

Abstract
It has been suggested that β2‐adrenergic receptor (β2‐AR)‐mediated signaling induced by catecholamines regulates the degradation of p53. However, the underlying molecular mechanisms were not known. In the present study, we demonstrated that catecholamines upregulated the expression of silent information regulator 1 (Sirt1) through activating β2‐AR‐mediated signaling pathway, since selective β2‐AR antagonist ICI 118, 551 and non‐selective β‐blocker proprenolol effectively repressed isoproterenol (ISO)‐induced Sirt1 expression. Catecholamines inhibited doxorubicin (DOX)‐induced p53 acetylation and transcription‐activation activities by inducing the expression of Sirt1. Knockdown of the Sirt1 expression by the specific siRNA remarkably blocked the inhibitory effects of ISO on DOX‐induced p53 acetylation. In addition, we demonstrated that catecholamines induced resistance of cervical cancer cells to chemotherapeutics both in vitro and in vivo and that β2‐AR was overexpressed in cervical cancer tissues. Our data suggest that the p53‐dependent, chemotherapeutics‐induced cytotoxicity in cervical cancer cells may be compromised by catecholamines‐induced upregulation of the Sirt1 expression through activating the β2‐AR signaling.
Keywords: Cervical cancer, chemoresistance, p53, Sirt1, β2‐AR
Cervical cancer is a primary cancer in females worldwide, with an estimated global incidence of approximately 500 000 new cases and approximately 250 000 deaths each year.1 Over the last few decades, concurrent chemoradiotherapy has been considered as the standard treatment for the patients with locally advanced cervical cancer. Neoadjuvant chemotherapy in combination with radical surgery has also been used as an alternative treatment for locally advanced cervical cancer.2 The efficacies of the nonspecific DNA damaging chemo‐ and radiotherapies in many instances are principally dependent on the functions of the p53 tumor suppressor.3 However, locally advanced cervical cancer is extremely difficult to treat and prognosis of the patients is poor. The leading cause of cervical cancer is latent human papillomavirus (HPV) infection. An important role of HPV in cervical carcinogenesis is functional inactivation of p53 by high‐risk HPV E6 protein through ubiquitin‐mediated proteasomal degradation.4 Inactivation of p53, by either mutations or degradation, may contribute to resistance to chemo‐ and radiotherapies.5
The p53 protein is a short‐lived protein and is normally maintained at low levels through the MDM2‐mediated ubiquitination and degradation pathway in unstressed mammalian cells. In response to various cellular stresses, such as DNA damage, p53 is transiently stabilized and activated, thereby regulating the expression of a wide variety of genes involved in apoptosis and growth arrest.6 The stability, DNA‐binding activity, and transcriptional activity of p53 are critically regulated by post‐translational modifications, including phosphorylation, acetylation, and sumoylation. Particularly, the acetylation of p53 plays an important role in stabilization and transcriptional activation of p53.7
Multiple lines of evidence indicate that chronic stress promotes tumor development and progression. Stress‐related neurotransmitters and hormones, such as norepinephrine (NE), epinephrine (EPI), and glucocorticoids (GCs), influence biobehaviors of tumor cells by modulating the expression of a variety of genes.8, 9 Several recent studies including ours demonstrate that stress‐related hormones affect the p53 functions.10 Our previous study shows that GCs upregulate HPV E6 protein, which downregulate p53 and p53‐dependent miR‐145 expression, resulting in the chemoresistance of cervical cancer cells.11 The study by Feng et al.10 demonstrates that chronic stress decreases p53 protein levels through the induction of serum‐ and glucocorticoid‐induced protein kinase 1 (SGK1), which in turn increases the MDM2 activity and attenuates the p53 function.
Modulation of the p53 activity by catecholamines (such as NE and EPI) was also reported recently. It was suggested that β2‐adrenergic receptor (β2‐AR)‐dependent signaling induced by catecholamines regulates the degradation of p53.12 In the present study, we demonstrate that catecholamines upregulate the expression of silent information regulator 1 (Sirt1), a member of the histone deacetylase family through activating β2‐AR and inhibits chemotherapeutics‐induced acetylation of p53, conferring chemoresistance of cervical cancer cells.
Materials and Methods
Cell culture and treatment
Human cervical cancer cell lines SiHa, CaSki, and HeLa are obtained from the American Type Culture Collection. For the treatment with the β2‐AR agonists, the cells were incubated overnight in a serum‐free medium and then treated with 0.625, 1.25, 2.5, 5 or 10 μM isoproterenol (ISO; Sigma, MO, USA) or EPI (Sigma) for the indicated time points. For the treatment with the β‐AR antagonists, the cells were treated with 10 μM propranolol (TOCRIS, Bristol, UK), 1 μM ICI 118,551 (TOCRIS) or 1 μM ATEN (TOCRIS) for 1 h before the β‐AR agonist stimulation.
Western blot
The following antibodies were used for immunoblotting: the antibodies against Sirt1 (Santa Cruz, CA, USA), p53 (Santa Cruz), acetylated‐p53 (Abcam, MA, USA), c‐Myc (Cell Signaling, MA, USA), caspase‐3 (Cell Signaling), caspase‐7 (Cell Signaling), and β‐actin (Sungene Biotech, Tianjin, China). All experiments were performed in duplicate.
Real‐time RT‐PCR
The Sirt1 mRNA expression was analyzed by real‐time RT‐PCR using the comparative threshold cycle method with β‐actin as an internal control. The experiments were performed three times independently.
Transient transfection
SiHa and CaSki cells were transfected with control Sirt1, c‐Myc or siRNA using Lipofectamine RNAiMAX (Invitrogen, CA, USA) according to the manufacturer's instructions. After transfection for 60 h, the cells were starved overnight and then treated with 2.5 μM ISO, 0.5 μM doxorubicin (DOX), or both.
Immunofluoresence microscopy
SiHa cells were starved overnight and then treated with 2.5 μM ISO and 0.5 μM DOX. After treatment for 48 h, the cells were washed with cold phosphate‐buffered saline (PBS) and fixed in 4% paraformaldehyde at 4°C for 10 min. Then cells were permeablized with methanol at −20°C for 10 min. After washing with PBS, the cells were blocked with 5% goat serum in PBS for 1 h, incubated with the mouse monoclonal antibody against Sirt1 and rabbit monoclonal antibody against acetylated p53 at 4°C overnight, and rinsed with PBS. The binding was detected by the green fluorescent AlexaFluor 488 conjugated to the goat anti‐mouse antibody (Invitrogen) and the red fluorescent AlexaFluor 594 conjugated to the goat anti‐rabbit antibody (Invitrogen) for 1 h. After washing with PBS, the cells were treated with the solution containing 1 μg/mL of DAPI (Sigma). The expression of Sirt1 and acetylated p53 were observed under a laser scanning confocal microscope (RADIANCE 2100; BioRad, CA, USA).
Apoptosis assay
To determine the effect of ISO on DOX‐induced apoptosis in cervical cancer cells, SiHa cells were treated with 0.5 μM DOX, 2.5 μM ISO or both for 48 h. Cell apoptosis was detected using Annexin V‐FITC detection kit (Sungene Biotech). The experiments were performed in duplicate.
Cell proliferation assay
SiHa, HeLa, and CaSki cells were cultured in 96‐well plates with an initial cell density of 3.6 × 103/well and then treated with β‐AR agonist (ISO or EPI), chemotherapy drugs [DOX or cis‐Dichlorodiamineplatinum (DDP)], or both. The cell proliferation activities were determined by CCK8 assays following the manufacturer's instructions. The experiments were performed in duplicate.
Luciferase assay
SiHa and CaSki cells were co‐transfected with the control siRNA or Sirt1 siRNA, p53 reporter (Promega, WI, USA) and pRL‐TK reporter plasmids using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. After transfection for 48 h, the cells were treated with 0.5 μM DOX, 2.5 μM ISO, or both. Then, the cells were lysed in the lysis buffer (Promega). Firefly and Renilla luciferase activities were measured with a dual luciferase assay kit (Promega) according to the manufacturer's instructions. The experiments were carried out in triplicate.
In vivo tumor model
Five to 6‐week‐old athymic female Balb/C nude mice were purchased from Beijing Vital River Laboratory Animal Technology. The mice were divided into two groups randomly and each group contained eight mice. The mice received PBS or ISO (10 mg/kg; Sigma) by intraperitoneal injections, commencing 2 day before inoculation of tumor cells. A total of 0.1 mL SiHa cell suspension (5 × 107 cells/mL) was injected subcutaneously in the upper right flank of the mice. DOX treatment was started 5 day after implantation of SiHa cells. The mice treated with PBS were further divided into two groups (four mice per group) randomly and DOX (10 mg/kg) or PBS was given to mice intravenously twice a week for 4 weeks. The mice treated with ISO were further grouped (four mice per group) randomly and cotreated with DOX (10 mg/kg, intravenous injection twice a week) and ISO (10 mg/kg, intraperitoneal injection daily) or PBS (intravenous injection twice a week) and ISO (10 mg/kg, intraperitoneal injection daily). At the end of the experiments, the mice were sacrificed and the primary tumors were dissected, weighted, and photographed.
Investigation was conducted in accordance with the ethical standards and national and international guidelines. The experiments were approved by the Ethics Committee of Institute of Basic Medical Sciences.
Immunohistochemistry
Immunohistochemical staining was performed as previously described.13 The expression of β2‐AR in cervical cancer tissue samples was analyzed by the rabbit polyclonal antibodies against β2‐AR (Abcam) and horseradish peroxidase‐labeled secondary antibody. Images were taken on an Olympus BX51 microscope (Olympus, Tokyo, Japan) using the Spot insight image capture system CCD camera.
Statistical analysis
Data were expressed as mean ± SD. Paired data were evaluated by Student's t‐test. For comparisons between multiple groups, one‐way anova followed by the Bonferroni post hoc test was used. A P < 0.05 was considered statistically significant.
Results
Catecholamines upregulate Sirt1 expression at both mRNA and protein levels
It was hypothesized that stress‐induced β‐adrenergic signaling might regulate the activity of Sirt1, which appears to control the cellular response to stress by deacetylation of some factors. We treated cervical cancer cell lines SiHa and CaSki with catecholamines (10 μM ISO or 1 μM EPI). Then the cells were harvested and lyzed. The expression of Sirt1 and c‐Myc was analyzed by Western blot after treatment for 0, 1, 3, 6, 9, and 12 h, respectively. The data in Figure 1(a–d) show that ISO and EPI upregulated the expression of Sirt1 in a time‐dependent manner. ISO stimulation also exhibited a dose‐dependent effect on upregulation of the Sirt1 expression (Fig. 1e,f).
Figure 1.
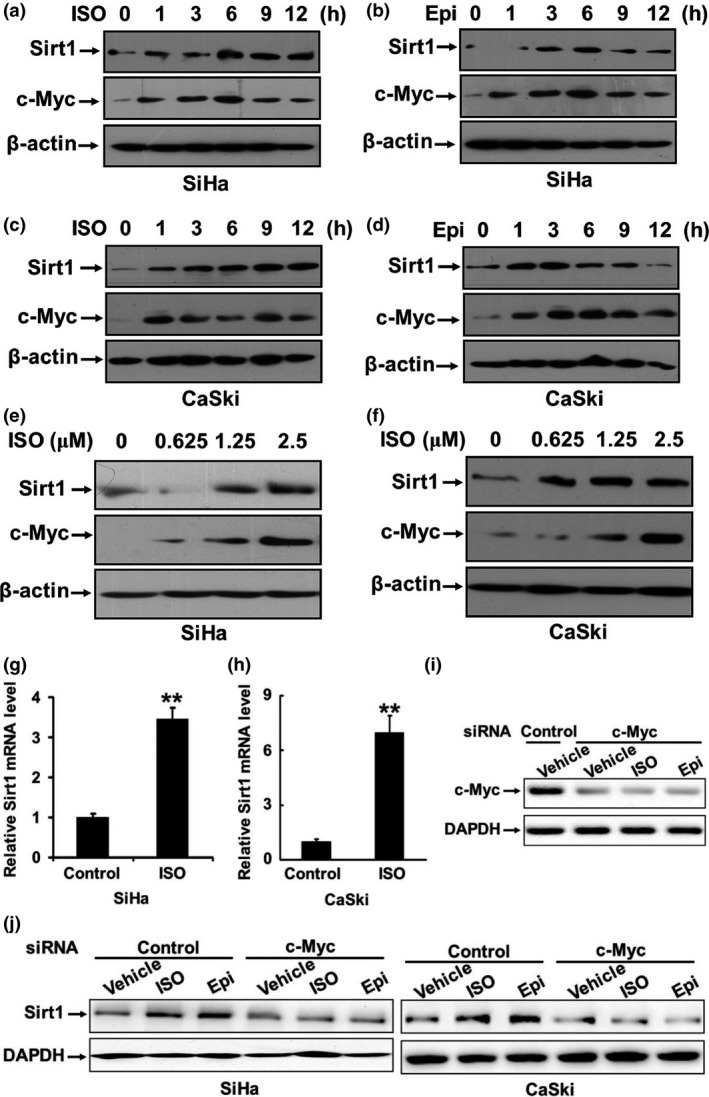
Catecholamines upregulate Sirt1 expression at both mRNA and protein levels. (a–d) SiHa and CaSki cells were starved overnight and then treated with 10 μM ISO or 1 μM EPI. The cells were harvested at the indicated time points. The expressions of Sirt1 and c‐Myc in the cell lysates were analyzed by Western blot. (e, f) SiHa and CaSki cells were treated with 0, 0.625, 1.25, 2.5, 5, or 10 μM ISO for 3 h. The expressions of Sirt1 and c‐Myc were analyzed by Western blot. (g, h) The total RNA was isolated from SiHa and CaSki cells that were treated with 10 μM ISO. The expression of the Sirt1 mRNA was assessed by real‐time RT‐PCR. (i) CaSki cells were transfected with the specific siRNA targeting c‐Myc or control siRNA. After transfection for 24 h, the cells were treated with ISO, EPI, or vehicle for 3 h. The expression of c‐Myc was analyzed by Western blot. (j) CaSki and SiHa cells were transfected with the specific siRNA targeting c‐Myc or control siRNA. After transfection for 24 h, the cells were treated with ISO, EPI, or vehicle for 3 h. The expression of Sirt1 was analyzed. **P < 0.01
To investigate whether upregulation of Sirt1 occurs at the transcription level, we isolated the total RNA from the SiHa and CaSki cells that were treated with 10 μM ISO and assessed the expression of the Sirt1 mRNA by real‐time RT‐PCR. As displayed in Figure 1(g,h), ISO stimulation resulted in a dramatic increase in the expression of Sirt1 at the mRNA level.
The previous study has demonstrated that c‐Myc binds to the Sirt1 promoter and regulates the Sirt1 expression at the transcriptional level.14 It was found that the expression of Sirt1 positively correlated with the level of c‐Myc in human hepatocellular carcinoma and colorectal cancer.15, 16 We observed that catecholamines induced the c‐Myc expression in a time‐ and dose‐dependent manner (Fig. 1a–f). Knockdown of c‐Myc by specific siRNA remarkably inhibited the upregulation of Sirt1 induced by ISO or EPI (Fig. 1i,j), suggesting that catecholamines may upregulate the Sirt1 transcription through promoting the expression of c‐Myc.
Catecholamines upregulate Sirt1 expression via activating of β2‐AR signaling pathway
It is known that the effects of catecholamines on the bio‐behaviors of tumor cells are mainly mediated through activation of β2‐AR. To determine whether the β2‐AR pathway is involved in catecholamine‐induced upregulation of the Sirt1 expression, SiHa and CaSki cells were pretreated with β2‐AR antagonist ICI 118, 551, β1‐AR inhibitor ATEN, or sympatholytic non‐selective beta blocker propranolol for 1 h and then treated with 2.5 μM ISO for 3 h. Figure 2 shows that these inhibitors effectively repressed ISO‐induced Sirt1 expression. However, the effect of ICI 118, 551 and proprenolol appeared to be stronger. The data indicate that catecholamines upregulate the Sirt1 expression mainly via activating the β2‐AR signaling pathway.
Figure 2.
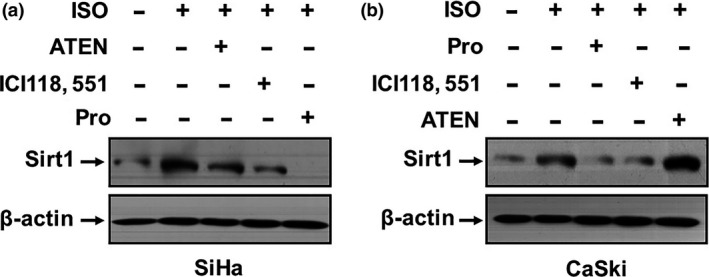
Catecholamines upregulate Sirt1 expression via activating of β2‐AR signaling pathway. (a) SiHa and (b) CaSki cells were pretreated with 1 μM ICI 118, 551, 1 μM ATEN, or 10 μM propranolol for 1 h and then with 2.5 μM ISO for 3 h. The expression of Sirt1 was analyzed by Western blot.
Catecholamines inhibit DOX‐induced p53 acetylation and transcription‐activation activities
Sirt1 is known as a NAD‐dependent deacetylase. The tumor suppressor p53 is the first non‐histone deacetylation target identified for Sirt1. The studies implicate that Sirt1 inactivates p53 via deacetylation in tumor cells, leading to downregulation of the p53 target genes.17, 18 To investigate whether upregulation of Sirt1 by catecholamines affects the p53 acetylation, we treated SiHa and CaSki cells, which express wildtype p53, with 0.5 μM DOX, an inducer of DNA double‐strand break, which causes p53 activation, for 18 h. Then the cells were stimulated with 2.5 μM ISO for 6 h. The data show that DOX significantly induced the expression and acetylation of p53. However, the effects of DOX on p53 were remarkably abrogated in the presence of ISO, whereas the expression of c‐Myc was markedly upregulated, especially in CaSki cells (Fig. 3a,b). We noticed that the expression of c‐Myc was also upregulated and Sirt1 slightly induced by DOX in SiHa cells. Ngan et al.19 reported that the level of c‐Myc was significantly associated with p53 in cervical cancer tissues. Our previous study also demonstrated that p53 overexpression resulted in elevation of c‐Myc in cervical cancer cells.11 It has been documented that c‐Myc and Sirt1 form a positive feedback loop, it may explain the upregulation of c‐Myc and Sirt1 by DOX in SiHa cells.20 Nevertheless, the expression and acetylation of p53 induced by DOX was clearly inhibited in the presence of ISO in both SiHa and CaSki cells.
Figure 3.
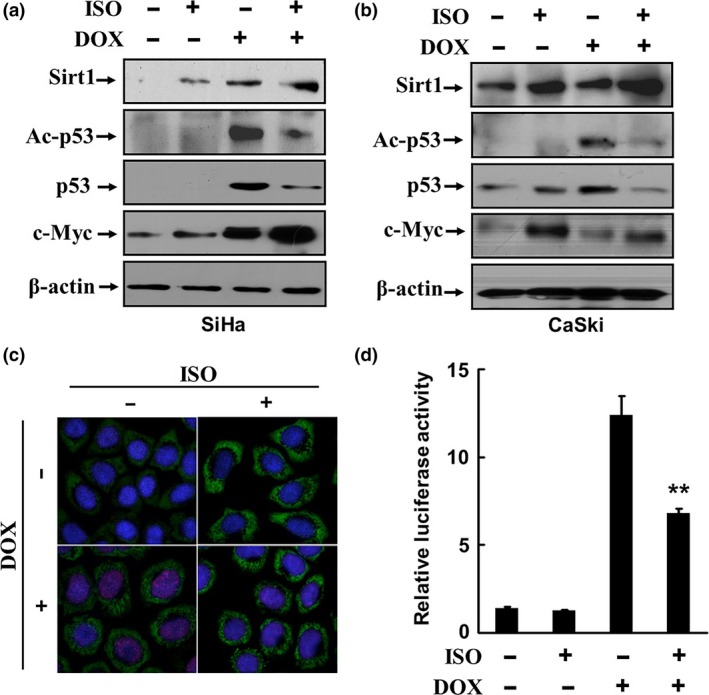
Catecholamines inhibit DOX‐induced p53 acetylation and transcription‐activation activities. SiHa and CaSki cells were treated with 0.5 μM DOX for 18 h and then with 2.5 μM ISO for 6 h. The expressions of Sirt1, c‐Myc, p53, and acetylated p53 were analyzed by Western blot (a, b). The expression of Sirt1 (green) and acetylation of p53 (red) in SiHa cells was detected by indirect immunofluorescence staining and cofocal microscopy (c). (d) SiHa cells were transfected with a luciferase reporter plasmid containing consensus p53 DNA binding sites and thymidine kinase promoter‐Renilla luciferase reporter plasmid. The transfected cells were treated with 0.5 μM DOX for 18 h and then stimulated with 2.5 μM ISO for 6 h. The activities of p53 were determined by luciferase assays. **P < 0.01, compared to the cells treated with DOX alone.
To further confirm the data, the expression of Sirt1 and acetylation of p53 in SiHa cells was detected using indirect immunofluorescence and cofocal microscopy. Figure 3(c) shows that the Sirt1 expression (green) was detectable in the cytoplasm, but acetylated p53 (red) was absent in the control cells. Treatment with 0.5 μM DOX resulted in a significant induction of p53 acetylation, as the nuclei were prominently stained in red. However, in the presence of ISO, the Sirt1 expression was significantly enhanced, but acetylated p53 almost disappeared.
To examine whether catecholamines affect the transcription‐activation activities of p53, SiHa cells were transfected with a luciferase reporter plasmid containing consensus p53 DNA binding sites and thymidine kinase promoter‐Renilla luciferase reporter plasmid. The transfected cells were treated with 0.5 μM DOX for 18 h and then stimulated with 2.5 μM ISO for 6 h. The activities of p53 were determined by luciferase assays. As shown in Figure 3(d), the treatment with DOX induced a remarkable increase of the p53 activities. However, the luciferase activities were dramatically decreased by approximately 50%, when ISO was added. These data suggest that upregulation of the Sirt1 expression induced by the adrenergic signaling plays a critical role in p53 acetylation and activation in response to DOX.
Catecholamines inhibit DOX‐induced p53 acetylation and transcription‐activation activities by upregulating Sirt1
To verify the role of Sirt1 in the repression of DOX‐induced p53 acetylation and activation by Sirt1, SiHa cells were transfected with either control siRNA or siRNA against Sirt1. Then the transfected cells were cotreated with 0.5 μM DOX and 2.5 μM ISO. Successful Sirt1 knockdown was confirmed by Western blot analysis (Fig. 4a,b). We observed that ISO potently inhibited DOX‐induced p53 acetylation, accompanied by marked upregulation of the Sirt1 expression in control cells. Treatment with the specific siRNA against Sirt1 completely abolished ISO‐induced Sirt1 expression and remarkably blocked the inhibitory effects of ISO on DOX‐induced p53 acetylation. We further determined the effect of ISO on the p53 transcription activity, which is important for p53‐dependent functions. SiHa and CaSki cells were treated with control or Sirt1 siRNA. After 24 h of incubation, the cells were cotransfected with p53 luciferase reporter plasmid. Then 0.5 μM DOX with or without 2.5 μM ISO was added to the transfected cells. Figure 4(c,d) demonstrate that ISO significantly inhibited DOX‐induced p53 activities, but silencing of the Sirt1 expression remarkably blocked the inhibitory effect of ISO on p53 transcription activation in both SiHa and CaSki cells. Together, these results confirm that catecholamines upregulate Sirt1, leading to suppression of the p53 acetylation and transcription activation activity.
Figure 4.
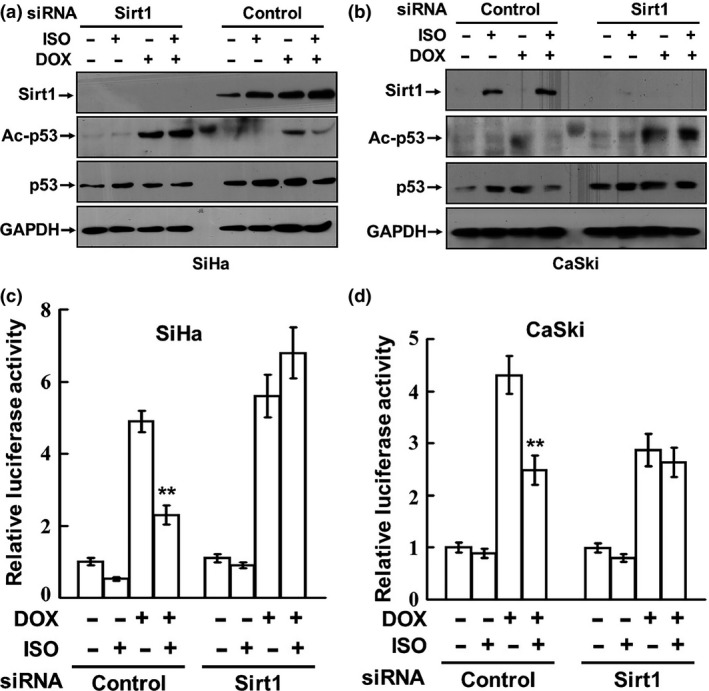
Catecholamines inhibit DOX‐induced p53 acetylation and transcription‐activation activities by upregulating Sirt1. (a, b) SiHa and CaSki cells were transfected with either control siRNA or siRNA against Sirt1. Then the transfected cells were cotreated with 0.5 μM DOX and 2.5 μM ISO. The expressions of Sirt1, p53, and acetylated p53 were analyzed by Western blot. (c, d) SiHa and CaSki cells were treated with control or Sirt1 siRNA. After 24 h of incubation, the cells were cotransfected with p53 luciferase reporter plasmid and thymidine kinase promoter‐Renilla luciferase reporter plasmid. Then 0.5 μM DOX with or without 2.5 μM ISO was added to the transfected cells. The luciferase activities were measured with a dual luciferase assay kit. **P < 0.01, compared to the cells treated with DOX alone.
Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro and in vivo
p53 is a very potent apoptosis inducer. Once activated in response to DNA‐damaging chemotherapeutic drugs, p53 triggers tumor cell apoptotic program. DOX induces cytotoxic activity in tumor cells mostly via p53‐dependent apoptosis. A functional p53 signaling pathway is critical for sensitizing tumor cells to DNA‐damaging agents.21 We observed that SiHa cells were very sensitive to the treatment of DOX and the rate of apoptosis was over 50% after drug administration. However, ISO prominently attenuated DOX‐induced cellular apoptosis, as determined by annexin V/FITC binding (Fig. S1a). A significant increase in cleavage of full‐length caspase‐3 and caspase‐7 was detected after treatment with DOX (Fig. S1b). However, DOX‐induced activation of caspase‐3 was remarkably inhibited by ISO or EPI. Treatment with DOX or DDP effectively inhibited the proliferation of SiHa cells, whereas catecholamines greatly antagonized the inhibitory effect of chemotherapeutic drugs, as determined by CCK8 assays (Fig. 5a–e). Similar data were obtained in HeLa and CaSki cells (Fig. S2). These data indicate that the catecholamines impair the sensitivity of cervical cancer cells to chemotherapeutics.
Figure 5.
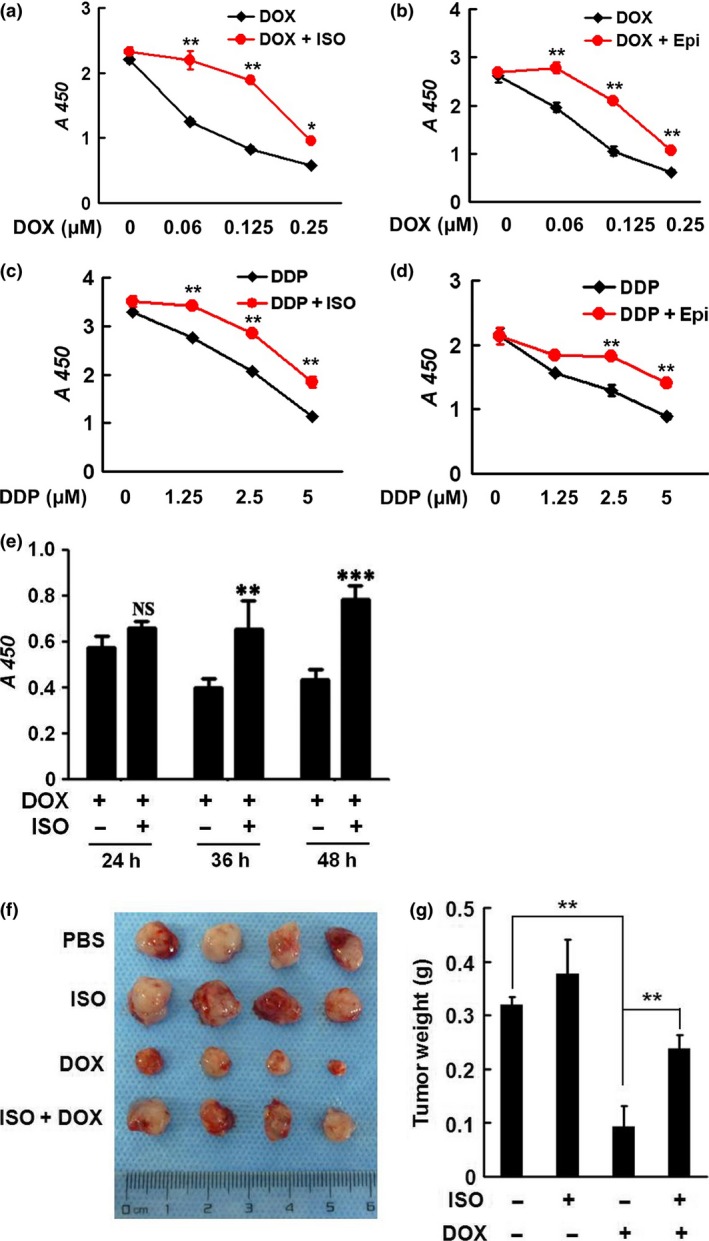
Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro and in vivo. (a–d) SiHa cells were cotreated with 2.5 μM ISO or 1 μM EPI and DOX or DDP. The cell proliferation activities were determined by CCK8 assays. (e) SiHa cells were treated with 0.5 μM DOX or 2.5 μM ISO. After treatment for 24, 36 or 48 h, the cell proliferative activities were determined by CCK8 assays. (f, g) Five to 6‐week‐old athymic female Balb/C nude mice received PBS or ISO (10 mg/kg) by intraperitoneal injections, commencing 2 day before inoculation of tumor cells. A total of 0.1 mL SiHa cell suspension (5 × 107 cells/mL) was injected subcutaneously in the upper right flank of the mice. DOX treatment (10 mg/kg) was started 5 day after implantation of SiHa cells. At the end of the experiments (4 weeks), the mice were sacrificed and the primary tumors were dissected, weighted, and photographed. **P < 0.01
To further confirm the antagonizing effects of catecholamines on chemotherapeutics, we established SiHa xenografts in nude mice and treated the mice with PBS or ISO (10 mg/kg) daily. When the tumors were palpable, the mice were administered with DOX (0.2 mg) once every 3 days in the presence or absence of ISO for 4 weeks. Figure 5(e,f) demonstrate that ISO significantly impaired the anti‐tumor efficacy of DOX in mice. The results confirm that the β2‐AR agonist confers resistance of cervical cancer cells to DOX in vivo.
β2‐AR is overexpressed in cervical cancer tissues
We collected 36 paraffin embedded cervical cancer tissue samples and analyzed the expression of β2‐AR by immunohistochemical staining. No staining was observed in the absence of the primary antibodies or with nonspecific immunoglobulin. In adjacent noncancerous tissues, the expression of β2‐AR was either weak or absent (Fig. 6a). However, the staining intensity was much stronger in 23/36 cervical cancer tissues (Fig. 6b), implicating that overexpression of β2‐AR may protect cervical cancer cells from chemotherapeutics‐induced cytotoxicity by mediating catecholamine signaling.
Figure 6.
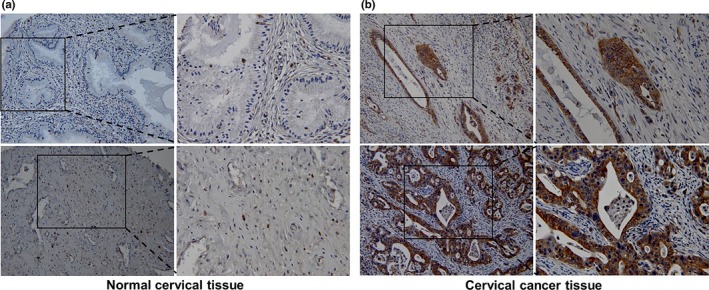
β2‐AR was overexpressed in cervical cancer tissues. Thirty‐six paraffin embedded cervical cancer tissue samples were collected. The expression of β2‐AR was analyzed by immunohistochemical staining.
Discussion
Sirt1 belongs to the family of type III histone deacetylase. The role of Sirt1 in tumorigenesis is highly controversial. It was originally considered as a tumor promoter, since it could repress p53‐dependent apoptosis in response to DNA damage by interacting with and deacetylating p53.22 Sirt1 was upregulated in several tumor cell lines and human cancers, such as acute myeloid leukemia and colorectal adenocarcinoma.23, 24 Upregulation of Sirt1 was associated with tumorigenesis, poor prognosis, and resistance to certain chemotherapeutic agents. However, the opposing roles regarding the tumor suppressing capability of Sirt1 were also reported. For example, Sirt1 was found to suppress the effect of TGF‐β signaling on MMP7 transcription through deacetylation of TGF‐β‐activated transcription factor Smad4 in oral squamous cell carcinoma cells, leading to reduced migration and metastasis of the tumor cells.25, 26 It was also shown that Sirt1 suppressed intestinal tumor formation in vivo. In ovarian cancer, the Sirt1 expression was correlated with a favorable prognosis.27 These conflicting roles in tumor promotion and progression for Sirt1 are likely due to complexity, diversity, and differential expression of the Sirt1 substrates in different type of cancer cells.28
In the present study, we show that catecholamines upregulate the expression of Sirt1 at both transcription and translation levels in cervical cancer cells. Activation of the β2‐AR pathway is involved in catecholamine‐induced elevation of the Sirt1 expression, because selective β2‐AR antagonist strongly abrogated the effect of catecholamines on the Sirt1 expression. The acetylase activity of Sirt1 can be self‐regulated. The Sirt1 C‐terminal region (aa 631–635) interacts with Sirt1 catalytic core domain.29 The N‐terminus (aa 190–244) of Sirt1 mediates allosteric activators for the Sirt1 catalytic core function. Therefore, maintenance of the Sirt1 activity partially relies on the expression level of Sirt1. When the cervical cancer cells were treated with DOX that induces DNA damage by intercalation of DNA double helices and inhibition of DNA‐topoisomerase I and II, the expression and acetylation of p53 were significantly induced. DNA damage is well known to activate p53‐mediated transcription through post‐translational mechanisms.21 Sirt1 functions as an NAD‐dependent p53 deacetylase and deacetylation impairs the function of p53. It was reported that Sirt1 repressed genotoxic stress‐induced p53 activation in acute myeloid leukemia.22 We observed that catecholamine stimulation remarkably inhibited DOX‐induced p53 expression and acetylation, concomitant with the upregulation of Sirt1 and c‐Myc. Several recent publications suggest the existence of a positive feedback loop between c‐Myc and Sirt1. It was suggested that c‐Myc transactivate the Sirt1 gene, resulting in inhibition of the p53 function. However, a negative feedback mechanism for the regulation of c‐Myc activity by Sirt1 was also documented. It has been demonstrated that c‐Myc and p53 generally function antagonistically toward each other.14, 20 We noticed that upregulation of Sirt1 in response to catecholamine stimulation was accompanied by elevation of the c‐Myc level and downregulation and deacetylation of p53, implicating that catecholamine may influence the expressions and functions c‐Myc and p53 simultaneously via induction of Sirt1. The deregulation of c‐Myc, which is found in approximately 50% of all human cancers, and inactivation of p53 are associated with stimulation of cellular proliferation and blockage of apoptosis, leading to resistance of cervical cancer cells to chemotherapeutics.
Inactivation of p53 is thought to be a major predictor of resistance to radiotherapy and chemotherapy in various human cancers. It was suggested that p53 promoted cellular apoptosis by transcriptionally activating a set of proapoptotic genes and repressing antiapoptotic genes in response to DNA‐damaging chemotherapeutics.21 We observed that Sirt1 upregulation induced by catecholamines not only inhibited the p53 acetylation, but also remarkably attenuated cellular apoptosis in response to DOX and antagonized the anti‐proliferative activities of chemotherapeutics. Knockdown of the Sirt1 expression effectively blocked the inhibitory effect of ISO on DOX‐induced p53 acetylation and transcription activity. The data suggest that catecholamines confer resistance of cervical cancer cells to chemotherapeutics by upregulating the Sirt1 expression and inhibiting the p53 functions.
We found that the expression of β2‐AR was very low or undetectable in noncancerous tissues but high in 23/36 cervical cancer tissues. The overexpression of β2‐AR may mediate enhanced catecholamine signaling and impair the sensitivity of cervical cancer cells to chemotherapeutics. DOX, an anthracycline antibiotic, is widely used for the treatment of a variety of cancers including cervical cancer. However, resistance is a major hurdle in successful treatment of cervical cancer. The previous study suggested that Sirt1 overexpression or treatment with Sirt1 activators reduced the anti‐proliferation activity of DOX and that inhibition of the Sirt1 signaling sensitized the antitumor activity of silybin against human lung adenocarcinoma cells.30, 31 Given that β2‐AR is overexpressed in tumor tissues and that the level of catecholamines is high in tumor microenvironment, the p53 transcription activation activity and p53‐dependent, chemotherapeutics‐induced apoptosis may be compromised in tumor. Thus, the aberrant expression of β2‐AR in cervical cancer may be a candidate biomarker for predicting the sensitivity to chemotherapeutic agents in clinical practice. Pharmacological intervention of the β2‐AR signaling pathway may be a potential strategy to rescue the function p53, enhance the efficacy of chemotherapeutics, and improve survival of cervical cancer patients.
Together, our findings demonstrate that catecholamine‐induced β‐adrenergic signaling upregulates the expression of Sirt1, resulting in deacetylation of p53 and inhibition of p53 transcriptional activity and DOX‐induced cytotoxicity in cervical cancer cells.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1.Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro.
Fig. S2.Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro.
Acknowledgments
National Key Technologies R&D Program for New Drugs (2013ZX09102056); National High‐Tech Research and Development Plan (863 Program, No. 2014AA020604); National Natural Science Foundation of China (No. 31370825, 81272232, 81402562, 81572845, and 31500702); Beijing Natural Science Foundation (No. 7162144, 7132163 and 7122124); China Postdoctoral Science Foundation (No. 2015T81095).
Cancer Sci 108 (2017) 1310–1317
Funding information
National Key Technologies R&D Program for New Drugs (2013ZX09102056); National High‐Tech Research and Development Plan (2014AA020604); National Natural Science Foundation of China (31370825, 81272232, 81402562, 81572845, and 31500702); Beijing Natural Science Foundation (7162144, 7132163, 7122124); China Postdoctoral Science Foundation (2015T81095).
Contributor Information
Ning Guo, Email: ning_guo@sina.com.
Ming Shi, Email: sm200@sohu.com.
References
- 1. Ginsburg O, Bray F, Coleman MP et al The global burden of women's cancers: a grand challenge in global health. Lancet 2016; 389: 847–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lapresa M, Parma G, Portuesi R, Colombo N. Neoadjuvant chemotherapy in cervical cancer: an update. Expert Rev Anticancer Ther 2015; 15: 1171–81. [DOI] [PubMed] [Google Scholar]
- 3. Beckta JM, Ahmad SF, Yang H, Valerie K. Revisiting p53 for cancer‐specific chemo‐ and radiotherapy: ten years after. Cell Cycle 2014; 13: 710–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narisawa‐Saito M, Kiyono T. Basic mechanisms of high‐risk human papillomavirus‐induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci 2007; 98: 1505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ding M, Zhang E, He R, Wang X. Newly developed strategies for improving sensitivity to radiation by targeting signal pathways in cancer therapy. Cancer Sci 2013; 104: 1401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duffy MJ, Synnott NC, McGowan PM et al p53 as a target for the treatment of cancer. Cancer Treat Rev 2014; 40: 1153–60. [DOI] [PubMed] [Google Scholar]
- 7. Reed SM, Quelle DE. p53 acetylation: regulation and consequences. Cancers 2014; 7: 30–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cole SW, Nagaraja AS, Lutgendorf SK et al Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 2015; 15: 563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi M, Liu D, Yang Z, Guo N. Central and peripheral nervous systems: master controllers in cancer metastasis. Cancer Metastasis Rev 2013; 32: 603–21. [DOI] [PubMed] [Google Scholar]
- 10. Feng Z, Liu L, Zhang C et al Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Natl Acad Sci USA 2012; 109: 7013–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi M, Du L, Liu D et al Glucocorticoid regulation of a novel HPV‐E6‐p53‐miR‐145 pathway modulates invasion and therapy resistance of cervical cancer cells. J Pathol 2012; 228: 148–57. [DOI] [PubMed] [Google Scholar]
- 12. Hara MR, Kovacs JJ, Whalen EJ et al A stress response pathway regulates DNA damage through β2‐adrenoreceptors and β‐arrestin‐1. Nature 2011; 477: 349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu D, Yang Z, Wang T et al β2‐AR signaling controls trastuzumab resistance‐dependent pathway. Oncogene 2016; 35: 47–58. [DOI] [PubMed] [Google Scholar]
- 14. Yuan J, Minter‐Dykhouse K, Lou Z. A c‐Myc‐SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol 2009; 185: 203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jang KY, Noh SJ, Lehwald N et al SIRT1 and c‐Myc promote liver tumor cell survival and predict poor survival of human hepatocellular carcinomas. PLoS ONE 2012; 7: e45119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kriegl L, Vieth M, Kirchner T, Menssen A. Up‐regulation of c‐MYC and SIRT1 expression correlates with malignant transformation in the serrated route to colorectal cancer. Oncotarget 2012; 3: 1182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaziri H, Dessain SK, Ng Eaton E et al hSIR2(SIRT1) functions as an NAD‐dependent p53 deacetylase. Cell 2001; 107: 149–59. [DOI] [PubMed] [Google Scholar]
- 18. Luo J, Nikolaev AY, Imai S et al Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107: 137–48. [DOI] [PubMed] [Google Scholar]
- 19. Ngan HY, Cheung AN, Liu SS et al Abnormal expression of pan‐ras, c‐myc and tp53 in squamous cell carcinoma of cervix: correlation with HPV and prognosis. Oncol Rep 2001; 8(3): 557–61. [PubMed] [Google Scholar]
- 20. Menssen A, Hydbring P, Kapelle K et al The c‐MYC oncoprotein, the NAMPT enzyme, the SIRT1‐inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA 2012; 109: E187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elabd S, Meroni G, Blattner C. TRIMming p53's anticancer activity. Oncogene 2016; 35: 5577–84. [DOI] [PubMed] [Google Scholar]
- 22. Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene 2014; 33: 1609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li L, Bhatia R. Role of SIRT1 in the growth and regulation of normal hematopoietic and leukemia stem cells. Curr Opin Hematol 2015; 22: 324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng F, Su L, Yao C et al SIRT1 promotes epithelial‐mesenchymal transition and metastasis in colorectal cancer by regulating Fra‐1 expression. Cancer Lett 2016; 375: 274–83. [DOI] [PubMed] [Google Scholar]
- 25. Simic P, Williams EO, Bell EL et al SIRT1 suppresses the epithelial‐to‐mesenchymal transition in cancer metastasis and organ fibrosis. Cell Rep 2013; 3: 1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen IC, Chiang WF, Huang HH et al Role of SIRT1 in regulation of epithelial‐to‐mesenchymal transition in oral squamous cell carcinoma metastasis. Mol Cancer 2014; 13: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jang KY, Kim KS, Hwang SH et al Expression and prognostic significance of SIRT1 in ovarian epithelial tumours. Pathology 2009; 41: 366–71. [DOI] [PubMed] [Google Scholar]
- 28. Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer 2010; 10: 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang H, Suh JY, Jung JW et al Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell 2011; 44: 203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shin DH, Choi YJ, Park JW. SIRT1 and AMPK mediate hypoxia‐induced resistance of non‐small cell lung cancers to cisplatin and doxorubicin. Cancer Res 2014; 74(1): 298–308. [DOI] [PubMed] [Google Scholar]
- 31. Liang Z, Yang Y, Wang H et al Inhibition of SIRT1 signaling sensitizes the antitumor activity of silybin against human lung adenocarcinoma cells in vitro and in vivo. Mol Cancer Ther 2014; 13: 1860–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1.Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro.
Fig. S2.Catecholamines induce resistance of cervical cancer cells to chemotherapeutics in vitro.