

Abstract
Endoplasmic reticulum stress (ERS) plays an important role in the pathogenesis and development of malignant tumors, as well as in the regulation of radiochemoresistance and chemoresistance in many malignancies. ERS signaling pathway protein kinase RNA‐like endoplasmic reticulum kinase (PERK)‐eukaryotic initiation factor‐2 (eIF2α) may induce aberrant activation of nuclear factor‐κB (NF‐κB). Our previous study showed that NF‐κB conferred radioresistance in lymphoma cells. However, whether PERK‐eIF2α regulates radioresistance in oropharyngeal carcinoma through NF‐κB activation is unknown. Herein, we showed that PERK overexpression correlated with a poor prognosis for patients with oropharyngeal carcinoma (P < 0.01). Meanwhile, the percentage of the high expression level of PERK in oropharyngeal carcinoma patients resistant to radiation was higher than in patients sensitive to radiation (77.7 and 33.3%, respectively; P < 0.05). Silencing PERK and eIF2α increased the radiosensitivity in oropharyngeal carcinoma cells and increased radiation‐induced apoptosis and G2/M phase arrest. PERK‐eIF2α silencing also inhibited radiation‐induced NF‐κB phosphorylation and increased the DNA double strand break‐related proteins ATM phosphorylation. NF‐κB activator TNF‐α and the ATM inhibitor Ku55933 offset the regulatory effect of eIF2α on the expression of radiation‐induced cell apoptosis‐related proteins and the G2/M phase arrest‐related proteins. These data indicate that PERK regulates radioresistance in oropharyngeal carcinoma through NF‐kB activation‐mediated phosphorylation of eIF2α, enhancing X‐ray‐induced activation of DNA DSB repair, cell apoptosis inhibition and G2/M cell cycle arrest.
Keywords: eIF2α, NF‐κB, oropharyngeal carcinoma, PERK, radiotherapy
Head and neck carcinoma is the sixth most common malignant tumor worldwide and the third most common tumor in developing countries. There are approximately 650 000 new onset cases and 350 000 cases of death annually.1 More than 90% of head and neck carcinomas are squamous cell carcinomas, and radiotherapy is one of the major treatment methods. Although current radiotherapy technology has undergone significant progress, over the previous 30 years, the 5‐year survival rate following radical radiotherapy has been approximately 50%. Therefore, increasing the radiosensitivity of head and neck carcinomas is an issue that urgently requires resolution.
Increasing evidence indicates that endoplasmic reticulum stress (ERS) plays an important role in the pathogenesis and development of malignant tumors, as well as the regulation of radio chemosensitivity and chemosensitivity. Protein kinase RNA‐like endoplasmic reticulum kinase (PERK) is a type I membrane protein in the endoplasmic reticulum (ER) and one of the important sensing proteins in ERS. The N‐terminus senses ERS signals and has a non‐ligand‐dependent dimerization domain. The C‐terminus has a serine/threonine protein kinase functional domain but lacks endonuclease activity. Upon ERS, PERK is activated to specifically phosphorylate eukaryotic initiation factor‐2 (eIF2α) at Ser51, thus inhibiting transcription and protein synthesis.2
Radiation causes DNA damage and the accumulation of misfolded proteins in the ER to induce ERS, thus activating the UPR. At the early stage of radiation‐induced ERS, the ER activates the self‐protection mechanism in cells and actively regulates stress‐responsive proteins, such as promoting the expression/activation of the survival molecule GRP78 to defend against stress‐induced damage. If the stimulation exceeds the cell repair capacity or if the ERS is extended, then the expression of ERS‐related pro‐apoptosis signals, such as PERK‐eIF2α‐ATF4‐CHOP/GADD153 and caspase‐12, will be initiated, and the balance between pro‐apoptosis proteins and anti‐apoptosis proteins (bcl‐2 family) will be disrupted, thus causing an increase in the expression of the BH3‐only proteins BIM, PUMS and NOXA, and inducing cytochrome C release to promote cell apoptosis.3 Therefore, ERS has bidirectional regulatory functions on the regulation of radiosensitivity and has cell type specificity. Inhibition of the ERS signaling pathway can increase radiosensitivity in nasopharyngeal carcinoma cells4 and breast cancer cells.5 However, this function exhibits tumor type specificity. In human colorectal cancer cells6 and oesophageal cancer cells,7 ERS activation can increase radiosensitivity. The only relevant research examining ERS and radiosensitivity in head and neck carcinoma was conducted by Liu et al.8 In contrast to our results, they show that HIV protease inhibitors increase radiosensitivity in head and neck squamous cell carcinoma SQ20B and FaDu cells by inducing ERS. They used the ERS activator thapsigargin as a positive control to indirectly confirm that ERS induction increased radiosensitivity in head and neck carcinoma cells, which might be associated with the specific functional pathway of the drugs.
Nuclear factor‐κB (NF‐κB) is a nuclear transcription regulatory factor with specific DNA binding sequences. It is extensively present in various types of cells and participates in cellular responses to various stimuli, such as growth factors and ionizing radiation. Our previous studies showed that NF‐κB is abnormally active in human lymphoma cells9 and regulates the radioresistance by regulating radiation‐induced DNA double‐strand break (DSB) repair,10 cell apoptosis11, 12 and cell cycle arrest.10, 11 The regulation of radioresistance in head and neck carcinoma by NF‐κB is similar to that in lymphoma cells.13 Interestingly, previous studies have shown that eIF2α phosphorylation abnormally activates NF‐κB,14, 15 and the specific mechanism may be associated with NF‐κB translocation from the cytoplasm to the nucleus to activate transcription after dissociating from IκB via eIF2α phosphorylation.
Based on previous study results, we hypothesized that PERK‐eIF2α activates the NF‐κB transcription factor to regulate downstream target genes, thus regulating radioresistance. To test this hypothesis, we first analyzed the association between PERK and the prognosis of oropharyngeal carcinoma after radical radiotherapy using oropharyngeal carcinoma tissue specimens, and then analyzed the effect of PERK on radiosensitivity in oropharyngeal carcinoma patients. Furthermore, PERK and eIF2α were silenced using siRNA transfection to analyze the regulation of radioresistance by PERK‐eIF2α through the NF‐κB signaling pathway in oropharyngeal carcinoma cells. In addition, the pathways and mechanisms underlying this function were investigated to identify new therapeutic targets to increase the radiotherapy efficacy in oropharyngeal carcinoma with high radioresistance.
Materials and Methods
Patients and specimens
Pathological tumor sections were obtained from 114 cases of oropharyngeal squamous cell carcinoma patients who received radical radiotherapy with or without concurrent chemotherapy in our hospital between 2005 and 2011.
In addition, 36 cases of HPV (−) oropharyngeal squamous cell carcinoma among patients who received radical radiotherapy between 2014 and 2016 were selected, including 18 patients with radioresistance and 18 patients with radiosensitivity. Patients with radioresistance were defined as those with residual lesions after 6 weeks of radiotherapy or local or regional lymph node recurrence in the lesions after 2 months of radiotherapy. Patients with radiosensitivity were defined as those with lesion disappearance after 6 weeks of radiotherapy or those without tumor and lymph node recurrence within 2 months.16 All recruited patients provided informed consent.
Immunohistochemistry
The method applied is described in detail in our previous study.17 PERK primary antibody was purchased from Abcam (1:100 dilution; Abcam, Cambridge, MA, USA). Two independent blinded investigators examined all tumor slides in a randomized manner. Cytoplasmic immunostaining of the tumor cells was considered a positive result. A semi‐quantitative scoring criterion for immunohistochemistry was used.18 Tumor samples with a final score ≤2 were considered to have weak staining; tumor samples with a final score of 3–4 were considered to have moderate staining; and tumor samples with a final score of 5–6 were considered to have intense staining.
Cell culture, transfection and reagents
The human oropharyngeal squamous cell carcinoma cell lines FaDu and Detroit562 were purchased from the ATCC (Manassas, VA, USA) and cultured in MEM containing 10% inactivated FBS, 100 U/mL penicillin and 100 μg/mL streptomycin.
On‐TargetPlus SMARTpool siRNA for PERK, IRE‐1, ATF‐6, eIF2α and ONTARGETplus non‐targeting siRNA #1 were purchased from Dharmacon (Thermo Fisher Scientific, Waltham, MA, USA). The cells were transfected with small interfering RNA (siRNA) using the DharmaFECT 1 transfection reagent from Dharmacon (Boulder, CO, USA).
The ataxia‐telangiectasia‐mutated (ATM) kinase inhibitor KU55933 and the NF‐κB activator tumor necrosis factor‐α (TNFα) were purchased from Sigma (Sigma Chemical, St Louis, MO, USA).
Construction of radioresistant FaDuR and Detroit562R cells
Radioresistant oropharyngeal carcinoma cells were constructed according to a standard method.19 FaDu and Detroid562 cells were inoculated onto six‐well plates at 1000 cells/cm2. After 24 h, the cells were subjected to 4 Gy radiation and then continuously cultured for 10–12 days to allow colony formation. Single‐cell clones were selected and continuously cultured. Irradiation was repeated four times according to the above method. FaDu cells received a total of 16 Gy radiation (4 Gy/time ×4 times), and Detroid562 cells received a total of 14 Gy radiation (4 Gy/time ×2 times + 3 Gy/time ×2 times). These radioresistant cells were denoted FaDuR and Detroit562R. Parental cells without irradiation were named FaDuP and Detroid562P.
Western blot analysis
Total cellular proteins and nuclear proteins were extracted using lysis buffer (Pierce, Rockford, IL, USA). The method applied has been described in detail in our previous study.9 Primary antibodies, including phospho‐eIF2α, phospho‐ATM, Bcl‐2, cleaved‐caspase3, DNA‐PK, Rad50, Nbs1, Mre11, Bcl‐xL, cleaved poly(ADP‐ribose) polymerase (PARP), phospho‐p65, p65, cyclin B, phosphor‐Cdc2 (Try15), β‐actin, histone H3 (1:1000; Cell Signaling Technology, Danvers, MA, USA), IRE‐1, ATF‐6,HIF‐1α and PERK (1:1000; Abcam), at 4°C overnight. Secondary antibodies, anti‐mouse or anti‐rabbit IgG antibody (1:1000 dilution; Cell Signaling Technology), were added and incubated at room temperature for 2 h. Target proteins on PVDF membranes were visualized with LumiGLO (Cell Signaling Technology) and captured using a DNR BioImaging System (DNR, Israel).
RNA preparation and real‐time quantitative PCR
RNA extraction was performed according to the protocol supplied with the RNeasy Mini reagent kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed using the M‐MLV reverse transcription reagent kit (Invitrogen, Carlsbad, CA, USA). Quantitative analyses of the mRNA expression levels of PERK and eIF2α were performed using the TaqMan analysis system (Applied Biosystems, Carlsbad, CA, USA).10
For PERK, the forward primer was 5′‐ CATCCAGCCTTAGCAAACCAGA‐3′ and the reverse was 5′‐AGGAACTGTTTCCATGCTTTCAC‐3′; for eIF2α, the forward primer was 5′‐GCGGTACCATGGTGAATGTGCGCTCCATT‐3′ and the reverse was 5′‐CGCTCGAGCTAGTCCTCTGTTTTGGCTTC‐3′; for p65, the forward primer was 5′‐ GCGAGAGGAGCACAGATACC‐3′ and the reverse was 5′‐CTGATAGCCTGCTCCAGGTC‐3′; and for β‐actin, the forward primer was 5′‐TGGCACCCAGCACAATGAA‐3′ and the reverse was 5′‐CTAAGTCATAGTCCGCCTAGAAGCA‐3′.
Colony survival experiment
Pre‐treated cells were inoculated onto six‐well plates at gradient concentrations ranging from 102 to 104. The cells were allowed to attach and were cultured overnight. The cells were irradiated using a SIEMENS linear accelerator (SIEMENS Medical Systems, Germany). The doses were 0, 2, 4 and 6 Gy. The dose rate was 2 Gy/min. The cells were continuously cultured, and the number of colonies in samples with more than 50 cells was counted after 10–14 days.
Detection of cell apoptosis and cell cycle phase using flow cytometry
Pre‐treated cells were washed with PBS, and experiments were performed according to the protocol supplied with the Annexin‐Green Apoptosis cell detection reagent kit (Cell Signaling Technology). The percentage of apoptotic cells was detected using a FACScan flow cytometer (FACSCalibur BD; BD Biosciences, San Jose, CA, USA). PI staining (Sigma Chemical) was used to determine the cell cycle phase using a flow cytometer. The specific experimental methods have been described previously.11
Immunofluorescence
Pre‐treated cells received 5 Gy radiation. Cells were collected after 1 h of radiation, evenly smeared onto slides, fixed in 4% paraformaldehyde, treated with 0.1% Triton X‐100 at room temperature for 15 min, and blocked in 5% BSA for 30 min, Slides were then incubated with primary anti‐γ‐H2AX antibody (1:500; Cell Singling Technology) at 4°C overnight.
Statistical analyses
The data from three independent experiments were expressed as the mean ± SD. The Kaplan–Meier method was used for the survival analyses. The χ2‐extract test was performed to analyze the correlation between PERK expression and tumor radiosensitivity. Comparisons between two groups were performed using the t‐test. P‐values <0.05 indicated statistical significance. SPSS 13.0 software (Chicago, IL, USA) was used to perform the statistical analyses.
Results
Expression of PERK in human oropharyngeal squamous cell carcinoma
Endoplasmic reticulum stress activation and high expression level of its central mediator GRP78 are associated with a poor prognosis of human head and neck squamous cell carcinoma.20, 21 To clarify the association between PERK and the prognosis of human oropharyngeal squamous cell carcinoma, immunohistochemistry was performed and the expression pattern of PERK was investigated in pathological sections from 114 cases of oropharyngeal squamous cell carcinoma patients who had undergone radical radiotherapy. The results showed that PERK was mainly localized in the cytoplasm of tumor cells (Fig. 1a). The Kaplan–Meier results showed that the 5‐year overall survival of patients with high PERK expression level was significantly lower than that of patients with moderate and low expression level (P < 0.01, Fig. 1b). However, PERK expression level did not significantly correlate with HPV (p16) expression (Spearman correlation analysis: r = 0.076, P = 0.438). The result showed that the overall survival of patients with oropharyngeal carcinoma had a significant correlation with PERK protein expression level.
Figure 1.
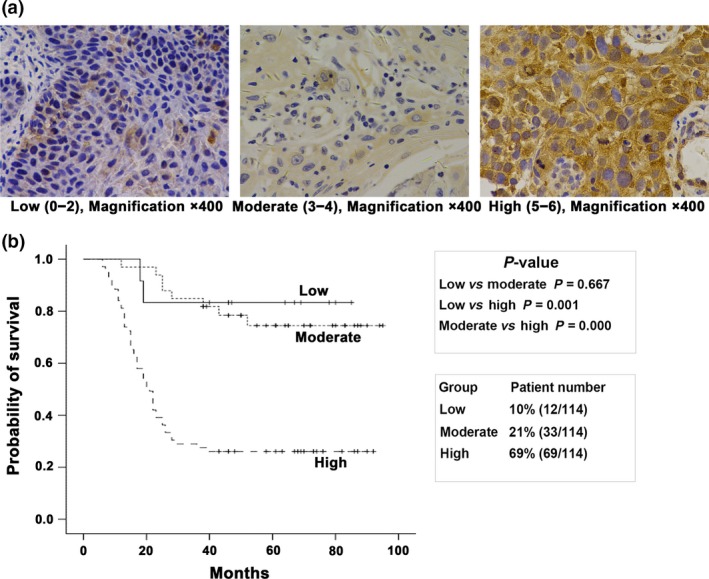
Expression of PERK protein in human oropharyngeal carcinoma samples. (a) Schematic diagram of high, moderate and low expression of PERK protein in the cytoplasm of oropharyngeal carcinoma samples. (b) The Kaplan–Meier analysis showed that overall survival of patients with oropharyngeal carcinoma had a significant correlation with PERK protein expression level.
To further confirm whether the association between PERK and the poor prognosis of oropharyngeal squamous cell carcinoma patients was caused by radioresistance, we selected 36 HPV (−) cases and divided them into a radioresistant group (18 cases) and a radiosensitive group (18 cases).16 The results showed that percentage of high level of PERK expression in the radioresistant group was 77.7%, compared with 33.3% in the radiosensitive group. The χ2‐exact test results showed that PERK overexpression was associated with radioresistance in patients with HPV (−) oropharyngeal squamous cell carcinoma (P < 0.05) (Table 1).
Table 1.
Expression of PERK in radioresistant and radiosensitive oropharyngeal carcinoma tissues
| n | Score | |||
|---|---|---|---|---|
| Low (0–2) | Moderate (3–4) | High (5–6) | ||
| Radioresistance | 18 | 0 (0.0%) | 4 (22.3%) | 14 (77.7%) |
| Radiosensitivity | 18 | 4 (22.2%) | 8 (44.4%) | 6 (33.3%) |
χ2‐extract test, P < 0.05.
Increased PERK‐eIF2α expression in radioresistant oropharyngeal carcinoma
The result of the colony survival assay showed significantly increased radioresistance in FaDuR compared to FaDuP (SF2 of 0.79 and 0.92). Similarly, Detroit562R demonstrated significantly greater radioresistance than Detroit562P (SF2 of 0.68 and 0.86; Fig. 2a).
Figure 2.

Expression of PERK‐eIF2α in radioresistant oropharyngeal carcinoma cells. (a) Colony formation results showing that the colony formation rates of FaDuR and Detroit562R were higher than those of FaDuP and Detroit562P after irradiation. (b) Western blot results showing that the expression levels of PERK and phospho‐eIF2α in radioresistant cells, FaDuR and Detroit562R were higher than those in FaDuP and Detroit562P. The bands were quantified using ImageJ software and normalized against a loading control. Changes in expression are shown in comparison to FaDuP and Detroit562P cells. NA, not applicable.
Previous studies showed elevated expression of GRP78 in head and neck cancer stem cells,21 and, to a certain extent, radioresistant cells have cancer stem‐like cells characteristics.19 We hypothesized that ERS might be activated in radioresistant oropharyngeal carcinoma cells. Therefore, we further investigated the expression level of PERK‐eIF2α protein. The results showed significantly higher expression levels in radioresistant FaDuR and Detroit562R cells than in parental FaDuP and Detroit562P cells (Fig. 2b). These results suggested that PERK‐eIF2α might be associated with radioresistance in oropharyngeal carcinoma.
X‐ray induced activation of PERK‐eIF2α in oropharyngeal carcinoma cell in a time‐dependent manner
Consistent with previous reports,22 our study showed that X‐ray irradiation activated PERK and phospho‐eIF2α proteins in a time‐dependent manner (Fig. 3a). The expression of PERK reached peak values in parental Detroit562P cells 20 min after irradiation and decreased 3 h after irradiation. However, the expression of phospho‐eIF2α increased 20 min after irradiation and reached peak values at 12 h, remained high for upto 48 h. This result suggested that X‐ray might induce eIF2α phosphorylation in human oropharyngeal carcinoma cells by activating PERK. Furthermore, in radioresistant Detroit562R cells, the expression of PERK increased 1 h after irradiation, peaking at 3 h, and persisted up to 48 h. These results suggested that PERK‐eIF2α activation induced by X‐ray was delayed in radioresistant cells. X‐ray‐induced PERK‐eIF2α activation in FaDuP and FaDuR cells was similar to that in Detroit562P and Detroit562R cells. The mRNA levels of PERK and eIF2α were also examined and the results showed higher levels in the irradiation group than that in the control group (P < 0.05, Fig. 3b). These results suggested that X‐ray irradiation regulated radioresistance in oropharyngeal carcinoma cells by inducing PERK and phospho‐eIF2α transcription.
Figure 3.

Expression of PERK‐eIF2α in oropharyngeal carcinoma cells after irradiation. (a) Western blot results showing that the expression of PERK‐eIF2α was activated in oropharyngeal carcinoma cells at different time points after 5 Gy irradiation. Bands were quantified using ImageJ software and normalized to a loading control. Fold changes are shown in comparison to the control lane. (b) Real‐time quantitative PCR results showing that the expression of PERK‐eIF2α mRNA increased 1 h after 5 Gy irradiation. Compared with the control group, *P < 0.05, #P < 0.01. NA, not applicable.
PERK increased radioresistance in oropharyngeal carcinoma cells through the induction of eIF2α phosphorylation
To confirm the role of PERK in regulation of radioresistance in oropharyngeal carcinoma, PERK was silenced in these cells by siRNA transfection. The results showed that PERK siRNA significantly suppressed PERK protein expression (Fig. 4a).
Figure 4.

Effects of PERK on radiosensitivity in human oropharyngeal carcinoma cells. (a) Western blot results showing that transfection of PERK siRNA effectively inhibited PERK protein expression and radiation‐induced PERK protein expression. (b) Colony formation results showing that PERK silencing reduced the colony formation rates after radiotherapy. (c) Western blot results showing that 12 h after 5 Gy irradiation, PERK silencing inhibited eIF2α phosphorylation and Bcl‐2 protein expression, and increased ATM phosphorylation and cleaved caspase‐3 protein expression. The bands were quantified using ImageJ software and normalized to a loading control. Fold changes are shown relative to the negative control lane without radiation. NA, not applicable.
The colony formation assay was performed to further confirm the regulatory role of PERK in radioresistance. As shown in Figure 4b, the PERK siRNA transfection group inhibited cell colony formation after radiation in comparison to the negative siRNA group (Fig. 4b). This result indicated that PERK silencing could increase radiosensitivity. The radiosensitizing ratio in FaDuP, FaDuR, Detroit562P and Detroit562R was 1.20, 1.11, 1.29 and 1.33, respectively.
To elucidate the mechanism underlying the regulation of radioresistance in oropharyngeal carcinoma cells by PERK, western blot was performed and the results showed that PERK silencing inhibited radiation‐induced eIF2α protein phosphorylation (Fig. 4c). Considering that DNA DSB repair was one of the major causes of radioresistance and ATM phosphorylation is a DNA DSB‐sensing protein,23 we investigated the effect of PERK siRNA on ATM phosphorylation. The results showed that PERK siRNA significantly increased ATM phosphorylation induced by radiation (Fig. 4c). DNA DSB are important for cell apoptosis; therefore, expression of the apoptosis‐related proteins Bcl‐2 and cleaved‐caspase 3 was detected by western blot. The results showed that PERK siRNA inhibited radiation‐induced expression of the anti‐apoptosis protein Bcl‐2 and increased the expression of the apoptosis marker protein cleaved‐caspase 3 (Fig. 4c). These results suggested that PERK increased radioresistance in oropharyngeal carcinoma cells through eIF2α protein phosphorylation induction, leading to DNA DSB repair and cell apoptosis inhibition.
Inhibition of eIF2α phosphorylation increased radiosensitivity in oropharyngeal carcinoma cells
To confirm the role of eIF2α phosphorylation induced by PERK in radioresistance, siRNA was transfected to silence eIF2α. The western blot results showed that eIF2α siRNA inhibited eIF2α phosphorylation (Fig. 5a). Colony formation revealed that compared with the negative siRNA, eIF2α siRNA transfection inhibited cell colony formation after radiation (Fig. 5b). The radiosensitizing ratio in FaDuP, FaDuR, Detroit562P and Detroit562R was 1.25, 1.29, 1.16 and 1.28, respectively. In contrast to PERK silencing, the radiosensitizing effect by eIF2α silencing in radioresistant cells, FaDuR, was stronger than that in parental cells, FaDuP. However, the fact that eIF2α phosphorylation remained detectable after PERK gene silencing suggested that other pathways might also be involved in the regulation of abnormal eIF2α phosphorylation.
Figure 5.

Effects of eIF2α phosphorylation on radiosensitivity in oropharyngeal carcinoma cells. (a) Western blot results showing that transfection of eIF2α siRNA effectively inhibited the cellular eIF2α phosphorylation level and radiation‐induced eIF2α phosphorylation. (b) Colony formation results showing that eIF2α silencing reduced colony formation rates after radiotherapy. (c) Western blot results showing that silencing of IRE and ATF‐6 did not affect eIF2α phosphorylation. Bands were quantified using ImageJ software and normalized to a loading control. Fold changes are shown in comparison to the negative control lane without radiation. NA, not applicable.
The effects of two other important regulatory proteins in the ERS pathway, IRE1α and ATF6, on eIF2α phosphorylation were further evaluated. The western blot results showed that IRE1α and ATF6 silencing using siRNA did not influence eIF2α phosphorylation (Fig. 5c,d), which suggested that eIF2α phosphorylation might also be regulated by mechanisms or pathways other than ERS. However, this hypothesis requires further investigation.
eIF2α phosphorylation increased radioresistance of oropharyngeal carcinoma cells through activation of the NF‐κB pathway
To investigate the mechanism underlying the regulation of radioresistance in oropharyngeal carcinoma cells by eIF2α phosphorylation, cell apoptosis was detected using Annexin V/PI staining. The results showed that inhibition of eIF2α phosphorylation could increase radiation‐induced apoptosis (Fig. 6a). In addition, the cell cycle distribution was detected by PI staining, and the results showed that G2/M arrest in irradiated cells after eIF2α silencing was significantly higher than that in the irradiation group (Fig. 6b).
Figure 6.

eIF2α regulated radioresistance in oropharyngeal carcinoma cells by activating the NF‐κB pathway. Cells were transfected with siRNA to silence eIF2α and irradiated with 5 Gy X‐ray. (a) Cells were collected after 24 h to detect apoptosis using Annexin V/PI staining followed by flow cytometry analysis. (b) Cells were collected after 48 h to identify the cell cycle using PI staining followed by flow cytometry. The results are expressed as the mean ± SD of three independent experiments. Compared with the IR group, *P < 0.05, #P < 0.01. (c) Cells were transfected with siRNA to silence eIF2α and irradiated with 5 Gy for 12 h. Nuclear and cytoplasm proteins were extracted to detect the expression of phospho‐p65 and its downstream protein HIF‐1α. Total protein was extracted to evaluate the expression of Rad50, Mre11, Nbs1 and phospho‐ATM proteins. Western blot results showed that eIF2α silencing inhibited radiation‐induced nuclear phospho‐p65 protein expression. Conversely, radiation inhibited cytoplasmic p65 protein phosphorylation, whereas eIF2α silencing reversed this function. These results indicated that eIF2α silencing inhibited radiation‐induced p65 nuclear translocation. In addition, eIF2α silencing inhibited the radiation‐induced HIF‐1α, Rad50 and Nbs1 expression, increased radiation‐induced phospho‐ATM expression, and did not affect Mre11 protein expression. (d) After eIF2α was silenced by siRNA transfection, the cells were irradiated (5 Gy, 1 h). The real‐time quantitative PCR results showed that eIF2α silencing decreased the radiation‐induced expression of p65 mRNA expression. Compared to the irradiated group, *P < 0.05 and # P < 0.01. (e) Immunofluorescence studies showed that after oropharyngeal carcinoma cells received 5 Gy radiation for 1 h, the γ‐H2AX foci in nucleus increased (the blue background indicates the cell nucleus, and light red dots indicate γ‐H2AX foci). In addition, the effect of radiation after eIF2α silencing was more evident than that of simple radiation. (f) The western blot results showed that eIF2α silencing inhibited radiation (5 Gy, 12 h)‐induced phospho‐cdc2, Cyclin B, Bcl‐2 and Bcl‐xL protein expression and increased radiation‐induced cleaved‐Caspase 3 and cleaved‐PARP protein expression. The above functions were inhibited by pretreatment of the oropharyngeal carcinoma cells with the NF‐κB activator TNFα (10 ng/mL) and the ATM inhibitor Ku55933 (10 μmol/L) for 12 h. Bands were quantified using ImageJ software and normalized to a loading control. Fold changes are shown compared with the negative control lane without radiation. NA, not applicable.
Previous studies have demonstrated that eIF2α phosphorylation can activate NF‐κB in human embryonic kidney cells.14, 15 NF‐κB is present as a homodimer or a heterodimer with members of the Rel protein family. The distribution and function of p50/p65 are most extensive, but only the terminus of p65 contains a transactivation domain and activates gene transcription; thus, it is the major component of active forms of NF‐κB. Therefore, we detect p65 protein, which served as the indicator of NF‐κB. As shown in Figure 6c, eIF2α silencing inhibited radiation‐induced nuclear phospho‐p65 protein expression. Conversely, radiation inhibited cytoplasmic p65 protein phosphorylation, whereas eIF2α silencing reversed this function. These results indicated that eIF2α silencing inhibited radiation‐induced p65 nuclear translocation. The real‐time quantitative PCR results showed that eIF2α silencing decreased p65 mRNA levels in oropharyngeal carcinoma cells treated with radiation (Fig. 6d), further suggesting that eIF2α participated in the regulation of radioresistance in human oropharyngeal carcinoma cells by regulating the aberrant activation of radiation‐induced NF‐κB. In addition, eIF2α silencing inhibited the radiation‐induced NF‐κB activation of the downstream hypoxia‐related protein HIF‐1α in oropharyngeal carcinoma cells (Fig. 6c).
Our previous result showed that NF‐κB could regulate the phosphorylation of the radiation‐induced DNA DSB‐related protein ATM in lymphoma cells.10 We speculated that inhibition of eIF2α phosphorylation could inhibit DNA DSB repair to increase radiosensitivity through inhibition of NF‐κB activation. The western blot results showed that eIF2α silencing inhibited radiation‐induced expression of the DNA DSB repair‐related proteins NBS1 and Rad50 and increased the expression of phosphorylated ATM, but it had no significant effect on Mre11 (Fig. 6c). In eukaryotic cells, DSB caused by exogenous chemical, physical and biological substances induced rapid phosphorylation of H2AX to form γ‐H2AX. The formation of γ‐H2AX is a marker of the DSB damage in DNA. Immunofluorescence studies clearly showed DNA DSB regions in the nucleus characterized by the formation of γ‐H2AX foci after radiation. In addition, the effect of radiation after eIF2α silencing was more evident than that of simple radiation (Fig. 6e).
We also previously showed that NF‐κB participated in the regulation of radioresistance in lymphoma cells by mediating cell apoptosis and cell cycle arrest.9, 10, 11 In addition, cell cycle arrest could be regulated by the DNA DSB marker protein ATM.10 Similarly, the present study demonstrated that eIF2α silencing suppressed radiation‐induced expression of the G2/M phase arrest‐related proteins cyclin B1 and phosphor‐Cdc2, inhibited radiation‐induced expression of the anti‐apoptosis proteins Bcl‐2 and Bcl‐xL, and increased radiation‐induced expression of the apoptosis‐related proteins cleaved Caspase‐3 and cleaved PARP. Further application of NF‐κB activator TNFα and ATM inhibitor Ku55933 could inhibit this function (Fig. 6d).
Discussion
In recent years, human papilloma virus (HPV) infection has become an important cause of oropharyngeal carcinoma. The rate of HPV infection in oropharyngeal squamous cell carcinoma reaches 70–80% in western countries24 but only 20% in China25 The prognosis of HPV (+) oropharyngeal carcinoma patients is better than that of HPV (−) patients by radiotherapy,26 suggesting that HPV (+) oropharyngeal squamous cell carcinoma has high intrinsic radiosensitivity.27 Patients with HPV (+) oropharyngeal carcinoma have high expression levels of p16.28 Consequently, detection of p16 expression can serve as a powerful marker of HPV infection.29 The ER is the major location of virus replication. Recent studies indicated that HPV could cause misfolded and unfolded proteins in infected cells to induce ERS and participate in the regulation of the survival and death of tumor cells.30 The regulation of ERS by HPV infection had tumor specificity. In addition, ERS was inhibited in HPV16 (+) genital epithelial keratinocytes.31 The results of the present study showed that PERK did not correlate with p16 expression in oropharyngeal carcinoma. This result might be associated with the smaller proportion of HPV‐positive cases in this study (36/151, 24.5%) and still requires further study. In addition, together with the epidemiological characteristics in the local area, HPV (−) oropharyngeal squamous cell carcinoma cells with a poor prognosis were selected in this study, and radioresistant cells were constructed to search for effective methods to increase radiosensitivity.
Increasing evidence indicates that ERS plays important roles in the oncogenesis and development of malignant tumors. ER homeostasis is abrogated by stimuli such as radiation, drugs and inflammatory responses, leading to an accumulation of misfolded proteins in the ER that causes the activation of cytoprotective signaling cascade reactions: the UPR. Therefore, UPR interference can be one of the major treatment methods for tumors. The UPR utilizes three transmembrane proteins that are localized in the ER as its effectors (i.e. PERK, IRE1α and ATF6). PERK activates eIF2α to increase ATF4 expression, which then binds to the promoter of GRP7832 to increase GRP78 and induce the reduction of nascent protein translation. GRP78 overexpression is associated with a poor prognosis of head and neck cancer.33 Together with the regulation of GRP78 by PERK, we hypothesized that PERK expression correlated with the prognosis of oropharyngeal carcinoma. The present results showed that PERK overexpression was associated with a poor prognosis of oropharyngeal carcinoma after radiotherapy (Fig. 1b). In addition, the levels of PERK expression were significantly higher in patients with oropharyngeal carcinoma with radioresistance than in those with radiosensitivity (Table 1). Our cytological studies showed that PERK inhibition could increase radiosensitivity by interfering with DNA damage/repair (Fig. 4). In contrast, Kim et al.33 showed that PERK activation could increase radiosensitivity in caspase‐3/7‐deficient breast cancer cells by inducing autophagy. Therefore, the regulation of radiosensitivity by PERK may be associated with different tumor types and pathways.
After ERS causes the accumulation of unfolded proteins in the ER, PERK autophosphorylates the cytoplasmic kinase domain to induce eIF2α phosphorylation. Phosphorylated eIF2a protein can inhibit the exchange between GDP and GTP in the translation initiation complex to block the assembly of the translation initiation complex eIF2‐GTP‐general tRNAMet, thus inhibiting protein translation and synthesis, reducing the influx of nascent proteins into the ER and further decreasing the increased accumulation of unfolded proteins.2 Early study results showed that eIF2α phosphorylation could activate NF‐κB,14 which was associated with ERS and nutritional deprivation. The specific underlying mechanism might be associated with NF‐κB translocation from the cytoplasm into the nucleus to activate transcription after dissociation from IκB by eIF2α phosphorylation; however, it was not associated with the degradation of IκBα by IKK phosphorylation in the canonical pathway.15 Jiang and Wek further showed that UV irradiation inhibited IκBα protein synthesis by phosphorylating eIF2α and, thus, inducing NF‐κB activation to regulate cell apoptosis. This process was not associated with the eIF2α downstream target factors ATF4 and CHOP.14 NF‐κB plays important roles in immune responses, cell proliferation, apoptosis, and radiotherapy and chemotherapy sensitivity in tumors by regulating more than 150 effector genes. Our previous studies have demonstrated that abnormal NF‐κB activation can induce the transcription of HIF‐1α, which can decrease radiotherapy‐induced apoptosis in lymphoma cells.9 Furthermore, van Uden et al.34 have reported that the HIF‐1α promoter contains an NF‐κB binding site, and this region plays an important role in the regulation of HIF‐1α transcription. Our study results also showed that eIF2α silencing increased radiosensitivity, inhibited radiation‐induced activation of NF‐κB and the downstream target gene HIF‐1α, and induced cell apoptosis and G2/M cell cycle arrest. In addition, after application of the NF‐κB activator TNFα, eIF2α silencing offset the effect of radiotherapy in the induction of the cell apoptosis and the G2/M phase arrest.
After exposure to ionizing irradiation, tumor cells will activate a series of stress factors to produce a DNA repair response to maintain cell proliferation and inhibit apoptosis, resulting in radioresistance. Loss of DNA repair ability is closely associated with increased radiosensitivity.35 The Mre11‐Rad50‐Nbs1 complex first senses DSB, plays important roles in the DNA damage/repair pathway, participates in DNA replication, maintains telomere stability, and regulates cell cycle check point signal transduction pathways.36, 37 Interference with Nbs1 and Rad50 expression can increase the radiosensitivity of head and neck carcinoma cells.38, 39 The Mre11‐Rad50‐Nbs1 complex can further activate ATM protein36, 37 ATM senses radiation‐induced DNA damage and transmits the DNA damage signaling to downstream target proteins through phosphorylation to activate the stress system and cause cell cycle arrest, thus completing DNA repair or promoting cell apoptosis.40 Our previous studies have revealed that ATM phosphorylation induces G2/M arrest in lymphoma cells to increase radiosensitivity.10 The present study provided results similar to those of previous studies. Silencing of eIF2α could inhibit radiation‐induced Rad50 and Nbs1, and induce ATM expression and cause G2/M phase arrest and cell apoptosis in oropharyngeal carcinoma cells after radiotherapy. Further application of the ATM inhibitor Ku55933 inhibited the effect of eIF2α phosphorylation on radiation‐induced expression of the G2/M phase arrest‐related proteins phospho‐cdc2 and cyclin B1 and the apoptosis‐related proteins Bcl‐2, Bcl‐xL, cleaved caspase3 and cleaved‐PARP. These results confirmed that through regulation of the DNA DSB repair proteins Rad50, Nbs1 and ATM, eIF2α caused cell cycle arrest and induced cell apoptosis to increase radiosensitivity.
In this study, we first demonstrated that PERK overexpression correlated with a poor prognosis of oropharyngeal carcinoma patients with radiotherapy. In addition, the expression level of PERK in patients with radioresistant oropharyngeal carcinoma was higher than that in patients with radiosensitivity. Studies in cell lines revealed an overexpression of PERK‐eIF2α in radioresistant cells. Furthermore, radiation induced PERK‐eIF2α activation in a time‐dependent manner, suggesting that PERK‐eIF2α overexpression was associated with radioresistance in tumor cells. We also showed that PERK was involved in the regulation of radioresistance in oropharyngeal carcinoma through phosphorylation of eIF2α. This mechanism may be mediated by the induction of NF‐κB pathway activation by eIF2α phosphorylation, thus activating radiation‐induced DNA DSB repair and interfering with cell apoptosis and cell cycle arrest.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81402521).
Cancer Sci 108 (2017) 1421–1431
Funding Information
National Natural Science Foundation of China (Grant/Award Number: 81402521).
References
- 1. Jemal A, Siegel R, Ward E et al Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000; 5: 897–904. [DOI] [PubMed] [Google Scholar]
- 3. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer 2004; 4: 966–77. [DOI] [PubMed] [Google Scholar]
- 4. Qi L, Wu P, Zhang X et al Inhibiting ERp29 expression enhances radiosensitivity in human nasopharyngeal carcinoma cell lines. Med Oncol 2012; 29: 721–8. [DOI] [PubMed] [Google Scholar]
- 5. Nagelkerke A, Bussink J, van der Kogel AJ, Sweep FC, Span PN. The PERK/ATF4/LAMP3‐arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother Oncol 2013; 108: 415–21. [DOI] [PubMed] [Google Scholar]
- 6. Drake TM, Ritchie JE, Kanthou C, Staves JJ, Narramore R, Wyld L. Targeting the endoplasmic reticulum mediates radiation sensitivity in colorectal cancer. Exp Mol Pathol 2015; 98: 532–9. [DOI] [PubMed] [Google Scholar]
- 7. Pang XL, He G, Liu YB, Wang Y, Zhang B. Endoplasmic reticulum stress sensitizes human esophageal cancer cell to radiation. World J Gastroenterol 2013; 19: 1736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu R, Zhang L, Yang J et al HIV protease inhibitors sensitize human head and neck squamous carcinoma cells to radiation by activating endoplasmic reticulum stress. PLoS ONE 2015; 10: e0125928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qiao Q, Nozaki Y, Sakoe K, Komatsu N, Kirito K. NF‐kappaB mediates aberrant activation of HIF‐1 in malignant lymphoma. Exp Hematol 2010; 38: 1199–208. [DOI] [PubMed] [Google Scholar]
- 10. Qiao Q, Jiang Y, Li G. Curcumin enhances the response of non‐Hodgkin's lymphoma cells to ionizing radiation through further induction of cell cycle arrest at the G2/M phase and inhibition of mTOR phosphorylation. Oncol Rep 2013; 29: 380–6. [DOI] [PubMed] [Google Scholar]
- 11. Qiao Q, Jiang Y, Li G. Curcumin improves the antitumor effect of X‐ray irradiation by blocking the NF‐kappaB pathway: an in‐vitro study of lymphoma. Anticancer Drugs 2012; 23: 597–605. [DOI] [PubMed] [Google Scholar]
- 12. Qiao Q, Jiang Y, Li G. Inhibition of the PI3K/AKT‐NF‐kappaB pathway with curcumin enhanced radiation‐induced apoptosis in human Burkitt's lymphoma. J Pharmacol Sci 2013; 121: 247–56. [DOI] [PubMed] [Google Scholar]
- 13. Quintana A, Aviles FX, Terra X et al Overexpression of the nuclear factor‐kappa B (p65) in association with local failure in patients with head and neck carcinoma undergong radiotherapy or chemoradiotherapy. Head Neck 2013; 35: 370–5. [DOI] [PubMed] [Google Scholar]
- 14. Jiang HY, Wek RC. GCN2 phosphorylation of eIF2alpha activates NF‐kappaB in response to UV irradiation. Biochem J 2005; 385: 371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang HY, Wek SA, McGrath BC et al Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF‐kappaB in response to diverse cellular stresses. Mol Cell Biol 2003; 23: 5651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. To EW, Chan KC, Leung SF et al Rapid clearance of plasma Epstein‐Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin Cancer Res 2003; 9: 3254–9. [PubMed] [Google Scholar]
- 17. Jiang Y, Han Y, Sun C et al Rab23 is overexpressed in human bladder cancer and promotes cancer cell proliferation and invasion. Tumour Biol 2016; 37: 8131–8. [DOI] [PubMed] [Google Scholar]
- 18. Hara A, Okayasu I. Cyclooxygenase‐2 and inducible nitric oxide synthase expression in human astrocytic gliomas: correlation with angiogenesis and prognostic significance. Acta Neuropathol 2004; 108: 43–8. [DOI] [PubMed] [Google Scholar]
- 19. Mihatsch J, Toulany M, Bareiss PM et al Selection of radioresistant tumor cells and presence of ALDH1 activity in vitro . Radiother Oncol 2011; 99: 300–6. [DOI] [PubMed] [Google Scholar]
- 20. Nagelkerke A, Sweep FC, Stegeman H et al Hypoxic regulation of the PERK/ATF4/LAMP3‐arm of the unfolded protein response in head and neck squamous cell carcinoma. Head Neck 2015; 37: 896–905. [DOI] [PubMed] [Google Scholar]
- 21. Wu MJ, Jan CI, Tsay YG et al Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol Cancer 2010; 9: 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li F, Zheng X, Liu Y et al Different roles of CHOP and JNK in mediating radiation‐induced autophagy and apoptosis in breast cancer cells. Radiat Res 2016; 185: 539–48. [DOI] [PubMed] [Google Scholar]
- 23. Shrivastav M, Miller CA, De Haro LP et al DNA‐PKcs and ATM co‐regulate DNA double‐strand break repair. DNA Repair 2009; 8: 920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chaturvedi AK, Engels EA, Pfeiffer RM et al Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011; 29: 4294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang H, Zhang B, Chen W, Zou SM, Xu ZG. Relationship between HPV‐DNA status and p16 protein expression in oropharyngeal squamous cell carcinoma and their clinical significance. Zhonghua Zhong Liu Za Zhi 2013; 35: 684–8. [In Chinese.] [PubMed] [Google Scholar]
- 26. Lassen P. The role of Human papillomavirus in head and neck cancer and the impact on radiotherapy outcome. Radiother Oncol 2010; 95: 371–80. [DOI] [PubMed] [Google Scholar]
- 27. Tuttle S, Hertan L, Daurio N et al The chemopreventive and clinically used agent curcumin sensitizes HPV (−) but not HPV (+) HNSCC to ionizing radiation, in vitro and in a mouse orthotopic model. Cancer Biol Ther 2012; 13: 575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chung CH, Gillison ML. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin Cancer Res 2009; 15: 6758–62. [DOI] [PubMed] [Google Scholar]
- 29. Lassen P, Overgaard J, Eriksen JG. Expression of EGFR and HPV‐associated p16 in oropharyngeal carcinoma: correlation and influence on prognosis after radiotherapy in the randomized DAHANCA 5 and 7 trials. Radiother Oncol 2013; 108: 489–94. [DOI] [PubMed] [Google Scholar]
- 30. Chu HH, Bae JS, Kim KM et al Expression of CHOP in squamous tumor of the uterine cervix. Korean J Pathol 2012; 46: 463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sudarshan SR, Schlegel R, Liu X. The HPV‐16 E5 protein represses expression of stress pathway genes XBP‐1 and COX‐2 in genital keratinocytes. Biochem Biophys Res Commun 2010; 399: 617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagelkerke A, Bussink J, Mujcic H et al Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3‐arm of the unfolded protein response. Breast Cancer Res 2013; 15: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Endoplasmic reticulum stress mediates radiation‐induced autophagy by perk‐eIF2alpha in caspase‐3/7‐deficient cells. Oncogene 2010; 29: 3241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia‐inducible factor‐1alpha by NF‐kappaB. Biochem J 2008; 412: 477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chavaudra N, Bourhis J, Foray N. Quantified relationship between cellular radiosensitivity, DNA repair defects and chromatin relaxation: a study of 19 human tumour cell lines from different origin. Radiother Oncol 2004; 73: 373–82. [DOI] [PubMed] [Google Scholar]
- 36. Lamarche BJ, Orazio NI, Weitzman MD. The MRN complex in double‐strand break repair and telomere maintenance. FEBS Lett 2010; 584: 3682–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Williams GJ, Lees‐Miller SP, Tainer JA. Mre11‐Rad50‐Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double‐strand breaks. DNA Repair 2010; 9: 1299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rhee JG, Li D, Suntharalingam M, Guo C, O'Malley BW Jr, Carney JP. Radiosensitization of head/neck squamous cell carcinoma by adenovirus‐mediated expression of the Nbs1 protein. Int J Radiat Oncol Biol Phys 2007; 67: 273–8. [DOI] [PubMed] [Google Scholar]
- 39. Chang L, Huang J, Wang K et al Targeting Rad50 sensitizes human nasopharyngeal carcinoma cells to radiotherapy. BMC Cancer 2016; 16: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang M, Miao ZH, Zhu H, Cai YJ, Lu W, Ding J. Chk1 and Chk2 are differentially involved in homologous recombination repair and cell cycle arrest in response to DNA double‐strand breaks induced by camptothecins. Mol Cancer Ther 2008; 7: 1440–9. [DOI] [PubMed] [Google Scholar]