at any one time. So, when and where is the best place to look for neuroinflammation? Is neuroinflammation part of the problem, or is it a self-defense mechanism without which disease would progress more quickly? What are the relevant cells to look at—the rare circulating monocytes that may be transiently entering the retina or the resident glial populations? There are new molecular markers, new molecular techniques, and new imaging modalities that would make answering these questions much easier now than even a few years ago. These tools should be used to study neuroinflammation in glaucoma. The preponderance of the basic science data supports the study of neuroinflammation as both a potential diagnostic tool and a potential therapeutic target. However, researchers and clinicians remain divided as to the relevance of neuroinflammation in glaucoma. We believe this may be due to the fact that the majority of evidence for a role of neuroinflammation in glaucoma has come primarily from a variety of different animal models – as opposed to human studies. While studies in animal models benefit from the ability to study onset and early progression of glaucoma, human studies to date have come from a small number of end-stage, post-mortem studies that might not truly reflect the full extent of inflammation in glaucoma. Therefore, more detailed assessment in early stages of human glaucoma using modern imaging modalities is required to bridge this divide.
2.2. Defining the sites of neuroinflammation in glaucoma
Neuroinflammation related to glaucoma can occur in many different sites, including (i) posterior compartments of the eye (e.g., retina, ONH); (ii) optic tract and brain (e.g., superior colliculus and lateral geniculate), and (iii) peripherally in the blood, bone marrow or other tissues. However, discussions focused on regions felt by the group to be most relevant to glaucoma: retinal ganglion cell (RGC) dysfunction/loss; the ONH, where many studies have shown critical events occur in glaucoma; the retina, where RGC soma, dendrites and synapses maybe affected by neuroinflammation, and the peripheral immune system. Recent studies have shown infiltration of leukocytes into the ONH and retina may be key events in glaucoma (Howell et al., 2012; Astafurov et al., 2014).
2.3. Defining neuroinflammatory cells in glaucoma
It has been long thought that the axons of RGCs are the first structures to be insulted during glaucoma pathogenesis, although the initial trigger that causes this insult is unknown. Mechanical alteration to the lamina cribrosa, changes in neurotrophic signaling, direct pressure on RGCs, and activation of glial cells (astrocytes and/or microglia) are all potential factors that drive this damage. Astrocytes (both in the ONH and retina), Müller glia, and microglia are the ‘resident cells’ that perform innate immune-like functions in the retina and/or ONH. In healthy tissues, astrocytes occupy discrete domains and help preserve a homeostatic environment for neurons to function, in part, through the maintenance of the neuro-vascular unit (Grieshaber et al., 2007; del Zoppo, 2010, 2012; Kur et al., 2012; da Fonseca et al., 2014). Activation of astrocytes is commonly termed reactive astrocytosis, which refers to a broad spectrum of changes from subtle changes in the morphology (thickening of astrocyte processes) to proliferation and migration of astrocytes to sites of injury where they can form a glial scar. Both subtle and severe astrocytosis, including the induction of inflammatory pathways, have been reported in ONH astrocytes during glaucoma, with some studies suggesting they may be among the earliest changes (Dai et al., 2012; Sun et al., 2013; Soto and Howell, 2014; Schneider and Fuchshofer, 2015). Recent data also show that ONH astrocytes are phagocytic (Nguyen et al., 2011; Mills et al., 2015). However, further work is needed to determine whether these changes are damaging or beneficial. Microglia are the main professional phagocytes of the central nervous system and survey the environment for perturbations, responding quickly to restore homeostasis. They are often considered first responders to infection and injury, activating a number of inflammatory pathways and inducing inflammatory responses in other cells such as astrocytes (discussed in section B4). Studies show that microglia increase in numbers and become activated in human and animal models of glaucoma, but it is unclear if these responses are protective, pathogenic, or both. There is some evidence that retinal and ONH microglia deactivation by minocycline at early stages of disease may result in reduced neurodegeneration (Shimazawa et al., 2005; Levkovitch-Verbin et al., 2006; Bosco et al., 2008; Levkovitch-Verbin et al., 2011; Huang et al., 2013; Levkovitch-Verbin et al., 2014a; Levkovitch-Verbin et al., 2014b). Also, macrophage (or monocyte) infiltration have been shown to be coincident with the onset of glaucoma in the DBA/2J mouse (Howell et al., 2012); furthermore, irradiated DBA/2J mice, which lack infiltrating monocytes, do not develop glaucoma. This observation raises the possibility that monocyte infiltration may be causative in glaucoma, although whether similar monocyte infiltration occurs in human glaucoma has not yet been substantiated. However, monocyte infiltration is a common event in other neurodegenerative diseases, suggesting it may also occur in human glaucoma. It is also worth noting the neuroinflammation occurs in the aging brain and involves multiple cell types, including astrocytes, microglia and macrophages ((Streit and Xue, 2014; Ward et al., 2015), Figure 2). Furthermore, given that aging is a major risk factor for glaucoma and neuroinflammation increases with age, the role of age-related neuroinflammation needs to be studied in glaucoma. Overall, the group felt that an important goal was to identify the neuroinflammatory cells in human glaucoma; then, animal models could be used to identify therapeutic strategies to target the cells for the treatment of glaucoma.
Figure 2. The major glial types in the optic nerve head in glaucoma.
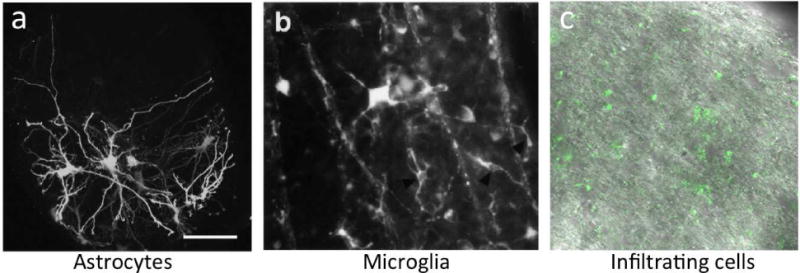
(A) GFP positive astrocytes in the glial lamina of mice with no glaucomatous neurodegeneration. From Lye-Barthel and Jakobs 2013 (Lye-Barthel et al., 2013), (B) Microglia responding to early neuron degeneration in the retina of a rat model of glaucoma. Adapted from Naskar et al, 2002 (Naskar et al., 2002), (C) Infiltrating, peripherally derived immune cells enter the optic nerve head during early stages of DBA/2J glaucoma. Adapted from Howell et al, 2012 (Howell et al., 2012).
2.4. Inflammatory pathways in glaucoma
There are a number of molecular pathways that are key regulators of neuroinflammation that in both humans and animal models have been implicated in the etiology of glaucoma. Here we focus on the complement cascade, the Toll-like receptors, and Tumor Necrosis Factor α pathway. In addition to these, interferons, interleukins, and various other pro- and anti-inflammatory cytokines (Wakefield and Lloyd, 1992; Tezel, 2008; Huang et al., 2009; Agarwal and Agarwal, 2012; Al-Gayyar and Elsherbiny, 2013; Križaj et al., 2014), and pathways involved in antigen presentation to T cells (Yang et al., 2001; Gramlich et al., 2015; Wong et al., 2015), such as β2-microglobulin and CD3, are likely involved in glaucoma, or at least in RGC damage in animal models of glaucoma.
The complement cascade is one of the key pathways in an evolutionary ancient immune system called the innate immune system, which helps to detect both pathogens and cellular damage. Various components of the complement system are increased in both human glaucoma (Stasi et al., 2006; Nikolskaya et al., 2009; Tezel et al., 2010; Doudevski et al., 2014) and animal models of glaucoma (Howell et al., 2008; Ren and Danias, 2010; Howell et al., 2011; Howell et al., 2013; Astafurov et al., 2014; Howell et al., 2014), as well as in the aging retina (Xu et al., 2009). There are three arms to the complement system that generally respond to different types of stimuli: the classical, alternative and lectin pathways. All pathways have multiple inputs and outputs and are subject to a high degree of regulation. To date, most evidence points to pronounced activation of the classical pathway in glaucoma (Stasi et al., 2006; Stevens et al., 2007; Fan et al., 2010; Howell et al., 2011; Astafurov et al., 2014). In the classical complement pathway, damaged or damaging stimuli, which can derive from dead or damaged cells, are tagged by the C1 complex. This then triggers activation of the C3 convertase, which produces the first of several complement peptides that can directly recruit blood leukocytes to the site of complement activation. C3 convertase activity also leads to activation of the C5 convertase, which produces further peptides that recruit leukocytes, but also the components that form the membrane attack complex (MAC) that lyses cells. An overly simplified view of the complement pathway in animal models of glaucoma is that the RGCs themselves sense the damage and respond by activating C1; the activation of C3 convertase requires glial cells such as astrocytes, which can, among other things, use the complement pathway to amplify the RGC distress signal to alert microglia and, if needed, to recruit monocytes. Interestingly, the complement pathway in glaucoma appears to be activated principally in two places—the ONH, the likely site of the blinding insult, and in the inner plexiform layer, where it may be involved in pruning RGC dendrites. The complement pathway plays a similar synaptic pruning function during normal CNS development (Stevens et al., 2007). Both in the ONH, but especially so in the inner plexiform layer, it is quite possible that activation of the complement pathway is not responsible for killing RGCs, but rather acts either to prevent further damage to RGCs or to maintain RGC function in the face of disease. This double-edged sword nature of the complement pathway may explain why in some animal models of glaucoma but not in others, complement components have a strong pathogenic role (Stasi et al., 2006). It is also worth noting that, unlike in human macular degeneration, where genome-wide association studies have provided strong evidence for a prominent role for the complement system, the same has not been true for similar studies in human glaucoma.
Toll like receptors (TLRs) have also been implicated in the pathogenesis of glaucoma in two ways: certain alleles of the TLR4 gene are associated with an increased risk of glaucoma, at least in some populations (Shibuya et al., 2008; Takano et al., 2012), and TLR4 and other TLRs are reported to be increased in glaucoma retinas (Luo et al., 2010). TLRs are one of the principal receptor systems to recognize Pathogen- and Damage- Associated Molecular Patterns (PAMPS and DAMPs). Different TLRs are tuned to respond to the presence of different stimuli that are not found in normal tissues other than when there is disease or damage; for example, TLR3 responds to microbial double-stranded RNA, and TLR4 responds to endogenous ligands induced by cell stress such as heat shock proteins (HSPs). Interestingly, one of the endogenous ligands for TLR4 is Tenascin-C, which is highly upregulated in glaucomatous ONH (Pena et al., 1999; Wallace et al., 2014; Wallace et al., 2015) and animal models of glaucoma (Johnson et al., 2007). Not surprisingly, astrocytes have a more limited set of damage-detecting TLRs than microglia and monocyte/macrophages. While there have been a few studies of TLRs in animal models of RGC injury, at this point it is unclear what role TLRs have in human glaucoma.
Tumor necrosis factor alpha (TNFα) is another neuroinflammation modulator for which there is strong evidence of involvement in glaucoma. Certain polymorphisms in TNFα confer an increased risk of glaucoma (Lin et al., 2003; Mossböck et al., 2006; Bozkurt et al., 2012; Wang et al., 2012; Xin et al., 2013; Dubey et al., 2015). Moreover, there is extensive data showing increased TNFα in vitreous, retina and optic nerve in glaucoma (Wakefield and Lloyd, 1992; Yuan and Neufeld, 2000; Tezel, 2008; Yang et al., 2011; Wiggs, 2015). Interestingly, the TNFα is produced by astrocytes and especially microglia (Sawada et al., 1989; Hostenbach et al., 2014). Generally, secretion of TNFα and a related protein FasL are thought to promote RGC cell death, through a variety of mechanisms (Wajant et al., 2003; Mac Nair et al., 2014; Cueva Vargas et al., 2015), which most likely involve direct killing of RGCs. To this, intravitreal injections of soluble murine TNFα induces oligodendrocyte and RGC death in a mouse model of glaucoma, and using specific TNFα blockers protects from RGC loss in a rat model of glaucoma (Nakazawa et al., 2006; Cueva Vargas et al., 2015). Why glia should have a mechanism killing RGCs is not obvious. Pharmacological inhibition of TNFα or FasL activity has been shown to be neuroprotective in animal models of glaucoma (Roh et al., 2012; Su et al., 2014), although these inhibitors have not provided protection in other neurodegenerative disorders. Given the wide availability of TNFα inhibitors approved for human use, they deserve further study in relation to human glaucoma; more studies, for example, are needed to determine precisely when and where TNFα is secreted in glaucoma.
Overall, although multiple neuroinflammatory pathways and genes have been identified in human and animal models of glaucoma (Figure 1), much work is still to be done to determine when they are activated and which ones are protective and which ones are pathogenic. As their roles become clearer, it should become clearer which ones may be therapeutic targets in human glaucoma.
3. Moving forward – recommended five-year goals
3.1: Determine the repertoire of neuroinflammatory cells and pathways in human glaucoma
The lack of assessment of neuroinflammation in human glaucoma is a key area that needs to be addressed. To achieve this, researchers need to properly utilize existing human and non-human primate glaucomatous tissues, using the best immunohistochemistry markers and microscopy. Improved availability and standardization of tissue sources, with as much associated data as possible, will aid these efforts. A significant challenge is to assess the range of neuroinflammatory cell types present in human glaucoma – particularly those cell types that may play a role early in disease but may be present in extremely low numbers. Often, tissue is obtained from patients with later stages of glaucoma, which may be less useful. New markers for distinguishing microglia from macrophages will be especially important in future studies.
3.2: Interrogation of genome-wide datasets to identify neuroinflammatory cells and genes in human glaucoma
During the last 10 years, the ability to identify genetic variants using new genetic tools has increased dramatically, and the cost of these experiments has dropped greatly. In glaucoma and a myriad of other likely-relevant human diseases, there is an increasing number of genome-wide datasets available, including genome-wide association studies, whole genome and exon sequences, and transcriptomes, which can be mined for genes that provide links between neuroinflammation and glaucoma. Accessibility of these datasets, and datasets from other neurodegenerative and immune-related diseases, will be important, as will be the platforms to mine the data. Where possible and appropriate, these datasets should be made widely accessible to interrogation by multiple researchers.
3.3: What are the triggers and drivers of neuroinflammation in glaucoma?
There are likely to be both local and peripheral inflammatory triggers, and these are likely to vary with age, genetics and the environment. There are many potential candidates, including cellular debris, nuclear DNA and mitochodrial DNA derived from cell debris, misfolded proteins, ATP release, proinflammatory cytokines, local microbial flora (oral, gastrointestinal, ocular) and peripheral inflammatory events. In addition, much work is still required to understand the neuroinflammatory genes (e.g. TNFα, TGFβ, APOE) and pathways (e.g., complement cascade, TLR family and interferon family).
3.4: Define the contribution of age-related neuroinflammation to glaucoma susceptibility and pathogenesis
Age is the major risk factor for glaucoma, and yet little is know about how the aging process increases risk. Neuroinflammatory responses have been shown to increase in the brain during aging, and, if the same is true in the ONH and retina, these responses may contribute to an increased susceptibility to glaucoma.
3.5: The role of the microbiome in glaucoma
Growing evidence suggests the microbiome plays a critical role in some complex diseases (Figure 3). This is likely to be the case in glaucoma, and a few studies are now being performed to explore this possibility. Animal models can play a role in this area, as it is known that individual animal colonies often respond very differently to the same stimulus or mutation, which may be due in large part because of differences in their microbiomes.
Figure 3. The human microbiome may affect glaucomatous neurodegeneration.
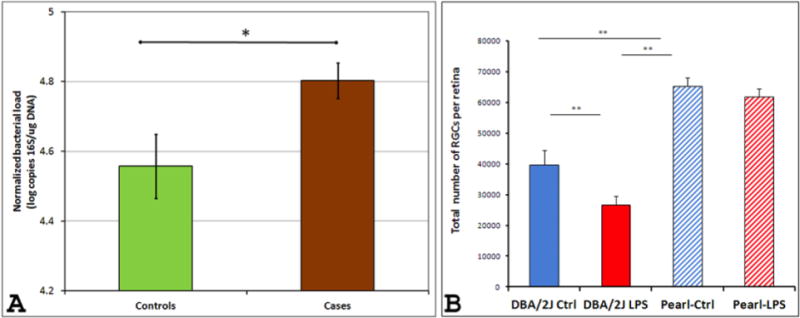
(A) Differences in oral bacterial load in a cohort of African-American patients with and without glaucoma. Normalized oral bacterial load (NOBL) of patients with glaucoma were higher than those of subjects without glaucoma (p<0.017, t-test). Although cases were significantly different from controls in age (p<0.008, t-test), gender (p<0.02 Chi-square) and diabetes status (p<0.021, Chi square), GLM ANOVA of NOBLs (using group, gender, diabetes status and age [above or below median value] as independent variables) revealed a significant effect of group status (whether a subject belonged to cases or controls) only (p<0.024) while all other parameters did not show a statistically significant effect (p>0.26 for all). B. Microbial components affect neurodegeneration in glaucoma animal models. Peripheral lipopolysaccharide (LPS) administration significantly accelerates glaucomatous pathology in DBA/2J mice (but not in the non-glaucomatous DBA/2J-Pe mice) as well as in the microbead-induced IOP elevation model of glaucoma in C57BL/6 mice (not shown here). LPS (60 μg) was injected into one hind footpad of 6-month old male DBA/2J and DBA/2J-Pe mice and retinal damage was assessed at 8 months of age. Computer-assisted counts of total RGCs per retina (n = 18 and n = 24 retinas for DBA/2J and DBA/2J+LPS, respectively; n = 4 and n = 10 retinas for DBA/2J-Pe and DBA/2J-Pe+LPS, respectively) show increased RGC loss in DBA/2J mice treated with LPS. Data are presented as mean ± SEM. Statistical differences were assessed by one-way ANOVA, followed by Tukey-Kramer post-hoc testing (**p<0.01). Adapted from Astafurov et al, 2014 (Astafurov et al., 2014).
3.6: Continued development of animal models relevant to ocular hypertension and glaucoma
At the Initiative in 2010, ‘Animal Models’ was a full session, and new models have emerged since then, including both genetic (MYOC) and inducible (mouse and rat bead models and various virus models). In addition to continued efforts to characterize and improve existing models, there is still a pressing need for the development of new models, especially new genetic animal models based on big-effect human mutations that greatly increase the risk of open angle glaucoma. No animal model is going to fully recapitulate all aspects of glaucoma, particularly given the heterogeneous nature of human glaucoma. However, it is important that models are used appropriately and characterized as extensively as possible.
Our idea of an appropriate species to model aspects of human glaucoma should not just be limited to mammals – a lot of valuable information, particularly relating to the function of genes and pathways relevant to glaucoma can be obtained from other organisms such as flies, worms, frogs and fish. The development of genome editing, particularly CRISPR/CAS9, has revolutionized our ability to manipulate genomes of potentially any species. Therefore, for the first time, genetic models relevant to glaucoma can be generated in many species with relative ease. Novel risk genes (such as ABCA1 and CDKN2A-AS) are being linked and associated with glaucoma through genome-wide association studies and exon and genome sequencing efforts, but little is known about how they contribute to glaucoma. Therefore, using genome editing, candidate genes can be manipulated in multiple species to understand how variations in these genes increase susceptibility to human glaucoma. Ultimately though, for preclinical studies, additional models in various mammals are still required. During the general discussion at the end of the meeting, it was exciting to hear from a number of investigators that efforts are underway to establish new glaucoma models in mammals with a lamina cribrosa that are easier and more cost effective to use than the larger non-human primates currently being used. These efforts should be encouraged. Overall, the group was highly positive that the next few years will bring novel insights into the role that neuroinflammation plays in glaucoma, and that, in so doing, we will come closer to being able to treat, and maybe even prevent, all vision loss due to glaucoma.
We describe the clinical relevance of inflammation in glaucoma
We define the sites of neuroinflammation in glaucoma
We discuss the roles of different cells and pathways in glaucoma
We discuss suggested 5 year goals as set about by the participants of the 2015 Lasker/IRRF Initiative session “Inflammation and Glaucomatous Neurodegeneration”
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This article summarizes the results of a targeted session on this topic at the March 2015 conference Astrocytes and Glaucomatous Neurodegeneration. This meeting was a follow-up to the 2010 meeting on the same topic, both of which were conducted as part of The Lasker/IRRF Initiative for Innovation in Vision Science. For more information about this conference, its participants and other review articles that originated from it see Tamm and Dowling, in press. A list of the other participants of the targeted session is provided at the end of this article.
List of participants
Discussion leaders: Nick Marsh-Armstrong, Gareth R Howell.
Scribe: Pete A Williams.
Discussion Participants: Alejandra Bosco (University of Utah, School of Medicine, Salt Lake City, Utah, USA), John Danias (Department of Ophthalmology, SUNY Downstate, New York, USA), Simon John (The Jackson Laboratory, Bar Harbor, Maine, USA), Adriana Di Polo (Department of Neuroscience and Centre de Recherche du Centre Hospitalier de l’Université de Montréal, Université de Montréal, Montréal, Québec, Canada), Markus H. Kuehn (Department of Ophthalmology and Visual Sciences, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA), Serge Przedborski (Departments of Pathology & Cell Biology, Columbia University Medical Center, New York, USA), Martin Raff (MRC Laboratory for Molecular Cell Biology, University College London, London, UK), Ian Trounce (Centre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, University of Melbourne, East Melbourne, Victoria, Australia)
References
- Agarwal R, Agarwal P. Glaucomatous neurodegeneration: an eye on tumor necrosis factor-alpha. Indian J Ophthalmol. 2012;60(4):255–61. doi: 10.4103/0301-4738.98700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airaksinen PJ, Heijl A. Visual field and retinal nerve fibre layer in early glaucoma after optic disc haemorrhage. Acta Ophthalmol (Copenh) 1983;61(2):186–94. doi: 10.1111/j.1755-3768.1983.tb01412.x. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MM, Elsherbiny NM. Contribution of TNF-α to the development of retinal neurodegenerative disorders. Eur Cytokine Netw. 2013;24(1):27–36. doi: 10.1684/ecn.2013.0334. [DOI] [PubMed] [Google Scholar]
- Astafurov K, Elhawy E, Ren L, Dong CQ, Igboin C, Hyman L, et al. Oral microbiome link to neurodegeneration in glaucoma. PLoS One. 2014;9(9):e104416. doi: 10.1371/journal.pone.0104416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltmr A, Duggan J, Nizari S, Salt TE, Cordeiro MF. Neuroprotection in glaucoma - Is there a future role? Exp Eye Res. 2010;91(5):554–66. doi: 10.1016/j.exer.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Baneke AJ, Lim KS, Stanford M. The Pathogenesis of Raised Intraocular Pressure in Uveitis. Curr Eye Res. 2015:1–13. doi: 10.3109/02713683.2015.1017650. [DOI] [PubMed] [Google Scholar]
- Bodh SA, Kumar V, Raina UK, Ghosh B, Thakar M. Inflammatory glaucoma. Oman J Ophthalmol. 2011;4(1):3–9. doi: 10.4103/0974-620X.77655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Crish SD, Steele MR, Romero CO, Inman DM, Horner PJ, et al. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS One United States. 2012:e43602. doi: 10.1371/journal.pone.0043602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49(4):1437–46. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- Bosco A, Romero CO, Ambati BK, Vetter ML. In vivo dynamics of retinal microglial activation during neurodegeneration: confocal ophthalmoscopic imaging and cell morphometry in mouse glaucoma. J Vis Exp. 2015;(99):e52731. doi: 10.3791/52731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. 2011;519(4):599–620. doi: 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozkurt B, Mesci L, Irkec M, Ozdag BB, Sanal O, Arslan U, et al. Association of tumour necrosis factor-alpha -308 G/A polymorphism with primary open-angle glaucoma. Clin Experiment Ophthalmol. 2012;40(4):e156–62. doi: 10.1111/j.1442-9071.2011.02595.x. [DOI] [PubMed] [Google Scholar]
- Chen SD, Wang L, Zhang XL. Neuroprotection in glaucoma: present and future. Chin Med J (Engl) 2013;126(8):1567–77. [PubMed] [Google Scholar]
- Choi F, Park KH, Kim DM, Kim TW. Retinal nerve fiber layer thickness evaluation using optical coherence tomography in eyes with optic disc hemorrhage. Ophthalmic Surg Lasers Imaging. 2007;38(2):118–25. doi: 10.3928/15428877-20070301-06. [DOI] [PubMed] [Google Scholar]
- Clark AF, Wilson K, de Kater AW, Allingham RR, McCartney MD. Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest Ophthalmol Vis Sci. 1995;36(2):478–89. [PubMed] [Google Scholar]
- Cueva Vargas JL, Osswald IK, Unsain N, Aurousseau MR, Barker PA, Bowie D, et al. Soluble Tumor Necrosis Factor Alpha Promotes Retinal Ganglion Cell Death in Glaucoma via Calcium-Permeable AMPA Receptor Activation. J Neurosci. 2015;35(35):12088–102. doi: 10.1523/JNEUROSCI.1273-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca AC, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C, et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362. doi: 10.3389/fncel.2014.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Khaw PT, Yin ZQ, Li D, Raisman G, Li Y. Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 2012;60(1):13–28. doi: 10.1002/glia.21242. [DOI] [PubMed] [Google Scholar]
- De Moraes CG, Liebmann JM, Ritch R. Predictive Factors Within the Optic Nerve Complex for Glaucoma Progression: Disc Hemorrhage and Parapapillary Atrophy. Asia Pac J Ophthalmol (Phila) 2012;1(2):105–12. doi: 10.1097/APO.0b013e31824a65f8. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ. The neurovascular unit, matrix proteases, and innate inflammation. Ann N Y Acad Sci. 2010;1207:46–9. doi: 10.1111/j.1749-6632.2010.05760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ. Aging and the neurovascular unit. Ann N Y Acad Sci. 2012;1268:127–33. doi: 10.1111/j.1749-6632.2012.06686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudevski I, Rostagno A, Cowman M, Liebmann J, Ritch R, Ghiso J. Clusterin and complement activation in exfoliation glaucoma. Invest Ophthalmol Vis Sci. 2014;55(4):2491–9. doi: 10.1167/iovs.13-12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drance SM. Disc hemorrhages in the glaucomas. Surv Ophthalmol. 1989;33(5):331–7. doi: 10.1016/0039-6257(89)90010-6. [DOI] [PubMed] [Google Scholar]
- Dubey SK, Hejtmancik JF, Krishnadas SR, Sharmila R, Haripriya A, Sundaresan P. Evaluation of Genetic Polymorphisms in Clusterin and Tumor Necrosis Factor-Alpha Genes in South Indian Individuals with Pseudoexfoliation Syndrome. Curr Eye Res. 2015:1–7. doi: 10.3109/02713683.2014.997884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Li X, Wang W, Mo JS, Kaplan H, Cooper NG. Early Involvement of Immune/Inflammatory Response Genes in Retinal Degeneration in DBA/2J Mice. Ophthalmol Eye Dis. 2010;1:23–41. doi: 10.4137/oed.s2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J, Piltz-Seymour JR. The significance of optic disc hemorrhages in glaucoma. J Glaucoma. 1997;6(1):62–4. [PubMed] [Google Scholar]
- Gramlich OW, Ding QJ, Zhu W, Cook A, Anderson MG, Kuehn MH. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol Commun. 2015;3:56. doi: 10.1186/s40478-015-0234-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber MC, Orgul S, Schoetzau A, Flammer J. Relationship between retinal glial cell activation in glaucoma and vascular dysregulation. J Glaucoma. 2007;16(2):215–9. doi: 10.1097/IJG.0b013e31802d045a. [DOI] [PubMed] [Google Scholar]
- Hostenbach S, Cambron M, D’haeseleer M, Kooijman R, De Keyser J. Astrocyte loss and astrogliosis in neuroinflammatory disorders. Neurosci Lett. 2014;565:39–41. doi: 10.1016/j.neulet.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, John SW. Mouse genetic models: an ideal system for understanding glaucomatous neurodegeneration and neuroprotection. Prog Brain Res. 2008;173:303–21. doi: 10.1016/S0079-6123(08)01122-9. [DOI] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011;121(4):1429–44. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, MacNicoll KH, Braine CE, Soto I, Macalinao DG, Sousa GL, et al. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol Dis. 2014;71:44–52. doi: 10.1016/j.nbd.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Ryan M, Graham LC, Smith RS, John SW. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J Neuroinflammation. 2013;10(1):76. doi: 10.1186/1742-2094-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Zhu X, Ryan M, Macalinao DG, Sousa GL, et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J Clin Invest. 2012;122(4):1246–61. doi: 10.1172/JCI61135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Parlier R, Shen JK, Lutty GA, Vinores SA. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS One. 2013;8(8):e71808. doi: 10.1371/journal.pone.0071808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Zhang SS, Zhang C. The two sides of cytokine signaling and glaucomatous optic neuropathy. J Ocul Biol Dis Infor. 2009;2(2):78–83. doi: 10.1007/s12177-009-9026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang YH, Kim YY, Kim HK, Sohn YH. Changes in retinal nerve fiber layer thickness after optic disc hemorrhage in glaucomatous eyes. J Glaucoma. 2014;23(8):547–52. doi: 10.1097/IJG.0000000000000083. [DOI] [PubMed] [Google Scholar]
- Jeoung JW, Park KH, Kim JM, Kang SH, Kang JH, Kim TW, et al. Optic disc hemorrhage may be associated with retinal nerve fiber loss in otherwise normal eyes. Ophthalmology. 2008;115(12):2132–40. doi: 10.1016/j.ophtha.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Johnson EC, Doser TA, Cepurna WO, Dyck JA, Jia L, Guo Y, et al. Cell proliferation and interleukin-6-type cytokine signaling are implicated by gene expression responses in early optic nerve head injury in rat glaucoma. Invest Ophthalmol Vis Sci. 2011;52(1):504–18. doi: 10.1167/iovs.10-5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EC, Jia L, Cepurna WO, Doser TA, Morrison JC. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2007;48(7):3161–77. doi: 10.1167/iovs.06-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Križaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, et al. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39(2):105–19. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MH. Immune phenomena in glaucoma and conformational disorders: why is the second eye not involved? J Glaucoma. 2014;23(8 Suppl 1):S59–61. doi: 10.1097/IJG.0000000000000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kur J, Newman EA, Chan-Ling T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog Retin Eye Res. 2012;31(5):377–406. doi: 10.1016/j.preteyeres.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurvinen L, Harju M, Saari J, Vesti E. Altered temporal peripapillary retinal flow in patients with disc hemorrhages. Graefes Arch Clin Exp Ophthalmol. 2010;248(12):1771–5. doi: 10.1007/s00417-010-1441-7. [DOI] [PubMed] [Google Scholar]
- Law SK, Choe R, Caprioli J. Optic disk characteristics before the occurrence of disk hemorrhage in glaucoma patients. Am J Ophthalmol. 2001;132(3):411–3. doi: 10.1016/s0002-9394(01)01009-1. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Kalev-Landoy M, Habot-Wilner Z, Melamed S. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol. 2006;124(4):520–6. doi: 10.1001/archopht.124.4.520. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Spierer O, Vander S, Dardik R. Similarities and differences between primary and secondary degeneration of the optic nerve and the effect of minocycline. Graefes Arch Clin Exp Ophthalmol. 2011;249(6):849–57. doi: 10.1007/s00417-010-1608-2. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Ilia P. Minocycline mechanism of neuroprotection involves the Bcl-2 gene family in optic nerve transection. Int J Neurosci. 2014a;124(10):755–61. doi: 10.3109/00207454.2013.878340. [DOI] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I. Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefes Arch Clin Exp Ophthalmol. 2014b;252(5):761–72. doi: 10.1007/s00417-014-2588-4. [DOI] [PubMed] [Google Scholar]
- Lin HJ, Tsai FJ, Chen WC, Shi YR, Hsu Y, Tsai SW. Association of tumour necrosis factor alpha −308 gene polymorphism with primary open-angle glaucoma in Chinese. Eye (Lond) 2003;17(1):31–4. doi: 10.1038/sj.eye.6700227. [DOI] [PubMed] [Google Scholar]
- Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci. 2010;51(11):5697–707. doi: 10.1167/iovs.10-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye-Barthel M, Sun D, Jakobs TC. Morphology of astrocytes in a glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2013;54(2):909–17. doi: 10.1167/iovs.12-10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Nair CE, Fernandes KA, Schlamp CL, Libby RT, Nickells RW. Tumor necrosis factor alpha has an early protective effect on retinal ganglion cells after optic nerve crush. J Neuroinflammation. 2014;11:194. doi: 10.1186/s12974-014-0194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EA, Davis CH, Bushong EA, Boassa D, Kim KY, Ellisman MH, et al. Astrocytes phagocytose focal dystrophies from shortening myelin segments in the optic nerve of Xenopus laevis at metamorphosis. Proc Natl Acad Sci U S A. 2015;112(33):10509–14. doi: 10.1073/pnas.1506486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossböck G, Weger M, Moray M, Renner W, Haller-Schober EM, Mattes D, et al. TNF-alpha promoter polymorphisms and primary open-angle glaucoma. Eye (Lond) 2006;20(9):1040–3. doi: 10.1038/sj.eye.6702078. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26(49):12633–41. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial responses in. Invest Ophthalmol Vis Sci. 2002;43(9):2962–8. [PubMed] [Google Scholar]
- Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch Ophthalmol. 1999;117(8):1050–6. doi: 10.1001/archopht.117.8.1050. [DOI] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente-Soriano FJ, et al. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A. 2011;108(3):1176–81. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–79. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- Nikolskaya T, Nikolsky Y, Serebryiskaya T, Zvereva S, Sviridov E, Dezso Z, et al. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med Genomics. 2009;2:24. doi: 10.1186/1755-8794-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overby DR, Bertrand J, Tektas OY, Boussommier-Calleja A, Schicht M, Ethier CR, et al. Ultrastructural changes associated with dexamethasone-induced ocular hypertension in mice. Invest Ophthalmol Vis Sci. 2014;55(8):4922–33. doi: 10.1167/iovs.14-14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena JD, Varela HJ, Ricard CS, Hernandez MR. Enhanced tenascin expression associated with reactive astrocytes in human optic nerve heads with primary open angle glaucoma. Exp Eye Res. 1999;68(1):29–40. doi: 10.1006/exer.1998.0577. [DOI] [PubMed] [Google Scholar]
- Qu J, Jakobs TC. The Time Course of Gene Expression during Reactive Gliosis in the Optic Nerve. PLoS One. 2013;8(6):e67094. doi: 10.1371/journal.pone.0067094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan VK, Morgan JT, Park SA, Weber D, Phinney BS, Murphy CJ, et al. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Invest Ophthalmol Vis Sci. 2015;56(8):4447–59. doi: 10.1167/iovs.15-16739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Danias J. A role for complement in glaucoma? Adv Exp Med Biol. 2010;703:95–104. doi: 10.1007/978-1-4419-5635-4_7. [DOI] [PubMed] [Google Scholar]
- Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 2012;7(7):e40065. doi: 10.1371/journal.pone.0040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989;491(2):394–7. doi: 10.1016/0006-8993(89)90078-4. [DOI] [PubMed] [Google Scholar]
- Schneider M, Fuchshofer R. The role of astrocytes in optic nerve head fibrosis in glaucoma. Exp Eye Res. 2015 doi: 10.1016/j.exer.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Seitz R, Ohlmann A, Tamm ER. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 2013;353(2):339–45. doi: 10.1007/s00441-013-1666-y. [DOI] [PubMed] [Google Scholar]
- Shibuya E, Meguro A, Ota M, Kashiwagi K, Mabuchi F, Iijima H, et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Invest Ophthalmol Vis Sci. 2008;49(10):4453–7. doi: 10.1167/iovs.07-1575. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Yamashima T, Agarwal N, Hara H. Neuroprotective effects of minocycline against in vitro and in vivo retinal ganglion cell damage. Brain Research. 2005;1053(1–2):185–94. doi: 10.1016/j.brainres.2005.06.053. [DOI] [PubMed] [Google Scholar]
- Siddique SS, Suelves AM, Baheti U, Foster CS. Glaucoma and uveitis. Surv Ophthalmol. 2013;58(1):1–10. doi: 10.1016/j.survophthal.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Soto I, Howell GR. The complex role of neuroinflammation in glaucoma. Cold Spring Harb Perspect Med. 2014;4(8) doi: 10.1101/cshperspect.a017269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasi K, Nagel D, Yang X, Wang RF, Ren L, Podos SM, et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2006;47(3):1024–9. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Xue QS. Human CNS immune senescence and neurodegeneration. Curr Opin Immunol. 2014;29:93–6. doi: 10.1016/j.coi.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Su W, Li Z, Jia Y, Zhuo Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS One. 2014;9(6):e99719. doi: 10.1371/journal.pone.0099719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Qu J, Jakobs TC. Reversible reactivity by optic nerve astrocytes. Glia. 2013;61(8):1218–35. doi: 10.1002/glia.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano Y, Shi D, Shimizu A, Funayama T, Mashima Y, Yasuda N, et al. Association of Toll-like receptor 4 gene polymorphisms in Japanese subjects with primary open-angle, normal-tension, and exfoliation glaucoma. Am J Ophthalmol. 2012;154(5):825–32.e1. doi: 10.1016/j.ajo.2012.03.050. [DOI] [PubMed] [Google Scholar]
- Taurone S, Ripandelli G, Pacella E, Bianchi E, Plateroti AM, De Vito S, et al. Potential regulatory molecules in the human trabecular meshwork of patients with glaucoma: immunohistochemical profile of a number of inflammatory cytokines. Mol Med Rep. 2015;11(2):1384–90. doi: 10.3892/mmr.2014.2772. [DOI] [PubMed] [Google Scholar]
- Tektas OY, Heinz C, Heiligenhaus A, Hammer CM, Luetjen-Drecoll E. Morphological changes of trabeculectomy specimens in different kinds of uveitic glaucoma. Curr Eye Res. 2011;36(5):442–8. doi: 10.3109/02713683.2011.566409. [DOI] [PubMed] [Google Scholar]
- Tezel G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res. 2008;173:409–21. doi: 10.1016/S0079-6123(08)01128-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004;15(2):80–4. doi: 10.1097/00055735-200404000-00003. [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X, Luo C, Kain AD, Powell DW, Kuehn MH, et al. Oxidative stress and the regulation of complement activation in human glaucoma. Invest Ophthalmol Vis Sci. 2010;51(10):5071–82. doi: 10.1167/iovs.10-5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler TA, Piltz-Seymour J. Optic disc hemorrhages in glaucoma and ocular hypertension: implications and recommendations. Curr Opin Ophthalmol. 2008;19(2):89–94. doi: 10.1097/ICU.0b013e3282f3e6bc. [DOI] [PubMed] [Google Scholar]
- Vohra R, Tsai JC, Kolko M. The role of inflammation in the pathogenesis of glaucoma. Surv Ophthalmol. 2013;58(4):311–20. doi: 10.1016/j.survophthal.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Wakefield D, Lloyd A. The role of cytokines in the pathogenesis of inflammatory eye disease. Cytokine. 1992;4(1):1–5. doi: 10.1016/1043-4666(92)90028-p. [DOI] [PubMed] [Google Scholar]
- Wallace DM, Murphy-Ullrich JE, Downs JC, O’Brien CJ. The role of matricellular proteins in glaucoma. Matrix Biol. 2014;37:174–82. doi: 10.1016/j.matbio.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Wallace DM, Pokrovskaya O, O’Brien CJ. The Function of Matricellular Proteins in the Lamina Cribrosa and Trabecular Meshwork in Glaucoma. J Ocul Pharmacol Ther. 2015;31(7):386–95. doi: 10.1089/jop.2014.0163. [DOI] [PubMed] [Google Scholar]
- Wang CY, Shen YC, Wei LC, Lin KH, Feng SC, Yang YY, et al. Polymorphism in the TNF-α(-863) locus associated with reduced risk of primary open angle glaucoma. Mol Vis. 2012;18:779–85. [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu L, Yang H. The prevalence of disc hemorrhage and papillary atrophy in Beijing Eye Study. Zhonghua Yi Xue Za Zhi. 2006;86(12):811–4. [PubMed] [Google Scholar]
- Ward RJ, Dexter DT, Crichton RR. Ageing, neuroinflammation and neurodegeneration. Front Biosci (Schol Ed) 2015;7:189–204. doi: 10.2741/S433. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp Eye Res. 2009;88(4):825–30. doi: 10.1016/j.exer.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Wiggs JL. Glaucoma Genes and Mechanisms. Prog Mol Biol Transl Sci. 2015;134:315–42. doi: 10.1016/bs.pmbts.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Huang P, Li W, Li Y, Zhang SS, Zhang C. T-helper1/T-helper2 cytokine imbalance in the iris of patients with glaucoma. PLoS One. 2015;10(3):e0122184. doi: 10.1371/journal.pone.0122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X, Gao L, Wu T, Sun F. Roles of tumor necrosis factor alpha gene polymorphisms, tumor necrosis factor alpha level in aqueous humor, and the risks of open angle glaucoma: a meta-analysis. Mol Vis. 2013;19:526–35. [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28(5):348–68. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Yang J, Patil RV, Yu H, Gordon M, Wax MB. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol. 2001;131(4):421–6. doi: 10.1016/s0002-9394(00)00862-x. [DOI] [PubMed] [Google Scholar]
- Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, et al. Neurodegenerative and inflammatory pathway components linked to TNF-α/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. 2011;52(11):8442–54. doi: 10.1167/iovs.11-8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000;32(1):42–50. [PubMed] [Google Scholar]
- Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. 2001;64(5):523–32. doi: 10.1002/jnr.1104. [DOI] [PubMed] [Google Scholar]