

Abstract
Short-lived radiolabeled tracers for positron emission tomography (PET) must be rapidly synthesized, purified, and formulated into injectable solution just prior to imaging. Current radiosynthesizers are generally designed for clinical use, and the HPLC purification and SPE formulation processes often result in a final volume that is too large for preclinical and emerging in vitro applications. Conventional technologies and techniques for reducing this volume tend to be slow, resulting in radioactive decay of the product, and often require manual handling of the radioactive materials. We present a fully-automated microfluidic system based on sweeping gas membrane distillation to rapidly perform the concentration and formulation process. After detailed characterization of the system, we demonstrate fast and efficient concentration and formulation of several PET tracers, evaluate residual solvent content to establish the safety of the formulated tracers for injection, and show that the formulated tracer can be used for in vivo imaging.
Keywords: Positron emission tomography (PET), radiopharmaceutical, formulation, membrane distillation, concentration, microfluidics, molecular imaging, rotary evaporation
Graphical Abstract
We present an automated microfluidic platform relying on sweeping gas membrane distillation for rapid concentration and formulation of PET tracers for preclinical imaging applications.
3 Introduction
Positron emission tomography (PET) is a 3D functional imaging modality used to monitor biological processes in vivo for both research and clinical applications. Depending on the particular radiolabeled ‘tracer’ injected before the scan, PET can provide in vivo measurements of the rates of various biological processes (e.g. glucose metabolism, DNA replication, or gene expression) based on the abundance of receptors or other cell surface markers [1]–[3]. In addition to widespread clinical use in diagnosing disease, monitoring response to therapy, and in establishing “precision medicine” [4]–[6], PET is also a valuable research and development tool. Measurements of processes such as apoptosis or hypoxia can be indispensable assays in the drug screening and development process [7]–[9], and radiolabeling a novel drug enables early in vivo preclinical evaluation of various properties such as metabolic stability, pharmacokinetics, biodistribution, target specificity, and clearance mechanisms [10]–[14].
Due to the short half-lives of PET radionuclides (e.g. 110 min for fluorine-18), the tracers must be synthesized just prior to imaging. The preparation of PET tracers is usually carried out inside radiation-shielded “hot cells” by automated radiosynthesizers that perform the needed chemical reactions, purify the crude product via high performance liquid chromatography (HPLC), and formulate the tracer for injection. Because most synthesizers are designed for clinical use, the output volume is often in the 10s of mL range. As a result these synthesizers are not suitable for preparing small amounts of research tracers in high enough concentration for small animal imaging or in vitro assays without the use of very high starting activities (1-5Ci). In vivo imaging in mouse tumor models typically requires 100-200 μCi in a volume of 100 μL or less (limited by low blood volume of the mouse) [15] which is a concentration of ∼1-2 mCi/mL. Similarly, existing and emerging platforms for in vitro cancer studies such as binding or uptake assays, drug response assays, enzyme activity assays, or kinetic modeling have reported the need for concentrations up to ∼1 mCi/mL [16]. Though these concentrations can be achieved by using large amounts of radioactivity in the synthesis and just discarding what is not needed, this approach increases radiation exposure and cost of the radioisotope and may not be practical for novel tracers where the yield is often very low. For example, if the uncorrected yield is ∼1%, then 100 mCi of the radioisotope would be needed to produce 1 mCi of the tracer. This is sufficient for a preclinical study of 10 mice if it can be concentrated down to ∼1 mL. On the other hand, if the final volume is 10 mL, then 10 mCi would need to be produced, initially requiring 1 Ci of the radioisotope. In this case, 1 mCi would be used and 9 mCi would be discarded: the extra activity is simply to increase the concentration. These numbers are even higher if the radiochemical yield is lower, if the probe must be transported a long distance, or if the probe must be used for imaging throughout the work day.
Instead, to avoid this issue, tracers can be concentrated after production as part of the reformulation process. After the radiosynthesis, the crude product volume is typically on the order of ∼1 mL, but the HPLC purification process results in volumes of 10-50 mL of mobile phase due to the high flow rates that are used. Mobile phases typically consist of mixtures of ethanol (EtOH) or acetonitrile (MeCN) in water, often with additives to control pH. EtOH-based mobile phases may by directly injected after sterile filtration if the EtOH content is sufficiently low (10% v/v for EtOH [17]). However if the mobile phase contains too much EtOH, or contains MeCN or other toxic solvents, these must be nearly completely removed (e.g. allowed injection limit of MeCN is 410 ppm [18]). In one approach, a rotary evaporator is used to evaporate off the mobile phase at elevated temperature, and then the dried residue is replaced with saline. However, rotovap systems are bulky and consume significant space in the hot cell, and due to the significant amount of water to be removed, this process can be rather slow especially in more compact systems [19], [20]. Alternatively, a solid-phase extraction (SPE) method can be used. In reversed-phase SPE, the collected HPLC fraction is first diluted with water to ensure the organic solvent content is sufficiently low (typically <5-10%), and then passed through the SPE cartridge to trap the PET tracer. The mobile phase is diverted to waste. After rinsing the cartridge, 1-2 mL of EtOH can then be used to release the tracer from the cartridge. For use in humans, this sample would be diluted with saline to reduce the EtOH content below the allowed limit; however, to avoid the volume increase an alternative is to directly evaporate the EtOH and then suspend the tracer in saline. While effective, the SPE process can be time consuming due to the large diluted sample volume, and the evaporation and resuspension in small volumes cannot be performed in most radiosynthesizers.
We developed a compact microfluidic device based on membrane distillation for rapid solvent removal to address this problem [21]. Compared to other microfluidic techniques such as using trapped air pockets to capture vapor from a liquid stream [22], or using droplet traps [23], atomization [24], or a sheath flow [25] to expose a large liquid surface area to a gas flow, implementations of membrane distillation have significantly faster evaporation speed and greater sample capacity [21], [26]. In this work, we present a second-generation chip with a 5-fold improved evaporation rate (3.4 versus 0.65 mL/min for water at 100°C), increased scope of mobile phases (now compatible with practically any PET tracer), and an improved modular construction that enables the chip material to be matched with the PET tracer for optimal performance. In addition, though we previously focused only on concentration, here the system performs simultaneous concentration and formulation for injection, and is now fully automated. The dramatic performance improvements, increased diversity of compatible tracers, simultaneous concentration and reformulation, and automation has translated our previous proof of concept system into mature technology suitable for routine use in the preparation of PET tracers. We present here a detailed characterization of the system, demonstration of concentration and formulation of several PET tracers, evaluation of residual solvent content to establish the safety of the formulated tracers for injection, and demonstration of in vivo imaging.
4 Methods
4.1 Microfluidic concentrator design and fabrication
The microfluidic device is designed to perform sweeping-gas membrane distillation, in which a fluid sample and a sweeping gas are separated by a hydrophobic porous membrane [26], [27]. The porous membrane prevents passage of the liquid as long as the contact angle is > 90°, but vapor generated as the aqueous sample is heated can pass through the membrane and into the sweeping gas stream. In distillation, the vapor carried away is condensed and collected for downstream use; however, in the case of sample concentration, the vapor is discarded, and the reduced volume of sample (containing the desired solute in more concentrated form) is collected.
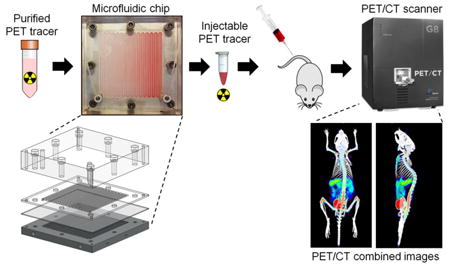
The design is based on preliminary work we previously reported [21] but with numerous improvements. The new device (Figure 1) measures 120 mm × 120 mm in lateral dimensions. The top layer (25.4 mm thick acrylic) provides optical transparency for visual monitoring of the concentration process and rigidity for clamping all layers together. The sample layer is made out of 2 mm thick plastic with a patterned serpentine channel (2.25mm wide × 0.05mm deep channels with 0.5mm spacing). The channel has rounded corners to avoid dead volumes that interfere with efficient sample recovery, and was designed with smaller channel width than previously used (4.5 mm) to avoid “sagging” of the membrane while still maintaining nearly identical evaporation surface area (∼57 cm2). The open side of the sample channel is in contact with a porous membrane (PTFE, 0.2 μm pore size; Sterlitech, Kent, WA, USA). The pore size was substantially smaller than our previous device (1.0 μm) and thus the device was expected to tolerate a wider range of operating pressures. The final layer is a 10 mm thick 6061 aluminum alloy block with a 3.5 mm deep serpentine channel matching the sample layer (CNC machined by Proto Labs, Inc., Maple Plain, MN, USA). This layer served to provide heat, and the channel carried the sweeping gas. The layers are clamped together using bolts; small deformation of the membrane layer ensures good sealing along channel walls to both the sample and gas flow layers.
Figure 1.
Schematic of the microfluidic concentrator chip. (A) Assembled chip with the ability to concentrate large volumes into ∼1 mL volume. (B) Exploded view of the chip showing acrylic compression layer, sample layer made of different plastics, a Teflon membrane with 0.2μm pore size and aluminum gas flow layer. (C) Sectional view A shows a cross-section along a channel during operation. (D) Sectional view B shows a cross-section through a channel during operation.
Prior work suggested that some PET tracers had adverse reactions (adsorption or absorption) with the chip [21]. Suspecting a material-dependence, we fabricated sample layers from a variety of materials, including: poly(methyl methacrylate) (PMMA), polytetrafluoroethylene (PTFE), glass-filled PTFE (glass-PTFE), polyetherimide (Ultem), cyclic olefin copolymer (COC) and polyether ether ketone (PEEK). These plastics were chosen for their chemical inertness, high temperature stability, and/or transparency. An initial proof-of-concept PMMA sample layer was fabricated by Aline Inc. (Rancho Dominguez, CA, USA) via laser ablation, and then sample layers of all materials were fabricated via CNC machining by Delmar Company (Lakeville, MN, USA). Holes through the acrylic top layer and sample layer provide fluid access to the inlet and outlet of the sample channel. Similarly, holes through the gas flow layer provide access to the inlet and outlet of the sweeping gas channel. To distinguish the two methods of fabricating PMMA layers, we refer to them as PMMA-machined and PMMA-ablated.
4.2 Automated concentrator system
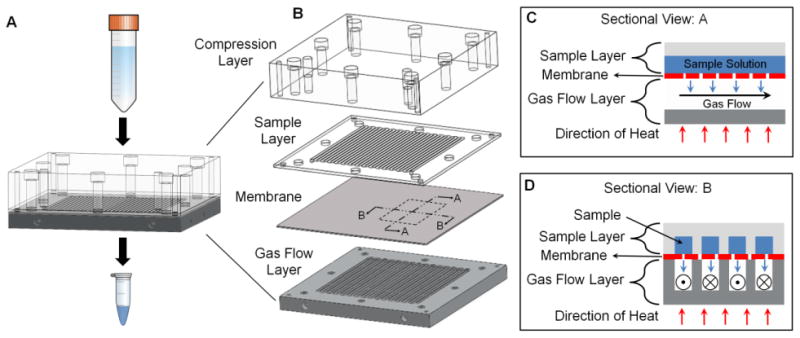
To automate its operation, the chip is integrated into the system shown in Figure 2. Reservoirs for the sample as well as the rinsing solution (e.g. saline, to help collect the sample after concentration) comprise 15mL conical tubes (352096, BD Biosciences, San Jose, CA, USA). Two holes were drilled in the cap of each tube, one for 1/8″ polyurethane tubing to pressurize the reservoir, and one for 1/16″ OD ETFE tubing (1517L, IDEX Health & Science, Oak Harbor, WA, USA) to deliver the reservoir contents. Tubing was sealed in place with a hot glue gun. The fluid delivery lines from the reservoirs are connected to the two inlet ports of an electronic 3-way valve (LVM105R, SMC Corporation, Japan). The output of this “sample inlet valve” is connected via 1/16″ OD ETFE tubing to the sample inlet of the concentrator chip. An electronic pressure regulator (ITV0010-2BL, SMC) connected to a nitrogen source supplies pressure to the reservoirs through independently controlled solenoid valves (S070B-5DG, SMC).
Figure 2.
Fluidic and electronic wiring diagram of the automated microfluidic concentration system.
The sample outlet from the chip is connected via 1/16″ OD ETFE tubing to another 3-way valve to choose whether the outlet is blocked (during evaporation) or connected to the sample collection reservoir. The collection vial is connected via a vacuum trap to a digitally controlled vacuum regulator (ITV2090, SMC) which in turn was connected to a central “house” vacuum. This vacuum source was capable of pressure as low as -90 kPa with a vacuum capacity of 1.9 L/min. The gas flow layer was connected via 1/4″ polyurethane tubing through an electronic pressure regulator (ITV2010, SMC) to the nitrogen source, and through a vacuum regulator (ITV2090, SMC) to the vacuum pump.
The regulators and valves were digitally controlled using a data acquisition module (NI USB-6009, National Instruments, Austin, TX, USA). A custom-built Darlington transistor array board was used to step up the voltage and current needed to drive the 3-way valves. Two tubing fluid sensors (OPB350W062Z, Optek Technologies, Carrollton, Texas, USA) are positioned on the 1/16″ tubing between the sample inlet valve and the concentration chip inlet (sensor 1) and on the 1/16″ tubing between the concentrator chip outlet and the sample collection valve (sensor 2). Analog voltage readings from the sensors are connected to the data acquisition module (NI USB-6009, National Instruments) for computer analysis. Comparison to a threshold value is used to determine if the tubing under the sensor is filled with air or liquid.
Heat was provided to the chip by 100W cartridge heaters (8376T27, McMaster Carr, Santa Fe Springs, CA, USA) inserted into holes drilled in the metal gas-flow layer. Thermal paste (OT-201-2, OMEGA Engineering, Inc., Stamford, CT, USA) was used to provide good thermal contact. For each cartridge heater, a K-type thermocouple (5TC-GG-(K)-30-(72), OMEGA Engineering, Inc., Stamford, CT, USA) was also inserted into the heating block near the heater. Feedback control of each heater-thermocouple pair was performed with an independent PID temperature controller (CN7500, OMEGA Engineering, Inc., Stamford, CT, USA). Control parameters were set by the “auto-tune” feature.
The heating power was selected based on the following considerations. To heat water from room temperature to 100°C and convert it to vapor form at a rate of ∼3 mL/min (maximum we have observed so far) requires ∼130W of power. Empirical testing showed that the theoretical minimum power was not sufficient to maintain the temperature of the heating block, presumably due to other thermal losses and the finite time needed for transport of heat from the cartridge heaters to the sample channel. Using two 100W heaters during a 100°C evaporation, we observed the measured temperature to be ∼6°C below the setpoint. On the other hand, with four 100 W heaters, the measured temperature matched the setpoint within ±1.5°C, and this configuration was used for all experiments. Further increased heating power (e.g., four 300W heaters) could heat the chip more quickly but gave similar temperature stability and was not used because the chip could be pre-heated prior to use.
4.3 Concentrator operation
The concentration process (described in Figure 3) was automated using a custom-written LabVIEW (National Instruments) program based on a finite state machine architecture. Prior to the concentration process, the chip is first heated to the desired temperature (∼5 min to reach steady state), the sample collection valve is closed, and the sample inlet valve is switched to the “sample” position. Sample is loaded into the chip by pressurizing the sample vial, to a pressure (Psample) that is constantly applied for the duration of the concentration process. Air initially in the system ahead of the sample readily escapes by passing through the membrane into the gas flow layer, allowing the sample to reach the chip. The sample advances until the sample channel is filled and sample begins to emerge from the outlet of the chip (due to compression of the remaining trapped air), triggering sensor 2. At this point, the sweeping gas flow is initiated (by applying Pgas_in and Pgas_out at the gas flow inlet and outlet, respectively). As solvent evaporates, additional space is created within the sample channel, allowing new sample to enter the chip. The solute becomes progressively more concentrated within the chip.
Figure 3.

Photographs showing system operation during the concentration process for a sample of diluted food dye. After initialization and pre-heating of the chip (1), the sample reservoir is pressurized to begin loading the sample (2). Once the chip is filled (3), the sweeping gas is activated to begin the concentration process (4). When the sample volume has been reduced to the point where it just fits within the chip (5), a timer is started and evaporation is continued to reduce the volume further (6). The concentrated sample is then collected (7), and any residual liquid in the chip (8) is recovered via rinsing. Each rinse step includes loading of rinse solution, partial evaporation to a set volume, and recovery of the concentrated rinse solution (9). After multiple rinsing steps, the chip is free of residual sample (10).
When the sample reservoir is exhausted, the trailing end of the sample passes through sensor 1 (i.e. liquid to air transition), and the concentrated sample volume matches the chip volume. The sample volume can be further reduced by continuing the heating process for an additional delay time to achieve a final volume smaller than the chip volume. This delay is needed because even though the designed chip volume is ∼0.29 mL, the collected volume without further evaporation is ∼2.75 mL. This discrepancy is presumably due to significant deflection of the membrane under the operating conditions. (Note, we explored the possibility of using a “laminated membrane”, i.e. thin 0.2 μm pore PTFE membrane bonded to a more rigid membrane with larger pores, and found the recovered volume to be ∼0.80 mL. However, evaporation speed and sample recovery were quite poor in initial tests and so this direction was not pursued further.) The actual delay time was varied, depending on the mode of operation. When performing partial solvent evaporation, a delay time of 50 s was used, resulting in a recovered volume from the chip of 0.4-0.5 mL (see Supplementary Information Section 1). On the other hand, when performing complete solvent evaporation, the delay time was typically set to 270 s (corresponding to ∼220 s for water to completely evaporate at 100°C plus a safety margin.)
When operating in partial evaporation mode, the concentrated sample is first collected, and then the chip is rinsed multiple times. In complete evaporation mode, there is no initial collection step and rinsing (described below) is performed directly after complete dryness is achieved.
To collect the concentrated sample, the sample collection valve opens after the delay time, and the sample is driven to the collection reservoir by the sample inlet pressure. During the collection process, the vacuum connected to the collection reservoir (Pvacuum) is turned on at -1.0 psi. The sample is collected for a set period of time that is empirically determined; for the concentrated sample volume resulting from 50s delay time, ∼4s was sufficient for collection, but we chose 14s to incorporate a safety margin. Next, the vacuum is ramped to – 8.8 psi (PVacuum_ramp) to recover any residual fluid trapped within the fluidic path. Residual fluid recovery is typically complete within ∼3s, but 9s was chosen to include a safety margin. We found it necessary to gradually ramp the vacuum pressure to prevent the fluid from breaking up in the tubing. A detailed video showing partial evaporation and collection of food dye in water can be seen in Supplemental Video 1.
To perform a rinse, the sample collection valve is closed, and the sample inlet valve is switched to the rinsing solution reservoir. The rinse solution reservoir is pressurized to Psample, and rinse solution is loaded just as initially performed for the sample. However, when it reaches sensor 2, the sample inlet valve is switched back to the empty sample reservoir (which is still pressurized at Psample). At this point, the total volume of rinse solution (including that in the chip, tubing, etc.) is ∼3 mL. The rinse solution is concentrated just like the initial sample, and when the trailing end of the rinse solution passes sensor 1, a timer is started for additional delay time. Finally, the concentrated rinse plug is ejected in the same fashion as the original sample. Multiple rinse steps can be performed to improve sample recovery.
4.4 Measuring Evaporation Rate
Evaporation rate was measured by placing a 5 mL Fisherbrand graduated serological pipet (13-678-11D, Fisher Scientific, Pittsburgh, PA, USA) inline between the sample reservoir and the chip. Connections were made via 1/4″ OD polyurethane tubing (TIUB07, SMC Pneumatics, Yorba Linda, CA). Evaporation rates were measured by observing the sample fluid meniscus as it passed through the graduated pipette. The amount of time it took for the fluid meniscus to move by 1 mL was recorded and used to calculate the evaporation rate. Evaporation rates were determined for five successive 1 mL increments and averaged.
For some experiments, we aimed to study the evaporation rate over a longer period of time (larger volume evaporated). For these cases, the sample reservoir was replaced with a larger conical tube (50mL Falcon conical tube; Corning Inc., Corning, NY, USA ), and a larger graduated pipette (50 mL Fisherbrand serological pipet, Fisher Scientific, Pittsburg, PA, USA) was used to monitor liquid movement. Though vertical orientation of the pipette adds a hydrostatic pressure to the sample pressure (∼0.4 psi in the worst case) this pressure was found to have negligible effect on evaporation rate.
4.5 Reagents
Ethanol (EtOH; 200 proof) was purchased from the UCLA Chemistry Department (Los Angeles, CA, USA). Anhydrous acetonitrile (MeCN), ammonium dihydrogen phosphate (NH4H2PO4), potassium carbonate (K2CO3), dimethyl sulfoxide (DMSO), and trifuoroacetic acid (TFA) were purchased from Sigma-Aldrich (Milwaukee, WI USA). Deionized water was obtained from a Milli-Q water purification system (EMD Millipore Corporation,Berlin, Germany). Food dye used in the experiments was purchased from Kroger (Cincinnati, OH, USA). Food dye was diluted with 18MΩ dionized water in the ratio of 1:50 v/v. Saline (0.9% w/v) was purchased from Hospira (Lake Forest, IL, USA). [18F]fluoride in [18O]H2O was obtained from the UCLA Biomedical Cyclotron (Los Angeles, CA, USA). 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (Kryptofix 222; K222) was purchased from ABX (Radeberg, Germany). Unless otherwise noted, all materials were used as received.
1-(2′-deoxy-2′-[18F]-β-D-fluoroarabinofuranosyl) cytosine ([18F]D-FAC), 2-deoxy-2-[18F]fluoro-5-ethyl-β-D-arabinofuranosyluracil ([18F]D-FEAU), (S)-N-((1-allyl-2-pyrrrolidinyl)methyl)-5-(3-[18F]fluoropropyl)-2,3-dimethoxybenzamide ([18F]fallypride), and 9-(4-[18F]fluoro-3-hydroxymethylbutyl)-guanine ([18F]FHBG) were synthesized using the ELIXYS radiosynthesizer (Sofie Biosciences, Culver City, CA, USA) as described previously [28], [29]. Synthesis of N-[2-(4-[18F]fluorobenzamido)ethyl]maleimide ([18F]FBEM) was synthesized on the ELIXYS radiosynthesizer by straightforward adaptation of literature methods [30]. All tracers and prosthetic groups were purified using semi-preparative HPLC, and the collected fraction of each (suspended in its respective mobile phase; see Table 1) was used directly without further formulation. [18F]D-FAC was formulated for use in imaging as described below.
Table 1.
Semi-preparative HPLC conditions used for the purification of example PET tracers. [18F]D-FAC and [18F]FHBG were purified using Phenomenex Gemini-NX column (10mm × 250mm); others used a Phenomenex Luna column (10mm × 250mm).
| Tracer | HPLC Mobile Phase (all ratios are v:v) |
Flow Rate (mL/min) | Purification Method |
|---|---|---|---|
| [18F]D-FAC | 1:99 EtOH/10mM NH4H2PO4 | 5.0 | Isocratic |
| [18F]FHBG | 5:95 MeCN/50mM NH4OAC | 5.0 | Isocratic |
| [18F]D-FEAU | 8:92 MeCN/water | 5.0 | Isocratic |
| [18F]FBEM | 20:80 MeCN/water | 5.0 | Isocratic |
| [18F]Fallypride | 60:40 MeCN/25mM NH4HCO2 with 1%TEA | 5.0 | Isocratic |
4.6 Determining Operating Conditions
In our previous work, we showed several constraints in operating parameters [21]. Briefly, Psample should be high enough that the chip remains full of sample during evaporation. In addition, the highest pressure in the gas flow layer must be less than the sample pressure, i.e. Pgas_in < Psample. Finally, the largest pressure difference across the membrane must not exceed the breakthrough pressure (BTP), i.e. Psample – Pgas_out < BTP. BTP is defined as the applied pressure at which sample (in liquid form) can directly leak through the pores of the membrane into the gas flow layer. It is closely related to the concept of capillary pressure, PC, which is the pressure needed to cause fluid to enter a pore with radius r, BTP ≈ PC = 2γcos(π-θ)/r, where γ is the surface tension of the sample (at an air interface) and θ is the contact angle of the liquid sample with respect to the membrane surface. (The implications of realistic pore geometries and size distribution is described in [31], [32].) Since γ and θ depend on the composition of the mobile phase and temperature, the BTP is a function of these variables as well. Spontaneous permeation of the membrane (i.e. ‘breakthrough’) always occurs when θ < 90°. For θ > 90°, permeation of the membrane only occurs when the fluid sample is pressurized against the membrane with a pressure exceeding the BTP.
First, measurements of contact angle for various solvent compositions and temperatures (see Supplementary Information Section 2) were made to determine the conditions under which θ > 90°, indicating the extent of the operating range in terms of solvent composition and temperature. Since it is important to operate under an adequate safety margin, more direct measurements of BTP were also determined for various solvent compositions and temperatures (see Supplementary Information Section 3). Once BTP was known, other operating parameters were selected to meet the above constraints.
4.7 Determining Sample Recovery Efficiency
The efficiency of sample recovery (i.e. amount of solute recovered compared to amount in initial sample) was quantitatively evaluated using radioactive solutions. Experiments used either a [18F]fluoride solution or a solution of an 18F-lableled PET tracer. [18F]fluoride solutions consisted of 0.3–1.25 mCi of [18F]fluoride, 2.25 mg K222 (0.6 mM final concentration) and 0.41 mg of K2CO3 (0.3 mM final concentration) in 10 mL ddH2O. The amount of solute directly corresponds to the amount of radioactivity, which was measured using a calibrated dose calibrator (CRC-25 PET, Capintec Inc., Ramsey, NJ). Labeled PET tracers were suspended in their respective HPLC mobile phase (Table 1) and had activity levels ranging from 0.2–1.0 mCi.
Radioactivity measurements were made of the original sample, the collected concentrated sample and of each five subsequent rinse steps. In the case of complete solvent evaporation method, the collected sample is obtained from an initial rinse step. Measurements were corrected for radioactive decay, and the fraction of initial radioactivity was calculated for each portion of the output volume.
4.8 Assessing thermal stability of tracers
Tracer stability at operating temperatures was assessed via analytical radio-HPLC as described in the Supplementary Information Section 4.
4.9 Determining residual organic solvent content
After partial or complete evaporation methods, the organic solvent content of several samples was assessed via gas chromatography mass spectrometry (GC-MS). Complete details are described in the Supplementary Information Section 5.
5 Results and Discussion
5.1 Scope of HPLC mobile phases
In the synthesis of PET tracers and radiolabeled prosthetic groups (for labeling biological molecules), HPLC purification methods nearly always use mixtures of EtOH in water or MeCN in water, sometimes with additives to buffer the pH to improve separation or stability. Examples of mobile phases for purification of several radiolabeled molecules are listed in Table 1.
We first examined contact angles (Supplementary Information, Figure S1) in order to determine the range of solvent compositions and temperatures for which θ > 90° (i.e. for which no spontaneous breakthrough would be expected). For mobile phases consisting of MeCN and water mixtures, we observed that θ > 90° for all compositions (from 0 to 100% MeCN in water, v/v) and all temperatures (RT to 80°C). For EtOH, concentrations up to 70% EtOH in water (v/v) can be sustained (θ > 90°) at all temperatures (RT to 80°C) without breakthrough, but compositions ≥ 80% EtOH (v/v) would not be suitable at any temperature. Unfortunately, for temperatures above 80 °C, droplets rapidly evaporated, preventing accurate contact angle measurement, and thus this method does not provide a good way to estimate the behavior under the most aggressive temperatures.
For both solvents, the effect of composition on wetting property appears to be more significant than the effect of temperature. This is expected because Eötvös rule describes a linear decrease in surface tension with increasing temperature [33], but surface tension has been observed to decrease superlinearly with increasing mole fraction of organic solvent [34].
While the contact angle gives an idea about spontaneous wetting, it does not indicate whether the BTP is sufficiently large for proper operation of the chip without breakthrough in practice. Using a specially designed test rig (see Supplementary Information Section 3), measurements of BTP for various solvent compositions and temperatures (up to 80°C) were made (see Table 2).
Table 2.
Breakthrough pressures measured with the membrane test rig for (A) MeCN and (B) EtOH. Pressures are in units of psi, and represent the average ± standard deviation of n=3 repeats.
|
A % MeCN in H2O (v/v) |
Temperature (°C) | ||||
|---|---|---|---|---|---|
| RT | 40 | 60 | 80 | 100 | |
| 0 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 10 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 20 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 30 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 40 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 50 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 60 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 70 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 80 | >13.5 | 12.2 ± 0.5 | 11.6 ± 1.4 | >13.5 | N.M. |
| 90 | 12.0 ± 0.2 | 10.6 ± 0.8 | 8.6 ± 0.5 | 8.1 ± 0.3 | N.M. |
| 100 | 7.3 ± 0.3 | 5.6 ± 0.2 | 3.3 ± 0.4 | 2.0 ± 0.4 | N.M. |
|
B % EtOH in H2O (v/v) |
Temperature (°C) | ||||
| RT | 40 | 60 | 80 | 100 | |
| 0 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 10 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 20 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 30 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 40 | >13.5 | >13.5 | >13.5 | >13.5 | N.M. |
| 50 | 11.5 ± 1.2 | 11.3 ± 0.7 | 12.0 ± 0.2 | >13.5 | N.M. |
| 60 | 7.9 ± 0.3 | 7.6 ± 0.3 | 7.6 ± 0.2 | 12.7 ± 0.5 | N.M. |
| 70 | 4.4 ± 0.2 | 4.1 ± 0.2 | 4.2 ± 0.3 | 5.4 ± 0.6 | N.M. |
| 80 | 1.8 ± 1.0 | 0.4 ± 0.4 | < 0.5 | 2.2 ± 0.8 | N.M. |
| 90 | < 0.5 | < 0.5 | < 0.5 | 3.9 ± 0.3 | N.M. |
| 100 | < 0.5 | < 0.5 | < 0.5 | 5.5 ± 0.2 | N.M. |
N.M. = not measured (see discussion in main text)
As expected, for samples containing either solvent (MeCN and EtOH), an increase in temperature or organic solvent composition results in decreased breakthrough pressure. For MeCN/water mixtures, BTP was very high (>13.5 psi) for all compositions up to 70% MeCN v/v (at all temperatures), and was moderately high (> 7.5 psi) for all compositions up to 90% MeCN v/v (at all temperatures). Moderate BTP values still allow considerable freedom in choice of operating conditions (Psample, Pgas_in, and Pgas_out). Only at 100% MeCN at elevated temperatures is the BTP low enough that reliable operation may be difficult. For EtOH/water mixtures, on the other hand, BTP was very high (>13.5 psi) for compositions up to 40% EtOH v/v at all temperatures tested, and was moderately high (>7.5 psi) for compositions up to 60% EtOH v/v at all temperatures. For all compositions ≥70% EtOH v/v, the BTP is low enough that reliable and efficient operation may be difficult. These results correspond well to the contact angle data.
Note that we observed some evidence that BTP could not be reliably measured in the test rig for temperatures ≥80°C. For example, starting with 80% MeCN at 80°C or 80% EtOH at 80°C, increased solvent composition would be expected to decrease the BTP, but the measurement actually increases. We hypothesize that the small surface area of the testing membranes (∼29 mm2) cannot transport the high vapor flux at high temperatures, leading to a buildup of pressure within the vicinity of the membrane. The vapor pressures of H2O, EtOH, and MeCN at 80°C are 6.9, 15.8, and 14.2 psi [35] respectively, and thus the vapor pressure of the tested mixtures will thus be >6.9 psi at 80°C (see Supplementary Information Section 6 Table S2). The build-up of vapor pressure counteracts the sample pressure, leading to an apparent increase in applied pressure needed to cause breakthrough. If sufficiently high, it can even push the sample away from the membrane, as was seen for samples at 100°C, making measurement of BTP impossible. Overall, these results suggest that the accuracy of this method of BTP measurement may be limited, especially when the vapor pressure is significant compared to the actual BTP (i.e. high organic solvent fractions and high temperatures).
5.2 Operating conditions
We chose a lower limit of BTP of ∼7.5 psi as a good compromise of compatibility with a wide range of solvent mixtures and temperatures as well as ensuring some room for optimizing the operating conditions. As a starting point, we selected Psample = 5.3 psi, Pgas_in = 4.5 psi, and Pgas_out = -0.25 psi. This ensures all the constraints described in Section 4.5 are met. Psample was sufficient to ensure the chip was always filled with sample under all conditions explored in this paper. The maximum pressure across the membrane is 5.6 psi, which is below 7.5 psi by a 35% safety margin. Pgas_in is below Psample by a ∼20% safety margin.
A limited exploration of gas-flow pressure differential and sample pressure was performed to determine the effect on evaporation rate performance (see Supplementary Information, Section 7). Over the range tested, Psample was found to have negligible effect on the evaporation rate. The gas-flow pressure differential did have a small effect, as was also observed in our previous work. Changing to a differential of 6.0 psi instead of 4.75 psi would increase evaporation rate by ∼ 5%, but if similar safety margins were used, the minimum BTP that could be tolerated would be ∼10 psi, which would exclude several solvent compositions; thus this change was not implemented.
Parameters related to sample collection were set to maximize sample recovery. Pvacuum ≤ -2.0 psi resulted in break-up of the sample in tubing during recovery, so Pvacuum was set to -1.0 psi. This was ramped up to Pvacuum_ramp = -8.8 psi, near the best vacuum that could be achieved with the pump used.
5.3 Evaporation rate
Using the above operating conditions, evaporation rates were extensively characterized. Maximum evaporation rates of water at 100°C exceeded 2 mL/min, compared to ∼0.6mL/min observed in our previous system [21]. We also performed concentration using a rotary evaporator according to parameters recommended by the manufacturer (see Supplementary Information Section 8). We found the evaporation rate at 80°C to be 1.03 ± 0.04 mL/min compared with 1.53 ± 0.02 mL/min achieved in the microfluidic chip at the same temperature.
The observed evaporation rate of the chip depended somewhat on the material that was used for the sample layer (Figure 4). Evaporation rates using PMMA-machined, PMMA-ablated, Ultem, and PEEK, were very similar across the temperature setpoints. The evaporation rate for glass-Teflon is significantly higher than the other materials. The differences may be related to the thermal conductivity of the sample layer materials. Thermal conductivities of PMMA, Ultem, and PEEK are all similar (0.19, 0.22, 0.25 W/mK, respectively), whereas the thermal conductivity of glass-PTFE is significantly higher (0.42 W/mK) [36]. Note that PTFE and COC turned out not to be suitable materials. For PTFE, significant deformation occurred at the inlet and outlet locations, most likely due to compressive forces from fluidic connections, interfering with sample loading and ejection. The COC material exhibited cracking and leaks, perhaps as a result of the elevated operating temperatures in combination with the compressive forces to hold the layers of the chip together.
Figure 4.
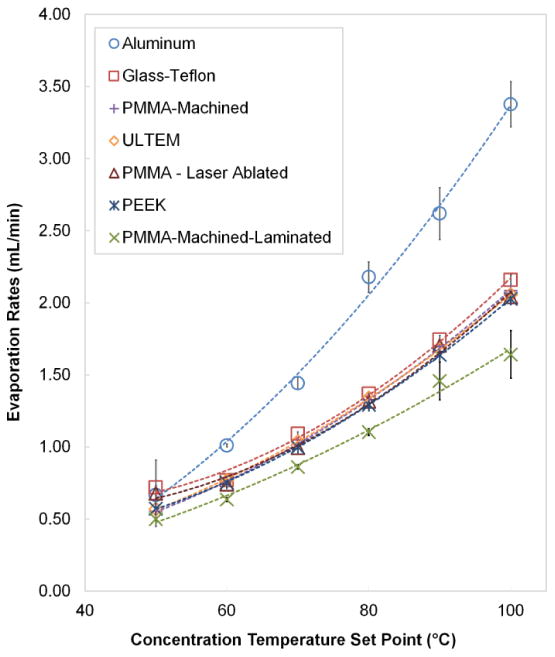
Evaporation rates of deionized water as a function of temperature for different chip materials. Data points show an average of 5 repeats and error bars represent standard deviation
In addition to comparing sample layer materials, different chip architectures were explored to compare the effect of heater placement (see Supplementary Information, Section 9). Most designs had similar or inferior performance with the exception of one design having a 3.2 mm thick 6061 aluminum alloy sample layer (thermal conductivity 167 W/m*K [37]) instead of a plastic sample layer. At 100°C, evaporation rate was 3.38±0.16 (n=5) mL/min compared to 2.02 ± 0.04 mL/min (n=5) for the PEEK sample layer, though the results are not directly comparable due to different arrangement of heaters. Because the opaqueness of the metal prevents convenient visualization, the plastic sample layers were used for all remaining experiments, but this result shows the potential improvement that may be possible by considering materials with high thermal conductivity.
5.4 Effect of solutes and solvents on evaporation rate
Radiolabeled molecules may be in HPLC mobile phase volumes as large as ∼50 mL prior to concentration and formulation using the microfluidic chip, leading to large increases (∼50×) in concentration of salts and additives that may be present in the mobile phase. Though an increase in solute concentration can lower the vapor pressure [38] (which would be expected to reduce the evaporation rate), we found that evaporation rates of 50 mL samples were remarkably constant throughout the entire evaporation process. Solutes became a factor during concentration only when the starting concentration of salts was 10× higher than normally used in HPLC mobile phases (see Supplementary Information Section 10).
The effect of organic solvents was also considered. Not surprisingly, increasing concentration of organic solvent in the mobile phase led to increase in evaporation rate (see Supplementary Information Section 10). For large sample volumes, we expected the evaporation rates to be initially higher and then decrease as the lower boiling point organic solvent was evaporated off leaving mostly water within the chip. However, we found the evaporation rates remained nearly constant.
5.5 Sample Recovery
After recovery of the concentrated sample, additional rinse steps are used to recover residual amounts of the sample. However, because rinse steps have the undesirable effect of increasing the final collected volume, we experimentally studied the effect of rinsing so the number of rinse steps could be optimized.
Using samples containing [18F]fluoride, the amount of radioactivity recovered in the initially ejected plug and five subsequent rinse plugs is plotted in Figure 5. For the majority of materials used as sample layers (glass-PTFE, Ultem, PEEK, PMMA-machined), 92-97% of [18F]fluoride is recovered from the initial ejection. Following the initial ejection with two sample rinses increases [18F]fluoride recovery for all materials (>97%). Subsequent rinses recover negligible amounts of activity.
Figure 5.
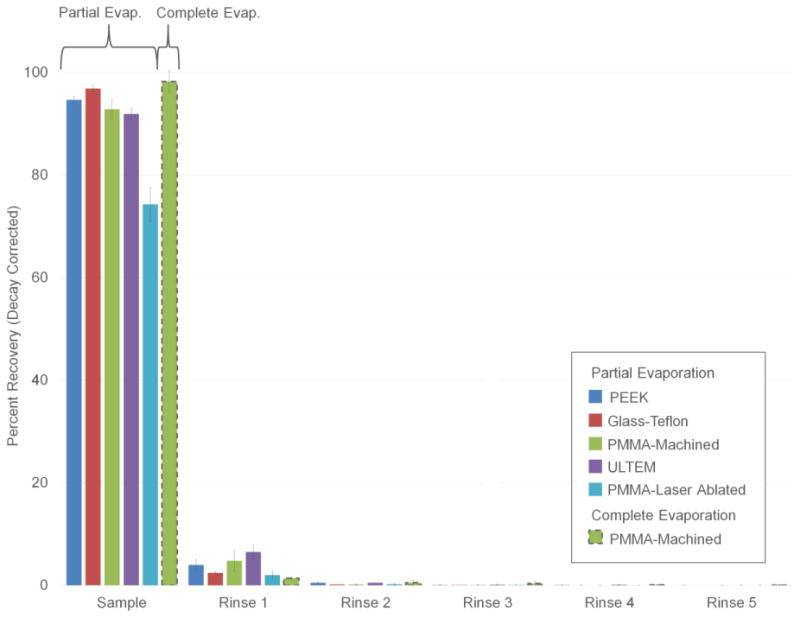
Sample recovery measurements using [18F]fluoride solutions. Results are shown for different sample layer materials. All data is for the partial evaporation method of operation, except PMMA-machined, for which both partial and complete evaporation modes were tested. Each data point represents n=3 repeats with error bars denoting standard deviation.
On the other hand, using PMMA-ablated, recovery following two rinses was poor (74 ± 3%, n=3). Since PMMA fabricated by CNC machining exhibited excellent recovery, this result is most likely explained by either higher surface roughness of the laser ablated surface (see Supplementary Information Section 11), or chemical modifications during laser ablation. Laser ablated PMMA was therefore not used in further experiments. In practice, it is likely that sample layers would be manufactured via injection molding and should be immune to this issue.
Sample recovery performance was also tested for cases where the system was operated in complete evaporation mode (Figure 5). A proof-of-concept experiment using PMMA-machined sample layer showed 98 ± 2% (n=3) recovery with the initial ejection and 100 ± 2% (n=3) recovery with two additional rinses.
5.6 In-chip measurements of breakthrough
Sample recovery experiments also provided a way to measure BTP in situ. As described above, measurements of contact angle or BTP using the test rig had limitations at high temperatures and could not make measurements at all >80°C. Loss due to breakthrough was monitored using 10 mL samples of different solvent compositions containing 0.6mM K2.2.2. and 0.3mM K2HCO3 spiked with ∼5-10 μL of a solution containing [18F]fluoride (∼0.5 mCi). Results are summarized in the Figure 6). We found that concentrations of MeCN up to 40% can be concentrated at 100°C with almost no sample loss, and only minor loss (<6%) when MeCN content is 40-80%. EtOH compositions up to 20% showed no apparent activity loss, and solutions with 30-40% EtOH showed minor (<7%) loss. Interestingly, the loss is not a binary effect, i.e. zero loss below breakthrough and 100% loss above breakthrough. This may be explained by slight non-uniformity in the membrane (e.g. where some regions have slightly larger pore size, with lower BTP), or by the pressure gradients in the system (i.e. such that BTP is exceeded only near the sample outlet, where the pressure across the membrane is greatest). In either case, breakthrough may only occur over a small fraction of the membrane area, limiting its effect. Since losses of 6-7% would be considered acceptable, we can conclude that mobile phases with up to 80% MeCN in water (v/v), or up to 40% EtOH in water (v/v) can be concentrated with the chip at all temperatures up to 100°C. This range is sufficient to cover all HPLC mobile phases used in the purification of PET tracers.
Figure 6.
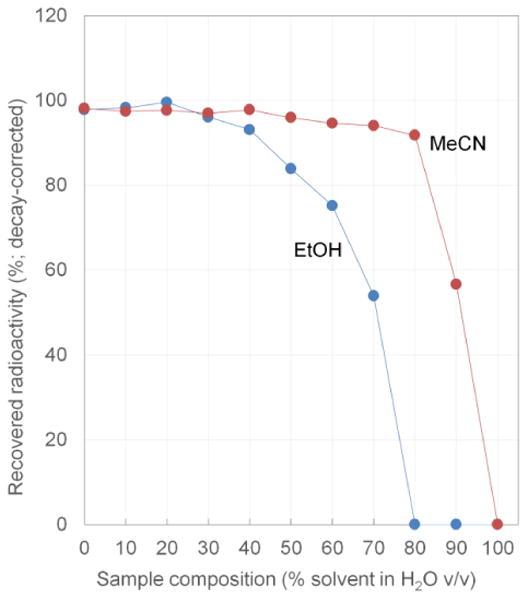
Sample recovery measurements using [18F]fluoride solutions enable in-chip measurement of the effects of breakthrough. The data indicate minimal loss of solute for mobile phases containing up to 80% MeCN v/v, or up to 40% EtOH v/v.
5.7 Concentration of PET tracers
5.7.1 Partial evaporation method
As a demonstration, batches of four PET tracers ([18F]D-FAC, [18F]FHBG, [18F]Fallypride and [18F]FEAU) as well as a prosthetic group for peptide and protein labeling ([18F]FBEM) were concentrated with the system. Initially, the partial evaporation method (100°C, plus 2 rinse steps) was explored (Figure 7).
Figure 7.
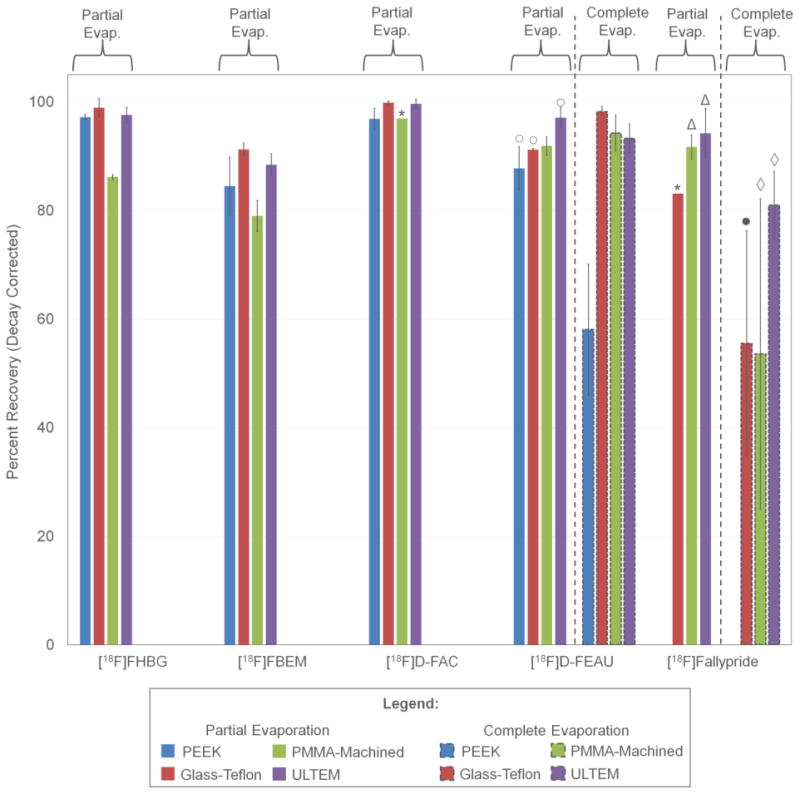
Percent recovery of different tracers on different sample layer materials concentrated with both partial and complete evaporation modes. Unless otherwise stated, each bar represents three repeats. (*) represents one repeat, (○) represents two repeats, (Δ) represents four repeats, (●) represents six repeats and (◊) represents nine repeats. Error bars represent standard deviation.
[18F]D-FAC was concentrated with recovery >96% for all chip materials. Concentration of [18F]FHBG using PEEK, Ultem and glass-Teflon exhibited >97% recovery, but recovery was significantly lower for PMMA-machined, i.e. 86.2 ± 0.4% (n=3). Recovery of concentrated [18F]Fallypride was 94.2 ± 4.6% (n=4) with the Ultem chip, slightly lower (91.7± 2.2%, n=4) for the PMMA-machined chip, and only 83.1% (n=1) for the glass-Teflon chip. (PEEK was not tested.) For [18F]FEAU, recovery in the Ultem chip was 97.1 ± 1.9% (n=2). Slightly lower recovery was seen in the PMMA-machined chip (91.1 ± 0.2%, n=3) and glass-Teflon chips (91.8 ± 1.7%, n =2), and significantly worse performance was observed for PEEK (87.8 ± 4.0%, n=2). After concentration, the prosthetic group [18F]FBEM was recovered with 91.2 ± 1.2% (n=3) recovery in the glass-PTFE chip. The PEEK and Ultem materials produced slightly lower recovery rates of 84.5 ± 5.3% and 88.4 ± 2.0%, respectively, and PMMA-machined performed the worst (recovery of 79.0 ± 2.9%, n=3). Partial evaporation could be completed at a rate of ∼2 mL/min (depending on conditions) plus a 4.0 min “overhead” for sample loading and final rinse steps. (More complete analysis of evaporation times is presented in the Supplementary Information Section 12).
In post-experiment analysis, residual radioactivity was found to be localized on the sample layer (rather than on the Teflon membrane) suggesting adverse tracer interaction is with the sample layer surface. Considering that the layers were fabricated using the same milling method resulting in similar surface finish, we hypothesize that loss of the tracer is due to chemical interaction with the surface rather than due to mechanical trapping on rough surfaces.
Even at the 100°C operating temperature, radio-HPLC analysis of tracers showed no significant difference in purity before and after concentration (see Supplementary Information Section 4) suggesting that tracers are stable under these conditions. Since high sample recovery was observed in all cases for one or more chip materials, we can also assume the tracers do not have significant volatility at 100°C.
We next examined the degree of organic solvent removal during the concentration process. Since MeCN and EtOH are significantly more volatile than water (MeCN bp=82°C; EtOH bp= 78.37°C), we suspected that these solvents would be preferentially lost compared to water. Figure 8 summarizes GC-MS measurements of the remaining organic solvent content after the concentration process as a function of starting solvent composition, evaporation temperature, and starting volume. Surprisingly, the starting sample volume (and hence total time to perform concentration process) has very minor effect on the final composition. We suspect the system rapidly reaches an “equilibrium” where the majority of the chip is filled with water (due to preferential removal of solvent), a small portion at the inlet is filled with the incoming sample solution, and a concentration gradient exists in between. Attainment of such a steady-state concentration gradient in the chip is consistent with the results of the 50 mL evaporation experiments described above that showed no change in evaporation rate throughout the concentration process. Temperature had a small effect on final solvent composition, with higher temperature resulting in lower final solvent amount. The starting composition had the dominant effect on the final composition after concentration. The concentration procedure seems to reduce the initial organic solvent fraction roughly 10× for MeCN mixtures and roughly 5× for EtOH mixtures. Since the allowable injection limit for EtOH is 10% v/v [17], adequate EtOH removal will occur after concentration of samples with mobile phase composition up to 50% v/v EtOH; thus the chip effectively performs formulation in addition to concentration. This encompasses the full range of EtOH-based mobile phases compatible with the chip. On the other hand, the allowed injection limit of MeCN is much lower (410 ppm or 0.041% v/v [18]). Since typical HPLC mobile phases contain > 0.41% MeCN, the amount of residual MeCN will be too high for injection and an additional formulation process will be needed.
Figure 8.
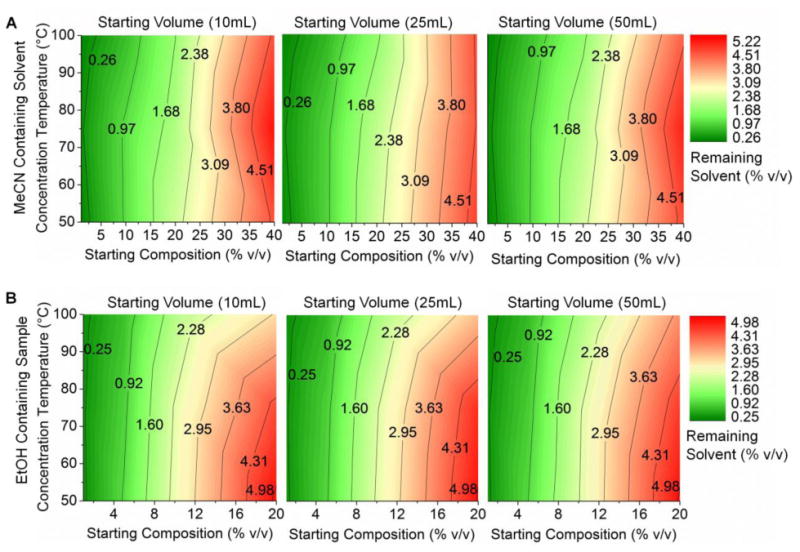
Contour plots of % residual solvent after concentration by partial evaporation of MeCN/water mixtures (A) and EtOH/water mixtures (B).
5.7.2 Complete evaporation method
One way that the composition of MeCN could be reduced more than 10× would be to extend the “delay time”, i.e. the additional evaporation time after the volume has already shrunk to the chip volume. Though we did not investigate the relationship between delay time and residual solvent amount, we studied the extreme case where the mobile phase is completely evaporated, leaving only a dry residue of the PET tracer.
To assess the ability to recover the tracer residue in an injectable solution such as saline, complete evaporation and recovery was characterized for two tracers (Figure 7). In the case of [18F]Fallypride, the best recovery was achieved with the Ultem chip (81.0 ± 6.3%, n=9). Recoveries in PMMA-machined and glass-PTFE were only 53.6 ± 28.6% (n=9) and 55.5 ± 20.7% (n=6), respectively. The low recovery and large variation suggests an adverse reaction of the tracer with the sample layer for these materials. For [18F]D-FEAU, PMMA-machined, glass-PTFE, and Ultem all yielded high recovery (>93%) with glass-PTFE being the best (98.2 ± 0.9%, n=3). Recovery with PEEK was significantly worse and exhibited high variability (58.1 ± 12.1%, n=3), suggesting an adverse interaction. Since the performance appears to be tracer-specific, the ability to easily switch the sample layer material to suit the tracer is a major advantage of the chip design. Complete evaporation could be completed at a rate of ∼2 mL/min (depending on conditions) plus a 9.0 min “overhead” for sample loading, drying of solvent, collection of solute, and final rinse steps. (More complete analysis of evaporation times is presented in the Supplementary Information Section 12).
For 3 samples of [18F]D-FEAU concentrated via the complete evaporation method, the residual MeCN concentrations in the first rinse measured via GC-MS were 700, 185, 87 ppm. Since, during operation, the initial rinse will be followed by two subsequent rinses, the final MeCN composition is therefore well below the allowed injection limit. As further assessment of tracer safety and stability, a [18F]D-FAC sample was injected into mice immediately after concentration via the partial evaporation method. (Detailed methods on mouse imaging can be seen in Supplementary Information Section 13). No adverse reactions were observed in mice and the biodistribution was very similar to that from [18F]D-FAC injection obtained via conventional concentration and formulation with rotary evaporation (see Supplementary Information Figure S11). Furthermore, the biodistribution is similar to literature reports, showing the expected high uptake in immune-related organs such as the thymus, bone/bone marrow, and spleen [39].
In addition to solvents in the mobile phase itself, the purified HPLC fraction can also contain small amounts of the reaction solvent and it is important to consider whether these can be adequately removed, especially high-boiling (bp = 189°C) solvents such as DMSO. To answer this question, we first measured the evaporation rate of pure DMSO in the device at 100°C with the PEEK sample layer and found it to be 0.87 ± 0.03 mL/min (n = 5). This relatively fast rate suggested that adequate DMSO removal should be possible in a reasonable time. Next, we measured residual DMSO content after performing microfluidic concentration. A 10 mL solution of DMSO (10,000 PPM in water) was concentrated using the complete evaporation method at 100°C using different delay times (270, 600, 870s) and then recovered with a water rinse. The normal delay time for complete evaporation (270s) resulted in a DMSO content of 1,400 PPM. Increased delay times of 600 and 870s resulted in remaining DMSO content of 800 PPM and 700 PPM, respectively. Operation at all delay times resulted in final compositions less than the injectable limit of 5000 PPM. Of course, the amount of solvent present in the purified fraction will depend on details of the purification protocol so measurements of residual solvents during synthesis optimization are recommended.
5.8 Conclusions
We have developed, optimized, and automated a compact microfluidic device to concentrate and formulate various types of radiolabeled molecules such as PET tracers as well as prosthetic groups used to convert fragile biological molecules into PET tracers. It is especially useful for tracers intended for preclinical or in vitro applications where the needed radioactivity concentration is high [40] but the total amount of radioactivity needed is low. The system, however, could also be used to perform formulation in clinical applications.
The system can readily be integrated with upstream (e.g. purification) and downstream (e.g. sterile filtration) processes via tubing connections, and operation is fully automated, ensuring straightforward operation and minimal operator exposure to radiation. The entire concentration and collection of a 10mL starting sample at 100°C can be completed in under 9 min for partial evaporation and under 14 min for complete evaporation. The small physical footprint of the complete system allows for relatively compact and lightweight shielding, or frees up valuable space inside existing radiation-shielded hot cells that would otherwise be needed for concentration and formulation systems. This microfluidic approach is ideally suited for integration with either conventional radiosynthesizers lacking integrated or automated formulation systems, or for use in conjunction with emerging compact microscale radiosynthesizers [41], [42]. Since the vast majority of conventional synthesizers are focused on clinical production and do not include a means to concentrate the final tracer, this microfluidic concentration system could be used to extend their functionality to the production of tracers for preclinical imaging.
Improvements in design resulted in significant performance increase compared to our previous proof-of-concept [21]. Depending on the architecture of the chip, evaporation rates of up to 3.4 mL/min for water were observed at 100°C operating temperature. By using a serpentine channel with rounded corners, the system is able to achieve high tracer recovery (>97%) with the use of two rinse plugs, resulting in a total sample volume after concentration of ∼1.0-1.5 mL. The range of compatible solvents was also greatly expanded. At 100°C operating temperature, the system can handle samples with up to 80% (v/v) MeCN in H2O, and up to 40% (v/v) EtOH in H2O without significant loss of the sample. While these limits include the majority of mobile phases used in the production of PET tracers [29], [43]–[47], these limits of organic solvent content could likely be further extended if needed, by decreasing operating temperatures (at the expense of evaporation rate) or by seeking membrane with even smaller pore dimensions.
In partial evaporation mode, the amount of EtOH can be adequately reduced to safely allow direct injection after concentration (for mobile phases containing up to 50% EtOH v/v). To directly use samples containing MeCN, the complete evaporation method must be used to ensure the residual MeCN is below the allowed limit. Concentration to dryness could also be used if the mobile phase contained other toxic solvents that needed to be removed, such as DMSO. This ability to perform formulation at the same time as concentration significantly streamlines the overall PET tracer production process.
The ability to customize sample layer material was valuable for ensuring the highest possible performance. Several tracers exhibited significant (20-50%) losses when partnered with an unsuitable chip material, presumably due to adsorption or other adverse interactions. Future investigation of materials with high thermal conductivity that are inert (or with protective inert coating) may provide a way to achieve the highest possible evaporation speed combined with high sample recovery.
In addition to the application described here, the microfluidic chip could also be used in related areas such as the concentration of radioisotopes prior to labeling. For example, there is considerable interest in isotopes that can be produced via generators rather than cyclotrons (e.g. 68Ga, 82Rb, and 62Cu) [48]. However, a challenge of 68Ga-labeling is that the output of the generator decreases over time, requiring larger and larger amounts of the eluent (HCl) to collect the desired amount of activity. While cartridge-based methods have been developed to concentrate gallium-68 into a consistent output volume [40], they are relatively slow, leading to considerable radioactive decay. Our microfluidic concentrator may provide a means to more rapidly concentrate the isotope and achieve a similar result. In addition, concentration of radiometals can also help improve the reaction kinetics between the chelator and metal, and increase apparent specific activity by enabling reduced precursor usage. It has been observed that the concentration of [68Ga]Ga3+ and [64Cu]Cu2+ has significant effect on the kinetics [49] and an automated means to concentrate these starting materials could lead to methods for improved labeling yields. More broadly, this microfluidic system can potentially be used for concentration or reformulation of any aqueous sample where compact size and high speed of liquid removal are important.
Supplementary Material
Acknowledgments
The authors thank Dr. Saman Sadeghi, Dr. Umesh Gangadharmath, and the staff of the UCLA Biomedical Cyclotron Facility for providing [18F]fluoride for these studies. The authors also thank Waldemar Ladno, Olga Sergeeva, and Theresa Falls at the UCLA Crump Institute Preclinical Imaging Technology Center for assistance with preclinical imaging, Seunghyun “Noel” Ha for assisting with tracer production, and Adrian Gomez and Alejandra Rios for their assistance with GC-MS. This work was supported in part by the Department of Energy Office of Biological and Environmental Research (DE-SC00001249), the National Cancer Institute (R21CA174611), the UCLA Foundation from a donation made by Ralph & Marjorie Crump for the UCLA Crump Institute for Molecular Imaging, an NIH CBI Grant (T32 GM008469), UCLA Graduate Division, and a UCLA Dissertation Year Fellowship.
Footnotes
Author Contributions: PHC, MVD, and WYT designed the microfluidic chip and automated system. PHC, and MVD designed experiments. PHC performed experiments. PHC and MVD analyzed results and data. JC synthesized tracers and aided in performing HPLC. JTL performed preclinical imaging and preclinical imaging data analysis. JPA and PHC performed GC measurements and analyzed GC data. PHC and MVD wrote the manuscript with input from JPA and JTL.
References
- 1.Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci. 2000 Aug;97(16):9226–9233. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobs A, et al. Positron Emission Tomography of Vector-Mediated Gene Expression in Gene Therapy for Gliomas. Lancet. 2001;358:727–729. doi: 10.1016/s0140-6736(01)05904-9. [DOI] [PubMed] [Google Scholar]
- 3.Herschman HR, et al. Seeing Is Believing: Non-Invasive, Quantitative and Repetitive Imaging of Reporter Gene Expression in Living Animals, Using Positron Emission Tomography. J Neurosci Res. 2000;59:699–705. doi: 10.1002/(SICI)1097-4547(20000315)59:6<699::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Greco M. Axillary Lymph Node Staging in Breast Cancer by 2-Fluoro- 2-deoxy-D-glucose–Positron Emission Tomography: Clinical Evaluation and Alternative Management. J Natl Cancer Inst. 2001;93(8):630–635. doi: 10.1093/jnci/93.8.630. [DOI] [PubMed] [Google Scholar]
- 5.Ciernik F, et al. Radiation Treatment Planning with an Integrated Positron Emission and Computer Tomography (PET/CT): A Feasibility Study. Int J Radiat Oncol Biol Phys. 2003;57(3):853–863. doi: 10.1016/s0360-3016(03)00346-8. [DOI] [PubMed] [Google Scholar]
- 6.Erdi Y, et al. Radiotherapy treatment planning for patients with non-small cell lung cancer using positron emission tomography (PET) Radiother Oncol. 2002;62:51–60. doi: 10.1016/s0167-8140(01)00470-4. [DOI] [PubMed] [Google Scholar]
- 7.Yagle K, et al. Evaluation of 18F-Annexin V as a PET Imaging Agent in an Animal Model of Apoptosis. J Nucl Med. 2005;46(4):658–666. [PubMed] [Google Scholar]
- 8.Sorger D, et al. [18F]Fluoroazomycinarabinofuranoside (18FAZA) and [18F]Fluoromisonidazole (18FMISO): A comparative study of their selective uptake in hypoxic cells and PET imaging in experimental rat tumors. Nucl Med Biol. 2003;30:317–326. doi: 10.1016/s0969-8051(02)00442-0. [DOI] [PubMed] [Google Scholar]
- 9.Yuan H, Schroeder T, Bowsher J, Hedlund L, Wong T, Dewhirst M. Intertumoral Differences in Hypoxia Selectivity of the PET imaging agent 64Cu(II)-Diacetyl-Bis(N4-mythylthiosemicarbaxone) J Nucl Med. 2006;47:989–998. [PubMed] [Google Scholar]
- 10.Seddon BM, Workman P. The role of functional and molecular imaging in cancer drug discovery and development. Br J Radiol. 2003 Dec;76(suppl_2):S128–S138. doi: 10.1259/bjr/27373639. [DOI] [PubMed] [Google Scholar]
- 11.Rudin M, Weissleder R. Molecular Imaging in Drug Discovery and Development. Nat Rev Drug Discov. 2003;2:123–131. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 12.Langer O, et al. Combined PET and Microdialysis for In Vivo Assessment of Intracellular Drug Pharmacokinetics in Humans. J Nucl Med. 2005;46(11):1835–1841. [PubMed] [Google Scholar]
- 13.Aboagye EO, Price PM, Jones T. In vivo pharmacokinetics and pharmacodynamics in drug development using positron-emission tomography. Drug Discov Today. 2001 Mar;6(6):293–302. doi: 10.1016/s1359-6446(01)01684-1. [DOI] [PubMed] [Google Scholar]
- 14.Hargreaves R, et al. Optimizing Central Nervous System Drug Development Using Molecular Imaging. Clin Pharmacol Ther. 2015 Jul;98(1):47–60. doi: 10.1002/cpt.132. [DOI] [PubMed] [Google Scholar]
- 15.Diehl KH, et al. A Good Practice Guide to the Administration of Substances and Removal of Blood, Including Routes and Volumes. Jounral Appl Toxicol. 2001;21:15–23. doi: 10.1002/jat.727. [DOI] [PubMed] [Google Scholar]
- 16.Vu NT, et al. A β-Camera Integrated with a Microfluidic Chip for Radioassays Based on Real-Time Imaging of Glycolysis in Small Cell Populations. J Nucl Med. 2011 May;52(5):815–821. doi: 10.2967/jnumed.110.078725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serdons K, Verbruggen A, Bormans G. The Presence of Ethanol in Radiopharmaceutical Injections. J Nucl Med. 2008 Dec;49(12):2071–2071. doi: 10.2967/jnumed.108.057026. [DOI] [PubMed] [Google Scholar]
- 18.ICH Expert Working Group. Impurities: Guideline for Residual Solvents Q3C(R5) 1997 [Google Scholar]
- 19.Eckert & Ziegler Eurotope GmbH. Modular-Lab PharmTracer. [Accessed: 04-Feb-2013];2013 [Online]. Available: http://www.ezag.com/home/products/radiopharma/radiosynthesis-technology/modular-lab-pharmtracer.html.
- 20.Trasis Pharmacy Instruments - All in One. Trasis Pharmacy Instruments.
- 21.Tseng WY, van Dam RM. Compact microfluidic device for rapid concentration of PET tracers. Lab Chip. 2014;14(13):2293. doi: 10.1039/c4lc00286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W, Wu LL, Li GP, Bachman M. A vapor-based microfluidic sample concentrator. Proceedings of the Fourteenth International Conference on Miniaturized Systems for Chemistry and Life Sciences; Groningen, The Netherlands. 2010. pp. 1208–1210. [Google Scholar]
- 23.i Solvas XC, Turek V, Prodromakis T, Edel JB. Microfluidic evaporator for on-chip sample concentration. Lab Chip. 2012 Sep;12(20):4049–4054. doi: 10.1039/c2lc40746a. [DOI] [PubMed] [Google Scholar]
- 24.Deadman B, Battilocchio C, Sliwinski E, Ley S. A prototype device for evaporation in batch and flow chemical processes. Green Chem. 2013;15:2050–2055. [Google Scholar]
- 25.Arima V, et al. Radiochemistry on chip: towards dose-on-demand synthesis of PET radiopharmaceuticals. Lab Chip. 2013 May;13(12):2328–2336. doi: 10.1039/c3lc00055a. [DOI] [PubMed] [Google Scholar]
- 26.Sharma NR, Lukyanov A, Bardell RL, Seifried L, Shen M. Microfluidics, BioMEMS, and Medical Microsystems VI. Vol. 6886. San Jose, CA, USA: 2008. Development of an evaporation-based microfluidic sample concentrator, in; pp. 68860R–9. [Google Scholar]
- 27.Alkhudhiri A, Darwish N, Hilal Nidal. Membrane distillation: A comprehensive review. Desalination. 2012;287:2–18. [Google Scholar]
- 28.Lazari M, et al. ELIXYS - a fully automated, three-reactor high-pressure radiosynthesizer for development and routine production of diverse PET tracers. EJNMMI Res. 2013 Dec;3(1):52. doi: 10.1186/2191-219X-3-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazari M, et al. Fully Automated Production of Diverse 18F-Labeled PET Tracers on the ELIXYS Multireactor Radiosynthesizer Without Hardware Modification. J Nucl Med Technol. 2014 Sep;42(3):203–210. doi: 10.2967/jnmt.114.140392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiesewetter DO, Jacobson O, Lang L, Chen X. Automated radiochemical synthesis of [18F]FBEM: A thiol reactive synthon for radiofluorination of peptides and proteins. Appl Radiat Isot. 2011 Feb;69(2):410–414. doi: 10.1016/j.apradiso.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng D, Kim J, Kim CJ. A degassing plate with hydrophobic bubble capture and distributed venting for microfludic devices. J Micromechanics Microengineering. 2006;16:419–424. [Google Scholar]
- 32.Meng D, Cubaud T, Ho CM, Kim CJ. A Methanol-Tolerant Gas-Venting Microchannel for a Microdirect Methanol Fuel Cell. J Microelectromechanical Syst. 2007;16(6):1403–1410. [Google Scholar]
- 33.Ali K, Shah AHA, Bilal S, Shah AHA. Surface tensions and thermodynamic parameters of surface formation of aqueous salt solutions: III. Aqueous solution of KCl, KBr and KI. Colloids Surf Physicochem Eng Asp. 2009;337:194–199. [Google Scholar]
- 34.Xu L, et al. Hydrophobic coating- and surface active solvent-mediated self-assembly of charged gold and silver nanoparticles at water–air and water–oil interfaces. Phys Chem Chem Phys. 2009 Aug;11(30):6490–6497. doi: 10.1039/b820970g. [DOI] [PubMed] [Google Scholar]
- 35.Wolfram Alpha LLC. WolframAlpha Computational Knowledge Engine. [Accessed: 15-Jan-2016];Wolfram|Alpha. [Online] Available: www.wolframalpha.com.
- 36.Professional Plastics. Thermal Properties of Plastic Materials [Google Scholar]
- 37.Trouve A, Minnich T. Thermal Properties Database. 2012 [Google Scholar]
- 38.Meranda D, Furter W. Elevation of the Boiling Point of Water by Salts at Saturation: Data and Correlation. J Chem Eng Data. 1977;22(3) [Google Scholar]
- 39.Radu CG, et al. Molecular imaging of lymphoid organs and immune activation using positron emission tomography with a new 18F-labeled 2′-deoxycytidine analog. Nat Med. 2008 Jul;14(7):783–788. doi: 10.1038/nm1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gebhardt P, Opfermann T, Saluz HP. Computer controlled 68Ga milking and concentration system. Appl Radiat Isot. 2010;68:1057–1059. doi: 10.1016/j.apradiso.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 41.Rensch C, et al. Microfluidics: A Groundbreaking Technology for PET Tracer Production? Molecules. 2013 Jul;18(7):7930–7956. doi: 10.3390/molecules18077930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keng PY, van Dam RM. Digital Microfluidics: A New Paradigm for Radiochemistry. Mol Imaging. 2015;14:579–594. doi: 10.2310/7290.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vallabhajosula S, Solnes L, Vallabhajosula B. A Broad Overview of Positron Emission Tomography Radiopharmaceuticals and Clinical Applications: What Is New? Semin Nucl Med. 2011 Jul;41(4):246–264. doi: 10.1053/j.semnuclmed.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Vallabhajosula S. 18F-Labeled Positron Emission Tomographic Radiopharmaceuticals in Oncology: An Overview of Radiochemistry and Mechanisms of Tumor Localization. Semin Nucl Med. 2007 Nov;37(6):400–419. doi: 10.1053/j.semnuclmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Lasne MC, et al. Chemistry of β +-Emitting Compounds Based on Fluorine-18. In: Krause W, editor. Contrast Agents II. Vol. 222. Heidelberg: Springer Berlin Heidelberg; 2002. pp. 201–258. [Google Scholar]
- 46.Vallabhajosula S. Positron Emission Tomography Radiopharmaceuticals for Imaging Brain Beta-Amyloid. Semin Nucl Med. 2011;41:283–299. doi: 10.1053/j.semnuclmed.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Kumar P, Bacchu V, Wiebe LI. The Chemistry and Radiochemistry of Hypoxia-Specific, Radiohalogenated Nitroaromatic Imaging Probes. Semin Nucl Med. 2015;45:112–135. doi: 10.1053/j.semnuclmed.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Fani M, Andre JP, Maecke HR. 68Ga-PET: a powerful generator-based alternative to cyclotron-based PET radiopharmaceuticals. Contrast Media Mol Imaging. 2008;3:67–77. doi: 10.1002/cmmi.232. [DOI] [PubMed] [Google Scholar]
- 49.Zeng D, Desai AV, Ranganathan D, Wheeler TD, Kenis PJA, Reichert DE. Microfluidic radiolabeling of biomolecules with PET radiometals. Nucl Med Biol. 2013 Jan;40(1):42–51. doi: 10.1016/j.nucmedbio.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.