

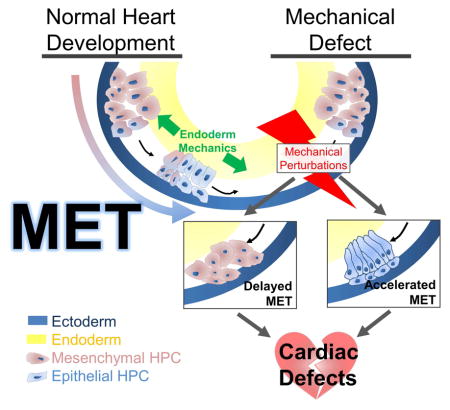
Summary
During early cardiogenesis, bilateral fields of mesenchymal heart progenitor cells (HPCs) move from the anterior lateral plate mesoderm to the ventral midline undergoing a mesenchymal-to-epithelial transition (MET) en route to form a single epithelial sheet. Through tracking of tissue level deformations in the heart forming region (HFR) as well as movement trajectories and traction generation of individual HPCs, we find the onset of MET correlates with a peak in mechanical stress within the HFR and changes in HPC migratory behaviors. Small molecule inhibitor treatments targeting actomyosin contractility reveal a temporally specific requirement of bulk tissue compliance to regulate heart development and MET. Targeting mutant constructs to modulate contractility and compliance in the underlying endoderm, we find MET in HPCs can be accelerated in response to microenvironmental stiffening and can be inhibited by softening. To test whether MET in HPCs was responsive to purely physical mechanical cues, we mimicked a high stress state by injecting an inert oil droplet to generate high strain in the HFR, demonstrating that exogenously applied stress was sufficient to drive MET. MET-induced defects in anatomy result in defined functional lesions in the larval heart implicating mechanical signaling and MET in the etiology of congenital heart defects. From this integrated analysis of HPC polarity and mechanics, we propose that normal heart development requires bilateral HPCs to undergo a critical behavioral and phenotypic transition on their way to the ventral midline and that this transition is driven in response to the changing mechanical properties of their endoderm substrate.
Keywords: heart development, mesenchymal-to-epithelial transition, biomechanics, congenital heart defects, endoderm development, actomyosin, ZO-1, mechanosensing, live-cell confocal imaging, microaspiration
Graphical abstract
Introduction
Physical mechanics are known to play a central role in shaping tissues, e.g. moving tissues into position, but their role in patterning cell behaviors and cell identity during development, growth, homeostasis, and disease is just now being explored [1, 2]. Physical forces can induce or guide gene expression, polarity and differentiation [3, 4]. Likewise, cell invasion and behaviors during cancer progression and regeneration are known to depend on mechanical cues within the microenvironment [5, 6]. In particular, heart regeneration studies have provided evidence that both substrate stiffness [7] and actomyosin contractility [8] guide cardiac cell fates, suggesting that mechanical cues may play an important role in heart formation, since many of the same cell biological processes contribute to assembly of the heart.
Heart progenitor cell (HPC) identities are established following gastrulation in a bilateral population of mesenchymal cells within the definitive mesoderm [9]. The two populations of HPCs then begin a large-scale movement from the anterior lateral plate mesoderm and undergo a mesenchymal-to-epithelial transition (MET; [10, 11]) before they merge on the ventral midline. In creating the architecture of the heart, HPCs undergo multiple transitions between mesenchymal and epithelial cell types [12]. Due to the precise timing of these transitions it has been proposed that small errors in the organization of the early heart field induce a cascade of defects leading to dramatic mutant phenotypes [13].
Classically, it was thought that HPCs actively migrate to the ventral midline [13]; however, recently it has been suggested that the underlying endoderm, while providing substrate and growth factors [14], also plays a mechanical role in HPC movements [15]. Since HPCs move in concert with the endoderm, it has been proposed that ventral convergence and active contraction of the endoderm drive HPC ventral displacement [15, 16] whereas autonomous HPC motility contributes minimally to their overall ventral movements [16–18]. Thus, endodermal convergent extension is crucial for proper heart formation, as endoderm deficient embryos exhibit abnormal extracellular matrix and disorganized myocardial epithelia [16].
Cardiac defects can also arise from defects in early HPC polarity [10], actomyosin contractility [19], and the microenvironment of the heart forming region (HFR; [20]). Even though these processes are likely involved in establishing the mechanical microenvironment of the HFR, the exact role of physical mechanics in early heart formation remains unclear. For instance, physical mechanical cues play a role in controlling cell phenotype during METs as well as epithelial-to-mesenchymal transitions (EMTs; [6]).
To understand whether mechanics plays a role in the phenotypic transitions during heart formation we turned to investigate the mechanical microenvironment in the HFR and its role in guiding MET within HPCs as they move to the ventral midline. This phenotypic transition can be modulated by global, autonomous and non-cell autonomous changes in cell contractility and mechanical compliance. Furthermore, as HPCs undergo MET they change their migratory behaviors and start actively exerting traction. Lastly, we find temporal dysregulation of MET in ventrally converging HPCs leads to profound changes in heart anatomy and physiological function. These findings support a model whereby successful heart development requires strict spatiotemporal regulation of mechanics to drive MET in HPCs.
Results
Heart progenitor cells undergo a mesenchymal-to-epithelial transition beginning at the mid tail-bud stage
To determine the timeline of MET progression in Xenopus laevis HPCs, we collected confocal transverse sections at three key stages of HPC movement to the ventral midline (Figure 1A and B). HPCs appear mesenchymal in the anterior lateral plate mesoderm in the early neurula (stage 15; Figure 1C) with little aPKC or ZO-1 (Figure 1D–E). By mid tailbud (stage 23), HPCs advance half-way to the ventral midline and apically localize aPKC (arrow in Figure 1D) but ZO-1 is only found perinuclearly (arrows Figure 1E), suggesting HPCs are preparing to form tight junctions [21]. By late tailbud (stage 28), the bilateral fields merge and HPCs form two distinct single-cell layers, with their apical surfaces at the interface between these two layers. Columnar HPCs in the deeper, dorsal-most layer express tropomyosin (Figure 1C; [22]), and are strongly polarized with apical aPKC and ZO-1 (arrows Figure 1D–E). Fibronectin fibrils are localized to the ectoderm-mesoderm and mesoderm-endoderm interfaces (Figure 1E) and become supplemented with dense fibrillin fibrils at stage 28 (Figure 1D). We find that stage 23 is the critical time point when MET is initiated in HPCs, approximately 7 hours before HPC fields merge.
Figure 1. MET progression in Xenopus heart progenitor cells.

(A) Xenopus embryos at three stages during HPC (red) movement to ventral midline. (B) Transverse schematic of HPCs and their microenvironment (blue, ectoderm; red, mesoderm; yellow, endoderm, brown, fibronectin; purple, fibrillin; orange, aPKC; green, ZO-1; dark red, tropomyosin). (C) Cell shapes by β-catenin localization and cardiomyocyte by tropomyosin expression. (D) aPKC appears on the apical surface of HPCs (white arrow) with fibrillin at the HPC basal surface. (E) Tight junction protein ZO-1 appears perinuclearly and nascently on the apical surface by stage 23 and strongly at HPC apical intercellular junctions by stage 28. Fibronectin appears at all germ layer interfaces. (F–I) Apical aPKC (F–G) and ZO-1 (H–I) during early heart tube formation. Immunofluorescence show epithelial individual markers and tropomyosin (upper panels) and normalized intensity with pseudocolor LUT (lower panels). (G and H) quantify apical polarity from 5 to 6 embryos per time point over 2 clutches. All scale bars are 50 μm. * indicates p < 0.05, ** indicates p < 0.01. See also Figure S1.
The first signs of MET appear at stage 23 and apicobasal polarity continues to increase as HPC fields merge at the ventral midline and then fold to form a heart tube. Just prior to HPC fields merging, cardiomyocytes can be identified by their expression of tropomyosin [22]. Using the tropomyosin channel as a mask to identify cardiomyocytes, we quantified apicobasal polarity of HPCs by the intensity of aPKC and ZO-1 localization (Fig 1F–I). Intensity along the apical surface was measured and normalized to expression in the endoderm (Figure S1). We see an approximately 50% increase in apical aPKC (Figure 1F–G) and ZO-1 (Figure 1H–I) localization between the time the HPCs merge on the ventral midline (stage 28) and form a heart tube (stage 32). Therefore, although we see the first signs of MET initiation at stage 23, apicobasal polarity progresses and propagates through stage 32.
Perturbations to embryo bulk mechanical properties can delay or accelerate MET
Since phenotypic transitions can depend on mechanical cues [6, 9], we wanted to test whether MET in HPCs was similarly dependent. To perturb bulk tissue mechanics, we incubated embryos with small molecule inhibitors that target actomyosin contractility as HPCs move to the ventral midline (from stage 20 to 28, or approximately 11 hours) and observed whole embryo defects (Figure 2A) including reduced anterioposterior (AP) length (Figure 2B) and higher incidence of cardiac and neural edemas (arrows, Figure 2A; Figure 2C). To confirm the effects of small molecule inhibitors on ventral tissue mechanical properties, we measured the compliance (the inverse of stiffness) of the ventral HFR at stage 23 using microaspiration (Figure 2D). The myosin II inhibitor blebbistatin induced a two-fold increase in tissue compliance (J(120), Figure 2E) whereas the myosin phosphatase inhibitor Calyculin A and ROCK inhibitor Y27632 produced a modest, although not statistically significant, 10 to 20% decrease and increase in tissue compliance, respectively. These findings implicate changes in tissue compliance as the cause of the observed cardiac defects.
Figure 2. Modulating bulk tissue mechanics can accelerate or delay MET in heart progenitor cells.

(A) Modulators of tissue compliance applied during stages of early heart development exhibit defects, including pericardial and neural edemas (see arrows; scalebar, 1 mm), (B) altered AP length, and (C) increased rates of edema per clutch (N = 30–35 embryos over 4 clutches). (D) Compliance measured by microaspriation of HFR. (E) Compliance at stage 22 confirms blebbistatin and Y27632 increase and calyculin A decreases compliance (N = 11–17 embryos per treatment over 3 clutches). (F) Transverse sections through HFR at stage 28 show changes in polarity (aPKC or ZO-1) within progenitor population (red). Lower panels show the epithelial marker masked using tropomyosin expression (scale bar, 50 μm). (G) Apical intensity after small molecule inhibitor treatment (N = 9–13 embryos over 4 clutches). (H) Representative lateral confocal sections of stage 39 tadpole hearts (scale bar, 100 μm). (I) Cardiac anatomy after stage-specific inhibitor treatments as shown in Figure S2 (N = 5 embryos per treatment per period). Error bars represent mean ± SEM. * indicates p < 0.05; ** indicates p < 0.01. See also Figures S1, S2 and S3A.
To test whether tissue compliance plays a role in driving MET we assessed apicobasal polarity in HPCs (Figure 2F). Calyculin A produced a 50% increase in aPKC and a modest increase in ZO-1 (Fig 2F–G). Y27632 and blebbistatin each produced a two-fold decrease in aPKC and ZO-1 (Fig 2F–G). To assess the effects of these treatments on late-stage heart development we washed out the inhibitors and cultured embryos until to the beating larval heart stage (+ 24 hours, stage 39). Small molecule modulators of tissue compliance and MET, particularly blebbistatin, produced abnormal cardiac anatomy and disrupted fibrillin organization (Fig 2H), indicating a malformed endocardium [22]. Importantly, none of the treatments prevented bilateral fields of HPCs from reaching the ventral midline or altered expression of cardiac markers (Figure 2F). Taken together, these results demonstrate that perturbing mechanical properties of the HFR inhibit MET and induce later cardiac defects but do not overtly alter the movements of these cells to the midline or their differentiation.
Having established a timeline of MET in HPCs (Figure 1), we wondered whether the stages of MET were sensitive to modulators of tissue mechanics. Following incubation with inhibitors for short time periods (Figure S2A), 3 hours and 45 minutes either prior to MET (stage 17 to 21), during MET (stage 21 to 24) or after MET (stage 24 to 27), inhibitors were washed out and embryos were raised to stage 39 when larval heart morphology, including size and shape was assessed (Figure S2B and S2C). Since cardiac morphology was highly sensitive to calyculin A prior to MET and blebbistatin during MET (Fig 2I) whereas modulating mechanics in the stages following MET did not significantly alter morphology, we propose that later defects in cardiac morphology are the result of precocious or accelerated MET. Thus, heart formation has stage-specific requirements for tissue compliance surrounding the onset of MET, suggesting that MET is dependent on temporally regulated mechanical cues.
Heart progenitor cells undergo changes in behavior as they move to the ventral midline
Since cell migratory behaviors are thought to change as a result of MET [23] and we observed a small number of cardiomyocytes excluded from the larval heart after actomyosin perturbations (Figure S3A, arrows), we tested the correlation between HPCs migratory behaviors and the timing of MET. At early neurula stages we observed mesenchymal behaviors in HPCs within HFR explants expressing moesin-GFP and cultured with Cy5-tagged mAb 4H2, a non-function blocking antibody to visualize Xenopus fibronectin live [24]. At this early time, HPCs extend bipolar lamellipodia and deform the surrounding fibronectin ECM (Figure 3A) like cells in dorsal mesoderm [25]. Next, we tracked individual nuclei expressing H2B-mCherry in ventral “windowed” embryos and used nkx2.5-GFP expression to positively identify HPCs (Fig 3B; Movie S1). HPC migration changes in both speed and direction in the time prior to MET (fast, anterior directed) compared to the later times (slow, posterior directed; Figure 3C and D). Surprisingly, migration speed, persistence and collectiveness are all lower after MET (Fig 3E), contrary to more conventional examples of epithelial migration [26].
Figure 3. Heart progenitor cells undergo behavioral transition during stages of mesenchymal-to-epithelial transition.

(A) Left panel: heart progenitor cells isolated from the anterior lateral plate mesoderm show mediolaterally oriented bipolar lamellopodial protrustions (see arrows) that deform fibronectin fibrils (left). Right panel: HPCs extend protrusion, pull on fibronectin fibril (red arrow), and move forward (green arrow). (B) Intravital windowed embryos with fluorescently labeled nuclei reveal cell movements (inset shows tissue organization of ectoderm (blue), mesoderm (pink), and HPCs (red)). At 24 h, expression of nkx2.5-GFP transgene allows retrospective identification of HPC tracks (scale bar = 100 μm). (C) Spider diagrams of nuclei migration trajectories over 4 hour period. (D) Nkx-2.5-GFP identified HPC movements from start to end (black circle). (E) Persistence, speed and collectiveness of HPC movements before MET (1 to 5 h) are higher than after MET (7 to 11 and 13 to 17 h). N = 252 cells for 1 to 5 h, 216 for 7 to 11 h, 411 for 13 to 17 h pooled from 3 embryos from 3 different clutches. * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001. (F) Schematic of traction force and motion correlation. (G) Correlation score between motion and bead displacement vectors on the left and membrane-MKO2 on the right. HPCs exhibit co-aligned motion and traction (+1, red) before transitioning to anticorrelated (−1, blue) (scale bar, 100 μm). Strip kymograph over 12 hour period (lower panel). (H) Percent of co-aligned HPCs decreases over MET (7 tissue isolates over 4 clutches, black; linear regression, blue; error bars, ± SEM). See also Movies S1 and S2.
Several studies have proposed that endoderm carries HPCs ventrally [15]. To investigate mechanical coupling between moving HPCs and their microenvironment we used traction-force microscopy (TFM). By tracking movements of cells and their deformable 2D extracellular matrix substrate, TFM has been used to estimate traction forces generated by single cells [27] and cell monolayers [28], but has not been widely applied to primary tissues or tissue explants. To evaluate coupling we adopted a “low-resolution” version of TFM to detect changes in the orientation of traction forces, e.g. traction vectors, rather than absolute force magnitudes. If HPC movements were dominated by active migration their traction vectors would be anti-correlated with their motion vectors; conversely, if HPCs were carried by endodermal movements, their traction vectors would be co-aligned with their motion vectors (Figure 3F). We found HPCs movement and tractions are co-aligned before MET (Figure 3G, red) and transition to anti-correlated after MET (Figure 3G, blue; Movie S2). By the time MET initiates (Figure 3H, green line), only 25% of HPCs are co-aligned. Thus, as MET progresses, HPCs become less dependent on the underlying endoderm for movement and instead actively direct traction forces to aid their movement toward the ventral midline; consequentially, both MET and coupling to the endoderm may contribute to the reduced HPC speed and collective behaviors.
Peak ventral elongation rates and bulk tissue stiffness coincide with MET onset
As HPCs move to the midline, dorsal and ventral tissues elongate four-fold [29], suggesting HPCs reside in a dynamic mechanical microenvironment. To determine whether mechanical changes in the HFR coincide with MET we tracked tissue movements of the embryo’s ventral anterior surface from late neurula stage (stage 17) using a custom image analysis program [30](Figure 4A; Movie S3). Rather than elongate anteroposteriorly at a constant rate like dorsal tissues, the HFR elongates in a complex manner (Figure 4B), initially shortening and then elongating (Figure 4C). From deformations observed in time-lapse sequences we calculated an instantaneous strain rate, which represents the percentage elongation per hour. Strain rates reach a peak of 11 % per hour at stage 23, which coincides with MET onset, before dropping to low rates consistent with larval growth (Figure 4D). To estimate stress experienced by HPCs we measured the mechanical compliance of the HFR with microaspiration. Compliance initially increases between stages 19 and 21 only to drop almost two-fold at stage 23 (Fig 4E). Combining compliance and strain rates, we see a sharp peak in mechanical stress at stage 23 (Figure 4F; see Supplemental Experimental Procedures). Thus, MET coincides with peak stiffness and strain rates in the HFR, suggesting that MET may be triggered in HPCs in response to high levels of mechanical stress.
Figure 4. Peak stress in ventral heart forming region at stage 23.

(A) Frames from time-lapse of ventral regions over 24 hours with tissue deformations superimposed (red grids; scale bar = 500 μm). (B) Anterioposterior (AP) length, instantaneous elongation rate (C), and the strain rate (D) indicates non-linear growth (thick black line is average of 4 embryos). (E) Compliance of HFR from stage 19 to 23 including fast-response (J(60)) and steady-state (J(120)) responses. (F) Anterioposterior stress index (Pa/hr) calculated from compliance and strain rate (error bars, ± SEM; * indicates p < 0.05; ** indicates p < 0.01). See also Movie S3.
Targeted injections to modulate endoderm contractility can accelerate or inhibit MET
Given the role of endoderm mechanics in driving HPC movements in chicken [15] and the dynamic changes in stress within the HFR in frog (Figure 4F) we wondered whether HPCs were responding directly to mechanical cues from the endoderm within the HFR. To test this we reduced compliance specifically in the anterior endoderm by modulating actomyosin contractility and tested whether MET was accelerated in HPCs. To modulate actomyosin contractility in the endoderm we used the 32-cell Xenopus fate map [31] and injected dorsal vegetal blastomeres targeting exogenous mRNA to either HPCs or cells in the anterior endoderm (Figure 5A and B). To reduce the compliance of endoderm, blastomeres were injected with constitutively active arhgef2-C55R, which can increase dorsal tissue stiffness two-fold [32]. Embryos expressing arhgef2-C55R in the anterior endoderm exhibited reduced AP lengths (Figure 5C) and cardiac edemas (Figure 5D), but no defects in cardiac tissue differentiation (nkx2.5-GFP, Fig 5B see arrows). Microaspiration at stage 23 revealed a 50% decrease in tissue compliance with arhgef2-C55R expression (Fig 5E). At stage 28, we found a 50% increase in MET by aPKC localization (Fig 5F) and a nearly two-fold increase in ZO-1 localization (Figure S4). The ROCK inhibitor Y27632, which operates downstream of arhgef2-C55R, was able to block accelerated MET with a two-fold decrease in aPKC localization (Figure 5F). By stage 39, tadpoles with arhgef2-C55R expressing endoderm displayed abnormal cardiac architecture, including smaller chambers, perturbed fibrillin structure and hypertrophy (Figure 5G), demonstrating not only that induced endoderm contractility perturbs MET, but that it is sufficient to generate cardiac defects.
Figure 5. Targeted injections to perturb microenvironmental stress via actomyosin contractility accelerate MET.

(A) Experiment schematic: 32-cell stage injected to target either HPCs (red) or the anterior endoderm (yellow). (B) Transgenic nkx2.5-GFP embryos co-injected with endoderm targeted arhgef-C55R mRNA and rhodamine dextran (scale bar, 1 mm). Cardiac differentiation proceeds but arhgef2-C55R reduces AP length (C) and increased rates of pericardial edemas per clutch (D; N = 23 control, 35 arhgef2-C55R, 3 clutches). (E) Compliance of arhgef-C55R reduced (N = 8–9 embryos over 2 clutches). (F) Upper: tropomyosin, aPKC, and rhodamine dextran. Lower: tropomyosin-masked aPKC expression in psuedocolor LUT (scale bar = 50 μm). Apical aPKC increased in arhgef2-C55R, while Y27632 treatment can reverse effect (N = 7–11 embryos over 3 clutches). (G) Representative confocal sections of stage 39 tadpole heart morphology (scale bar = 100 μm). Error bars, ± SEM. See also Figures S1, S3B, S4 and S5.
To further confirm the role of endoderm mechanics in inducing HPC MET, we targeted injections of a constitutively active form of the myosin binding subunit (MBS, MYPT1, or formally PPP1R12A) which we hypothesized would soften tissues. MBS-T695A cannot be phosphorylated at one of two inhibitory sites, and can induce a two-fold increase in myosin phosphatase activity compared to the wild-type [33]. MBS-T695A expressed in the anterior endoderm produced whole embryo defects (Figure S4A), including reduced AP length (Figure S4B) and increased rates of cardiac edemas (Figure S4C). Microaspiration of the HFR revealed compliance increased in MBS-T695A embryos by 30% (Figure S4D). Similar to what we observed in blebbistatin and Y27632 treated embryos, we found aPKC intensity decreased by approximately 40% with increased compliance in the endoderm (Figure S4E and F). Calyculin A, which inhibits myosin phosphatase activity downstream of MBS-T695A, was able to rescue many of these defects, including the reduced AP length (Figure S4B) and localization of aPKC (Figure S4E and F).
Next we tested whether HPC autonomous mechanics might also play a role in regulating MET. Arhgef2-C55R targeted to HPCs displayed stronger aPKC localization than neighboring non-expressing HPCs (Figure S5A, arrow) and significant correlation between levels of arhgef2-C55R expression and aPKC (Figure S5A) suggesting a graded increase in apicobasal polarity with increasing levels of HPC contractility. Furthermore, as with the case of increased contractility in endoderm, HPCs expressing arhgef2-C55R treated with Y27632 showed little evidence of MET (Figure S5A). By stage 39, tadpoles with arhgef2-C55R expressing cardiomyocytes displayed abnormal cardiac structure, with defects centered around cells expressing arhgef2-C55R (Figure S5B). Thus, mechanical perturbations targeted to either endoderm or HPCs confirm results obtained with small molecule inhibitors and identify a direct role for cell contractility and mechanics in regulating early MET.
External mechanical tension can induce MET
Since cell contractility is a key contributor to junction formation between epithelial cells [34], we wanted to test whether MET in HPCs could be driven purely by exogenous mechanical stress. To increase stress within the heart forming region we injected a 70 nl droplet of biologically inert mineral oil (~500 μm diameter) into the anterior cavity of the archenteron of just after neural tube formation (stage 21; Movie S4). Upon injection, the droplet produces a 5% stretch in the ventral ectoderm surface area (Fig 6A) and remains nearly spherical (Figure S6A; Movie S5). Droplet-injected embryos continue to elongate normally but develop cardiac edemas with high frequency (~80%; Fig 6B); embryos wounded with the microinjection needle (Sham control; Figure 6B), or injected with a smaller 12 nl droplet (140 μm diameter) that produces no deformation, exhibit much lower rates of cardiac edema (Figure S6B–C). MET increases within HPCs in large droplet-injected embryos (Fig 6C) with a 50% increase in apical aPKC and a twofold increase in ZO-1 similar to the case of embryos with stiffer endoderm (Fig 5F). Thus, we were able to show that physically-induced stress is sufficient to accelerate MET in HPCs without manipulating actomyosin contractility.
Figure 6. Exogenously applied stress can accelerate MET in HPCs.

(A) Biologically inert mineral oil injected into foregut (blue outline). Sagittal and transverse views pre-injection (upper) and post-injection (lower) with forces indicated by yellow arrows. Right: grid-tracked stretch of 5% from pre- to post-injection. (B) Embryos injected with oil show no defects in AP length (N = 13–14 embryos over 2 clutches) but a high frequency of ventral edemas per clutch (N = 2 clutches; scale bar = 1 mm). (C) Confocal transverse sections of HFR show tropomyosin and aPKC or ZO-1 (green) and lower panels show tropomyosin-masked polarity in pseudocolor LUT (scale bar = 50 μm). Apical aPKC and ZO-1 increases after oil injection (N = 11 to 13 embryos over 3 clutches; Error bars represent ± SEM’ * indicates p < 0.05 and ** indicates p < 0.01). See also Figures S1 and S6A–C and Movies S4 and S5.
Perturbing MET has physiological consequences in the larval beating heart
We were surprised by the specific and robust defects in cardiac architecture when MET was transiently disrupted and wondered if perturbations in the timing of MET translated to physiological defects in cardiac function. To assess cardiac function we visualized heart beat once the larval heart begins to drive circulation (stage 42) after a range of MET perturbations including: modulation of contractility from stages 20 to 28, endoderm-targeted injection of arhgef2-C55R, and stress-inducing mineral oil injections. Using hemoglobin contrast subtraction angiography (HCSA; [35]), we visualized blood flow through the ventricle (Movie S6; Figure 7A and 7B; arrows indicate individual trabeculae) and measured ejection fraction (EF), heart rate (HR), end-diastolic area (Aed), end-systolic area (Aes), and ejected area (Aej; Figure 7C and Figure S6D–H). Control embryos have a mean EF of 91%, comparable to previous reports [35]. Blebbistatin produced the most severe physiological defects with stationary blood islands forming superficial to the primitive gut (data not shown); while these embryos exhibited a regular heartbeat they had no blood flow, likely indicating a failure to connect to the systemic circulation. Both calyculin A and arhgef2-C55R yielded significantly larger Aes indicating that ventricular contraction was unable to efficiently pump blood out of the heart. Oil injected embryos presented highly variable heart morphologies, ranging from fairly normal to fully bifid hearts, with highly variable function. Y27632 produced mild defects with lower EF, lower HR and higher Aes but none were statistically significant. Thus, perturbations in MET in the heart progenitor cells produce highly specific defects in cardiac architecture and distinctive defects in cardiac function.
Figure 7. Specific lesions to larval heart function in response to MET perturbations.

(A) RGB image of 3-day old tadpole heart with ventricle outlined in white and outflow tract outlined in yellow. Hemoglobin contrast subtraction angiography (HCSA) and multi-frame, time-projection of HCSA over one cardiac cycle shows individual trabeculae (arrows; scale bar = 100 μm). (B) Time varying hemoglobin blush reveals typical cardiac cycle dynamics, with end-diastolic volume (Aed) and end-systolic volume (Aes) indicated on graph and table (C; pooled from 3 clutches; ejection fraction, EF; heart rate, HR; end-diastolic area, Aed; end-systolic area, Aes; area ejected, Aej). * indicates p < 0.05; ** indicates p < 0.01. Treatments increasing compliance (decreased cell contractility, blue) and treatments decreasing compliance (increased cell contractility) or stress (red). (D) Schematic of model of mechanical coupling between endoderm development, heart progenitor cell development, MET, and cardiogenesis. (E) Endogenous apical polarity increases over time (data redrawn from Figure 1F–I) and changes with mechanical perturbations at stage 28 (data redrawn from Figs. 2, 5, 6). Regression line (aPKC: y = 0.77t − 1.34; R2 = .446; ZO-1: y = 0.92t − 1.96; R2 = .567). (F) Apical polarity decreases with compliance (regression fit to data from arhgef2-C55R, calyculinA, control and Y27632 data (y = −27.22*J(60) + 4.99; R2 =.843). See also Figure S6D–H and Movie S6.
Discussion
On their way to the ventral midline HPCs traverse a dynamically changing mechanical microenvironment marked by fibronectin remodeling [10, 16, 20], changing tissue stiffness [19, 36], and tension from ventral convergent extension [15, 19, 29]. While MET is not a prerequisite for HPC differentiation or arrival at the ventral midline, the proper timing of MET is both sensitive to the mechanical environment of the HFR and required for proper heart structure (Figure 2H–I, Figure 5G, Figure S2) and function (Figure 7C and Figure S6D–H). The role of mechanical cues in driving MET and subsequent defects in heart anatomy and function is supported by global-, HFR-specific, endoderm-specific, and HPC-specific mechanical perturbations. Based on alterations in HPC MET under experimental perturbation (Figure 7E) we find HFR tissue compliance and apicobasal polarity display an inverse relationship (Figure 7F) except at very high levels of compliance where HPCs completely fail to polarize (Figure 7F; purple line).
The epithelium formed by heart progenitor cells is unique in both its anatomy and its contribution to the larval heart and suggests that MET occurs at a critical moment as bilateral fields of HPCs assemble into the medial plate. Progenitor cells initially move as a bilayer, with the apical surface of the epithelium assembling along the middle of the two layers (Figure 1B). The deep layer gives rise to endocardial cells and cardiomyocytes [37] and the superficial layer likely contributing to pericardium [38]. The formation of apical junctions between two layers of mesoderm is a likely first step in pericardial coelom formation [37] as separation of inner and outer mesoderm layers isolates precardiac mesoderm from body wall mesoderm [11]. Furthermore, because this epithelialization is transient and mature cardiomyocytes do not form an epithelium [38], the function and timing of MET likely plays a key role in establishing the structural foundation of the vertebrate heart.
Establishing a causal link between transcriptional regulation of HPC MET and how those changes alter HPC behaviors to establish organization within the heart fields will require a detailed mechanistic analysis. Much is known about HPC movements, including their multiphasic nature [16] and coordination with endoderm [15, 17, 18], however, little is known about the process of MET and its implications for later heart development. Several molecular pathways have been implicated in establishing HPC polarity, including PRKCI and Crumbs [39], hand2 and fibronectin [20] and Slit and Robo [40]. Mechanical signaling must also play a critical role, and because stiffness gradients can generate cytoskeletal polarity and directional migration in mesenchymal cell culture [41], it is tempting to speculate that the mediolateral stiffness gradient generated by endoderm convergent extension during chicken cardiogenesis [19] causes HPCs to experience higher stiffness or tension as they approach the ventral midline. Thus, the biomechanical processes that shape the ventral endoderm are not only required to position HPCs, but can also establish the apicobasal polarity and epithelial identity in HPCs (Figure 5, S4), ultimately allowing for proper heart organization, structure and function.
At present, we do not know how HPCs sense changes in mechanics although there are a number of likely candidates [1]. Cilia can play mechanosensory roles in kidney and vasculature development with variety of ciliopathies associated with cardiac defects [42]; filopodia-sensed tension may shape outflow tract morphogenesis [43]. Yet another possibility is that mechanical cues are sensed by focal adhesions to fibronectin mediated by α5β1 integrin since both fibronectin and α5β1 integrin are required for movements of the HPC and endoderm [13, 20] and appear to polarize cardiomyocytes along the mesoderm-endoderm interface [37]. Fibronectin null-mutants in Zebrafish disrupt both mechanical signaling and migration [13]; however, when force transduction is decoupled from signaling, pericardial edemas form that are similar to those found after MET is disrupted [44]. Future mechanistic studies are warranted in order to test the role of putative mechanosensors used by HPCs in the activation of MET.
Early mechanical events during development can play a critical role in shaping organs and enabling their physiological function. In the forming heart, myosin II contractility and tissue mechanics plays a role in directing where and when HPC MET occurs. Failure of MET leads to disorganized heart fields prior to merging, disrupted looping and failure to integrate the heart with the systemic circulation. Either precocious or accelerated HPC MET results in significantly larger end-systolic area without a change in ejection fraction (Figure 7C), which is a common feature of congenital heart defects [45] including mitral or aortic valve insufficiency [46] or defects to the ventricular wall strength, including hypertrophy [47]. Since precocious or accelerated MET produces a distinct phenotype, future studies may uncover the relationship between genetic mutations known to cause congenital heart defects and causative defects in mechanical signaling in early heart development.
Experimental Procedures
Xenopus embryos
Xenopus laevis embryos were obtained via standard methods and cultured to stage 14 [48] when heart progenitor cells are located within the anterior lateral plate mesoderm (Figure S7A). Ex vivo tissue isolates consisting of the ventral mesoderm and the bilateral fields of anterior lateral plate mesoderm are microsurgically removed from the embryo (Figure S7B) while intravital “windowed” embryos are generated by microsurgically removing the ventral ectoderm (Figure S7C). Microsurgically manipulated tissues can be cultured for more than 48 hours, at which time they build chambers and express cardiac markers similar to those observed in vivo (Figure S7E). Additional details are found in the Supplemental Experimental Procedures.
Microscopy and microaspiration
Live fluorescent imaging and fixed immunofluorescence imaging was performed using a laser-scanhead mounted on an inverted compound microscope (Leica SP5). Images were then imported into ImageJ [49] for image analysis, including quantification of apical intensity, cardiac anatomy and HCSA analysis [35]. For nuclei tracking, tissue deformation mapping and traction force microscopy, custom image analysis algorithms were created utilizing the Insight Toolkit (ITK). Microaspiration was performed using a custom dual-reservoir microaspirator apparatus previous described [50] and illustrated in Figure 2D. In brief, a suction pressure was applied to the ventral HFR of the embryo as displacement into a microchannel was tracked. From the displacement data, a power law model for creep compliance implemented in MATLAB (Mathworks) was used to obtain time-dependent compliance measurements for each embryo. Material within a 150 uM depth contributes to the measurement [50], so the measured compliance reflects a composite mechanical property of all three germ layers within the HFR. Additional details are found in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We would like to thank Callie Miller, Joe Shawky, Deepthi Vijayraghavan, Holley Lynch, Takehiko Ichikawa and other members of the group for their comments and discussions. This work was supported by the National Science Foundation (CBET-1547790) and the National Institutes of Health (R01HD044750; R56HL13495). TRJ was supported in part by the Cardiovascular Bioengineering Training Program (NIH NHLBI T32 HL076124). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation or the National Institutes of Health. We thank the National Xenopus Resource (RRID:SCR_013731) for providing Nkx-2.5:GFP transgenic frogs.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and six movies and can be found with this article online at XXXXX.
Author Contributions
Conceptualization, T.R.J. and L.A.D.; Methodology, T.R.J., H.Y.K., C.S. and L.A.D.; Investigation, T.R.J. and U.L.B.; Resources, C.S.; Formal analysis, visualization and writing –original draft, T.R.J.; Validation, T.R.J. and U.L.B; Writing – Review & Editing, T.R.J., H.Y.K., U.L.B., C.S. and L.A.D.; Supervision and funding acquisition, L.A.D.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller CJ, Davidson LA. The interplay between cell signalling and mechanics in developmental processes. Nat Rev Genet. 2013;14:733–744. doi: 10.1038/nrg3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 3.Weber GF, Bjerke MA, DeSimone DW. A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev Cell. 2012;22:104–115. doi: 10.1016/j.devcel.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 5.Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, Webster JD, Hoover S, Simpson RM, Gauldie J, et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010;70:5706–5716. doi: 10.1158/0008-5472.CAN-09-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HY, Jackson TR, Davidson LA. On the role of mechanics in driving mesenchymal-to-epithelial transitions. Semin Cell Dev Biol. 2016 doi: 10.1016/j.semcdb.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kshitiz Hubbi ME, Ahn EH, Downey J, Afzal J, Kim DH, Rey S, Chang C, Kundu A, Semenza GL, et al. Matrix rigidity controls endothelial differentiation and morphogenesis of cardiac precursors. Sci Signal. 2012;5:ra41. doi: 10.1126/scisignal.2003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morikawa Y, Zhang M, Heallen T, Leach J, Tao G, Xiao Y, Bai Y, Li W, Willerson JT, Martin JF. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci Signal. 2015;8:ra41. doi: 10.1126/scisignal.2005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120:1351–1383. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Yelon D, Stainier DYR. Hand2 Regulates Epithelial Formation during Myocardial Differentiation. Current Biology. 2005;15:441–446. doi: 10.1016/j.cub.2004.12.083. [DOI] [PubMed] [Google Scholar]
- 11.Linask KK, Knudsen KA, Gui YH. N-cadherin-catenin interaction: necessary component of cardiac cell compartmentalization during early vertebrate heart development. Dev Biol. 1997;185:148–164. doi: 10.1006/dbio.1997.8570. [DOI] [PubMed] [Google Scholar]
- 12.Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–1808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trinh LA, Stainier DYR. Fibronectin regulates epithelial organization during myocardial migration in zebrafish. Developmental Cell. 2004;6:371–382. doi: 10.1016/s1534-5807(04)00063-2. [DOI] [PubMed] [Google Scholar]
- 14.Nascone N, Mercola M. An inductive role for the endoderm in Xenopus cardiogenesis. Development. 1995;121:515–523. doi: 10.1242/dev.121.2.515. [DOI] [PubMed] [Google Scholar]
- 15.Varner VD, Taber La. Not just inductive: a crucial mechanical role for the endoderm during heart tube assembly. Development (Cambridge, England) 2012;139:1680–1690. doi: 10.1242/dev.073486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye D, Xie H, Hu B, Lin F. Endoderm convergence controls subduction of the myocardial precursors during heart-tube formation. Development (Cambridge, England) 2015;142:2928–2940. doi: 10.1242/dev.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aleksandrova A, Czirok A, Kosa E, Galkin O, Cheuvront TJ, Rongish BJ. The endoderm and myocardium join forces to drive early heart tube assembly. Developmental Biology. 2015;404:40–54. doi: 10.1016/j.ydbio.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aleksandrova A, Czirók A, Szabó A, Filla MB, Hossain MJ, Whelan PF, Lansford R, Rongish BJ. Convective tissue movements play a major role in avian endocardial morphogenesis. Developmental biology. 2012;363:348–361. doi: 10.1016/j.ydbio.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Varner VD, Taber LA. Why is cytoskeletal contraction required for cardiac fusion before but not after looping begins? Phys Biol. 2015;12:016012. doi: 10.1088/1478-3975/12/1/016012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garavito-Aguilar ZV, Riley HE, Yelon D. Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development (Cambridge, England) 2010;137:3215–3220. doi: 10.1242/dev.052225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottardi CJ, Arpin M, Fanning AS, Louvard D. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc Natl Acad Sci U S A. 1996;93:10779–10784. doi: 10.1073/pnas.93.20.10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolker SJ, Tajchman U, Weeks DL. Confocal imaging of early heart development in Xenopus laevis. Developmental biology. 2000;218:64–73. doi: 10.1006/dbio.1999.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson LA, Dzamba BD, Keller R, Desimone DW. Live imaging of cell protrusive activity, and extracellular matrix assembly and remodeling during morphogenesis in the frog, Xenopus laevis. Dev Dyn. 2008;237:2684–2692. doi: 10.1002/dvdy.21600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson LA, Marsden M, Keller R, Desimone DW. Integrin alpha5beta1 and fibronectin regulate polarized cell protrusions required for Xenopus convergence and extension. Curr Biol. 2006;16:833–844. doi: 10.1016/j.cub.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 26.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nature reviews. Molecular cell biology. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 27.Sabass B, Gardel ML, Waterman CM, Schwarz US. High resolution traction force microscopy based on experimental and computational advances. Biophysical journal. 2008;94:207–220. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vincent R, Bazellieres E, Perez-Gonzalez C, Uroz M, Serra-Picamal X, Trepat X. Active Tensile Modulus of an Epithelial Monolayer. Phys Rev Lett. 2015;115:248103. doi: 10.1103/PhysRevLett.115.248103. [DOI] [PubMed] [Google Scholar]
- 29.Larkin K, Danilchik MV. Ventral cell rearrangements contribute to anterior-posterior axis lengthening between neurula and tailbud stages in Xenopus laevis. Dev Biol. 1999;216:550–560. doi: 10.1006/dbio.1999.9495. [DOI] [PubMed] [Google Scholar]
- 30.Kim Y, Hazar M, Vijayraghavan DS, Song J, Jackson TR, Joshi SD, Messner WC, Davidson LA, LeDuc PR. Mechanochemical actuators of embryonic epithelial contractility. Proc Natl Acad Sci U S A. 2014;111:14366–14371. doi: 10.1073/pnas.1405209111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dale L, Slack JM. Fate map for the 32-cell stage of Xenopus laevis. Development (Cambridge, England) 1987;99:527–551. doi: 10.1242/dev.99.4.527. [DOI] [PubMed] [Google Scholar]
- 32.Zhou J, Kim HY, Wang JH, Davidson LA. Macroscopic stiffening of embryonic tissues via microtubules, RhoGEF and the assembly of contractile bundles of actomyosin. Development. 2010;137:2785–2794. doi: 10.1242/dev.045997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579:6611–6615. doi: 10.1016/j.febslet.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 34.Yamada S, Nelson WJ. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell-cell adhesion. J Cell Biol. 2007;178:517–527. doi: 10.1083/jcb.200701058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deniz E, Jonas S, Khokha M, Choma MA. Endogenous contrast blood flow imaging in embryonic hearts using hemoglobin contrast subtraction angiography. Opt Lett. 2012;37:2979–2981. doi: 10.1364/OL.37.002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Kim HY, Davidson La. Actomyosin stiffens the vertebrate embryo during crucial stages of elongation and neural tube closure. Development (Cambridge, England) 2009;136:677–688. doi: 10.1242/dev.026211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linask KK. N-cadherin localization in early heart development and polar expression of Na+,K(+)-ATPase, and integrin during pericardial coelom formation and epithelialization of the differentiating myocardium. Dev Biol. 1992;151:213–224. doi: 10.1016/0012-1606(92)90228-9. [DOI] [PubMed] [Google Scholar]
- 38.von Gise A, Pu WT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circulation research. 2012;110:1628–1645. doi: 10.1161/CIRCRESAHA.111.259960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohr S, Bit-Avragim N, Abdelilah-Seyfried S. Heart and soul/PRKCi and nagie oko/Mpp5 regulate myocardial coherence and remodeling during cardiac morphogenesis. Development. 2006;133:107–115. doi: 10.1242/dev.02182. [DOI] [PubMed] [Google Scholar]
- 40.Medioni C, Astier M, Zmojdzian M, Jagla K, Semeriva M. Genetic control of cell morphogenesis during Drosophila melanogaster cardiac tube formation. J Cell Biol. 2008;182:249–261. doi: 10.1083/jcb.200801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J Cell Biol. 2012;199:669–683. doi: 10.1083/jcb.201205056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duncan AR, Khokha MK. Xenopus as a model organism for birth defects-Congenital heart disease and heterotaxy. Semin Cell Dev Biol. 2016;51:73–79. doi: 10.1016/j.semcdb.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Francou A, Saint-Michel E, Mesbah K, Kelly RG. TBX1 regulates epithelial polarity and dynamic basal filopodia in the second heart field. Development. 2014;141:4320–4331. doi: 10.1242/dev.115022. [DOI] [PubMed] [Google Scholar]
- 44.Rozario T, Dzamba B, Weber GF, Davidson LA, DeSimone DW. The physical state of fibronectin matrix differentially regulates morphogenetic movements in vivo. Dev Biol. 2009;327:386–398. doi: 10.1016/j.ydbio.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abualsaud AO, Lowe BS, Guo K, Marelli AJ, Kaouache M, Guo L, Jutras L, Martucci G, Therrien J. Cardiac output as a predictor in congenital heart disease: Are we stating the obvious? Int J Cardiol. 2016;210:143–148. doi: 10.1016/j.ijcard.2016.02.071. [DOI] [PubMed] [Google Scholar]
- 46.Hundley WG, Li HF, Willard JE, Landau C, Lange RA, Meshack BM, Hillis LD, Peshock RM. Magnetic resonance imaging assessment of the severity of mitral regurgitation. Comparison with invasive techniques. Circulation. 1995;92:1151–1158. doi: 10.1161/01.cir.92.5.1151. [DOI] [PubMed] [Google Scholar]
- 47.Song W, Dyer E, Stuckey DJ, Copeland O, Leung MC, Bayliss C, Messer A, Wilkinson R, Tremoleda JL, Schneider MD, et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC Gene) that causes apical hypertrophy in man and mouse. J Biol Chem. 2011;286:27582–27593. doi: 10.1074/jbc.M111.252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nieuwkoop PD, Faber J. Normal tables of Xenopus laevis (Daudin) Amsterdam: Elsevier North-Holland Biomedical Press; 1967. [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Dassow M, Strother JA, Davidson LA. Surprisingly simple mechanical behavior of a complex embryonic tissue. PLoS One. 2010;5:e15359. doi: 10.1371/journal.pone.0015359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.