

Abstract
Paclitaxel is not as effective for neuroblastoma as most of the front‐line chemotherapeutics due to drug resistance. This study explored the regulatory mechanism of paclitaxel‐associated autophagy and potential solutions to paclitaxel resistance in neuroblastoma. The formation of autophagic vesicles was detected by scanning transmission electron microscopy and flow cytometry. The autophagy‐associated proteins were assessed by western blot. Autophagy was induced and the autophagy‐associated proteins LC3‐I, LC3‐II, Beclin 1, and thioredoxin‐related protein 14 (TRP14), were found to be upregulated in neuroblastoma cells that were exposed to paclitaxel. The inhibition of Beclin 1 or TRP14 by siRNA increased the sensitivity of the tumor cells to paclitaxel. In addition, Beclin 1‐mediated autophagy was regulated by TRP14. Furthermore, the TRP14 inhibitor suberoylanilide hydroxamic acid (SAHA) downregulated paclitaxel‐induced autophagy and enhanced the anticancer effects of paclitaxel in normal control cancer cells but not in cells with upregulated Beclin 1 and TRP14 expression. Our findings showed that paclitaxel‐induced autophagy in neuroblastoma cells was regulated by TRP14 and that SAHA could sensitize neuroblastoma cells to paclitaxel by specifically inhibiting TRP14.
Keywords: Autophagy, drug resistance, inhibitor, neuroblastoma, paclitaxel
Neuroblastoma (NB) is an aggressive malignant tumor commonly seen in childhood, and the long‐term survival rate of high‐risk patients is still poor.1, 2 NB is a chemotherapy‐sensitive tumor, and chemotherapy plays an important role in the treatment of patients with NB.3 The effective chemotherapy regimens for NB primarily include cyclophosphamide, ifosfamide, adriamycin, vincristine, etoposide, and platinum. However, few effective drugs are available for patients who are resistant to front‐line chemotherapy.4 Paclitaxel (PTX), which is a cytotoxic agent that can bind to the spindle microtubules of tumor cells, causes mitotic arrest and cell death,5 and has been widely used to treat many adult tumors.6, 7, 8, 9 In a phase II investigational window study, an objective response was documented in 25% of children with NB who received PTX alone.10 This finding suggests that PTX is not as effective as most front‐line chemotherapeutics in the treatment of NB. The sensitization of NB to paclitaxel would provide more choices in clinical practice.
Autophagy is a physiological process that occurs during metabolic stress, such as in times of energy deficiency and hunger.11 It is also closely associated with tumor progression.12 Recent studies have found that autophagy is one of the modalities of cell death that occurs after chemotherapy and that it is one of the mechanisms underlying chemoresistance in tumor cells.13, 14, 15, 16 The Beclin 1 gene, which was the first gene reported to be associated with autophagy in mammalian cells, is homologous to the autophagy‐related Atg6 gene in yeast.17 Currently, studies on the Beclin 1 protein are primarily focused on autophagy regulation.18 Unfortunately, the association between autophagy and NB sensitivity to chemotherapy has yet to be investigated.
Thioredoxin‐related protein 14 (TRP14) is a disulfide reductase with a molecular mass of 14 kDa that was recently found to be expressed in the human HeLa cell line; however, this protein is also expressed in multiple types of human tissues.19 A crystal structure study showed that the sequence similarity between TRP14 and human thioredoxin (Trx) 1 is only 37%, but these two proteins have similar spatial topological structures and active centers. Thus, TRP14 belongs to the Trx family.20 The function of TRP14 has not yet been fully elucidated, but TRP14 may participate in the regulation of the tumor necrosis factor alpha (TNF‐α) signaling pathway,21 which is associated with the process of autophagy. We hypothesize that TRP14 plays a role in Beclin1‐mediated autophagy in NB.
Suberoylanilide hydroxamic acid (SAHA) is a broad‐spectrum hydroxamic histone deacetylase (HDAC) inhibitor that may inhibit Trx activity by increasing the expression level of the endogenous Trx suppressor thioredoxin‐binding protein‐2.22 SAHA can also slow cell growth and induce cell differentiation and apoptosis.23 Furthermore, SAHA may be used in combination with various chemotherapeutics to promote chemotherapy sensitivity and produce a synergistic effect in tumors.24, 25 Whether SAHA can inhibit TRP14‐induced autophagy has not been investigated. The present study explores the regulatory mechanism that underlies PTX‐induced autophagy and whether SAHA can sensitize neuroblastoma to paclitaxel through the inhibition of TRP14‐mediated autophagy.
Methods
Ethics approval
Ethics approval was obtained from the Institutional Review Board of Sun Yat‐Sen University in China, and the study was conducted according to the principles of the Declaration of Helsinki.26
Materials
SK‐N‐SH and QDDQ‐NM cells, in which MYCN was amplified and expressed at normal levels, respectively, were supplied by the Cell Bank of the Chinese Academy of Sciences (Beijing, China). The methyl thiazolyl tetrazolium (MTT), monodansylcadaverine (MDC) and Alexa Fluor 430‐A were purchased from Sigma (San Francisco, CA, USA). The microplate reader and MetaMorph offline 7.7.8.0 software packagewere purchased from Bio‐Rad (Hercules, CA, USA). The transmission electron microscope was purchased from NIKON (TKY, Japan). The Beclin 1 siRNA and TRP14 siRNA were both purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and the suberoylanilide hydroxamic acid was purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
Cells were cultured in DMEM‐F12 medium supplemented with 10% fetal calf serum. The cells were maintained in a 5% CO2 incubator under saturated humidity at 37°C and were passaged as previously described.27 Cells in the logarithmic growth phase were used for the experiment.
MTT assay
After digestion and centrifugation, SK‐N‐SH and QDDQ‐NM cells were seeded in a 96‐well plate where each well contained 100 μL culture solution and 1 × 104 cells. The plates were maintained in an incubator overnight in an atmosphere of CO2 to achieve full cell attachment. PTX was added at a final concentration of 0, 2, 5, 10, 50, or 100 nM in triplicate. After 24 h of cell culture, 20 μL methyl thiazolyl tetrazolium (MTT) was added to each well and the cells were allowed to incubatefor 4‐6 h, after which the culture solution was discarded. A total of 150 μL dimethyl sulfoxide was then added to each well. The plate was subjected to vibration on an oscillator for 10 min to fully dissolve the formazan. The absorbanceat 570 nm was then measured in a microplate reader.
Western blot
The cells in each group were collected, rinsed with PBS, and centrifuged before the addition of lysis buffer to extract the protein. The protein purity was evaluated by the Bradford method.28 After albuminous degeneration, an equal amount of protein (20 μg) from each group was separated by SDS‐PAGE. The separated proteins were transferred onto PDVF membranes and blocked with skim milk powder for 4 h. The primary antibody was added at an appropriate diluted concentration; the blots were incubated overnight, rewarmed for 1 h, and then rinsed with TBS‐T three times for 15 min each time. Then, the second antibody was added at an appropriate diluted concentration; the blots were incubated for 2 h, rinsed with TBS‐T, stained, and exposed.
Scanning transmission electron microscopy
SK‐N‐SH and QDDQ‐NM cells in the logarithmic growth phase were seeded in a 6‐well plate at a density of 1 × 105 cells/well for overnight culture. After they were cultured with PTX (10 nM) for 24 h, the cells were collected and fixed in 2.5% glutaraldehyde and 1% osmic acid before elution with an ethanol gradient. After infiltration, embedding, and aggregation, ultrathin sections (50 nm) were obtained and double‐stained with uranyl acetate and lead citrate. Finally, the cell ultrastructure was observed under a transmission electron microscope and was video‐recorded.
Flow cytometry and confocal microscopy
Flow cytometric analysis was performed to identify apoptotic cells as described.29 To quantify the cells with autophagic vacuoles, the fluorochrome MDC was used to stain the vacuoles. Staining was detected by flow cytometry, which measured the absorbance at 488 nm as previously described.30 To further determine the autophagic vacuoles, LysoTracker staining method was used and detected by confocal microscopy as previously described.31
RNA transference
Tumor cells were seeded in a 6‐well plate at a density of 4 × 105 cells/well. After 24 h of culture, tumor cells were transfected with two small interfering RNA (siRNA) to produce NB cells with siBeclin 1a and siBeclin 1b, as previously described.32 Similarly, tumor cells were transfected with siRNA to produce NB cells with siTRP14a and TRP14b. After transfection for 72 h, the cells in each group were harvested, and the expression of Beclin 1 and TRP14 was detected by western blot.
SAHA exposure
Tumor cells in the logarithmic growth phase were seeded in a 96‐well plate at a density of 1.0 × 104 cells/well, after which the plates were maintained in an incubator overnight. The experiment was conducted after cell attachment occurred. The cells were exposed to PTX at doses of 0, 2, 5, 10, 50, or 100 nM and 0.25 μmol/L SAHA, either alone or in combination. After 24 h of culture, the viability of the SK‐N‐SH and QDDQ‐NM was evaluated by MTT assay. The expression of LC3‐I, LC3‐II, Beclin 1, and TRP14 in the cells in each group was investigated by western blot. To further determine the inhibitory effect of SAHA on TRP14, the viability of TRP14low cells was reevaluated by MTT assay after exposure to PTX and SAHA, either alone or in combination.
Transduction
SK‐N‐SH and QDDQ‐NM cells were seeded in a 24‐well plate at a density of 1 × 104 cells/well in 500 μL complete culture solution. When cell confluence reached 60–80%, the cells were transducted according to the lentivirus transduction manual; the purpose of the transduction was to upregulate the Beclin 1 and TRP14 genes and to establish Beclin 1high and TRP14high tumor cells, as previously described.33 The empty vector LacZ was used as a control construct. Beclin 1high and TRP14high NB cells were treated with 10 nM PTX and 0.25 μmol/L SAHA. After 24 h of culture, the expression of LC3‐I, LC3‐II, Beclin 1, and TRP14 was detected by western blot. To determine the effect of SAHA on the chemotherapy sensitivity of Beclin 1high and TRP14high cells to PTX, these cells were treated with PTX at doses of 0, 2, 5, 10, 50, or 100 nM and SAHA, either alone or in combination. Cell viability was then evaluated by MTT assay.
Statistical analysis
Data are given as the means ± SEM. For statistical comparisons, a one‐way ANOVA followed by Tukey's test was performed using SPSS software (SPSS GmbH Software, Munich, Germany). P < 0.05 was considered significant.
Results
PTX induced the expression of autophagy‐associated proteins in NB cells
First, the viability of tumor cells treated with PTX was analyzed by MTT assay. As the PTX dose increased, the viability of both SK‐N‐SH and QDDQ‐NM cells decreased, which demonstrates dose dependence (Fig. 1a). Flow cytometry also showed that apoptosis was enhanced as the PTX dose increased (Fig. 1b). However, PTX at a concentration as high as 100 nM could not completely inhibit the growth of NB cells, and the SK‐N‐SH and QDDQ‐NM cells still had viability rates of 43.7% and 45.6%, respectively. Similarly, the apoptosis was not further enhanced concurrent with the increased PTX dose at high levels. A western blot showed that the levels of LC3‐I, LC3‐II, Atg 5, Beclin 1, TRP14 and LC3‐II‐to‐LC3‐I ratio in NB cells were increased after PTX exposure in a dose‐dependent manner (Fig. 1c–e).
Figure 1.
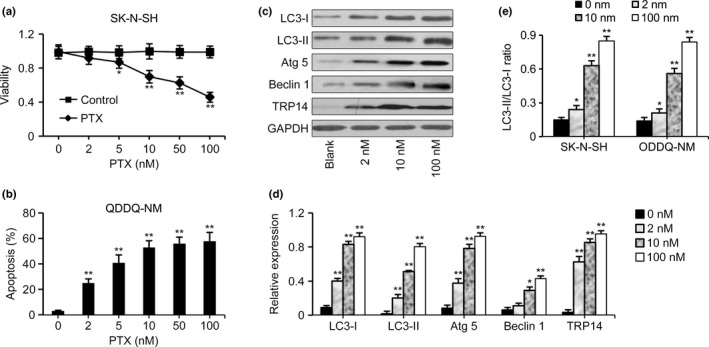
Expression of autophagy‐associated proteins induced by paclitaxel (PTX). (a) An MTT assay showed the viability of SK‐N‐SH cells exposed to PTX at the doses of 2, 5, 10, 50 and 100 nM. As the PTX dose increased, cell viability decreased. However, neuroblastoma (NB) cells still had viability rates of 43.7% even when they were treated with PTX at a concentration as high as 100 nM. (b) The apoptosis of QDDQ‐NM cells was not further enhanced concurrent with the increased PTX dose at high levels. (c, d) Expression of autophagy‐associated proteins in SK‐N‐SH cells as detected by western blot. The expression of LC3‐I, LC3‐II, Atg 5, Beclin 1, and TRP14 in NB cells was significantly upregulated after PTX exposure in a dose‐dependent manner. (e) Increased LC3‐II‐to‐LC3‐I ratio in QDDQ‐NM cells exposed to PTX. The data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, two‐tailed t test.
PTX induced vacuole formation and increased autophagic flux in NB cells
To further analyze the autophagy induced by PTX in NB cells, the formation of autophagic vesicles was evaluated by scanning transmission electron microscopy. Large vacuoles with a double‐membrane were observed in the cytoplasm of SK‐N‐SH and QDDQ‐NM cells after PTX exposure (Fig. 2a). LysoTracker and MDC+staining found that the percentage of cells with vacuoles was higher in NB cells exposed to PTX (10 nM) compared with cells that were not exposed to PTX (Fig. 2b–d). To determine the increased cellular autophagic flux, the expression of LC3‐II and p62 proteins was detected by western blot. We found that the expression of LC3‐II was consistent with that of p62 in NB cells exposed (or not) to PTX, with or without the addition of the lysosomal inhibitor BafA1 (Fig. 2e,f). However, the increased expression levels of LC3‐II and p62 that resulted from BafA1 treatment were higher in NB cells exposed to PTX compared with cells that were not exposed to PTX (Fig. 2e,f). Thus, the autophagic vacuoles induced by PTX exposure in NB cells were increased due to the increased cellular autophagic flux and were not increased due to the inhibition of the degradation of the autophagic vacuoles.
Figure 2.
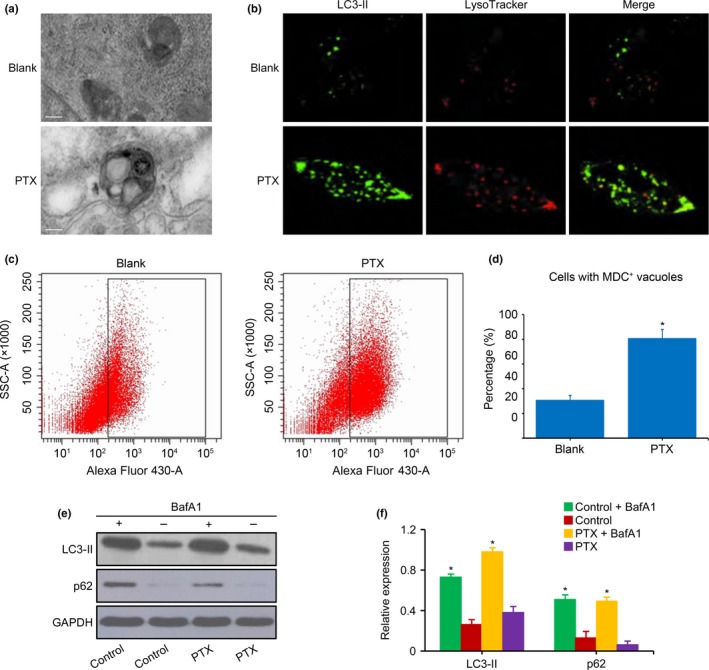
Vacuole and autophagic flux in neuroblastoma (NB) cells exposed to paclitaxel (PTX). (a) Representative ultrastructure of SK‐N‐SH cells was analyzed by transmission electron microscopy. Large vacuoles with a double‐membrane were observed in the cytoplasm of cells exposed to PTX (10 nM), whereas the membranes, organelles, and nuclei were normal in cells not exposed to PTX. (b) Cells with vacuoles detected by LysoTracker staining in QDDQ‐NM cells exposed to PTX (10 nM). (c,d) Quantification of the autophagosomes in SK‐N‐SH cells as determined by flow cytometry. The percentage of NB cells with MDC + vacuoles was higher after exposure to PTX (10 nM) than in NB cells that were not exposed to PTX. (e,f) LC3‐II and p62 in QDDQ‐NM cells measured by western blot. The increased levels of LC3‐II and p62 expression that resulted from BafA1 treatment were higher in NB cells exposed to PTX (10 nM) compared with cells that were not exposed to PTX. The data represent the mean ± SEM of three independent experiments. Scale bar is 2 μm. *P < 0.01, two‐tailed t‐test.
Inhibition of autophagy increased the sensitivity of tumor cells to PTX
To elucidate the underlying mechanisms of the autophagy induced by PTX, the expression of Beclin 1 and TRP14 was interrupted by specific siRNAs. Western blot showed that compared with the nontargeting siRNA control, the expression of Beclin 1 was significantly decreased in SK‐N‐SH and QDDQ‐NM cells treated with siBeclin 1. However, the expression of TRP14 was unchanged (Fig. 3a). In NB cells treated with siTRP14, the expression of both Beclin 1 and TRP14 was significantly decreased (Fig. 3b). When either Beclin 1 or TRP14 was downregulated, the expression of LC3‐I, LC3‐II and Atg 5 was inhibited in tumor cells exposed to PTX (Fig. 3a,b), and the sensitivity of cells to PTX was significantly increased (Fig. 3c,d). Additionally, the autophagy inhibitor 3‐methyladenine (3‐MA) exerted similar effect as siBeclin 1 and siTRP14 on the increased sensitivity of cells to PTX (Fig. 3c,d).
Figure 3.
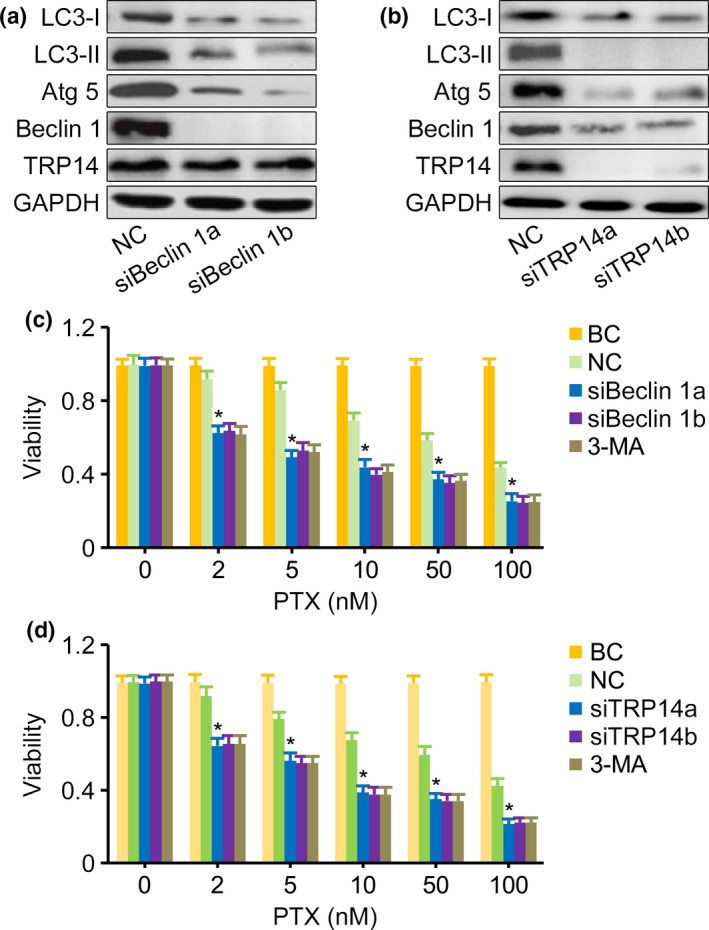
Downregulation of both Beclin 1 and TRP14 inhibited autophagy. (a) Western blot showed that the expression of Beclin 1, LC3‐I, LC3‐II and Atg 5 was significantly decreased in siBeclin 1 SK‐N‐SH cells exposed to paclitaxel (PTX) (10 nM) compared with cells in which Beclin 1 was not downregulated. (b) Western blot showed that the expression of TRP14, Beclin 1, LC3‐I, LC3‐II and Atg 5 was significantly decreased in siTRP14 QDDQ‐NM cells exposed to PTX (10 nM) compared with those in which TRP14 was not downregulated. (c) MTT assay showed that the viability of siBeclin 1‐treated SK‐N‐SH cells that were exposed to PTX was significantly decreased compared with the viability of cells in which Beclin 1 was not downregulated. SiRNAs alone had no effect on the growth of cells treated with siBeclin. (d) MTT assay showed that the viability of siTRP14‐treated QDDQ‐NM cells that were also exposed to PTX was significantly decreased compared with the viability of cells in which TRP14 was not downregulated. SiRNAs alone had no effect on the growth of cells treated with siTRP14. GAPDH was used as the loading control. NC, nontargeting siRNA control. BC, blank control, Beclin 1low or siTRP14low cells without PTX exposure. 3‐MA, 3‐methyladenine. The results are presented as the mean ± SEM of three independent experiments. *P < 0.01, two‐tailed t‐test.
SAHA inhibited TRP14‐induced autophagy
To determine the role of TRP14 in PTX‐induced autophagy, TRP14 was downregulated by the inhibitor SAHA, and autophagy in PTX‐treated NB cells was reevaluated. Compared with PTX exposure alone, the levels of LC3‐I, LC3‐II, Atg 5, Beclin 1, TRP14 and LC3‐II‐to‐LC3‐I ratio did not increase in SK‐N‐SH and QDDQ‐NM cells exposed to SAHA alone or in those exposed to a combination of SAHA and PTX (Fig. 4a,b). SAHA alone could slightly inhibit the viability of these tumor cells. However, the viability of SK‐N‐SH and QDDQ‐NM cells exposed to PTX at the doses of 2, 5, 10, 50 or 100 nM combined with SAHA was significantly lower than the viability of cells treated with PTX or SAHA alone (Fig. 4f). Concurrently, the apoptosis was enhanced in NB cells exposed to PTX plus SAHA compared to PTX alone (Fig. 4e). SAHA alone or combined with PTX exerted no effect on cellular autophagic flux (Fig. 4c,d).
Figure 4.
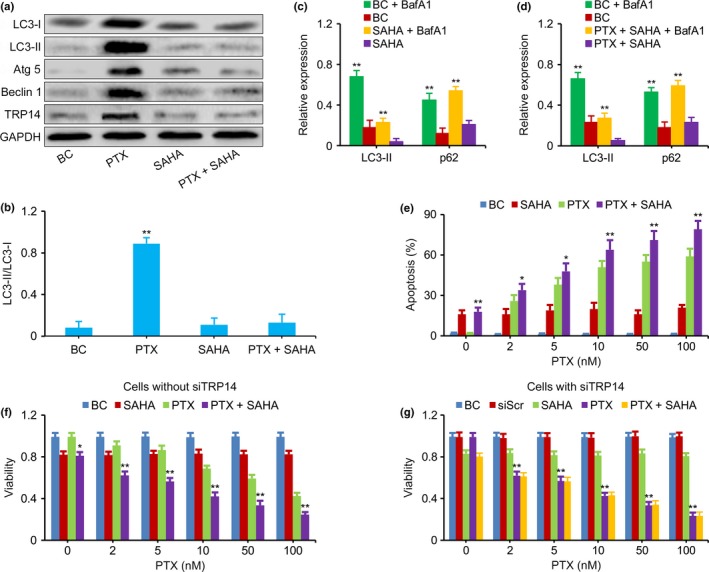
Inhibition of TRP14 increased the antitumor effects of paclitaxel (PTX). (a,b) Western blot showed that the expression of LC3‐I, LC3‐II, Atg 5, Beclin 1, TRP14 and LC3‐II‐to‐LC3‐I ratio was not increased in SK‐N‐SH cells exposed to SAHA alone or SAHA combined with PTX; this is the opposite result to that obtained in cells exposed to PTX alone. GAPDH was used as the loading control. (c,d) LC3‐II and p62 in QDDQ‐NM cells measured by western blot. The increased levels of LC3‐II and p62 expression that resulted from BafA1 treatment were similar in cells exposed to SAHA (with or without PTX) compared with cells that were not exposed to SAHA (with or without PTX). (e) The apoptosis was enhanced in QDDQ‐NM cells exposed to PTX plus SAHA compared to PTX alone. (f) MTT assay showed that the viability of SK‐N‐SH cells treated with PTX combined with SAHA was lower than that of cells treated with PTX or SAHA alone at the doses of 2, 5, 10, 50 and 100 nM. (g) MTT assay showed that the viability of siTRP14‐treated QDDQ‐NM cells that were exposed to PTX alone at each PTX dose level was similar to that of cells exposed to PTX plus SAHA. BC, blank control, neuroblastoma (NB) cells without PTX and SAHA exposure. The results are presented as the mean ± SEM of three independent experiments. *P < 0.01, **P < 0.01, two‐tailed t test.
To functionally determine whether the inhibition of TRP14 was important for the sensitization of NB cells to PTX after SAHA treatment, the anti‐tumor effects of PTX alone or those of PTX combined with SAHA were reevaluated in NB cells treated with siRNA against TRP14. The viability of TRP14low cells exposed to PTX alone was similar to that of cells exposed to PTX plus SAHA, which indicates that SAHA sensitized NB cells to PTX via the inhibition of TRP14 (Fig. 5g).
Figure 5.
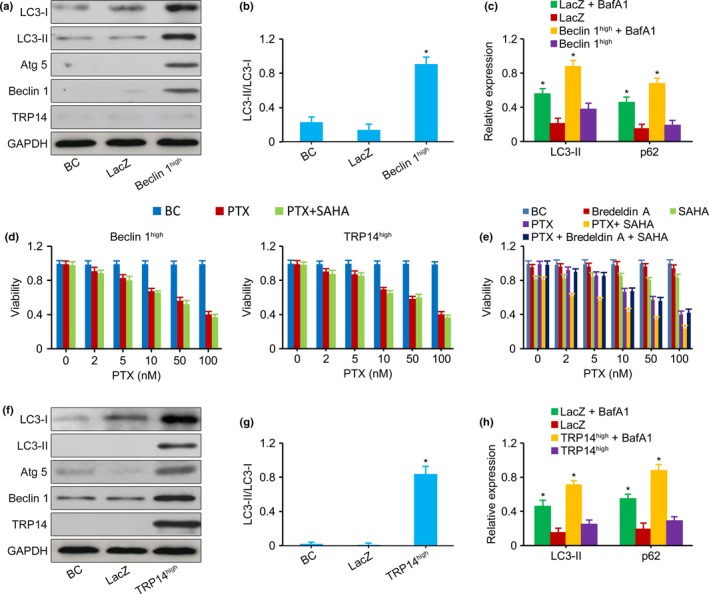
Upregulation of Beclin 1 or TRP14 Reversed the Inhibition of Autophagy by SAHA. (a,b) Western blot showed that the levels of LC3‐I, LC3‐II, Atg 5 and LC3‐II‐to‐LC3‐I ratio were increased in Beclin 1high SK‐N‐SH cells. (c) LC3‐II and p62 in Beclin 1high QDDQ‐NM cells measured by western blot. The increased levels of LC3‐II and p62 expression that resulted from BafA1 treatment were higher in Beclin 1high cells compared with LacZ control. (d) MTT assay showed that the viability of Beclin 1high SK‐N‐SH cells and TRP 14high QDDQ‐NM cells exposed to PTX alone was similar to that of cells exposed to PTX plus SAHA. (e) MTT assay showed that SAHA could not sensitize ordinary SK‐N‐SH cells to PTX when the cells were treated with a pro‐autophagic drug Bredeldin A followed by PTX plus SAHA. (f,g) Western blot showed that the levels of LC3‐I, LC3‐II, Atg 5 and LC3‐II‐to‐LC3‐I ratio were increased in TRP14high QDDQ‐NM cells. (h) LC3‐II and p62 in TRP14high SK‐N‐SH cells measured by western blot. The increased levels of LC3‐II and p62 expression that resulted from BafA1 treatment were higher in TRP14high cells compared with LacZ control. LacZ, an empty vector used as a control construct. BC, blank control, Beclin 1high or siTRP14high cells that were not exposed to PTX and SAHA. Bredeldin A, a pro‐autophagic drug. The results are presented as the mean ± SEM of three independent experiments.*P < 0.01, two‐tailed t‐test.
Upregulation of Beclin 1 or TRP14 reversed the inhibition of autophagy by SAHA
To elucidate whether SAHA inhibited autophagy via the TRP14/Beclin 1/LC3‐II pathway, the Beclin 1 and TRP14 genes were upregulated via plasmid transfection. Western blot showed that SK‐N‐SH and QDDQ‐NM cells, in which the Beclin 1 and TRP14 genes were upregulated, highly expressed the Beclin 1 and TRP14 proteins and increased the levels of LC3‐I, LC3‐II, Atg 5 and LC3‐II‐to‐LC3‐I ratio (Fig. 5a,b,f,g). Beclin 1high and TRP14high increased cellular autophagic flux (Fig. 5c). The viability of Beclin 1high or TRP14high cells exposed to PTX alone was similar to that of cells exposed to PTX plus SAHA (Fig. 5d). Thus, upregulation of Beclin 1 or TRP14 reversed the SAHA‐induced inhibition of autophagy. To further support the definitive role of autophagy in the response of NB cells to PTX, ordinary NB cells were pretreated with a pro‐autophagic drug Bredeldin A followed by PTX plus SAHA. SAHA could not sensitize NB cells to PTX in this setting (Fig. 5e).
Discussion
Autophagy is one of the underlying mechanisms of tumor resistance to chemotherapy.13, 14, 15, 16, 34 In the present study, NB showed moderate sensitivity to PTX, as demonstrated in vivo in a previous study.10 Notably, the tumor cells still had a viability rate of 43.7–45.6% at a high PTX dose of 100 nM and the apoptosis was not further enhanced concurrent with the increased PTX dose at high levels. We further demonstrated that PTX resistance resulted from autophagy in NB cells. The levels of the autophagy‐associated proteins LC3‐I, LC3‐II, Atg 5, Beclin 1 and LC3‐II to LC3‐I ratio were significantly increased in tumor cells exposed to PTX. Increased numbers of autophagosomes and increased autophagic flux were also observed in the PTX‐exposed cells. Similar findings have been noted in lung cancer35, 36 and breast cancer cells.37 However, the opposite result was obtained here in that PTX induced mitotic arrest and significantly inhibited the formation of autophagosomes in tumor cells.38 These findings indicate that the induction of autophagy by PTX varies among different cell types.
To demonstrate the key role of Beclin 1 in the regulation of PTX‐induced autophagy, Beclin 1 expression was interrupted in NB cells using specific siRNA. We found that LC3‐1, LC3‐II and Atg 5 in PTX‐exposed NB cells were downregulated and that the sensitivity of NB cells to PTX was increased. The Beclin 1 gene has been shown to be located on chromosome 17q21, and the Beclin 1 protein forms a complex by combining with the class III PI3K catalytic subunit to mediate other autophagy‐associated proteins positioned on the autophagosome.39 Moreover, Beclin 1 contains a BH3 protein region, which could combine with either Bcl‐2 or Bcl‐XL to inhibit apoptosis.40 Thus, this protein exerts its functions at an important intersection of the autophagy and apoptosis pathways. A previous study also demonstrated that the inhibition of Beclin 1 can increase the chemotherapeutic sensitivity of liver cancer.41
Surprisingly, the newly defined protein TRP14 was concurrently upregulated along with Beclin 1. When TRP14 expression was inhibited by siRNA, LC3‐I, LC3‐II, Atg 5 and Beclin 1 were not upregulated in PTX‐exposed cells, and the sensitivity of NB cells to PTX increased at each dosage level. This finding confirmed that Beclin 1‐induced autophagy is regulated by TRP14. TRP14 contains Trx‐like active center proteins, functions as a disulfide reductase and peroxidase, and maintains redox homeostasis in cells.19 In addition, TRP14 is involved in the regulation of the TNF‐α signal transduction pathway. TNF‐α may activate c‐Jun N‐terminal kinase and p38 mitogen‐activated protein kinases, and may enhance the activation of TNF‐α‐induced caspases and the subsequent apoptotic process.42 Thus, TRP14 may regulate the expression of the Beclin 1 gene and autophagy via the TNF‐α pathway.
SAHA is a novel HDACI that increases the acetylation of histones by binding to HDACs. This leads to abnormal gene transcription, which further induces apoptosis in cancer cells. SAHA has been shown to exert synergistic antitumor effects in some tumors when given with cis‐platinum43 or tamoxifen,25 but the anticancer mechanism through which SAHA exerts its effects is still unknown. We found that PTX and SAHA have additive antitumor effects in NB. Interestingly, SAHA could inhibit autophagy and reverse resistance to PTX in common control SK‐N‐SH and QDDQ‐NM cells, but not in Beclin 1high, TRP14high cells or common NB cells pretreated with a autophagy inducer, which further indicates that TRP14 regulates PTX‐induced autophagy via the Beclin 1/LC3‐II pathway.
In conclusion, our findings showed that PTX‐induced autophagy in NB cells is regulated by TRP14 and that the TRP14 inhibitor SAHA can sensitize NB cells to PTX. TRP14 is therefore a novel treatment target in NB and is worthy of further investigation.
Disclosure Statement
The authors declare no conflict of interest.
Abbreviations
- BafA1
Bafilomycin A1
- HDAC
hydroxamic histone deacetylase
- MDC
monodansylcadaverine
- MTT
methyl thiazolyl tetrazolium
- NB
neuroblastoma
- PTX
paclitaxel
- SAHA
suberoylanilide hydroxamic acid
- TNF‐α
tumor necrosis factor alpha
- TRP14
thioredoxin‐related protein 14
- Trx
thioredoxin
Acknowledgments
This work was supported by a grant from the Science and Technology Planning Project of Guangdong Province, China (2012B031800460 and 2013B021800069). The funders had no role in study design, datacollection and analysis, decision to publish, or preparation of the manuscript. We are grateful to Dr. Youjian He at our center for the study design.
Cancer Sci 108 (2017) 1485–1492
Funding Information
Science and Technology Planning Project of Guangdong Province, China, (2012B031800460, 2013B021800069).
References
- 1. Cheung NK, Cheung IY, Kushner BH et al Murine anti‐GD2 monoclonal antibody 3F8 combined with granulocyte‐macrophage colony‐stimulating factor and 13‐cis‐retinoic acid in high‐risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 2012; 30: 3264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kreissman SG, Seeger RC, Matthay KK et al Purged versus non‐purged peripheral blood stem‐cell transplantation for high‐risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 2013; 14: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoo SY, Kim JS, Sung KW et al The degree of tumor volume reduction during the early phase of induction chemotherapy is an independent prognostic factor in patients with high‐risk neuroblastoma. Cancer 2013; 119: 656–64. [DOI] [PubMed] [Google Scholar]
- 4. Saulnier Sholler GL, Bond JP, Bergendahl G et al Feasibility of implementing molecular‐guided therapy for the treatment of patients with relapsed or refractory neuroblastoma. Cancer Med 2015; 4: 871–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang HL, Ruan L, Zheng LM, Whyte D, Tzeng CM, Zhou XW. Association between class III β‐tubulin expression and response to paclitaxel/vinorebine‐based chemotherapy for non‐small cell lung cancer: a meta‐analysis. Lung Cancer 2012; 77: 9–15. [DOI] [PubMed] [Google Scholar]
- 6. Kubota T, Okano Y, Sakai M et al Carboplatin plus weekly paclitaxel with bevacizumab for first‐line treatment of non‐small cell lung cancer. Anticancer Res 2016; 36: 307–12. [PubMed] [Google Scholar]
- 7. Schneider BP, O'Neill A, Shen F et al Pilot trial of paclitaxel‐trastuzumab adjuvant therapy for early stage breast cancer: a trial of the ECOG‐ACRIN cancer research group (E2198). Br J Cancer 2015; 113: 1651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fountzilas G, Papakostas P, Dafni U et al Paclitaxel and gemcitabine vs. paclitaxel and pegylated liposomal doxorubicin in advanced non‐nasopharyngeal head and neck cancer. An efficacy and cost analysis randomized study conducted by the Hellenic Cooperative Oncology Group. Ann Oncol 2006; 17: 1560–7. [DOI] [PubMed] [Google Scholar]
- 9. Bang YJ, Im SA, Lee KW et al Randomized, double‐blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol 2015; 33: 3858–65. [DOI] [PubMed] [Google Scholar]
- 10. Kretschmar CS, Kletzel M, Murray K et al Response to paclitaxel, topotecan, and topotecan‐cyclophosphamide in children with untreated disseminated neuroblastoma treated in an upfront phase II investigational window: a pediatric oncology group study. J Clin Oncol 2004; 22: 4119–26. [DOI] [PubMed] [Google Scholar]
- 11. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang SY, Yu QJ, Zhang RD, Liu B. Core signaling pathways of survival/death in autophagy‐related cancer networks. Int J Biochem Cell Biol 2011; 43: 1263–6. [DOI] [PubMed] [Google Scholar]
- 13. Riz I, Hawley TS, Hawley RG. KLF4‐SQSTM1/p62‐associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015; 6: 14814–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. García‐Cano J, Ambroise G, Pascual‐Serra R et al Exploiting the potential of autophagy in cisplatin therapy: a new strategy to overcome resistance. Oncotarget 2015; 6: 15551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng X, Zhao H, Li Y et al Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR‐ABL‐positive chronic myeloid leukemia. Autophagy 2015; 11: 355–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belounis A, Nyalendo C, Le Gall R et al Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 2016; 16: 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang RC, Wei Y, An Z et al Akt‐mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338: 956–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He W, Wang Q, Xu J et al Attenuation of TNFSF10/TRAIL‐induced apoptosis by an autophagic survival pathway involving TRAF2‐ and RIPK1/RIP1‐mediated MAPK8/JNK activation. Autophagy 2012; 8: 1811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeong W, Yoon HW, Lee SR, Rhee SG. Identification and characterization of TRP14, a thioredoxin‐related protein of 14 kDa. New insights into the specificity of thioredoxin function. J Biol Chem 2004; 279: 3142–50. [DOI] [PubMed] [Google Scholar]
- 20. Woo JR, Kim SJ, Jeong W et al Structural basis of cellular redox regulation by human TRP14. J Biol Chem 2004; 279: 48120–5. [DOI] [PubMed] [Google Scholar]
- 21. Jeong W, Jung Y, Kim H, Park SJ, Rhee SG. Thioredoxin‐related protein 14, a new member of the thioredoxin family with disulfide reductase activity: implication in the redox regulation of TNF‐alpha signaling. Free Radic Biol Med 2009; 47: 1294–303. [DOI] [PubMed] [Google Scholar]
- 22. Marks PA, Jiang X. Histone deacetylase inhibitors in programmed cell death and cancer therapy. Cell Cycle 2005; 4: 549–51. [DOI] [PubMed] [Google Scholar]
- 23. Kelly WK, Marks PA. Drug insight: histone deacetylase inhibitors–development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol 2005; 2: 150–7. [DOI] [PubMed] [Google Scholar]
- 24. Jin JS, Tsao TY, Sun PC, Yu CP, Tzao C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol Oncol Res 2012; 18: 713–20. [DOI] [PubMed] [Google Scholar]
- 25. Lee YJ, Won AJ, Lee J et al Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen‐resistant MCF‐7 breast cancer cells. Int J Med Sci 2012; 9: 881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013; 310: 2191–4. [DOI] [PubMed] [Google Scholar]
- 27. Zhen Z, Sun X, He Y, Cai Y, Wang J, Guan Z. The sequence of drug administration influences the antitumor effects of bevacizumab and cyclophosphamide in a neuroblastoma model. Med Oncol 2011; 28: S619–25. [DOI] [PubMed] [Google Scholar]
- 28. Khaghani‐Razi‐Abad S, Hashemi M, Pooladi M, Entezari M, Kazemi E. Proteomics analysis of human oligodendroglioma proteome. Gene 2015; 569: 77–82. [DOI] [PubMed] [Google Scholar]
- 29. Qu X, Qing L. Abrin induces HeLa cell apoptosis by cytochrome c release and caspase activation. J Biochem Mol Biol 2004; 37: 445–53. [DOI] [PubMed] [Google Scholar]
- 30. Demishtein A, Porat Z, Elazar Z, Shvets E. Applications of flow cytometry for measurement of autophagy. Methods 2015; 75: 87–95. [DOI] [PubMed] [Google Scholar]
- 31. Mani J, Vallo S, Rakel S et al Chemoresistance is associated with increased cytoprotective autophagy and diminished apoptosis in bladder cancer cells treated with the BH3 mimetic (‐)‐Gossypol (AT‐101). BMC Cancer 2015; 15: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu X, Tan D, Hou Z et al The effect of miR‐338‐3p on HBx deletion‐mutant (HBx‐d382) mediated liver‐cell proliferation through CyclinD1 regulation. PLoS ONE 2012; 7: e43204. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Liao Y, He X, Qiu H et al Suppression of the epithelial‐mesenchymal transition by SHARP1 is linked to the NOTCH1 signaling pathway in metastasis of endometrial cancer. BMC Cancer 2014; 14: 487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herman‐Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res 2006; 66: 5828–35. [DOI] [PubMed] [Google Scholar]
- 35. Xi G, Hu X, Wu B et al Autophagy inhibition promotes paclitaxel‐induced apoptosis in cancer cells. Cancer Lett 2011; 307: 141–8. [DOI] [PubMed] [Google Scholar]
- 36. Liu F, Liu D, Yang Y, Zhao S. Effect of autophagy inhibition on chemotherapy‐induced apoptosis in A549 lung cancer cells. Oncol Lett 2013; 5: 1261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ajabnoor GM, Crook T, Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF‐7 breast cancer cells. Cell Death Dis 2012; 3: e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Veldhoen RA, Banman SL, Hemmerling DR et al The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013; 32: 736–46. [DOI] [PubMed] [Google Scholar]
- 39. Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005; 1: 46–52. [DOI] [PubMed] [Google Scholar]
- 40. Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl‐XL‐Beclin 1 peptide complex: beclin 1 is a novel BH3‐only protein. J Biol Chem 2007; 282: 13123–32. [DOI] [PubMed] [Google Scholar]
- 41. Daniel F, Legrand A, Pessayre D, Vadrot N, Descatoire V, Bernuau D. Partial Beclin 1 silencing aggravates doxorubicin‐ and Fas‐induced apoptosis in HepG2 cells. World J Gastroenterol 2006; 12: 2895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jeong W, Chang TS, Boja ES, Fales HM, Rhee SG. Roles of TRP14, a thioredoxin‐related protein in tumor necrosis factor‐alpha signaling pathways. J Biol Chem 2004; 279: 3151–9. [DOI] [PubMed] [Google Scholar]
- 43. Asgar MA, Senawong G, Sripa B, Senawong T. Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int J Oncol 2016; 48: 409–20. [DOI] [PubMed] [Google Scholar]