

Abstract
The proliferation of prostate cancer cells is controlled by the androgen receptor (AR) signaling pathway. However, the function of AR target genes has not been fully elucidated. In previous studies, we have identified global AR binding sites and AR target genes in prostate cancer cells. Here, we focused on Claudin 8 (CLDN8), a protein constituting tight junctions in cell membranes. We found one AR binding site in the promoter region and two functional androgen‐responsive elements in the sequence. Reporter assay revealed that transcriptional activation of the CLDN8 promoter by androgen is dependent on these androgen‐responsive elements. Furthermore, CLDN8 mRNA is induced by androgen time‐dependently and the induction is blocked by AR inhibitor, suggesting that AR is involved in the transcriptional activation. In addition, our functional analyses by overexpression and knockdown of CLDN8 mRNA indicate that CLDN8 promotes prostate cancer cell proliferation and migration. Claudin 8 was overexpressed in prostate cancer clinical samples compared to benign tissues. Furthermore, we found that CLDN8 regulates intracellular signal transduction and stabilizes the cytoskeleton. Taken together, these results indicate that CLDN8 functions as an AR downstream signal to facilitate the progression of prostate cancer. Claudin 8 may be a novel molecular target for prostate cancer therapy.
Keywords: Androgen receptor, cell migration, cell proliferation, CLDN8, prostate cancer
The AR is a main driver in the development and progression of prostate cancer.1, 2, 3 Androgen receptor belongs to the nuclear receptor superfamily and functions as a ligand‐dependent transcriptional factor. Following androgen stimulation, AR binds to AREs with co‐regulators on the genome and regulates the transcription of its target genes.4, 5, 6, 7 The majority of primary prostate cancers are androgen‐dependent, and therefore androgen deprivation therapy is initially effective for inhibiting the growth of prostate cancer by suppressing AR activity. Although initial hormone therapy is so effective, prostate cancers often obtain resistance to androgen deprivation therapy as CRPC. Establishment of CRPC is associated with enhanced AR signaling, hypersensitivity to androgen, intratumoral steroidogenesis, and AR amplifications, mutations, and splicing variants.8, 9, 10, 11, 12, 13, 14, 15, 16 Thus, identification of AR downstream signaling events is critical for understanding the progression to CRPC.
Recent analysis using ChIP techniques have shown that the AR‐regulated gene network is deeply involved in the prostate cancer progression. Global ChIP analyses combined with DNA microarray (ChIP‐chip) or deep sequencing (ChIP‐seq)17, 18, 19, 20 have defined various androgen‐regulated genes in the vicinity of ARBSs with histone modifications, as we previously reported.6, 21, 22, 23, 24, 25
Recent studies have reported that androgen regulates the expression of Cldn8 26, 27 in prostate of mice. Claudin 8 belongs to the CLDN superfamily, located in the cell membrane and associated with tight junctions of cell adhesion.28 Claudin 8 is associated with several signaling pathways implicated in the epithelial environment and cell proliferation.29 Interestingly, previous analyses have reported that CLDN8 is overexpressed in several human cancer cells and in prostate cancer.30, 31 However, the functions of CLDN8 and its regulatory mechanism by androgen in human prostate cancer cells have not been well elucidated.
In the present study, we reported the regulatory mechanism of an androgen‐regulated gene, CLDN8, by AR and its functional role in prostate cancer cells. By global analysis of ARBSs, we found an evident ARBS in the promoter region of CLDN8. We further clarify the oncogenic mechanisms in prostate cancer by CLDN8 overexpression and knockdown. Our study presents the first evidence that CLDN8 is directly regulated by AR and functions to promote prostate cancer cell proliferation and migration.
Materials and Methods
Cell culture and reagents
Human prostate cancer LNCaP and VCaP cells were purchased from the ATCC (Manassas, VA, USA). LNCaP cells were grown in RPMI‐1640 supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin. VCaP cells were maintained in DMEM supplemented with 10% FBS. Dihydrotestosterone (Wako, Tokyo, Japan) was used for androgen treatment. Before androgen treatment, these cells were cultured in phenol red‐free medium containing 5% charcoal‐stripped FBS for 72 h. Anti‐androgen medicine bicalutamide was purchased from Sigma (St. Louis, MO, USA). Transfection was carried out using X‐tremeGene HP DNA Transfection Reagent (Roche Applied Science (Basel, Switzerland)), according to the manufacturer's instructions. Small interfering RNA was transfected using RNAi MAX (Invitrogen, Carlsbad, CA, USA). Antibodies used in the present study were anti‐Flag (M2; Sigma), anti‐CLDN8 (183738; Abcam, Cambridge, UK), anti‐Akt (Ser473; 9271), ERK1/2 (9101), total Akt (9272) and total ERK1/2 (9102; all Cell Signaling Technology, Danvers, MA, USA).
Quantitative RT‐PCR
Total RNA was extracted using ISOGEN reagent (Nippon Gene, Toyama, Japan). First‐strand cDNA was synthesized using the PrimeScript RT reagent kit (Takara Bio, Kyoto, Japan) according to manufacturer's protocol. The primer sequences for CLDN8 and GAPDH are shown below. The mRNA was quantified by real‐time PCR using SYBR Green PCR mix (Applied Biosystems, Framingham, MA, USA) and the ABI Prism 7000 system (Applied Biosystems). The evaluation of relative differences of PCR product amounts among the treatment groups was carried out by the Ct method, using GAPDH as an internal control. The primer sequences of CLDN8 were: forward, 5′‐TCTGCAGTAGGACATAGAAACCCCTAA‐3′; and reverse, 5′‐CGTTTAGGGGTTTCTATGTCCTACTGC‐3′.
Luciferase assay
Luciferase reporter constructs containing the CLDN8 promoter region with ARE and ARE mutation were generated using pGL3 promoter plasmid (Promega, Madison, WI, USA). Cells were plated in 24‐well culture plates at a density of 30 000 cells/well in phenol red‐free medium containing 5% charcoal‐stripped serum. Cells were transfected with plasmids using X‐tremeGene HP DNA Transfection Reagent (Roche Applied Science) and, 24 h later, were treated with DHT or vehicle (0.1% ethanol) for 24 h. Luciferase activity was determined by the Dual Luciferase Assay Kit (Promega) according to the manufacturer's instructions. A Renilla luciferase reporter Tk‐pRL was co‐transfected as a control for evaluating transfection efficiency.
Electrophoresis mobility shift assay
LNCaP cells were incubated with DHT (100 nm) or vehicle in 15‐cm dishes for 48 h. Nuclear extracts were collected by Hypotonic Buffer(20 mm HEPES [pH 7.9], 10 mm KCl, 1 mm EDTA, 1 mm EGTA, 0.65% NP‐40, and 1 mm DTT) and RIPA buffer (25 mm Tris–HCl [pH 7.6], 150 mm NaCl, 1.0% NP‐40, 1.0% sodium deoxycholate, and 0.1% SDS). Oligonucleotides were synthesized for ARE (1 and 2) and ARE mutation. The oligonucleotide sequences for CLDN8 ARE and CLDN8 ARE mutations were: CLDN8 ARE1, TCTGCAGTAGGACATAGAAACCCTAAA; CLDN8 ARE1 mutation, TCTGCAGTAGGAAATAGAAACACTAAA; CLDN8 ARE2, TAAACGCAAGACAATTTGAACTTTCTT; and CLDN8 ARE2 mutation, TAAACGCAAGAAAATTTTAACTTTCTT. Electrophoresis mobility shift assay was carried out using the DIG Gel Shift Kit, 2nd Generation (Roche Applied Science), according to the manufacturer's instructions.
Cell proliferation assay
A total of 3000 cells were seeded in 96‐well plates and cultured in RPMI supplemented with 10% FBS. The MTS assay was carried out using cell titer reagent (Promega) according to the manufacturer's instructions. Each time point was undertaken in quadruplicate, and each experiment was carried out at least three times.
Cell migration assay
The cell migration assay was carried out using the Cell Culture Insert with an 8.0‐μm pore size PET filter (BD Biosciences, Billerica, MA, USA). Before the assay, the lower surface of the filter was immersed for 30 min in 10 μg/mL fibronectin (Sigma) diluted with PBS. RPMI‐1640 medium containing 10% FBS was added to the lower chamber. Subsequently, the same number of cells per well were suspended in RPMI‐1640 medium containing 10% FBS and added to the upper chamber. After incubation for 24 h at 37°C in a humid 5% CO2 atmosphere, the cells on the upper surface of the filter were completely removed by wiping with cotton swabs. The cells on the lower surface of the filter were fixed in methanol for 30 min, washed with PBS, and then incubated with Giemsa solution (Muto Pure Chemicals, Tokyo, Japan) for 30 s. The cells on the lower surface were counted in at least five fields at a magnification of ×200 under a microscope.
Small interfering RNA
We purchased negative control siRNA (Sigma) and siRNAs targeting CLDN8 from Sigma Genosys Japan (Tokyo, Japan). These two siRNA sequences were: siCLDN8 #1, 5′‐GCCAUCCUUGGCAUGAAAUGCACCA‐3′; and siCLDN8 #2, 5′‐UGGAGAGUGUCGGCCUUCAUUGAAA‐3′. Cells were transfected with RNA using RNAi MAX (Life Technologies, Waltham, MA, USA) 48–72 h before each experiment.
Immunohistochemistry
Immunohistochemical analysis was carried out using the streptavidin–biotin amplification method using a peroxidase catalyzed signal amplification system (Dako, Santa Clara, CA, USA). Catalyzed signal amplification was carried out according to the manufacturer's instructions. The tissue sections were deparaffinized and pretreated by heating in a microwave oven for antigen retrieval. After blocking the endogenous peroxidase with 0.3% H2O2, the sections were incubated in 10% BSA for 30 min. Application of the anti‐CLDN8 antibody was followed by sequential 60‐min incubation. The antigen–antibody complexes were visualized with DAB solution. Immunohistochemical assessment was evaluated for the proportion of positive cells to total cells (score 0, none; score 1, <1/100; score 2, 1/100 to 1/10; score 3, 1/10 to 1/3; score 4, 1/3 to 2/3; and score 5, >2/3) and the staining intensity (score 0, none; score 1, weak; score 2, moderate; score 3, strong) of positively stained cells. The total scores of immunoreactivity (0–8) were obtained as the sum of the proportion of positively stained cells and the stained intensity values. A total score of 7 or more was evaluated as high immunoreactivity, and 6 or less was evaluated low immunoreactivity. We obtained 13 prostate cancer tissue samples, which included benign regions, from biopsies undertaken at Itabashi Hospital, Nihon University (Tokyo, Japan). The study was approved by the ethics committee of Nihon University, and written informed consent was obtained from each patient.
Immunocytochemistry
Twelve‐millimeter circular coverslips (Matsunami, Tokyo, Japan) in 24‐well plates were used for plating cells. After incubation with 4% paraformaldehyde in PBS for 10 min at room temperature, cells were permeabilized with 0.5% Triton X‐100/PBS for 5 min. After cells were washed in PBS, blocking with 5% normal goat serum/PBS was carried out for 30 min. Cells were reacted with rabbit polyclonal anti‐CLDN8 antibody and mouse monoclonal anti‐Flag M2 antibody (Sigma) in 5% normal goat serum/PBS at 4°C overnight. After washing three times with PBS, cells were reacted with anti‐mouse IgG with Alexa Fluor 546 and anti‐rabbit IgG with Alexa Fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) in goat serum/PBS for 1 h. Cells were washed three times with PBS. Nuclei were counterstained with DAPI. Coverslips were mounted in 10% glycerol. Cells were visualized using an Olympus confocal laser microscope FV10. For phalloidin staining (Acti‐stain 488 phalloidin; Cytosleleton, Denver, CO, USA), we treated cells with phalloidin (100 nm) for 30 min according to the manufacturer's protocol. We observed the slide after DAPI staining as described above.
Western blot analysis
Cells were lysed with NP‐40 lysis buffer (150 mm NaCl, 1% NP40, and 50 mm Tris–HCl [pH 8.0]) for immunoblotting. Western blot analysis was carried out as described.20, 23
Statistical analysis
Statistical differences between the results of each group and its corresponding control were evaluated using one‐way anova with a Bonferroni post hoc test, Student's t‐test, or Wilcoxon's signed rank test. A P‐value < 0.05 was considered to be statistically significant. Statistical procedures were undertaken using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA) or Excel (Microsoft, Redmond, WA, USA).
Results
Androgen‐regulated CLDN8 identified by ChIP‐seq analysis
We previously screened global AR direct target genes using ChIP‐seq analysis in two AR‐positive prostate cancer cells, LNCaP and VCaP.32 In the promoter region of the CLDN8 gene, we identified strong AR bindings and the peak point is located approximately 154 bp 5′‐upstream relative to the TSS of CLDN8 in both cell lines (Fig. 1). In addition, in the distal enhancer region, several AR binding peaks could be found, suggesting multiple AR binding regions cooperate to regulate CLDN8 gene expression.
Figure 1.
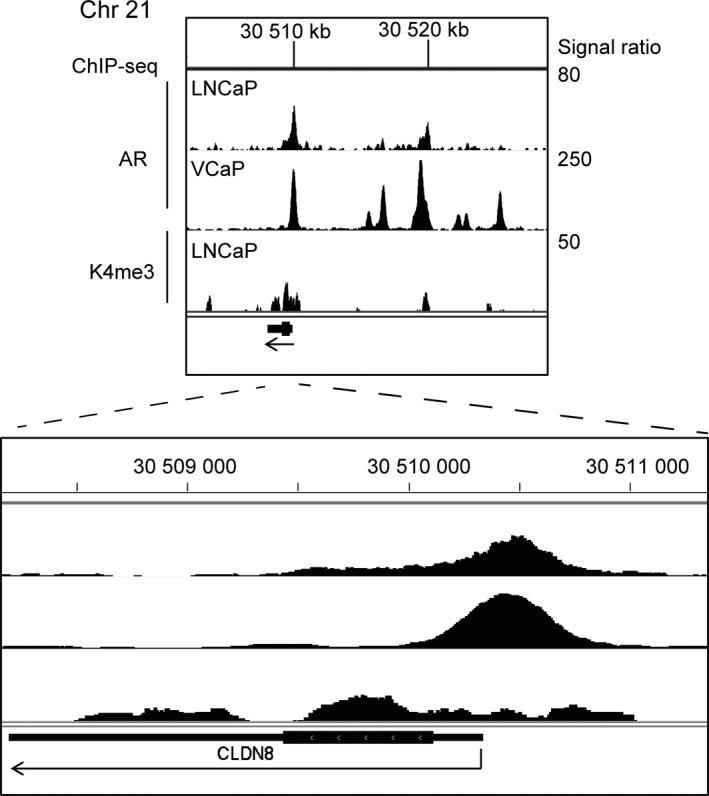
Androgen receptor (AR) binds to the promoter region of CLDN8 on chromosome (Chr) 21. Identification of AR binding site (ARBS) in the vicinity of CLDN8 on chromosome 21. LNCaP or VCaP prostate cancer cells were treated with 10 nm dihydrotestosterone. ChIP was carried out using AR and histone H3 lysine (K4) antibody for ChIP‐seq analysis.32 Genomic view of ChIP‐seq results in the CLDN8 gene location is shown using Integrative Genomics Viewer version 2.3. Arrows indicate the direction of transcription. Signal ratios (ChIP/input) are shown on the right.
Next, to examine whether this AR binding region functions to enhance transcription dependent on AR, we cloned the ARBS genomic region (−2042 to +500 bp relative to TSS) to luciferase vector (pGL3‐basic vector) and undertook a reporter assay (Fig. 2a). By luciferase reporter assay, we observed the transcriptional activation by androgen in the cells transfected with CLDN8‐ARBS luciferase vector (Fig. 2b). These results indicate the transcriptional induction of CLDN8 by AR binding in the promoter region.
Figure 2.
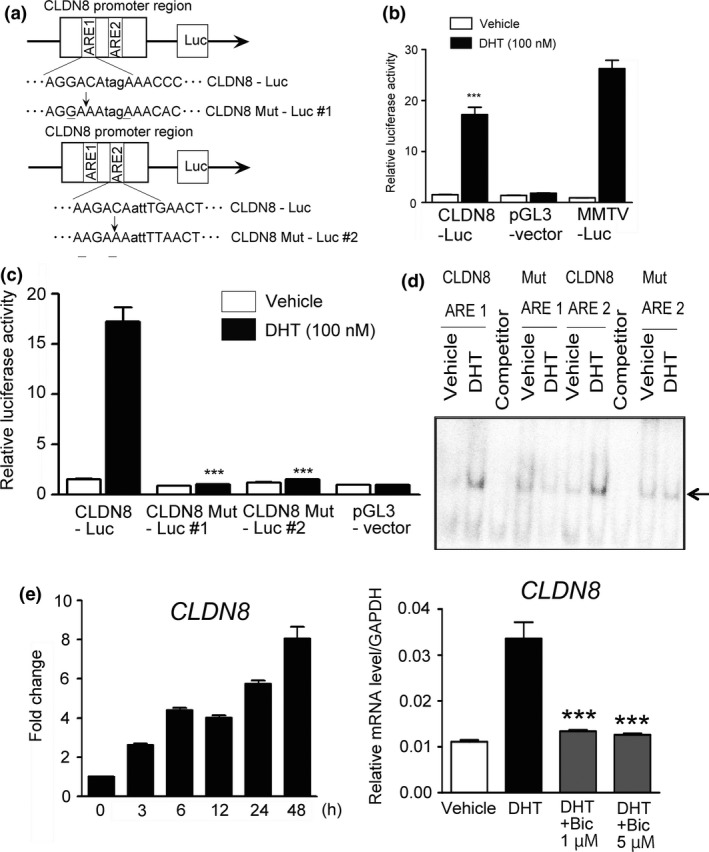
Androgen‐mediated transcriptional activation dependent on androgen response element (ARE) sequences. (a) We constructed luciferase reporter plasmids containing the CLDN8 promoter region. A genomic fragment of CLDN8 promoter region containing two ARE sequences and the fragment with mutation in each ARE sequence as shown were cloned into pGL3‐vector. We designated them as CLDN8‐Luc and CLDN8 Mut‐Luc #1 and #2. (b) LNCaP prostate cancer cells were transfected with luciferase vector. Cells were treated with vehicle or 100 nm dihydrotestosterone (DHT) for 24 h (n = 3). MMTV luciferase reporter gene containing ARE sequences was used as a positive control. pGL3‐vector was used as a negative control. Bar = SD. ***P < 0.001, compared with vehicle. (c) Luciferase activity of two ARE sequences in the CLDN8 promoter region is enhanced by DHT. LNCaP cells were transfected with each luciferase vector. Cells were treated with vehicle or 100 nm DHT for 24 h (n = 3). ***P < 0.001, compared with CLDN8‐Luc with DHT treatment. (d) Androgen receptor binds to ARE sequences. LNCaP cells were treated with 100 nm DHT or vehicle for 24 h. Nuclear extracts were used for EMSA. We used the ARE sequences located at the peaks from ChIP‐seq analysis and their mutated sequences. Unlabeled oligonucleotides were used as competitors. (e) CLDN8 mRNA is induced by DHT and repressed by bicalutamide. Left, cells were treated with 100 nm DHT for the indicated time. Right, cells were treated with 100 nm DHT. After 12 h of incubation, cells were treated with 1 or 5 μm bicalutamide for 24 h. Expression level of CLDN8 mRNA was measured by quantitative RT‐PCR. Results are presented as mean and SD. ***P < 0.001, compared with DHT.
Transcription of CLDN8 is activated by two important AREs dependent on AR
We found the genomic fragment of the CLDN8 promoter region contains two ARE‐like sequences. Two fragments of CLDN8‐ARBS with mutation in the ARE sequences were cloned into pGL3 vector (CLDN8 Mut‐Luc #1 and #2) (Fig. 2a). Luciferase activity was evaluated using LNCaP cells transfected with these reporter vectors in the absence or presence of DHT treatment for 24 h. In the cells transfected with CLDN8‐Luc construct, we observed increased transcriptional activity by DHT, whereas in the cells with both CLDN8 Mut‐Luc #1 and #2 constructs we could not (Fig. 2c). To validate whether this ARE‐dependent transcriptional activation is related to AR association with the ARE sequence, we undertook EMSA using CLDN8 AREs (ARE#1 and #2) and two CLDN8 ARE mutation oligonucleotides (Mut ARE#1 and #2). In this EMSA with LNCaP cell nuclear lysates, we observed that the CLDN8 ARE sequence (Fig. 2d) forms a complex with protein in the nuclear lysate androgen‐dependently, and this complex formation is inhibited by the competitor (unlabeled) oligonucleotides. Moreover, Mut AREs did not form androgen‐dependent complexes. These results suggest the importance of ARE sequences associating with AR to activate transcription.
CLDN8 mRNA is overexpressed in prostate cancer cells
Next, we investigated the transcriptional regulation of CLDN8 mRNA levels by androgen and expression in prostate cancer tissues. We found that CLDN8 mRNA expression was upregulated by DHT relative to vehicle control time‐dependently. These phenomena were inhibited by treatment with anti‐androgenic agent, bicalutamide (Fig. 2e), suggesting this induction is dependent on AR. We then analyzed CLDN8 expression in prostate cancer tissues using the Oncomine database of genome‐wide expression profiles of clinical cancer samples (www.oncomine.org) . In 11 analyses, CLDN8 expression was significantly overexpressed in prostate cancer tissues compared with benign tissues. In none of the analyses in this database was underexpression in cancers relative to normal tissues observed (Fig. 3a). Moreover, we carried out immunohistochemical analyses of prostate cancer specimens using anti‐CLDN8 antibody. Claudin 8 was significantly expressed in cancerous tissues compared with normal tissues (P = 0.0039) (Fig. 3b). These results indicate that CLDN8 expression is significantly upregulated by AR in prostate cancer and may be involved in the development of cancer.
Figure 3.

CLDN8 mRNA is induced by androgen and overexpressed in clinical prostate cancer tissues. (a) Representative analysis of CLDN8 mRNA expression in prostate cancer using the Oncomine database. CLDN8 mRNA levels were increased in prostate cancer tissue compared to normal prostate tissue. Bar = log2 median‐centered ratio in benign tissue versus cancer datasets, Oncomine database. (b) Immunohistochemistry of CLDN8 in prostate cancer tissues. Left, summary of cases with high or low CLDN8 expression in both benign and cancerous (Pca) regions. Representative view of CLDN8 protein expression in benign regions (middle panel) and cancerous regions (right panel). Immunohistochemistry was carried out with anti‐CLDN8 antibody (n = 13). P‐value obtained by Wilcoxon signed rank test. Bar = 50 μm.
Claudin 8 functionally promotes prostate cancer cell proliferation and migration of LNCaP cells
For further analyses, we established two LNCaP clones stably overexpressing CLDN8 (CLDN8 #1 and #2) (Fig. 4a). We first carried out cell migration assays to show that the migration of stably CLDN8‐overexpressing cells is enhanced compared with control cells (Fig. 4b). In addition, the MTS assay showed that CLDN8‐overexpressing cells show a higher cell proliferation rate than control cells (Fig. 4c). These results revealed that CLDN8 promotes cell migration and proliferation in prostate cancer cells.
Figure 4.
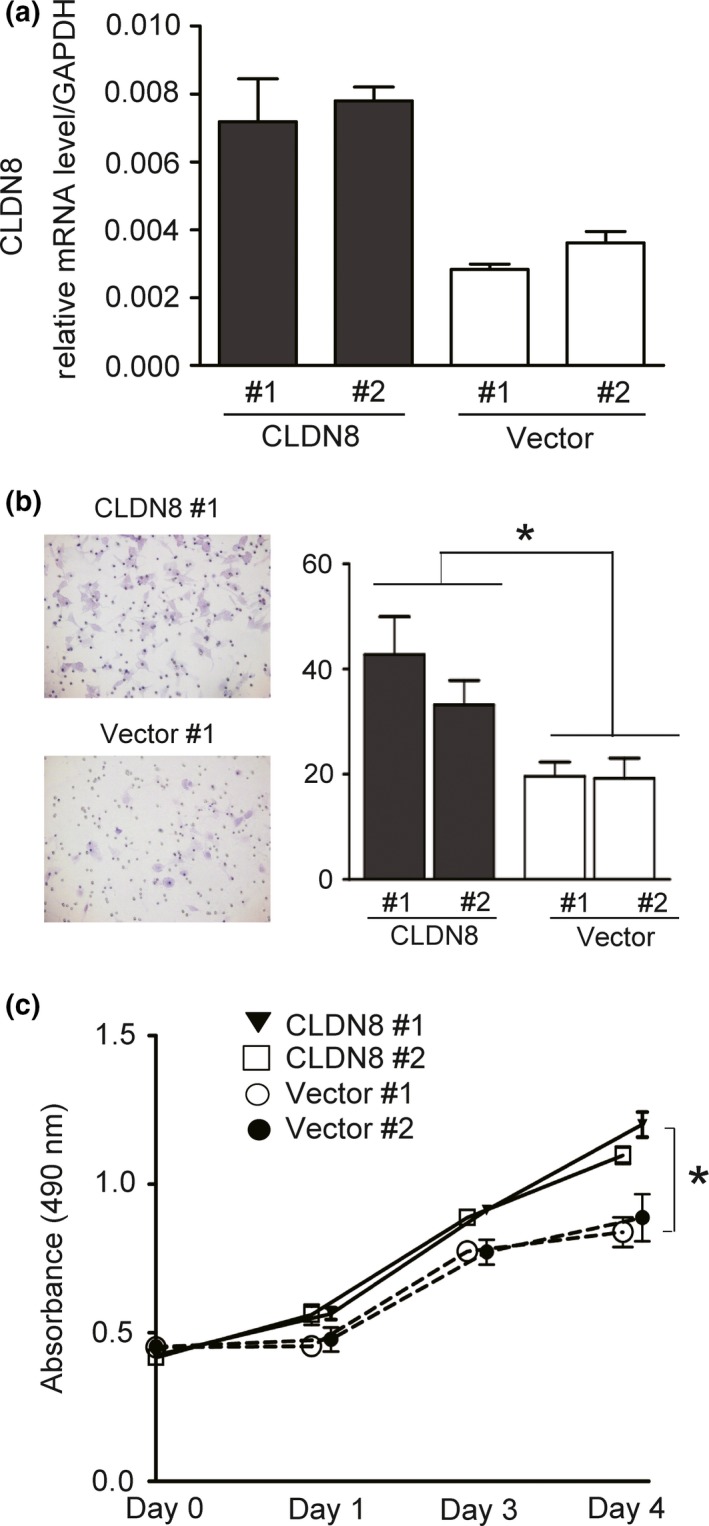
Overexpression of CLDN8 promotes proliferation and migration. (a) LNCaP prostate cancer cells were transfected with pcDNA3‐CLDN8 or control vector. We established two clones of LNCaP cells that stably expressed CLDN8 and control clones. Quantitative RT‐PCR was carried out by the Ct method. GAPDH was used as an internal control. (b) Overexpression of CLDN8 promotes cell migration. A total of 3000 cells were seeded in a cell culture insert. Migrated cells were stained with Giemsa. The number of cells in five representative fields was counted and quantified. Bar = SD. *P < 0.05, compared with vector controls. (c) CLDN8‐overexpressing cells show a higher cell proliferation rate than control cells. Cell proliferation assay was carried out in LNCaP cells stably expressing CLDN8 or vector controls by MTS assay. Bar = SD. *P < 0.05, compared with vector controls.
Furthermore, we examined the functions of CLDN8 by silencing endogenous CLDN8 expression using siRNA. We designed two siRNAs and confirmed that both siRNAs targeting CLDN8 reduced the level of endogenous CLDN8 expression compared with that of control siRNA‐transfected cells (Fig. 5a). Cell migration assays showed that the number of invaded cells was significantly decreased in LNCaP cells treated with siCLDN8 compared to those with siControl (Fig. 5b). We also examined the impact on the growth ability of prostate cancer cells by inhibiting CLDN8. The MTS assay showed that knockdown of CLDN8 significantly repressed cell proliferation (Fig. 5c). These results indicate that CLDN8 may be a new prostate cancer treatment target.
Figure 5.

Knockdown of CLDN8 reduces growth and migration of LNCaP prostate cancer cells. (a) Verification of the effect of siRNAs targeting CLDN8 in LNCaP cells. Cells were transfected with control siRNA (siControl) or siRNAs targeting CLDN8 (siCLDN8 #1 and #2). After 48 h of incubation, quantitative RT‐PCR analysis was undertaken. Results are presented as the mean and SD. ***P < 0.001, compared with siControl. (b) Knockdown of CLDN8 inhibits cell migration of prostate cancer cells. A total of 5000 cells were seeded in a cell culture insert. Migration assay was carried out in LNCaP cells. Bar = SD. ***P < 0.001, compared with siControl. (c) Knockdown of CLDN8 cells repress cell proliferation. LNCaP cells were transfected with siControl or siCLDN8 #1 or #2. MTS assay was carried out to analyze cell proliferation. Bar = SD. ***P < 0.001, compared with siControl.
Claudin 8 regulates intracellular signal transduction and cytoskeleton stability
We further analyzed the mechanism of CLDN8 action in pro‐proliferative and migration stimulatory effects. Through a PDZ‐binding motif at their C‐terminus, CLDNs directly bind to PDZ domain‐containing peripheral membrane proteins such as ZO‐1, ‐2, and ‐3. Furthermore, the C‐terminals of ZO1 have a binding affinity to actin filaments to function as cross‐linkers between CLDNs and F‐actin.33 We speculate that CLDN8 expression in prostate cancer is required for both the formation of tight junction and cell cytoplasmic actin formation. Therefore, we explored whether CLDN8 is involved in actin distribution or cell shape by immunofluorescence and analyzed the localization of CLDN8. To analyze the specificity of CLDN8 antibody, we overexpressed Flag‐tagged CLDN8 and transfected with LNCaP cells to detect exogenous CLDN8 protein. We observed CLDN8 staining in the cell membrane with Flag and CLDN8 without non‐specific staining in other cells (Fig. 6a). We also observed weak endogenous CLDN8 protein signals in the cell membrane by CLDN8 antibody (Fig. 6b). Flag‐tagged CLDN8 could be detected by anti‐Flag antibody and this band disappeared following siCLDN8 treatment, suggesting the specificity of siCLDN8 #1 and #2 (Fig. 6c). We then analyzed cell shape by phalloidin staining, which binds to F‐actin filaments. In this analysis, F‐actin was stained densely, particularly in the cell surface regions of cell–cell connections. However, knockdown of CLDN8 reduced actin staining and cell–cell adhesion, suggesting the important role of CLDN8 in the formation of tight junction in prostate cancer cells. In addition, cell shape was impaired and more rounded types of cells were observed (Fig. 6d).
Figure 6.
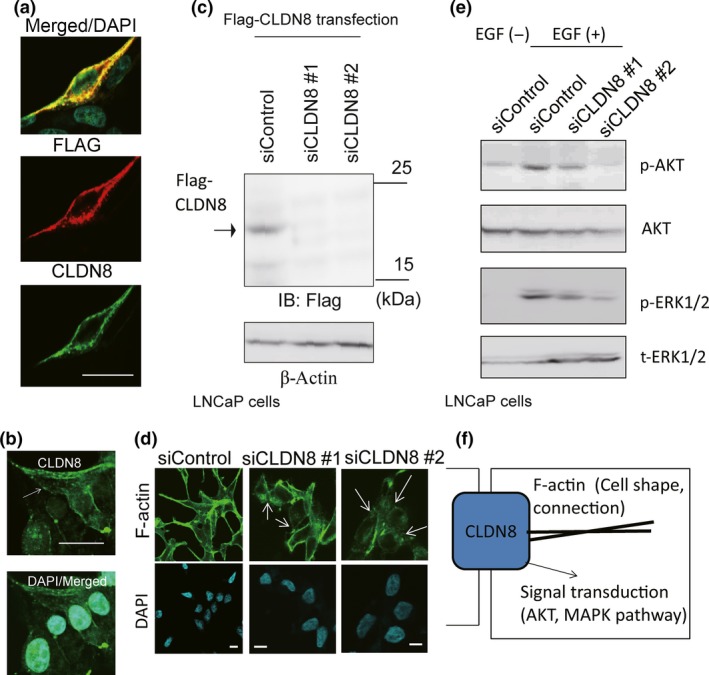
Knockdown of CLDN8 inhibited the stability of the cytoskeleton in LNCaP cells and collapsed tight junctions. (a) Overexpressed Flag‐CLDN8 was detected by anti‐CLDN8 antibody. Cells were transfected with Flag‐CLDN8 plasmid. After 48 h of incubation, immunocytochemical analysis was undertaken with anti‐Flag and anti‐CLDN8 antibody. Bar = 10 μm. (b) Analysis of localization of endogenous CLDN8 in LNCaP cells detected by anti‐CLDN8 antibody. Bar = 10 μm. (c) Verification of the effect of siRNAs targeting CLDN8 in LNCaP cells. Cells were transfected with Flag‐CLDN8 plasmid. Next day, control siRNA (siControl) or siRNAs targeting CLDN8 (siCLDN8 #1 and #2) were transfected. After 48 h, Western blot analysis was carried out to detect Flag‐CLDN8 expression by anti‐Flag antibody. IB, immunoblot. (d) Confocal microscopic changes in F‐actin cytoskeleton organization in LNCaP cells. Cells were treated with siControl or siCLDN8 #1 or #2 for 72 h. Arrows indicate the collapse of tight junctions and cell adhesions. Bar = 10 μm. (e) Knockdown of CLDN8 inhibits protein kinase B (Akt) and MAPK activation in LNCaP cells. Cells were treated with 10 ng/mL epidermal growth factor (EGF) for 10 min. Western blot analysis was undertaken to detect phosphorylated (p‐)Akt (Ser473), p‐ERK1/2, and total Akt and ERK1/2. (f) Summary of CLDN8 function in prostate cancer cells. CLDN8 is involved in cytoskeleton organization by regulating tight junction stability. In the cellular signal transduction, CLDN8 positively regulates Akt and MAPK phosphorylation pathways to promote cell proliferation or migration.
Downstream signals of CLDN8 were also investigated. It is well known that activation of the CLDN family is associated with the MAPK pathway.34 In addition, a recent study has shown reduction of CLDN5, 7, and 18 showed sustained phosphorylated Akt and promoted cell proliferation.35 We then hypothesized that Akt or MAPK activation is the downstream target of CLDN8. We treated LNCaP cells with epidermal growth factor (EGF) and analyzed the downstream activation of Akt and ERK phosphorylations. Using Western blot analysis, we found that knockdown of CLDN8 inhibits phosphorylation of both Akt and ERK, indicating the role of CLDN8 in these downstream signaling (Fig. 6e). Taken together, we consider that both cell shape formation by organizing actin filament and cell adhesion and intracellular signal transduction, such as Akt and MAPK, were modulated by CLDN8 upregulation with androgen treatment (Fig. 6f).
Discussion
Our study showed that CLDN8 is an androgen‐regulated gene in AR‐positive prostate cancer cells. Furthermore, the present study indicated that CLDN8 has two functional AREs in an ARBS located in the promoter region, upstream of the TSS. Following DHT stimulation, AR certainly binds to this region and induces transcriptional activity of CLDN8. In the previous study of AR ChIP‐seq in another cell line (PC3) transfected with exogenous AR,27 CLDN8 was also identified as the AR direct target. The results are in line with the experimental findings obtained in LNCaP cells in our study, validating the AR‐dependent regulation of CLDN8 in prostate cancer.
As the first evidence, we showed that CLDN8 functionally promotes prostate cancer cell proliferation and migration in prostate cancer cells. This result was validated by knockdown of endogenous CLDN8 in LNCaP cells and observed reduced cell growth and repressed cell migration by CLDN8 silencing. These phenomena indicated that CLDN8 could be a new therapeutic target gene because CLDN8 was dramatically upregulated in human prostate cancer tissues in several clinical cohorts, including ours. Claudin proteins are four transmembrane protein components of tight junction strands.36 Tight junctions are mainly constituted of claudins, occludins, and junction adhesion molecules.37 Tight junctions create barriers to some mediators, molecules, solutes, ions, and cytokines. A limitation of our study is the lack of appropriate antibodies to detect endogenous CLDN8 protein by Western blotting, although the antibody we used in immunocytochemistry could detect overexpressed Flag‐CLDN8 specifically in LNCaP cells.
The members of the CLDN superfamily have been identified as CLDN1–27 in several mammals.38 The CLDNs are reported to be aberrantly expressed in several tumors. Both CLDN3 and CLDN4 are especially overexpressed in primary human prostate cancer tissues and are associated with cell proliferation.39 In particularly, CLDN4‐targeted therapy using Clostridium perfringens enterotoxin affected antitumor formation by inducing cytotoxicity.40 These results suggest the possibility of a new cancer therapy targeting CLDN molecules.41, 42
Mechanistically, CLDNs have a short cytoplasmic N‐terminus, two extracellular loops, and a C‐terminal cytoplasmic domain,43 forming tight junction strands and thus play a role as the backbone of the tight junction. The first extracellular loop, in which there is a wide variation in the position and number of charged amino acids depending on each CLDN, are the major determinant of the paracellular charge and size selectivity by modulating paracellular channels for selective ions between neighboring cells.43 The C‐terminal cytoplasmic tail of CLDNs is required for their stability and downstream targeting. The PDZ‐binding motif at their C‐terminus binds to PDZ domain‐containing proteins ZO‐1, ‐2, and ‐3, which link between CLDNs and F‐actin.33
In this study, we showed that CLDN8 is involved in actin filament distribution and cell shape by modulating cell adhesion. Moreover, inhibition of CLDN8 inactivated Akt and ERK1/2 phosphorylation, indicating both signals were targets of CLDN8. Thus, our findings indicate that CLDN8 modulates the intracellular phosphorylation pathway and cell shape with tight junction formation required for cell proliferation and migration. However, the precise mechanism that links Akt and CLDN8 should be investigated in the future. Interestingly, Meng et al.26 reported that CLDN8 transcripts were significantly decreased following castration, and decreasing CLDN8 results in collapse of the tight junction barrier. Loss of the tight junction barrier leads to loss of immune privilege, and induces inflammation and autoimmune response. It is well known that inflammatory response and immune mechanisms are closely involved in cancer development and progression. This androgen‐dependent barrier of tight junction systems composed by CLDN8 may maintain cellular homeostasis and cytoskeleton organization.
In summary, we identified a novel regulatory mechanism of CLDN8 by AR. Increased CLDN8 expression promoted prostate cancer cell proliferation and migration. Furthermore, siRNA targeting CLDN8 significantly decreased cell growth and migration. These results suggest that CLDN8 plays a critical role in prostate cancer progression. In addition, CLDN8 could have the efficacy to be a new therapeutic target. Further studies investigating CLDN8 molecular function would contribute to the understanding of prostate cancer biology.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- Akt
protein kinase B
- AR
androgen receptor
- ARBS
androgen receptor binding site
- ARE
androgen response element
- CLDN8
Claudin 8
- CRPC
castration‐resistant prostate cancer
- DHT
dihydrotestosterone
- PDZ
PSD‐95/Dlg/ZO‐1
- TSS
transcriptional start site
- ZO
zonula occludens
Acknowledgments
We thank Drs. T. Fujimura, Y. Yamada, and J. Kumagai (Department of Urology, University of Tokyo, Tokyo, Japan) for critical advice. This work was supported by grants from: the Cell Innovation Program, P‐DIRECT, and P‐CREATE from Ministry of Education, Culture, Sports, Science and Technology, Japan (to S.I.); the Japan Society for the Promotion of Science (grant nos. 15K20116, 15K15581, and 15K15353 to D.A., K.T., and S.I.); the Program for Promotion of Fundamental Studies in Health Sciences, National Institute of Biomedical Innovation of Japan (to S.I.); Takeda Science Foundation (to S.I. and K.T.); the 2016 Research Grant of 60th Anniversary Memorial Fund from Nihon University Medical Alumni Association (to D.A.); the Princess Takamatsu Cancer Research Fund (to K.T.); and the Terumo Foundation for Life Sciences and Arts (to K.T.).
Cancer Sci 108 (2017) 1386–1393
Funding Information
Cell Innovation Program, P‐DIRECT, Ministry of Education, Culture, Sports, Science and Technology, Japan, Japan Society for the Promotion of Science (grant nos. 15K20116, 15K15581, and 15K15353), Program for Promotion of Fundamental Studies in Health Sciences, National Institute of Biomedical Innovation of Japan, Takeda Science Foundation, Nihon University Medical Alumni Association, Princess Takamatsu Cancer Research Fund, Terumo Foundation for Life Sciences and Arts.
References
- 1. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev 2004; 25: 276–308. [DOI] [PubMed] [Google Scholar]
- 2. Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J Cell Biochem 2006; 99: 333–44. [DOI] [PubMed] [Google Scholar]
- 3. Debes JD, Tindall DJ. The role of androgens and the androgen receptor in prostate cancer. Cancer Lett 2002; 187: 1–7. [DOI] [PubMed] [Google Scholar]
- 4. Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell 2002; 9: 601–10. [DOI] [PubMed] [Google Scholar]
- 5. Wang Q, Li W, Liu XS et al A hierarchical network of transcription factors governs androgen receptor‐dependent prostate cancer growth. Mol Cell 2007; 27: 380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obinata D, Takayama K, Urano T et al Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int J Cancer 2012; 130: 1021–8. [DOI] [PubMed] [Google Scholar]
- 7. Takayama K, Inoue S. Transcriptional network of androgen receptor in prostate cancer progression. Int J Urol 2013; 20: 756–68. [DOI] [PubMed] [Google Scholar]
- 8. Chen CD, Welsbie DS, Tran C et al Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004; 10: 33–9. [DOI] [PubMed] [Google Scholar]
- 9. Waltering KK, Helenius MA, Sahu B et al Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res 2009; 69: 8141–9. [DOI] [PubMed] [Google Scholar]
- 10. Locke JA, Guns ES, Lubik AA et al Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration‐resistant prostate cancer. Cancer Res 2008; 68: 6407–15. [DOI] [PubMed] [Google Scholar]
- 11. Guo Z, Dai B, Jiang T et al Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006; 10: 309–19. [DOI] [PubMed] [Google Scholar]
- 12. Sun S, Sprenger CC, Vessella RL et al Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 2010; 120: 2715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thadani‐Mulero M, Portella L, Sun S et al Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res 2014; 74: 2270–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang KH, Li R, Kuri B et al A gain‐of‐function mutation in DHT synthesis in castration‐resistant prostate cancer. Cell 2013; 154: 1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu Z, Chen S, Sowalsky AG et al Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res 2014; 20: 1590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antonarakis ES, Lu C, Wang H et al AR‐V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371: 1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takayama K, Kaneshiro K, Tsutsumi S et al Identification of novel androgen response genes in prostate cancer cells by coupling chromatin immunoprecipitation and genomic microarray analysis. Oncogene 2007; 26: 4453–63. [DOI] [PubMed] [Google Scholar]
- 18. Takayama K, Tsutsumi S, Katayama S et al Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome‐wide androgen receptor signaling in prostate cancer cells. Oncogene 2011; 30: 619–30. [DOI] [PubMed] [Google Scholar]
- 19. Obinata D, Takayama K, Urano T et al ARFGAP3, an androgen target gene, promotes prostate cancer cell proliferation and migration. Int J Cancer 2012; 130: 2240–8. [DOI] [PubMed] [Google Scholar]
- 20. Takayama K, Tsutsumi S, Suzuki T et al Amyloid precursor protein is a primary androgen target gene that promotes prostate cancer growth. Cancer Res 2009; 69: 137–42. [DOI] [PubMed] [Google Scholar]
- 21. Murata T, Takayama K, Katayama S et al miR‐148a is an androgen‐responsive microRNA that promotes LNCaP prostate cell growth by repressing its target CAND1 expression. Prostate Cancer Prostatic Dis. 2010; 13: 356–61. [DOI] [PubMed] [Google Scholar]
- 22. Obinata D, Takayama K, Fujiwara K et al Targeting Oct1 genomic function inhibits androgen receptor signaling and castration‐resistant prostate cancer growth. Oncogene 2016; 35: 6350–6358. [DOI] [PubMed] [Google Scholar]
- 23. Takayama K, Horie‐Inoue K, Katayama S et al Androgen‐responsive long noncoding RNA CTBP1‐AS promotes prostate cancer. EMBO J 2013; 32: 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takayama K, Misawa A, Suzuki T et al TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat Commun 2015; 6: 8219. [DOI] [PubMed] [Google Scholar]
- 25. Obinata D, Ito A, Fujiwara K et al Pyrrole‐imidazole polyamide targeted to break fusion sites in TMPRSS2 and ERG gene fusion represses prostate tumor growth. Cancer Sci 2014; 105: 1272–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meng J, Mostaghel EA, Vakar‐Lopez F, Montgomery B, True L, Nelson PS. Testosterone regulates tight junction proteins and influences prostatic autoimmune responses. Horm Cancer. 2011; 2: 145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sutinen P, Malinen M, Heikkinen S, Palvimo JJ. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res 2014; 42: 8310–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lal‐Nag M, Morin PJ. The claudins. Genome Biol 2009; 10: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsukita S, Yamazaki Y, Katsuno T, Tamura A, Tsukita S. Tight junction‐based epithelial microenvironment and cell proliferation. Oncogene 2008; 27: 6930–8. [DOI] [PubMed] [Google Scholar]
- 30. Rohan S, Tu JJ, Kao J et al Gene expression profiling separates chromophobe renal cell carcinoma from oncocytoma and identifies vesicular transport and cell junction proteins as differentially expressed genes. Clin Cancer Res 2006; 12: 6937–45. [DOI] [PubMed] [Google Scholar]
- 31. Hewitt KJ, Agarwal R, Morin PJ. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 2006; 6: 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takayama K, Suzuki T, Fujimura T et al CtBP2 modulates the androgen receptor to promote prostate cancer progression. Cancer Res 2014; 74: 6542–53. [DOI] [PubMed] [Google Scholar]
- 33. Ito M, Furuse M, morita K, Kubota K, Saitou M, Tsukita S. Direct bindings of three tight junction‐associated MAGUKs, ZO‐1, ZO‐2 and ZO‐3, with the COOH terminal of Caludins. J Cell Biol 1999; 147: 1351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu Z, Ding L, Hong H, Hoggard J, Lu Q, Chen YH. Claudin‐7 inhibits human lung cancercell migration and invasion through ERK/MAPK signaling pathway. Exp Cell Res 2011; 317: 1935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akizuki R, Shimobaba S, Matsunaga T, Endo S, Ikari A. Claudin‐5, ‐7, and ‐18 suppress proliferation mediated by inhibition of phosphorylation of Akt in human lung squamous cell carcinoma. Biochim Biophys Acta 2017; 1864: 293–302. [DOI] [PubMed] [Google Scholar]
- 36. Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four‐transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA 1999; 96: 511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells 1998; 3: 569–73. [DOI] [PubMed] [Google Scholar]
- 38. Gunzel D. Claudins: vital partners in transcellular and paracellular transport coupling. Pflugers Arch 2017; 469: 35–44. [DOI] [PubMed] [Google Scholar]
- 39. Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin‐3, a tight junction integral membrane protein. FEBS Lett 2000; 476: 258–61. [DOI] [PubMed] [Google Scholar]
- 40. Maeda T, Murata M, Chiba H et al Claudin‐4‐targeted therapy using Clostridium perfringens enterotoxin for prostate cancer. Prostate 2012; 72: 351–60. [DOI] [PubMed] [Google Scholar]
- 41. Saitoh Y, Suzuki H, Tani K et al Tight junctions. Structural insight into tight junction disassembly by Clostridium perfringens enterotoxin. Science 2015; 347: 775–8. [DOI] [PubMed] [Google Scholar]
- 42. Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res 2005; 65: 9603–6. [DOI] [PubMed] [Google Scholar]
- 43. Chiba H, Osanai M, Murata M, Kojima T. Transmembrane proteins of tight junctions. Biochim Biophys Acta 2007; 1778: 588–600. [DOI] [PubMed] [Google Scholar]