

Abstract
The hormone ouabain has been shown to enhance the cystic phenotype of autosomal dominant polycystic kidney disease (ADPKD). Among other characteristics, the ADPKD phenotype includes cell de-differentiation and epithelial to mesenchymal transition (EMT). Here, we determined whether physiological concentrations of ouabain induces EMT in human renal epithelial cells from patients with ADPKD. We found that ADPKD cells respond to ouabain with a decrease in expression of the epithelial marker E-cadherin and increase in the expression of the mesenchymal markers N-cadherin, α smooth muscle actin (αSMA) and collagen-I; and the tight junction protein occludin and claudin-1. Other adhesion molecules, such as ZO-1, β-catenin and vinculin were not significantly modified by ouabain. At the cellular level, ouabain stimulated ADPKD cell migration, reduced cell-cell interaction, and the ability of ADPKD cells to form aggregates. Moreover, ouabain increased the transepithelial electrical resistance of ADPKD cell monolayers, suggesting that the paracellular transport pathway was preserved in the cells. These effects of ouabain were not observed in normal human kidney (NHK) cells. Altogether these results show a novel role for ouabain in ADPKD, inducing changes that lead to a partial EMT phenotype in the cells. These effects further support the key role that ouabain has as a factor that promotes the cystic characteristics of ADPKD cells.
Keywords: Na, K-ATPase signaling, polycystic kidney disease, adhesion molecule
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder that occurs in 1:400-1:1,000 individuals worldwide [1]. ADPKD is initiated by mutations in either the PKD1 or PKD2 genes, which encode for polycystin-1 and polycystin-2 (PC1, PC2) respectively [2-4]. The main manifestations of ADPKD appear in the kidney, with the formation of numerous epithelial-lined cysts that develop throughout the nephron and predominantly in collecting duct cells. Cysts progressively expand, impair renal function, and lead to end stage renal disease in 50% of the affected individuals by age 60 [5, 6]. Many patients with ADPKD require dialysis or undergo transplant therapy [6, 7].
ADPKD cystic epithelial cells have been shown to display an undifferentiated phenotype and to undergo epithelial to mesenchymal transition (EMT) changes as part of their phenotype. Thus, in ADPKD kidneys there is upregulation of EMT-related genes and increased fibrosis [8-14]. In addition, it has been suggested that ADPKD renal epithelial cells show an abnormal response to injury, signaling a “wounded status,” which initiates a futile wound-healing program that exacerbates the progression of the disease [15]. This injury response has been shown to be associated with conversion into an EMT phenotype [16, 17]. In addition, PC1 has been reported to directly interact with a number of proteins that are involved in EMT. For instance, PC1 localizes at the junctions of renal epithelial cells grown in vitro [18, 19], and interacts with components of focal adhesion complexes and the extracellular matrix (reviewed in [20]). Moreover, the altered expression of PC1 in MDCK cells has been shown to promote EMT-like characteristics, such as cytoskeletal changes, rearrangement of cell adhesion proteins, and altered cell migration [21].
Although ADPKD is a genetic disorder, environmental factors and hormones have been shown to significantly affect the severity of the disease [22, 23]. We have previously shown that the hormone ouabain, a steroid produced in the adrenal glands [24], is one of those effectors. Ouabain activates signaling events that lead to changes in cell metabolism, adhesion, and growth in a cell type specific manner [25-29]. These effects are mediated by the binding of ouabain to its receptor, Na,K-ATPase (NKA), a plasma membrane protein complex also involved in the active exchange of Na+ and K+ across the cell surface [30]. The mechanism of action of ouabain does not only involve changes in intracellular ion concentrations that result from inhibition of NKA ion transport activity, but it also depends on the stimulation of a cascade of secondary messengers in the target cells that depend on the function of NKA as a cell signal transducer [31-33]. We have previously shown that the NKA of epithelial cells derived from the cysts of patients with ADPKD exhibit an affinity for ouabain, which is significantly higher than that of normal human kidney epithelial cells (NHK cells) [27]. In ADPKD cells, physiological concentrations of ouabain (3 nM), promote the activation of extracellular regulated kinase (ERK) signaling and increase the proliferation of ADPKD cells. Moreover, ouabain stimulates the cAMP induced transepithelial secretion of fluid by ADPKD cell monolayers, enhances the growth of ADPKD microcysts cultured in a 3-D collagen matrix, and promotes tubular expansion in embryonic kidneys from ADPKD mice grown in vitro [26, 34]. In contrast, ouabain does not affect proliferation and fluid secretion by normal human kidney (NHK) cell monolayers, or induce tubular dilation in wild type mouse kidney in metanephric organ culture [27, 35]. Increased cell proliferation and fluid secretion are hallmarks of the pathogenesis of ADPKD. Therefore, ouabain is a circulating factor that can potentially influence the progression of ADPKD [26, 27, 35].
Interestingly, signal transduction pathways that are known to promote EMT, such as EGFR, SRC, PI3K, ERK1/2, and PKC have been shown to be activated by the binding of ouabain to NKA in different cell types [32, 36-40]. In addition, ouabain has been shown to alter the content of cell junctional proteins and to affect cell-to-substrate adhesion of MDCK cells, [28, 41-43]. Interestingly, marinobufagenin another member of the cardiotonic steroid family of compounds (to which ouabain belongs), induces fibrosis and EMT like effects in the kidney [44]. It is therefore plausible that ouabain could enhance the ADPKD phenotype, not only by activating cell proliferation and cAMP dependent fluid secretion, but perhaps also by promoting EMT. In the present study, we determined whether physiological concentrations of ouabain stimulate EMT in NHK and ADPKD cells. Our results suggest that ouabain induces changes in ADPKD cells that correspond to a partial EMT phenotype and that this may contribute to enhance the cystic properties of ADPKD cells.
Materials and Methods
Cell cultures
A protocol for the use of human kidney tissues was approved by our institutional review board at Kansas University Medical Center (KUMC). Primary cultures of ADPKD and NHK cells were derived from discarded human kidneys with the assistance of the PKD Biomaterials and Cellular Models Core at KUMC.
ADPKD kidneys (14 males, 15 females) were obtained from the surgery department at the Kansas University Medical Center and hospitals participating in the Polycystic Kidney Research Retrieval Program through the PKD Foundation (Kansas City, MO). The average age of the ADPKD patients at the time of the nephrectomy was 53 years (with a range of 42-73 years), and most patients were at or near end stage renal disease (ESRD). These kidneys were most likely from PKD1 patients because 85% of the ADPKD cases are caused by PKD1 mutations, and individuals with PKD2 mutations have a milder phenotype and later onset of ESRD (median age, approximately 54 years for PKD1 mutations versus 74 years for PKD2 mutations) [45]. Normal human kidneys (7 males, 4 females; average age, 44 years) that were unsuitable for transplantation, because of abnormalities in the vasculature or presenting poor perfusion characteristics, were obtained from the Midwest Transplant Network (Kansas City, KS). Kidneys were obtained at the time of surgery, sealed in a sterile bag, immediately submerged in ice and shipped overnight to the PKD Biomarkers and Biomaterials Core at KUMC for generation of the primary cell cultures. Primary cell cultures were prepared as previously described [46]. Briefly, ADPKD cell cultures were obtained from multiple surface cysts ranging in size; NHK cells were cultured from sections of the cortex and outer medulla. Primary cultures of ADPKD and NHK cells stained for Dolichos biflorus lectin, indicating the cultures are enriched in collecting duct cells [47]. Cells were seeded and grown in DMEM/F12 supplemented with 1% heat-inactivated fetal bovine serum (FBS), 100 IU/ml penicillin G and 0.1 mg/ml streptomycin, 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml sodium selenite (ITS). All experiments were done with ADPKD and NHK cells obtained from at least three different patients.
Protein extraction and Immunoblots
ADPKD and NHK cells were treated with 3 nM ouabain at 70% confluency for 24 or 48 hr in low FBS (0.002%) media. Untreated cells served as controls. Cells were then washed with ice-cold PBS and lysed with RIPA buffer (1% NP-40, 0.25% sodium deoxycholate, 1 mM NaVO3, 1 mM NaF, 150 mM NaCl, 1 mM EDTA, 50 mM Tris, and protease inhibitor cocktail (Sigma, St. Louis, MO). Cleared lysates were stored at -80°C. To obtain membrane extracts, CNM kit (Biochain Institute, Inc, Hayward, CA) was used following the manufacturer's instructions. Protein concentration was measured using the dye binding assay from Bio-Rad (Hercules, CA). Total protein (20 μg) and membrane extracts (10 μg) were electrophoresed on a 10% polyacrylamide gel and transferred to nitrocellulose membrane. Immunoblots were probed with antibodies against occludin, claudin-1, ZO-1, E-cadherin (ThermoFisher Scientific, Waltham, MA), vinculin (Millipore, Temecula, CA), αSMA (Sigma, St. Louis, MO), N-cadherin, vimentin, pSMAD3 (Cell Signaling, Danvers, MA), TGFp, fibronectin, and snail (Santa Cruz Biotechnology Inc., Dallas, TX), following protocols described previously [27]. After detection using chemiluminescence, the images were scanned and quantified for band intensity using the Gel-Pro software. The protein levels were normalized against tubulin levels and expressed as density units relative to the untreated controls.
Cell aggregation assays
ADPKD and NHK cells were suspended at 25,000 cells/mL in 50 mL conical tubes media containing 0.002% FBS with or without ouabain (3 nM). Cells were incubated in the tube with loosened cap at 37°C and 5% CO2 for 1 h in a shaking incubator at 100 rpm to allow the cells to form aggregates. Cell aliquots (500 μL) were collected at 0, 30 and 60 min and placed on ice. After addition of 500 μL phosphate buffered saline (PBS), 0.4% Tween-20, cells were labeled with the nuclear dye DAPI. The amount of cell clusters was quantified using flow cytometry (LSRII flow cytometer, Beckton Dickinson, Franklin Laks, NJ).
Cell Migration Assays
ADPKD or NHK cells were plated on 6 well plates and grown until confluency. Cells were then switched to medium with 0.002% FBS for 24 h and treated in the absence and presence of 3 nM ouabain. Then, a scratch was made on the cell monolayer using a pipette tip and healing of the wound in the monolayer was monitored using an inverted microscope connected to an imaging system (Celigo imaging cytometer; Nexcelom, Biosciences LOC, Lawrence MA) at 0, 4 and 8 h after the scratch. Images were analyzed using ImageJ software.
Immunofluorescence analysis
ADPKD and NHK cells were grown until they achieved confluency on transwell inserts plates and then incubated in 0.002% FBS for 24 h. Ouabain (3 nM) was applied to the basolateral side of the cells for additional 24 hr. Cell monolayers were washed three times with ice-cold PBS and treated for 20 min with methanol at −20°C. After three additional washes with PBS, samples were blocked for 1 hr with 0.5% BSA, and incubated for 1 h at 37°C with the corresponding specific primary antibody. Samples were washed 3 times as above and samples were incubated with FITC conjugated goat anti mouse or anti rabbit antibodies (1:1000) depending on the source of the primary antibody. After 3 washes with PBS, samples were mounted on glass slides with Slow Fade Gold Antifade reagent containing DAPI (ThermoFisher Scientific, Waltham, MA), and viewed on a Nikon Eclipse 80i equipped with digital camera.
Transepithelial Electrical Resistance (TER) measurements
Transepithelial electrical resistance (TER) was measured using Transwell permeable supports (0.4 μm, 12 well, Corning, NY). Cells were seeded on Transwell filters at a density of 120,000 cells/cm2. Cells were grown until they achieved confluency, time at which they were switched to medium with 0.002% FBS for 24 h and ouabain was applied at a concentration of 3 nM to the basolateral side of the cells. Cultures were further maintained in culture for 24, 48 and 72 hr, then TER was measured by using the automated ohmmeter system, cellZscope (NanoAnalytics, Munster, Germany).
Statistical Analysis
All the experiments were performed in triplicates with cystic epithelial cells obtained from at least three different ADPKD patients. Statistical significance of the differences between ouabain treated and untreated controls was determined by Student's T-test. ANOVA was used for determining the significance for Dextran assays. Statistical significance was defined as P<0.05.
Results
Ouabain alters the expression of EMT markers in ADPKD cells
Changes resembling EMT, which may contribute to the cystic phenotype, have been reported in ADPKD [48]. Based on our findings that ouabain enhances other aspects of ADPKD cystogenesis, we tested whether ouabain could induce EMT changes in ADPKD cells. Initially, we examined the expression of common markers of EMT after 24 h treatment of the cells with or without 3 nM ouabain. Immunoblot analysis of EMT related proteins in NHK cells showed that ouabain had no significant effect on expression levels of E-cadherin, N-cadherin, αSMA, collagen-I, vinculin, or fibronectin (Fig. 1A-F). In contrast, ouabain treatment decreased the epithelial marker E-cadherin and increased the mesenchymal markers N-cadherin, αSMA, and collagen-I in ADPKD cells (Fig. 1A-D). The expression of other mesenchymal markers, such as vimentin and fibronectin where not changed in ADPKD cells with ouabain treatment (Fig. 1E and 1F). In addition, to determine if ouabain has delayed effects on this last EMT markers, we performed extended experiments in which we treated the cells with ouabain for 48 h. This, similar the experiments at 24 h, showed no changes in vimentin and fibronectin (supplemental Fig. 1). Altogether, these results show that ouabain is able to induce protein expression changes that are commonly found in EMT. The lack of effect for some EMT markers, even after 48 h incubation with ouabain, suggests that not all EMT markers are regulated by ouabain and that this hormone is unable to induce a full EMT switch of the ADPKD cells.
Figure 1. Ouabain effect on the expression of EMT markers in NHK and ADPKD cells.
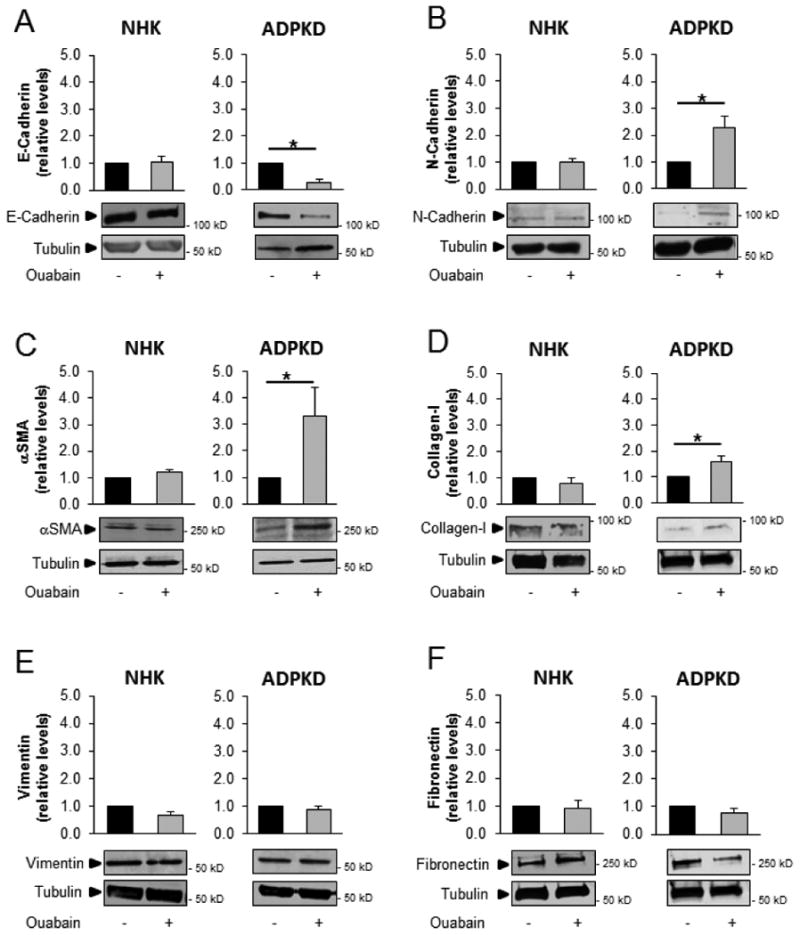
Cultured ADPKD and NHK cells were treated without and with 3 nM ouabain for 24 h. Cells were lysed and proteins of interest were identified by immunoblot. (A) E-cadherin, (B) N-cadherin, (C) αSMA, (D) Collagen-I, (E) vimentin, and (F) fibronectin. Lower panels show representative blots, while the top panels show the mean ± SEM of the densitometric analysis of 3 different experiments. Values are expressed relative to the corresponding untreated controls. Statistical significant differences are shown relative to control values, with asterisks indicating P<0.05.
Ouabain modifies cell-cell adhesion properties in ADPKD
In order to investigate if ouabain influences cellular events that are associated with EMT in ADPKD cells, we studied the effect of ouabain on the adhesion properties of the cells. ADPKD and NHK in suspension were treated with and without 3 nM ouabain and then were allowed to form aggregates under incubation in a shaker. The number of 2n and 4n nuclei, indicative of aggregated cells, was evaluated by flow cytometry. Ouabain did not significantly change cell aggregation in NHK cells (Fig. 2A). In contrast, ouabain reduced the ability of ADPKD cells to form cell aggregates in ADPKD after 30 min of incubation; and this effect persisted at 60 min (Fig. 2B). These data suggest that ouabain affects the cell to cell attachment properties of ADPKD, but not NHK cells.
Figure 2. Ouabain effect on cell-cell attachment of NHK and ADPKD cells.
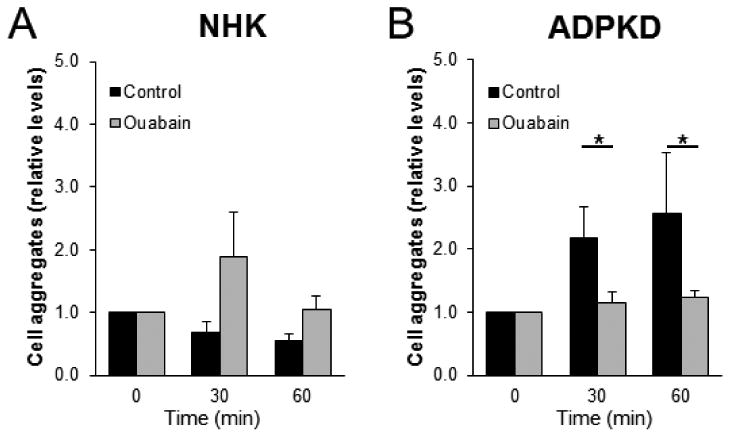
(A) NHK and (B) ADPKD cells were incubated in suspension on a shaking incubator and were treated with and without 3 nM ouabain. After the indicated times, the number of cell aggregates formed was determined, based on the number of 2n and 4n nuclei, measured by flow cytometry. Bars represent mean densitometry values ± SEM of 3 determinations using cells from different NHK and ADPKD kidneys. Asterisks indicate statistical significance, with P<0.05.
Ouabain enhances migration of ADPKD cells
Another functional consequence of EMT is the enhanced ability of cells to migrate [49, 50]. Therefore, we tested if ouabain was able to alter the migration rate of ADPKD and NHK cells, using wound healing assays. To achieve this, confluent cell monolayers were pretreated with ouabain for 24 h and the culture was scratched with a micropipette tip to inflict a wound. Repair of the wound in the presence and absence of ouabain was followed over time by taking serial images at 0, 4, and 8 h after the scratch was performed. In the presence of ouabain, a significant enhancement of migration occurred at 8 h after the scratch was inflicted in ADPKD cell monolayers, but not in NHK cell monolayers (Fig. 3A-D). This suggests that ouabain is able to enhance cell migration, a characteristic of EMT, in ADPKD cells.
Figure 3. Ouabain effects on NHK and ADPKD cell migration.
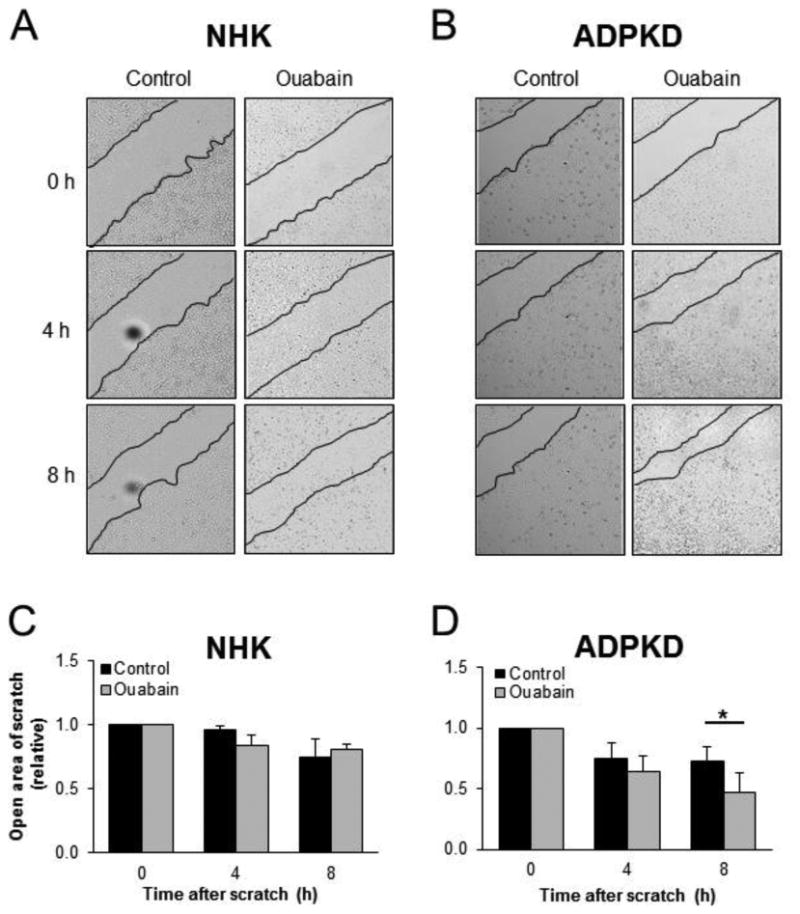
(A-D) Cell migration/wound healing assays. Confluent cell monolayers were pretreated with ouabain for 24 h. Then a scratch was made in the monolayer with a pipette tip and healing of the wound was monitored by taking images at the indicated times. Representative images of NHK (A) and ADPKD (B) cell monolayers. Black lines delineate the edge of the wound in the culture. Panels C (NHK) and D (ADPKD) are measurements of the open wound, relative to the corresponding untreated controls. Bars indicate mean ± SEM. Cells from 4 separate kidneys were assayed, in duplicate determinations. Asterisks indicate statistical significance with respect to 0 time, with P<0.05.
Ouabain selectively modulates tight junctions in ADPKD cells
Tight junctions between epithelial cells maintain the structure of the cell monolayer and form a semipermeable diffusion barrier that restricts the passive diffusion of molecules, according to charge and size (reviewed in [51]). Tight junctions are dynamic structures formed by multi-transmembrane protein complexes, which include claudins and occludins, as well as scaffold proteins, such as ZO proteins [52]. Since we found ouabain to induce relaxation of cell-cell adhesion and increase cell migration, we hypothesized that ouabain may alter the expression of tight junction proteins. In addition, changes in tight junction proteins are part of the phenotypic changes of EMT [49, 50]. To test this, we cultured NHK and ADPKD cells for 24 h with 3 nM ouabain, after which whole cell lysates were prepared and a series of tight junction proteins were analyzed by immunoblot and immunocytochemistry. Ouabain did not change the overall level of tight junction proteins in NHK cells (Fig. 4A-C). In contrast, ouabain differentially affected tight junction protein expression in ADPKD cells. While claudin-1 and ZO-1 were not significantly modified (Fig. 4A, and 4C), occludin levels were elevated (Fig. 4B). Immunofluorescence analysis of these proteins showed that ouabain did not change the plasma membrane localization of tight junction proteins in either NHK or ADPKD cells (Fig. 5A and B). In addition, the amount of claudin-1 and ZO-1 in membrane fractions prepared from the cells did not vary in ADPKD cells, but the membrane levels of occludin were elevated (Fig. 5C-E). Longer incubation of the cells with ouabain (48 h) showed similar results, with no effect in tight junction proteins in NHK cells; and elevation of occludin-1 in ADPKD cells. However, after 48 h of treatment, ouabain was able to increase claudin-1 in the ADPKD cells (supplemental Fig. 2). These results show that ouabain is a selective regulator of tight junction protein expression in ADPKD cells, upregulating the expression of both occludin and claudin-1 with different time kinetics.
Figure 4. Ouabain effect on expression of NHK and ADPKD cell tight junction proteins.
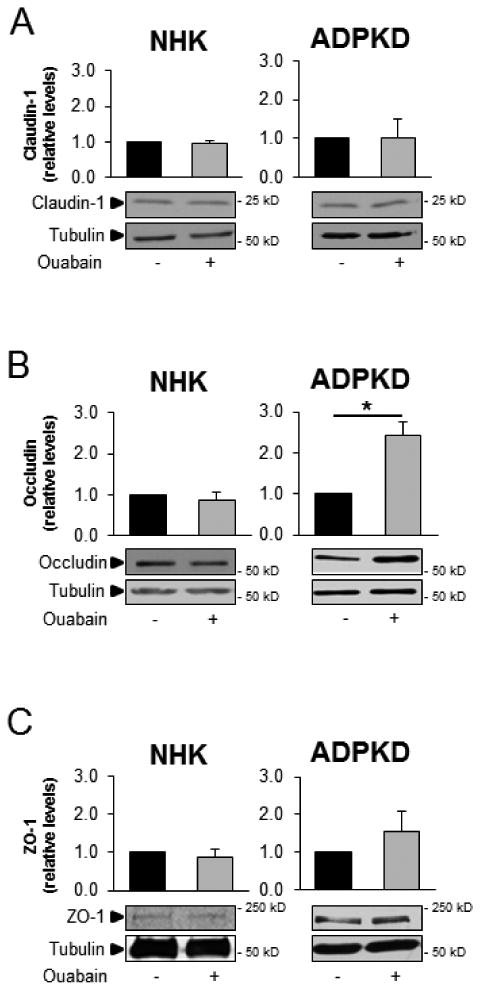
NHK and ADPKD cells were treated without and with ouabain (3 nM) for 24 h and expression of tight junction proteins (A) claudin-1, (B) occludin, and (C) ZO-1 were analyzed from cell lysates by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different NHK and ADPKD kidneys. Values are expressed relative to untreated controls. Asterisks indicate statistical significance with respect to untreated controls, with P<0.05. Panels below each graph show representative immunoblots.
Figure 5. Ouabain effect on the localization of cell tight junction proteins in NHK and ADPKD cells.
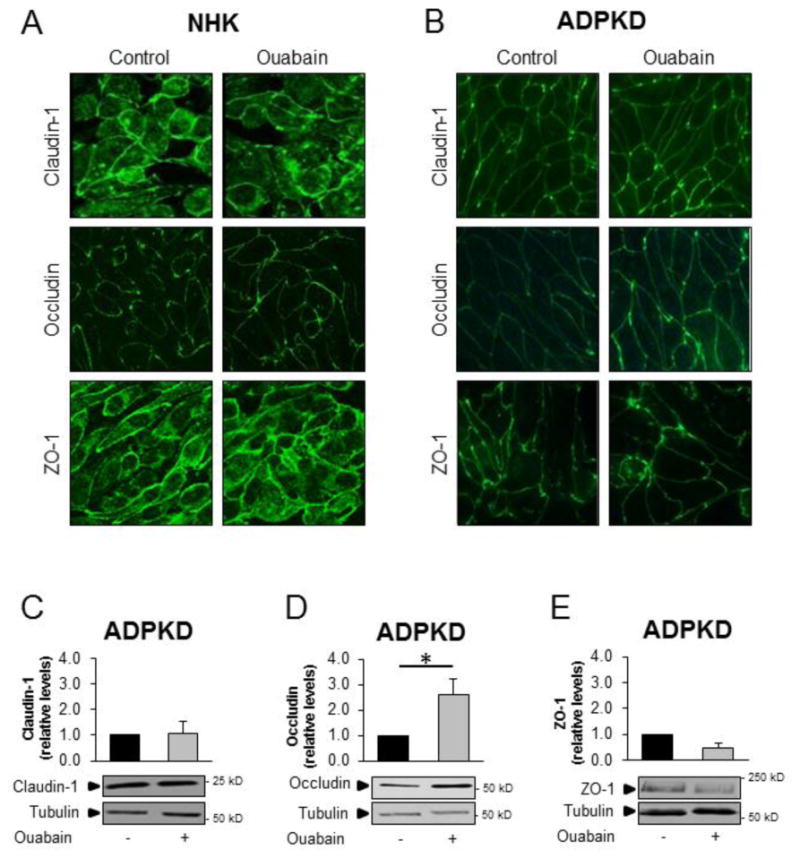
NHK and ADPKD cells were treated without and with ouabain (3 nM) for 24 h and tight junction protein expression was visualized by immunocytochemistry and by immunoblot of cell membrane fractions. A,B) Claudin-1, occludin, and ZO-1 in NHK and ADPKD cells respectively. C-D) Representative immunoblot and densitometry analysis of claudin-1, occludin, and ZO-1 in membrane fractions isolated from ADPKD cells. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different ADPKD kidneys. Values are expressed relative to untreated controls.
Ouabain modulates adherens junctions in ADPKD cells in a selective manner
In addition to tight junction proteins, we explored the effects of ouabain on the expression levels and cellular localization of several adherens junction proteins, including E-cadherin, β-catenin, and vinculin in NHK and ADPKD cells. Ouabain did not cause significant changes of the expression levels of E-cadherin, β-catenin or vinculin in NHK cells (Fig. 1A, 6A and B). In ADPKD cells however, ouabain decreased the expression levels of E-cadherin (Fig. 1A), but did not affect the expression of β-catenin and vinculin (Fig. 6A and B). Therefore, similar to what occurred with tight junctions, ouabain selectively regulated the expression of adherens proteins in ADPKD cells, specifically decreasing the levels of E-cadherin. Ouabain did not change the cellular localization of E-cadherin, β-catenin or vinculin in NHK cells (Fig. 7A). Similarly, ouabain did not alter the cellular localization (Fig. 7B) or plasma membrane levels (Fig. 7D and E) of β-catenin or vinculin in ADPKD cells; however, it reduced the amount of E-cadherin at the cell surface, as shown by immunocytochemistry (Fig. 7B) and immunoblot analysis of this protein in plasma membrane fractions from ADPKD cells (Fig. 7C). Longer incubation time with ouabain (48 h) did not change the overall levels of β-catenin or vinculin in ADPKD cells (supplemental Fig. 3). In conclusion, ouabain specifically alters E-cadherin expression in ADPKD cells, but not the other adherens proteins tested.
Figure 6. Ouabain effect on the expression of NHK and ADPKD cell adherens junction proteins.
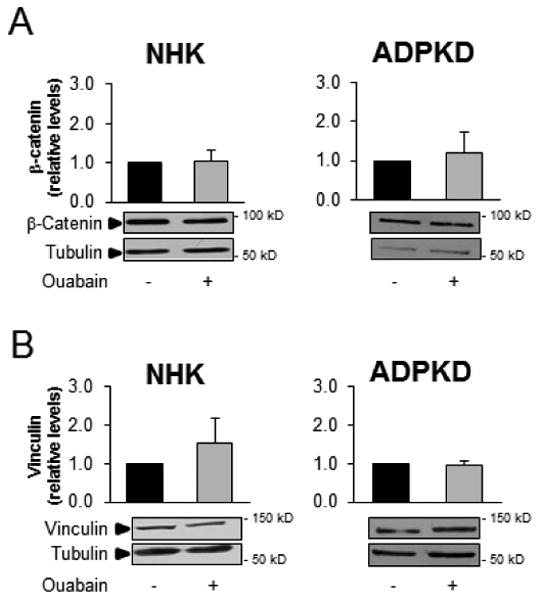
NHK and ADPKD cells were treated without and with 3 nM ouabain for 24 h and the expression of the adherens junction proteins (A) β-catenin and (B) vinculin were analyzed by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different NHK and ADPKD kidneys. Values are expressed relative to untreated controls. Asterisks indicate statistical significance with respect to untreated controls, with P<0.05. Panels below each graph show representative immunoblots.
Figure 7. Ouabain effect on the localization of cell adhesion proteins in NHK and ADPKD cells.
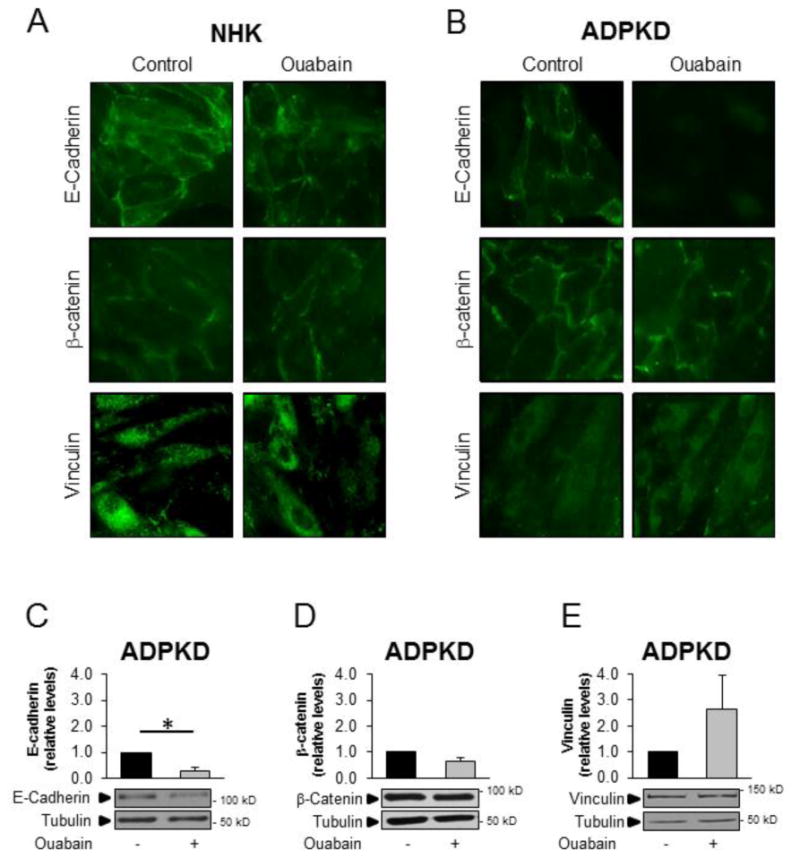
NHK and ADPKD cells were treated without and with 3 nM ouabain for 24 h and expression of the adhesion molecules E-cadherin, β-catenin and vinculin was analyzed by immunocytochemistry and by immunoblot of cell membrane fractions. A,B) E-cadherin, β-catenin and vinculin localization in NHK and ADPKD cells respectively. C-E) Representative immunoblot and densitometry analysis of E-cadherin, (3-catenin and vinculin in membrane fractions isolated from ADPKD cells. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different ADPKD kidneys. Values are expressed relative to untreated controls.
TER is altered in response to ouabain in ADPKD cells
Junctional complexes control the diffusion of ions and hydrophilic solutes across the paracellular route [52]. The ability of ADPKD cells to form cysts suggests that their junctional complexes function as a selective barrier to maintain the solute gradients across the epithelium that is necessary for passive water movement into the lumen of the cyst. However, some reports have shown that ADPKD is accompanied by alterations in the apical junctional complexes of the cells [21, 53, 54]. We tested whether ouabain modifies the transepithelial electrical resistance (TER) of NHK or ADPKD cells. To achieve this, we treated NHK and ADPKD cells without and with 3 nM ouabain for different times. We found that ouabain did not significantly changed TER in NHK cells (Fig. 8A). In contrast, ouabain enhanced TER in ADPKD cells, an effect which was observed at 48 and 72 h after the addition of ouabain (Fig. 8B). These results suggest that ouabain enhances the capacity of ADPKD cells to maintain the tightness to the epithelium, which is needed to support the structure of the cysts.
Figure 8. Ouabain effect on TER in NHK and ADPKD cells.
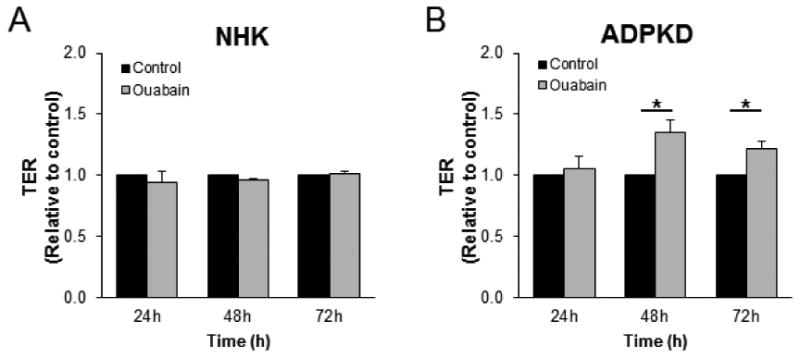
A, B) Cells were grown to confluency on Transwell culture inserts and were treated with and without ouabain (3 nM) for the indicated times. TER was measured using an automated ohmmeter system. Bars represent the mean ± SEM of cells obtained from 3 separate kidneys. *P<0.05.
The TGFβ-Smad3 signaling is activated by ouabain in ADPKD cells
Transforming growth factor β (TGFβ) is a key factor in the development of EMT and fibrosis in several kidney diseases [13, 50, 55, 56], including ADPKD [12, 13, 57]. TGFβ stimulates the TGFβ receptor (TGFβR), which in turn activates the transcription factors Smad2 and Smad3. Together with Smad4, the Smad2/3 complex translocates into the nucleus and binds to regulatory elements on the DNA, leading to increased transcription of key genes which promote EMT [55]. We found that ouabain increased TGFβ levels in ADPKD cells treated with 3 nM ouabain for 24 h, compared to untreated cells (Fig. 9A). In addition, the expression of Snail, a TGFβ target gene which governs EMT and fibrosis [58, 59], was increased in response to ouabain (Fig. 9B). These effects on TGFβ and Snail were not found in NHK cells (Fig. 9A and 9B). When Smad3 signaling was analyzed, we found that ouabain activated the phosphorylation of Smad3 in ADPKD cells within 15 minutes of ouabain application. In contrast, ouabain did not promote Smad3 phosphorylation in NHK cells (Fig. 9C). The involvement on the TGFβ pathway in the response of ouabain in EMT was confirmed by using the TGFβ/Smad inhibitor, SB431542 (Selleckchem, Houston, TX). The inhibitor was added, along with ouabain for 24 h at a final concentration of 5 μM. SB431542 abrogated the reduction of E-cadherin and the elevation of N-cadherin caused by ouabain in ADPKD cells (Fig. 9D). Altogether, these results indicate that the EMT changes induced by ouabain in ADPKD cells are mediated through the TGFβ pathway.
Figure 9. Ouabain effect on the TGFβ signaling pathway in NHK and ADPKD cells.
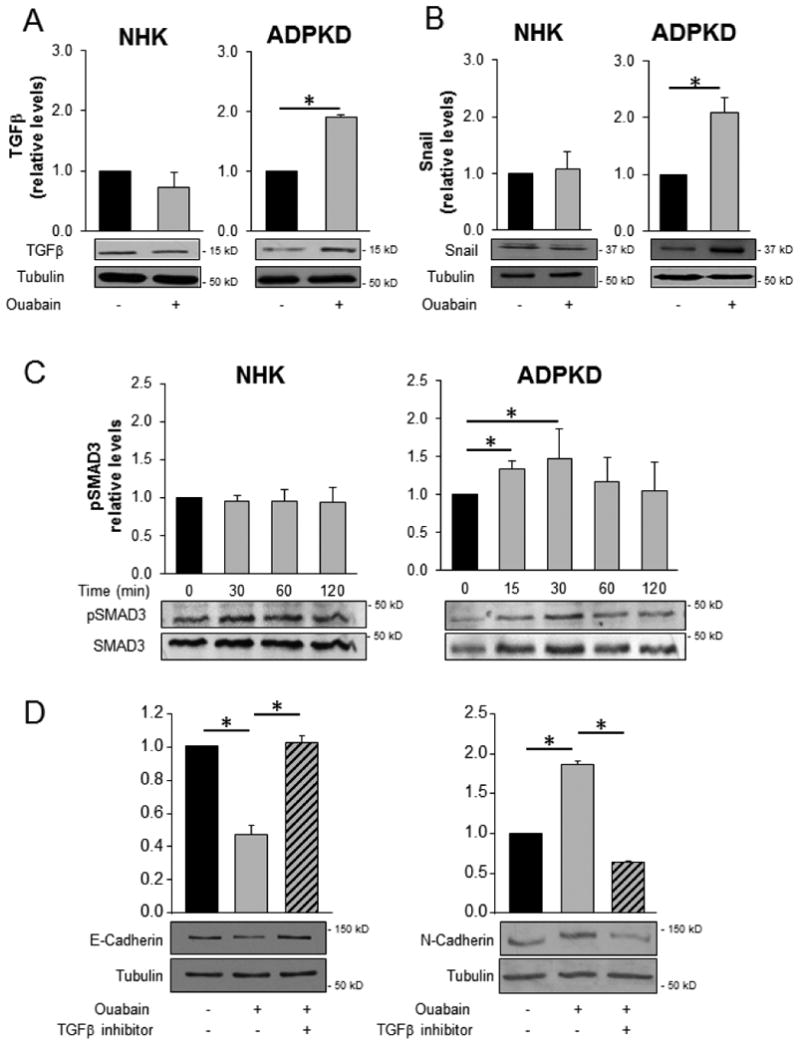
A, B) NHK and ADPKD cells were treated without and with 3 nM ouabain for 24 h and the relative levels of TGFβ were determined by immunoblot. Upper panels show the densitometric values, while bottom panels display representative immunoblots. C, D) NHK and ADPKD cells were treated with or without 3 nM ouabain and total and phosphorylated Smad3 (P-Smad3) levels were determined at the indicated time points. Bars represent P-Smad3 levels as a fraction of total Smad3 and relative to the untreated controls. D) ADPKD cells were treated for 24 h without and with 3 nM ouabain, and in the absence or presence of the TGFβ/Smad inhibitor SB431542. The levels of E-cadherin and N-cadherin were determined by immunoblot and quantified by the densitometric analysis of the corresponding bands on the blot. In all cases, bars represent the mean ± SEM of 3 determinations using cells from different ADPKD kidneys. Asterisks indicate statistical significance with respect to untreated controls, with P<0.05.
Discussion
Ouabain is a factor that enhances certain phenotypic features of ADPKD cells, including an exacerbation of ADPKD cell proliferation and an increase in cAMP-mediated fluid secretion [60]. Ouabain and other cardiotonic steroids have been shown to induce EMT and modify cell junctional complexes in some epithelial cells in culture [28, 61, 62]. In this work, we found that, in contrast to NHK cells, ouabain exerts a series of effects on ADPKD cells that are compatible with EMT. This shows a novel effect for ouabain in ADPKD and is of high relevance to the disease, since by accentuating the de-differentiated state of the cystic cells, ouabain will contribute to exacerbate ADPKD. We found that ouabain modified the expression of several major markers of the mesenchymal phenotype, decreasing E-cadherin and increasing N-cadherin, αSMA, and collagen-I protein levels in ADPKD cells. While this supports the idea that ouabain is pushing ADPKD cells towards an EMT phenotype, we did not observe a complete transition of the cells to a mesenchymal phenotype, since markers, such as vimentin and fibronectin appeared not to be affected. While the classic definition of EMT includes the complete transition of the cells from an epithelial to a mesenchymal state [50], recent studies in epithelial and cancer cells have recognized the existence of intermediate or “metastable” stages of EMT, suggesting that cells can display a wide range of EMT patterns [63-65]. Our observations indicate that ouabain induces a partial or metastable EMT status in ADPKD cells. An incomplete EMT state has been shown in renal cells under conditions of stress or wound repair after injury [66-69]. It is important to note that our experiments were performed on primary cell cultures. It is possible that when placed under in vitro conditions, the cells exhibit some degree of spontaneous de-differentiation that is not present in the in vivo environment. This could result in relatively higher background levels of expression of EMT makers, which may make it more difficult to detect changes in EMT-related gene expression. In any case, our results clearly show a specific response in ADPKD cells in favor of a higher mesenchymal phenotype of the cells. Moreover, the increase in collagen-I that ouabain induces in ADPKD cells, suggests that ouabain may contribute to fibrotic changes in ADPKD. Fibrosis is associated with the progression of ADPKD and is responsible for the deleterious consequences of ADPKD on renal function [70]. Therefore, the effect on collagen-I may represent another action by which ouabain contributes to ADPKD progression.
We also found that ADPKD, but not NHK cells respond to ouabain with selected changes in expression of proteins involved in cell tight and adherens junctions. Ouabain reduced E-cadherin and increased occludin in ADPKD cells. Changes in E-cadherin have been reported in ADPKD, probably due to disruption of the PC1/E-cadherin/β-catenin complex, which these proteins normally form [71-74]. The effects of ouabain therefore appear to exacerbate the already abnormally low level of E-cadherin of ADPKD cells. Our immunocytochemical analysis showed that ouabain not only decreased E-cadherin amounts globally, but also at the plasma membrane of the ADPKD cells. Since we did not find E-cadherin increased in intracellular stores, it appears that ouabain either decreases the overall synthesis of E-cadherin, or causes its rapid degradation. The ouabain stimulated increase in the expression of occludin in ADPKD cells was unexpected, since occludin is commonly decreased in EMT [49]. Claudin-1 was not significantly affected by ouabain at 24 h of treatment; however, it was increased at 48 h. These changes in occludin and claudin-1 may represent a compensatory mechanism that allows the ADPKD epithelium to retain its structural integrity despite the lower E-cadherin amounts in the cells; and to maintain functional junctional complexes, which are necessary for the accumulation of fluid within the cysts. In addition, the incomplete EMT change caused by ouabain may also contribute to maintain tight junction functionality in the ADPKD epithelium, since cells with a complete EMT phenotype are incapable of generating an electrical gradient across the cell monolayer [26, 34]. In support of this, we found that ouabain increases TER of the ADPKD cell monolayers. Coincidentally, the increase in claudin-1 induced by ouabain occurs relatively late (48 h), when we detect the changes in TER. Interestingly, exogenous overexpression of claudin-1 has been shown to increase TER in MDCK cells [75]. At present, how the ouabain dependent changes in TER correlate with the composition of ADPKD cell tight junction proteins is unclear. Further studies will be needed to ascertain the structure function relationship of tight junctional complexes in these cells. [76]. Changes in tight junction proteins in response to ouabain treatment, with an increase in TER values have also been reported by Larre et al. [43]. However, these authors did not define the structural basis for how TER changes correlated with regulation of different junctional proteins (reviewed in [51]).
The decrease in E-cadherin and the metastable EMT state that ouabain promotes could explain the higher mobility and the reduced capacity that the ouabain treated ADPKD cells have for forming cell-cell aggregates. Due to the different nature of the experiments, the changes in cell adhesion molecules and in cell-cell aggregation need to be determined at different times (24 h vs 30-60 min). It is possible that small changes in tight and adherens proteins occur early after ouabain addition. However, these changes would have been difficult to detect, due to the limitation of the immunochemical approaches that we used. Similar to our studies, ouabain induces loss of tight junction complexes and substrate detachment in MDCK cells. However, this response was obtained at relatively high concentrations of ouabain (1 μM) [77], and this effect was not seen when lower concentrations of ouabain (10-50 nM) were used [43]. The lower ouabain concentration needed to elicit an effect on tight junctions in ADPKD cells compared to MDCK cells may depend on the different affinity that ADPKD cells have for ouabain, which is much higher than that of MDCK cells [27]. Another dissimilarity, is that MDCK cells responded to high ouabain doses with changes in a different subset of adhesion proteins than the ones we find modulated by low concentrations of ouabain in ADPKD cells. This could perhaps be attributed to differences in the cell type used in each of the studies. In any case, it appears clear that ouabain causes a remodeling of cell junctions that contributes to enhance the dedifferentiated cystic phenotype of the ADPKD epithelium and promotes cell mobility, which will help ADPKD cells to continue dividing and proliferating to increase cyst size. Also, this remodeling of the cell junctions allows the ADPKD epithelium to preserve its TER properties, which is necessary to maintain the structure of the cysts.
We have previously shown that ouabain causes an internalization of both the α and the β subunits of NKA from the surface to the cytoplasm of ADPKD cells [27]. The NKA β subunit is known to function as a cell-cell adhesion molecule, which interacts with other Na,K-ATPase β polypeptides expressed at the plasma membrane of neighboring cells (reviewed in [42, 78]). Interestingly, decreased expression of NKA has also been suggested as a marker of EMT [79]. Therefore ouabain-induced endocytos is of NKA subunits (an specifically the β polypeptide) may be an additional mechanism that could explain the decrease in cell-cell adhesion promoted by ouabain in ADPKD cells.
We previously found that human ADPKD cells have elevated TGFβ expression and aberrant expression of matrix molecules involved in fibrosis [80]. Here, we show that in ADPKD cells, ouabain increases the expression of TGFβ, leading to phosphorylation of SMAD3 and elevated levels of Snail protein. Moreover, the TGFβ/Smad inhibitor SB431542 blocked ouabain effects on E- and N-cadherin, supporting the idea that ouabain effects on EMT are mediated via the TGFβ/Smad pathway in ADPKD cells. Several studies in kidneys of ADPKD patients have shown that target genes of the TGFβ pathway are upregulated compared to normal controls [12, 13, 48], with SMAD2/3 rather than SMAD 1/5/8 being activated [12, 13]. Therefore, the effect of ouabain on this pathway agrees with an enhancement on the ADPKD phenotype. TGFβ is a known driver of EMT, yet the targets of TGFβ signaling depend on the cell type (reviewed in [81]). This may explain why in ADPKD cells we find that some, but not all markers of EMT are affected by ouabain.
Different from ADPKD cells, NHK cells do not respond to ouabain with changes in expression of EMT markers and adhesion proteins, nor they show modification in their cell-cell aggregation, migration and TER characteristics. This may depend on the lower affinity that the NKA of normal kidney cells have for ouabain, as we have previously reported [27]. This is important, since at the ouabain amounts used in this work, which are within the levels found to be circulating in blood, normal kidney cells will not be significantly affected. While in theory higher concentrations of ouabain could be expected to induce responses in NHK cells, this appears not to be the case. We have shown that higher ouabain amounts have a negative effect not only in NHK, but also in ADPKD cells, with concentrations of 0.1 μM and higher slowing the growth of these cells. This effect is presumably due to extensive inhibition of Na,K-ATPase activity and cell toxicity [27].
In conclusion, we found a novel effect for ouabain in ADPKD cells, promoting in them specific transformations that resemble those of a partial EMT state. Importantly, these changes are consistent with an enhancement of an abnormal phenotype of ADPKD cells, further supporting the role of ouabain as a factor that promotes ADPKD progression.
Supplementary Material
Supplemental Figure 1. Expression of vimentin and fibronectin in ADPKD cells after treatment with ouabain for 48 h. ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of vimentin (A) and fibronectin (B) were analyzed from cell lysates by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations. Values are expressed relative to untreated controls. Panels below each graph show representative immunoblots.
Supplemental Figure 2. Expression of tight junction proteins in NHK and ADPKD cells after treatment with ouabain for 48 h. NHK and ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of tight junction proteins (A) claudin-1, (B) occludin, and (C) ZO-1 were analyzed from cell lysates by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different NHK and ADPKD kidneys. Values are expressed relative to untreated controls. Asterisks indicate statistical significance with respect to untreated controls, with P<0.05. Panels below each graph show representative immunoblots.
Supplemental Figure 3. Expression of β-catenin and vinculin in ADPKD cells after treatment with ouabain for 48 h. ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of β-catenin (A) and vinculin (B) were analyzed from cell lysates by immunoblot. Bars show mean densitometry levels ± SEM. Values are expressed relative to untreated controls. Panels below each graph show representative immunoblots.
Highlights.
Ouabain promotes EMT and the mesenchymal phenotype of ADPKD cells.
In contrast, ouabain does not affect normal human kidney cells.
Ouabain effects in ADPKD cells are essential to exacerbate the ADPKD phenotype.
Acknowledgments
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases, NIH grant DK081431. We thank the PKD Research Biomaterials and Cellular Models Core at the University of Kansas Medical Center and hospitals participating in the Polycystic Research Retrieval Program for providing human normal and ADPKD kidneys from which primary cells were obtained.
Footnotes
Conflict of Interest: No conflict of interest, financial or otherwise related with this work, are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iglesias CG, et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983;2(6):630–9. doi: 10.1016/s0272-6386(83)80044-4. [DOI] [PubMed] [Google Scholar]
- 2.Hanaoka K, et al. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408(6815):990–4. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 3.Kimberling WJ, et al. Linkage heterogeneity of autosomal dominant polycystic kidney disease. N Engl J Med. 1988;319(14):913–8. doi: 10.1056/NEJM198810063191405. [DOI] [PubMed] [Google Scholar]
- 4.Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–39. doi: 10.1159/000421651. [DOI] [PubMed] [Google Scholar]
- 5.Harris PC, et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2006;17(11):3013–9. doi: 10.1681/ASN.2006080835. [DOI] [PubMed] [Google Scholar]
- 6.Hateboer N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353(9147):103–7. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 7.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–37. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns WC, Kantharidis P, Thomas MC. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs. 2007;185(1-3):222–31. doi: 10.1159/000101323. [DOI] [PubMed] [Google Scholar]
- 9.Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidneycollecting duct epithelial cells. American Journal of Physiology-Renal Physiology. 2008;294(5):F1238–F1248. doi: 10.1152/ajprenal.00326.2007. [DOI] [PubMed] [Google Scholar]
- 10.Rastaldi MP, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney International. 2002;62(1):137–146. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 11.Roxburgh SA, et al. Recapitulation of embryological programmes in renal fibrosis–the importance of epithelial cell plasticity and developmental genes. Nephron Physiol. 2006;103(3):139–48. doi: 10.1159/000092453. [DOI] [PubMed] [Google Scholar]
- 12.Hassane S, et al. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J Pathol. 2010;222(1):21–31. doi: 10.1002/path.2734. [DOI] [PubMed] [Google Scholar]
- 13.Chea SW, Lee KB. TGF-beta mediated epithelial-mesenchymal transition in autosomal dominant polycystic kidney disease. Yonsei Med J. 2009;50(1):105–11. doi: 10.3349/ymj.2009.50.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schieren G, et al. Gene profiling of polycystic kidneys. Nephrol Dial Transplant. 2006;21(7):1816–24. doi: 10.1093/ndt/gfl071. [DOI] [PubMed] [Google Scholar]
- 15.Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol. 2007;293(5):F1423–32. doi: 10.1152/ajprenal.00275.2007. [DOI] [PubMed] [Google Scholar]
- 16.Jiang YS, et al. Epithelial-mesenchymal transition of renal tubules: divergent processes of repairing in acute or chronic injury? Med Hypotheses. 2013;81(1):73–5. doi: 10.1016/j.mehy.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 17.De Chiara L, Crean J. Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney. J Clin Med. 2016;5(1) doi: 10.3390/jcm5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibraghimov-Beskrovnaya O, et al. Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum Mol Genet. 2000;9(11):1641–9. doi: 10.1093/hmg/9.11.1641. [DOI] [PubMed] [Google Scholar]
- 19.Ibraghimov-Beskrovnaya O, et al. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc. Natl Acad Sci U S A. 1997;94(12):6397–402. doi: 10.1073/pnas.94.12.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drummond IA. Polycystins, focal adhesions and extracellular matrix interactions. Biochim Biophys Acta. 2011;1812(10):1322–6. doi: 10.1016/j.bbadis.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boca M, et al. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol Biol Cell. 2007;18(10):4050–61. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowley BD, Jr, et al. Gender and the effect of gonadal hormones on the progression of inherited polycystic kidney disease in rats. Am J Kidney Dis. 1997;29(2):265–72. doi: 10.1016/s0272-6386(97)90039-1. [DOI] [PubMed] [Google Scholar]
- 23.Qian F, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979–87. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 24.Schoner W. Ouabain, a new steroid hormone of adrenal gland and hypothalamus. Exp Clin Endocrinol Diabetes. 2000;108(7):449–54. doi: 10.1055/s-2000-8140. [DOI] [PubMed] [Google Scholar]
- 25.Chorvatova A, et al. Effect of ouabain on metabolic oxidative state in living cardiomyocytes evaluated by time-resolved spectroscopy of endogenous NAD(P)H fluorescence. J Biomed Opt. 2012;17(10):101505. doi: 10.1117/1.JBO.17.10.101505. [DOI] [PubMed] [Google Scholar]
- 26.Jansson K, et al. Endogenous concentrations of ouabain act as a cofactor to stimulate fluid secretion and cyst growth of in vitro ADPKD models via cAMP and EGFR-Src-MEK pathways. Am J Physiol Renal Physiol. 2012;303(7):F982–90. doi: 10.1152/ajprenal.00677.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen AN, Wallace DP, Blanco G. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J Am Soc Nephrol. 2007;18(1):46–57. doi: 10.1681/ASN.2006010086. [DOI] [PubMed] [Google Scholar]
- 28.Larre I, et al. Contacts and cooperation between cells depend on the hormone ouabain. Proc Natl Acad Sci U S A. 2006;103(29):10911–6. doi: 10.1073/pnas.0604496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61(1):9–38. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aperia A, et al. Na+-K+-ATPase, a new class of plasma membrane receptors. Am J Physiol Cell Physiol. 2016;310(7):C491–5. doi: 10.1152/ajpcell.00359.2015. [DOI] [PubMed] [Google Scholar]
- 31.Xie Z, Askari A. Na(+)/K(+)-ATPase as a signal transducer. Eur J Biochem. 2002;269(10):2434–9. doi: 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- 32.Xie JX, Li X, Xie Z. Regulation of renal function and structure by the signaling Na/K-ATPase. IUBMB Life. 2013;65(12):991–8. doi: 10.1002/iub.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pierre SV, Xie Z. The Na,K-ATPase receptor complex: its organization and membership. Cell Biochem Biophys. 2006;46(3):303–16. doi: 10.1385/cbb:46:3:303. [DOI] [PubMed] [Google Scholar]
- 34.Jansson K, et al. Ouabain Regulates CFTR-Mediated Anion Secretion and Na,K-ATPase Transport in ADPKD Cells. J Membr Biol. 2015;248(6):1145–57. doi: 10.1007/s00232-015-9832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen AN, et al. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. Am J Physiol Renal Physiol. 2011;301(4):F897–906. doi: 10.1152/ajprenal.00095.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kometiani P, Liu L, Askari A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol Pharmacol. 2005;67(3):929–36. doi: 10.1124/mol.104.007302. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, et al. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J Biol Chem. 2011;286(37):32394–403. doi: 10.1074/jbc.M110.207597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu L, et al. Association of PI3K-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am J Physiol Cell Physiol. 2007;293(5):C1489–97. doi: 10.1152/ajpcell.00158.2007. [DOI] [PubMed] [Google Scholar]
- 39.Jansson K, et al. Endogenous concentrations of ouabain act as a cofactor to stimulate fluid secretion and cyst growth of in vitro ADPKD models via cAMP and EGFR-Src-MEK pathways. American Journal of Physiology-Renal Physiology. 2012;303(7):F982–F990. doi: 10.1152/ajprenal.00677.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian J, et al. Changes in Sodium Pump Expression Dictate the Effects of Ouabain on Cell Growth. Journal of Biological Chemistry. 2009;284(22):14921–14929. doi: 10.1074/jbc.M808355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Souza WF, et al. Ouabain-induced alterations of the apical junctional complex involve alpha1 and beta1 Na,K-ATPase downregulation and ERK1/2 activation independent of caveolae in colorectal cancer cells. J Membr Biol. 2014;247(1):23–33. doi: 10.1007/s00232-013-9607-y. [DOI] [PubMed] [Google Scholar]
- 42.Rajasekaran AK, Rajasekaran SA. Role of Na-K-ATPase in the assembly of tight junctions. Am J Physiol Renal Physiol. 2003;285(3):F388–96. doi: 10.1152/ajprenal.00439.2002. [DOI] [PubMed] [Google Scholar]
- 43.Larre I, et al. Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci U S A. 2010;107(25):11387–92. doi: 10.1073/pnas.1000500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fedorova LV, et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol. 2009;296(4):F922–34. doi: 10.1152/ajprenal.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei Y. Practical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118(1):c19–30. doi: 10.1159/000320887. [DOI] [PubMed] [Google Scholar]
- 46.Reif GA, et al. Tolvaptan inhibits ERK-dependent cell proliferation, Cl(-) secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol. 2011;301(5):F1005–13. doi: 10.1152/ajprenal.00243.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi T, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63(6):1983–94. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 48.Song X, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18(13):2328–43. doi: 10.1093/hmg/ddp165. [DOI] [PubMed] [Google Scholar]
- 49.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1(2):a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steed E, B M, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010;20(3):142–9. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Charron AJ, et al. Compromised cytoarchitechture and polarized trafficking in autosomal dominant polycystic kidney disease cells. J Cell Biol. 2000;149:111–124. doi: 10.1083/jcb.149.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu AS, et al. Tight junction composition is altered in the epithelium of polycystic kidneys. J Pathol. 2008;216(1):120–8. doi: 10.1002/path.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344) doi: 10.1126/scisignal.2005189. re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Connor JW, Gomez EW. Biomechanics of TGFbeta-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clin Transl Med. 2014;3:23. doi: 10.1186/2001-1326-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamura T, et al. Growth factor gene expression in kidney of murine polycystic kidney disease. J Am Soc Nephrol. 1993;3(7):1378–86. doi: 10.1681/ASN.V371378. [DOI] [PubMed] [Google Scholar]
- 58.Boutet A, et al. Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis? Cell Cycle. 2007;6(6):638–42. doi: 10.4161/cc.6.6.4022. [DOI] [PubMed] [Google Scholar]
- 59.Boutet A, et al. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25(23):5603–13. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blanco G, Wallace DP. Novel role of ouabain as a cystogenic factor in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2013;305(6):F797–812. doi: 10.1152/ajprenal.00248.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larre I, L A, Contreras RG, Balda MS, Matter K, Flores-Maldonado C, Ponce A, Flores-Benitez D, Rincon-Heredia R, Padilla-Benavides T, Castillo A, Shoshani L, Cereijido M. Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci. 2010;107(25):11387–92. doi: 10.1073/pnas.1000500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fedorova LV, R V, El-Okdi N, Shidyak A, Kennedy DJ, Vetteth S, Giovannucci DR, Bagrov AY, Fedorova OV, Shapiro JI, Malhotra D. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol. 2009;296(4):F922–34. doi: 10.1152/ajprenal.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10(17):2865–73. doi: 10.4161/cc.10.17.17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ribeiro AS, Paredes J. P-Cadherin Linking Breast Cancer Stem Cells and Invasion: A Promising Marker to Identify an “Intermediate/Metastable” EMT State. Front Oncol. 2014;4:371. doi: 10.3389/fonc.2014.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savagner P. Epithelial-mesenchymal transitions: from cell plasticity to concept elasticity. Curr Top Dev Biol. 2015;112:273–300. doi: 10.1016/bs.ctdb.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 66.Rastaldi MP, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62(1):137–46. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- 67.Huang S, Susztak K. Epithelial Plasticity versus EMT in Kidney Fibrosis. Trends Mol Med. 2016;22(1):4–6. doi: 10.1016/j.molmed.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ovadya Y, Krizhanovsky V. A new Twist in kidney fibrosis. Nat Med. 2015;21(9):975–7. doi: 10.1038/nm.3938. [DOI] [PubMed] [Google Scholar]
- 69.Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121(2):468–74. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norman J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD) Biochim Biophys Acta. 2011;1812(10):1327–36. doi: 10.1016/j.bbadis.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huan Y, van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest. 1999;104(10):1459–68. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geng L, et al. Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation. Biochim Biophys Acta. 2000;1535(1):21–35. doi: 10.1016/s0925-4439(00)00079-x. [DOI] [PubMed] [Google Scholar]
- 73.Roitbak T, W C, Harris PC, Bacallao R, aW NA, Ness SA. A Polycystin-1 Multiprotein Complex Is Disrupted in Polycystic Kidney Disease Cells. Molecular Biology of the Cell. 2004;15:1334–1346. doi: 10.1091/mbc.E03-05-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Streets AJ, et al. Homophilic and heterophilic polycystin 1 interactions regulate E-cadherin recruitment and junction assembly in MDCK cells. J Cell Sci. 2009;122(Pt 9):1410–7. doi: 10.1242/jcs.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCarthy KM, et al. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J Cell Sci. 2000;113 Pt 19:3387–98. doi: 10.1242/jcs.113.19.3387. [DOI] [PubMed] [Google Scholar]
- 76.Balda MS, et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134(4):1031–49. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Contreras RG, et al. Relationship between Na(+),K(+)-ATPase and cell attachment. J Cell Sci. 1999;112(Pt 23):4223–32. doi: 10.1242/jcs.112.23.4223. [DOI] [PubMed] [Google Scholar]
- 78.Vagin O, Tokhtaeva E, Sachs G. The role of the beta1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J Biol Chem. 2006;281(51):39573–87. doi: 10.1074/jbc.M606507200. [DOI] [PubMed] [Google Scholar]
- 79.Rajasekaran SA, et al. Na,K-ATPase subunits as markers for epithelial-mesenchymal transition in cancer and fibrosis. Mol Cancer Ther. 2010;9(6):1515–24. doi: 10.1158/1535-7163.MCT-09-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wallace DP, et al. Periostin induces proliferation of human autosomal dominant polycystic kidney cells through alphaV-integrin receptor. Am J Physiol Renal Physiol. 2008;295(5):F1463–71. doi: 10.1152/ajprenal.90266.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Expression of vimentin and fibronectin in ADPKD cells after treatment with ouabain for 48 h. ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of vimentin (A) and fibronectin (B) were analyzed from cell lysates by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations. Values are expressed relative to untreated controls. Panels below each graph show representative immunoblots.
Supplemental Figure 2. Expression of tight junction proteins in NHK and ADPKD cells after treatment with ouabain for 48 h. NHK and ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of tight junction proteins (A) claudin-1, (B) occludin, and (C) ZO-1 were analyzed from cell lysates by immunoblot. Bars denote mean densitometry levels ± SEM of 3 determinations using cells from different NHK and ADPKD kidneys. Values are expressed relative to untreated controls. Asterisks indicate statistical significance with respect to untreated controls, with P<0.05. Panels below each graph show representative immunoblots.
Supplemental Figure 3. Expression of β-catenin and vinculin in ADPKD cells after treatment with ouabain for 48 h. ADPKD cells were treated without and with ouabain (3 nM) for 48 h and expression of β-catenin (A) and vinculin (B) were analyzed from cell lysates by immunoblot. Bars show mean densitometry levels ± SEM. Values are expressed relative to untreated controls. Panels below each graph show representative immunoblots.