

Summary
Gastric acid secretion by parietal cells requires trafficking and exocytosis of H-K-ATPaserich tubulovesicles (TVs) toward apical membranes in response to histamine stimulation via cAMP elevation. Here, we found that TRPML1 (ML1), a protein that is mutated in type IV Mucolipidosis (ML-IV), is a tubulovesicular channel essential for TV exocytosis and acid secretion. Whereas ML-IV patients are reportedly achlorhydric, transgenic overexpression of ML1 in mouse parietal cells induced constitutive acid secretion. Gastric acid secretion was blocked and stimulated by ML1 inhibitors and agonists, respectively. Organelle-targeted Ca2+ imaging and direct patch-clamping of apical vacuolar membranes revealed that ML1 mediates a PKA-activated conductance on TV membranes that is required for histamine-induced Ca2+ release from TV stores. Hence, we demonstrated that ML1, acting as a Ca2+ channel in TVs, links transmitter-initiated cyclic nucleotide signaling with Ca2+-dependent TV exocytosis in parietal cells, providing a regulatory mechanism that could be targeted to manage acid-related gastric diseases.
Keywords: Tubulovesicle, cAMP, TRPML1, Membrane trafficking, Ca2+ release, Exocytosis
eTOC Blurb
Acid secretion from the parietal cells of the stomach is essential for food digestion. Sahoo et al. identified TRPML1 as a histamine-activated Ca2+ channel in the tubulovesicles required for gastric acid secretion. Synthetic agonists and inhibitors of TRPML1 may be developed to control acid secretion and treat acid-related gastric diseases.
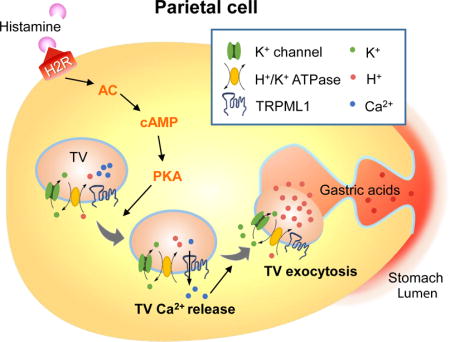
Introduction
Acid secretion in the stomach is mediated by parietal cells that are filled with vesicular and tubular organelles known as tubulovesicles (TVs), which carry the H+/K+-ATPase responsible for H+ pumping (Hersey and Sachs, 1995; Yao and Forte, 2003). Upon stimulation with gastric transmitters (secretagogues), parietal cells undergo striking morphological changes, including SNARE-dependent membrane remodeling and actin-dependent cytoskeleton reorganization (Forte and Zhu, 2010). Subsequently, TVs are translocated to fuse with apically-directed canaliculi, resulting in a large, deeply-invaginated secretory surface that is connected to the stomach lumen (Forte et al., 1977). The primary secretagogue is histamine, produced by enteroendocrine cells, which activates the Gs-coupled type 2 histamine (H2) receptor to initiate a cAMP-dependent signaling cascade (Chew et al., 1980; Malinowska et al., 1988). Although some types of regulated exocytosis, including synaptic neurotransmitter release, are Ca2+-dependent (Thorn et al., 2016), it remains uncertain whether Ca2+ is involved in histamine-triggered TV exocytosis (Chew and Brown, 1986; Negulescu et al., 1989; Yao and Forte, 2003). Acetylcholine, which also acts as a gastric secretogogue, has been shown to induce large releases of Ca2+ from endoplasmic reticulum (ER) Ca2+ stores (Forte and Zhu, 2010; Negulescu et al., 1989). Meanwhile, histamine has been reported to induce very small Ca2+ increases in the gastric parietal cells of some species, and no detectable Ca2+ responses in other species (Chew and Brown, 1986; Courtois-Coutry et al., 1997; Negulescu et al., 1989; Yao and Forte, 2003). In other processes for which Ca2+-dependence has been debated, organelle-targeted Ca2+ imaging methods with very high detection sensitivity have implicated non-ER Ca2+ stores are essential for the cellular response (Xu et al., 2015).
Transient Receptor Potential Mucolipin-1 (ML1, aka TRPML1 or MCOLN1) is the principle Ca2+ release channel in the lysosome; it regulates membrane fusion/fission and the transport of lysosomes in response to cellular cues (Xu and Ren, 2015). Notably, mutations in the gene that encodes ML1 in humans cause type IV mucolipidosis (ML-IV), a neurodegenerative disease with an additional gastric phenotype of constitutive achlorhydria, which is recapitulated in ML1 knockout (KO) mice (Chandra et al., 2011; Schiffmann et al., 1998; Venugopal et al., 2007). The aim of the present study was to investigate whether ML1 has a direct role in gastric acid secretion. To address this question, we examined how gastric acid secretion is altered in genetically engineered mouse models and affected by direct, pharmacological manipulations of ML1 channel activity.
Results
Synthetic inhibitors of ML1 block gastric acid secretion
To test the physiological role of ML1 in proton secretion, we isolated stomachs, glands, and parietal cells from wild-type (WT) and ML1 KO mice (Venugopal et al., 2007). The gastric corpus glands were pathologically enlarged in ML1 KO mice (Fig. 1A, S1A-E), suggestive of a hypertrophic phenotype. Compared with glands from WT controls, ML1 KO mouse glands had increased corpus height (Fig. S1D, E), abnormal parietal cell morphology, number, and proliferation (Fig. S1F, G, H, I), increased mucosal thickness (Fig. S1F), reduced expression of αβ heteromeric H+/K+-ATPase (Fig. S1J, L), and increased expression of lysosome-associated membrane protein 1 (Lamp1, Fig. S1J, K). Patch-clamping of vacuolin-1-enlarged vacuoles (Dong et al., 2010) in parietal cells within cultured corpus glands revealed whole-endolysosome ML1-mediated currents (IML1) in WT, but not ML1 KO, parietal cells (Fig. 1B, 1C).
Fig. 1. ML1 involvement in histamine-stimulated acid secretion.
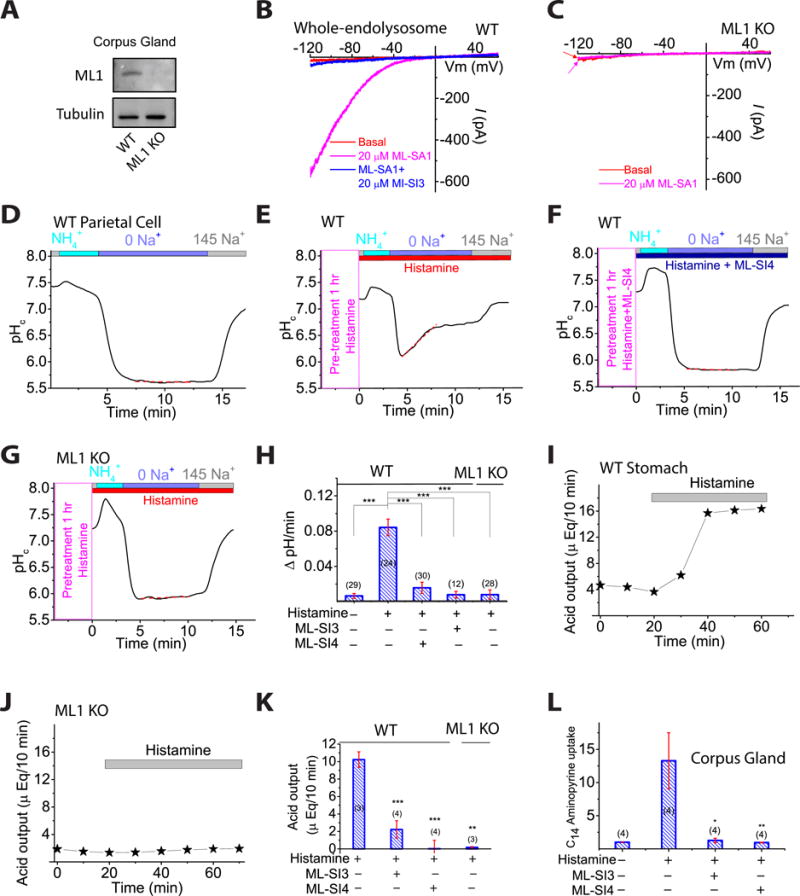
(A) Corpus gland immunoblots. (B, C) Whole-endolysosomal IML1 was activated by TRPML agonists (ML-SA1/3/5; 1–20 μM) and inhibited by ML1 antagonists ML-SI3/4 (10–20 μM) in WT (B), but not ML1 KO parietal cells (C). (D–G) Histamine (100 μM + 20 μM IBMX) and ML-SI4 (10 μM) effects on proton secretion, indexed by NIPR observed while cytoplasmic pH (pHc) was under H+/K+-ATPase control (see also Fig. S1O) relative to responses in 0 Na+ (red dotted lines). (H) NIPR rates (n = 10–30 cells) under resting and histamine-stimulated conditions. (I, J) Whole-stomach acid contents following histamine administration (1 mg/kg, IP). (K) Histamine-induced acid increases in the presence of ML-SI3 (20 μM) or ML-SI4 (10 μM). n = 3–4 mice/group. (L) Effect of ML-SIs on histamine-stimulated [14C] aminopyrine accumulation in gastric glands (n = 4 mice per experiment; normalized to basal output). ML-SI3 (20 μM) and ML-SI4 (10 μM) were applied 30 min before histamine (100 μM + 20 μM IBMX). Panels H, K, and L show means ± SEMs from ≥3 experiments. *P < 0.05, **P < 0.01, ***, P < 0.001 one-way ANOVA, Bonferroni’s post-hoc analysis.
As a quantitative measure of H+/K+-ATPase-dependent proton secretion, we determined the rate of Na+-independent cytoplasmic pH (pHc) recovery (NIPR) by measuring the fluorescence intensity of the pH-sensitive dye BCECF (Pasham et al, 2013) (Fig. S1O). NIPR was virtually absent under resting conditions (Fig. 1D, 1H) in both WT (0.006 ± 0.003 ΔpH/min, n = 29 cells) and ML1 KO parietal cells. Histamine stimulation increased NIPR by ∼1,400% to 0.084 ± 0.009 ΔpH/min (n = 24) in WT cells, but did not affect NIPR in ML1 KO parietal cells (0.008 ± 0.005 ΔpH/min, n = 28, Fig. 1E, 1G, 1H). Conversely, there was no detectable histamine-induced NIPR in the presence of omeprazole, a specific H+/K+-ATPase inhibitor commonly used to treat gastric diseases (Shin et al, 2009) (Fig. S1P). Strikingly, brief exposure of WT parietal cells to ML-SI3 or ML-SI4 (10–20 μM), two structurally-independent ML1 inhibitors (Zhang et al., 2016), also abolished histamine-induced NIPR (Fig. 1F, 1H).
We next investigated the role of ML1 in acid secretion in vivo. Intra-peritoneal (IP) injection of histamine resulted in a rapid increase in whole-stomach acid contents in WT, but not ML1 KO mice (Fig. 1I and 1J). This histamine induction of stomach acid secretion was attenuated markedly in the presence of intra-gastric ML-SI3 or ML-SI4 (Fig. 1K and Fig. S1Q). Furthermore, in an ex vivo acid secretion assay, in which luminal acidification traps protonated C14-aminopyrines in enzymatically-isolated glands (Mettler et al, 2007), pre-incubation with ML-SI3 or ML-SI4 blocked histamine-induced C14-aminopyrine uptake (Fig. 1L). These results, from three independent acid secretion assays, suggest that ML1 functions as a direct physiological regulator of gastric acid secretion and provide counterevidence to the prior suggestion that secondary or developmental defects underlie the achlorhydria phenotype of ML1 KO mice and ML-IV patients (Chandra et al, 2011).
ML1 agonists induce gastric acid secretion independent of histamine
Incubation of WT parietal cells with the ML1 agonist ML-SA1 or the more potent structurally-independent agonist ML-SA5 (Shen et al, 2012; Zhang et al., 2016) resulted in an NIPR that was ∼1,000% of that observed in untreated WT cells (Fig. 2A–2C). Conversely, ML-SA treatment did not affect NIPR in ML1 KO parietal cells. ML-SA-triggered NIPR in WT cells was blocked completely by omeprazole (Fig. S1R). In addition, acid secretion was also increased markedly by ML-SA5 in both stomachs and isolated glands obtained from WT, but not ML1 KO, mice (Fig. 2D–2G). Taken together, these results suggest that ML1 activation is sufficient to induce acid secretion both in vitro and in vivo.
Fig. 2. ML1 activation induces gastric acid secretion.
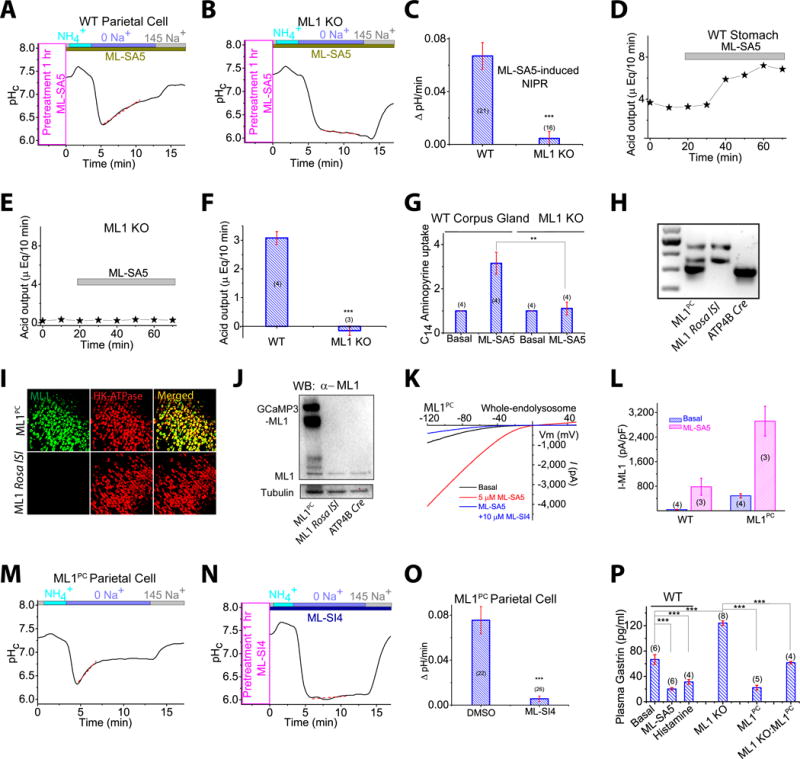
(A, B) ML-SA5 (10 μM, applied 1 h prior) potentiation of NIPR in WT (A), but not ML1 KO (B) parietal cells. (C) NIPR of ML-SA5-treated WT and ML1 KO parietal cells. (D, E) Representative traces of ML-SA5 effects on whole-stomach acid contents in vivo. (F) Average acid secretory rates (n = 3–4 mice/group). (G) ML-SA5 effects on [14C]-aminopyrine incorporation into isolated glands. (H) Genotype verification of ML1 Rosa loxSTOPlox (lSl), ATP4BCre, and ML1 Rosa lSl: ATP4B-Cre (ML1PC) mice. Three DNA bands, each amplified by specific PCR primers, represent WPRE (upper), ROSA (middle), and ATP4BCre (lower) (see Fig. S2A). (I) Immunohistochemical detection of GCaMP3-ML1 and H+/K+-ATPase α subunit in corpus tissues. Scale bar, 100 μm. (J) Immunoblot demonstration of GCaMP3-ML1 expression in ML1PC corpus glands. (K) Whole-endolysosome IML1 in ML1PC parietal cells. (L) IML1 current densities in parietal cells. (M, N) ML-SI4 (10 μM) annulment of constitutive NIPR in ML1PC parietal cells. (O) ML-SI4 effects on ML1PC -cell NIPR. (P) Plasma gastrin levels of overnight-fasted WT (control, histamine, and ML-SA5), ML1 KO, ML1PC, and ML1 KO: ML1PC mice. Quantitative data are means ± SEM from ≥3 experiments. *P < 0.05, **P < 0.01, ***, P < 0.001 one-way ANOVA, Bonferroni’s post-hoc analysis (G, P) or Student’s t-test (C, F, O).
Transgenic ML1 overexpression in parietal cells causes constitutive secretion of gastric acid
To investigate the in vivo role of ML1 in acid secretion, we generated a targeted transgenic mouse model, in which a floxed (fl) allele carrying the GCaMP3-ML1 transgene (Shen et al, 2012) was integrated into the ROSA26 locus behind a loxSTOPlox cassette (Fig. S2A). Crossing ROSA-loxSTOPlox-GCaMP3-ML1 (abbreviated ML1 ROSA-lSl) mice with a parietal-cell-specific Cre line (ATP4B Cre; see ref (Syder et al, 2004)) generated parietal cell-specific ML1 overexpression (ML1 ROSA-lSl:ATP4B Cre or ML1PC) mice (Fig. 2H and S2B). We observed multifold increases in both basal and ML-SA-activated ML1 currents, relative to WT levels, in ML1PC parietal cells (Fig. 2K, 2L), consistent with increased transcription and protein expression of ML1 in the corpus glands and parietal cells of ML1PC mice (Fig. 2I, 2J, and S2C, S2D).
Notably, a high NIPR rate was observed in ML1PC parietal cells even under resting conditions without histamine (0.076 ± 0.010 ΔpH/min, n = 22; Fig. 2M, 2O). This constitutive NIPR, which was as large as histamine-stimulated NIPR in WT cells, was blocked completely by ML-SIs (Fig. 2N, 2O) or omeprazole, but not by histamine receptor antagonists (Fig. S2E). Plasma gastrin levels, a negative indicator of stomach acidity related to acid-hormone feedback inhibition (Trudeau and McGuigan, 1971), was elevated in ML1 KO mice (Fig. 2P). Conversely, plasma gastrin levels were lower in ML1PC mice and in WT mice that were administered with ML-SA5 via oral gavage than in controls (Fig. 2P). Taken together, these results suggest that the hyper-chlorohydric phenotype of ML1PC mice is caused by histamine-independent acid secretion induced by ML1 upregulation.
Localization of ML1 proteins in the TVs
We next investigated the subcellular localization of ML1 with dual-STED super-resolution imaging. Double immunohistochemistry analyses revealed that GCaMP3-ML1 (detected by anti-GFP antibodies) was co-localized partially with Lamp1 (Fig. S3A). However, ML1 immunoreactivity was observed mostly in Lamp1-negative compartments in the parietal cells (Fig. S3A). Indeed, both endogenous ML1 (detected by anti-ML1 antibodies) and GCaMP3-ML1 were co-localized mostly (60–80%) with α or β subunit of H+/K+-ATPase (Fig. 3A–C), which are TV markers (Courtois-Coutry et al, 1997). In contrast, Lamp1 and H+/K+-ATPase were rarely colocalized with each other (Fig. S3B). Moreover, the subcellular localization of GCaMP3-ML1 was confirmed by immuno-gold electron microscopy wherein gold particles were localized in the limited membranes of TV-like vesicles in ML1PC cells (Fig. 3D). These results suggest that within parietal cells, ML1 is present in H+/K+-ATPase-resident TVs as well as in Lamp1-resident late endosomes and lysosomes.
Fig. 3. TV-localized ML1 mediate Ca2+ release from TVs.
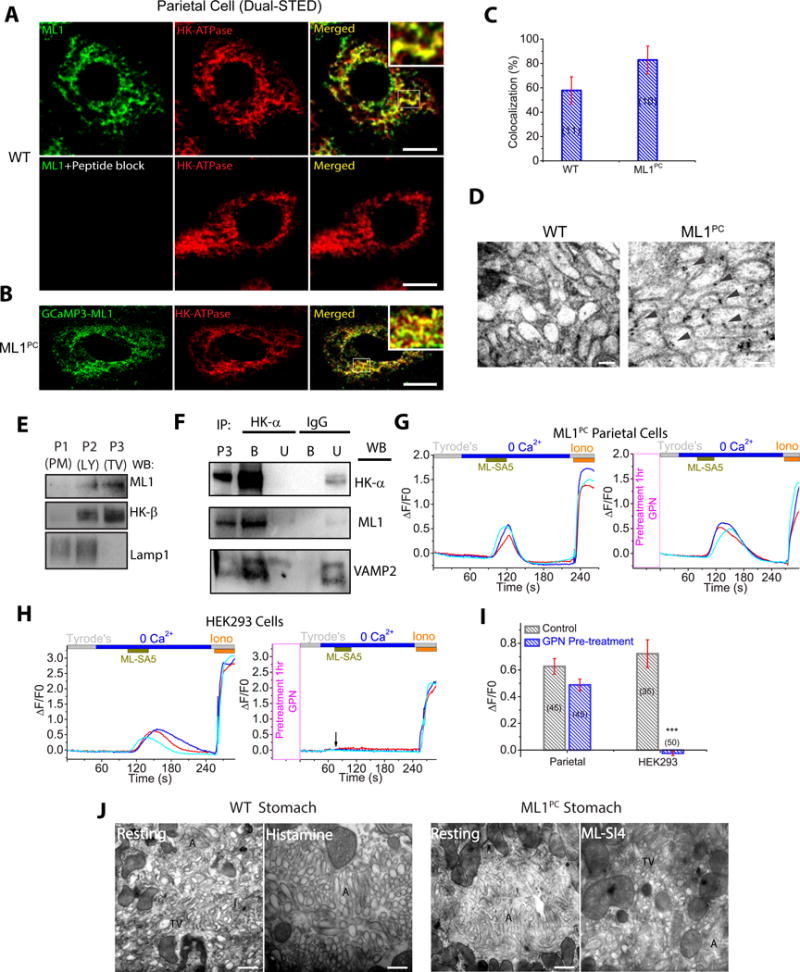
(A) Dual-STED images of WT corpus tissues immuno-labeled with anti-HK-α and anti-ML1. A pre-incubation of anti-ML1 with ML1 epitope peptide confirmed the specificity of the ML1 antibody. Scale bar, 5 μm. (B) Dual-STED images of ML1PC corpus tissues immuno-labeled with anti-HK-α and anti-GFP (recognizing GCaMP3-ML1). Note that constitutive TV exocytosis in ML1PC cells was blocked by ML-SI4 for this particular experiment. (C) Quantitative co-localization analysis of ML1 and H/K-ATPase based on randomly-selected images, as shown in A & B. (D) Immuno-gold electron microscopy images of WT and ML-SI4-treated (to prevent constitutive TV exocytosis) ML1PC parietal cells. Scale bar, 0.1 μm. (E) Gradient centrifugation purification of TVs from WT parietal cells. P1 (3,200 ×g), P2 (20,000 ×g), and P3 (100,000 ×g) pellets represent plasma membrane (PM)-rich, lysosome/mitochondria (LY), and H+/K+-ATPase-rich fractions (TV), respectively. (F) The expression of ML1, H+/K+-ATPase, and VAMP2 in TVs that were immunoisolated by anti-HK-α from P3 TV-derived membranes in E. IgG was used as a negative control. Western blot analyses were performed in both bound (B) immunoisolated vesicles and unbound (U) supernatants using P3 (see E) as a comparison. Shown were representative blots of three separate experiments. (G, H) Effects of GPN pretreatment on ML-SA5-induced Ca2+ release (GCaMP3 fluorescence, F480, in zero Ca2+ solution) from ML1PC parietal cells (G) or GCaMP3-ML1-transfected HEK293 cells (H). Cells were pretreated with ML-SI4 for 30 min to prevent TV exocytosis. (I) Quantitation of ML-SA5-induced GCaMP3 Ca2+ responses under control and GPN-pretreated conditions (mean ± SEM, n = 30–50 cells from ≤4 coverslips/experiment). ***P < 0.001 Student’s t-test. (J) TEM images of free TVs and apical canaliculi (A) in parietal cells. Scale bar, 0.5 μm.
In TV-derived membrane fractions isolated by cellular fractionation (Suda et al, 2011), which are enriched with H+/K+-ATPase but devoid of Lamp1, both endogenous ML1 and GCaMP3-ML1 were abundantly detected (Fig. 3E and S3C). When the TV-derived membranes were further immunoisolated by using anti-H+/K+-ATPase-α (Calhoun and Goldenring, 1997; Lapierre et al., 2007), the resulted high-purity TVs also contained abundant ML1 proteins (Fig. 3F). Consistently, co-immunoprecipitation analyses revealed that both endogenous and recombinant ML1 proteins were in the same protein complex with α or β subunits of H+/K+-ATPase (Fig. S3D, 3E). Collectively, these results suggest that ML1 is targeted predominantly to the TVs of parietal cells.
Activation of ML1 induces Ca2+ release from TVs
Like lysosomes, TVs are also intracellular Ca2+ stores (Tsunoda et al, 1988). In GCaMP3-ML1-expressing parietal cells isolated from ML1PC mice, application of ML-SA1 or ML-SA5 (1–10 μM) in a “zero” Ca2+ external solution (free [Ca2+] < 10 nM) evoked robust Ca2+ release evidenced by GCaMP3 fluorescence changes (Fig. S3F) in time-lapse confocal microscopy (ΔF/F0; Fig. 3G, 3I). ML-SA5-induced Ca2+ release was also observed in GCaMP3-ML1–transfected HEK293 cells (Fig. 3H, 3I). However, when cells were pretreated with GPN, a membrane-permeable dipeptide that depletes lysosome Ca2+ stores (Berg et al., 1994), ML-SA5-induced Ca2+ responses were abolished in HEK293 cells, but preserved in parietal cells (Fig. 3G–3I). Hence, unlike other cell types wherein ML1 is localized exclusively to lysosomes, in parietal cells, ML1 is expressed in GPN-inaccessible TVs. Taken together, these results establish TVs as intracellular Ca2+ stores in which ML1 is functionally present.
ML1 channel activity is necessary and sufficient for TV exocytosis
To investigate the role of ML1 in TV trafficking and exocytosis, we conducted immunofluorescence analysis on live glands, in which cells with fragmented vs. continuous phalloidin (actin) staining are considered to be in the “resting” and “stimulated” state, respectively (Zavros et al., 2008). In WT glands, the majority of parietal cells were in a “resting” state (Fig. S3G, 3H). Upon histamine stimulation, most parietal cells switched to a “stimulated” configuration (Fig. S3G, 3H). On the other hand, in ML1PC glands, the majority of parietal cells appeared to be “stimulated” even in the absence of histamine stimulation (see Fig. S3G, 3H).
To further investigate the role of ML1 in TV exocytosis, we employed transmission electron microscopy (TEM). Consistent with previous studies (Forte et al., 1977), numerous free TVs were observed throughout the cytosol of parietal cells in stomach sections from WT mice (Fig. 3J and S3I, 3K). Following IP histamine treatment, extensive canalicular invaginations were observed with minimal free TVs in WT specimens (Fig. 3J and S3I, 3K). Strikingly, similar canalicular structures were observed in resting ML1PC parietal cells, again with few free cytosolic TVs (Fig. 3J and S3J, 3K). Following administration of ML-SIs to ML1PC mice, relatively little canalicular extension was observed whereas abundant free TVs were seen throughout the cytosol of parietal cells (Fig. 3J and S3J, 3K). These results suggest that the ML1PC phenotype is associated with elevated TV exocytosis, and that ML1 blockage in ML1PC tissues leads to TV reformation/biogenesis (Forte and Zhu, 2010).
In cultured parietal cells, upon secretagogue stimulation, apical canalicular membranes are engulfed into the cell to form multiple actin-wrapped vacuoles known as vacuolar apical compartments (VACs), which remain separate from the basolateral membrane and free TVs in the cytosol (Nakada et al., 2012). Hence, total VAC membrane area provides a quantitative measurement of TV exocytosis (Nakada et al., 2012). In resting WT cells, small (diameter < 2 μm) VACs were observed occasionally with a total surface area < 20 μm2 (Fig. 4A, 4B). VACs formed within 10–20 min after bath application of histamine (Fig. 4A, 4B and Movie S1 and S2) and then fused together to generate one or a few large VACs (up to 8 μm in diameter; total VAC surface area > 50 μm2; Fig. 4A, 4B, S4A and S4B). Histamine-stimulated VAC formation was abolished by ML-SIs (Fig. 4A and 4B). In contrast, VAC formation induced by the acetylcholine receptor agonist carbachol was insensitive to ML-SIs (Fig. S4C, S4D). No histamine-induced VAC formation was observed in ML1 KO parietal cells (Fig. S4E, S4F). Hence, ML1 was found to be necessary for histamine-dependent TV exocytosis. On the other hand, ML-SA5 treatment was sufficient to induce VAC formation in WT cells (Fig. 4A, 4B and Movie S3). Furthermore, large VACs were observed in resting ML1PC parietal cells, but their presence was decreased sharply by ML-SI4 (Fig. 4C, 4D and S4G), but not by histamine or muscarinic receptor blockers (Fig. S4H). Taken together, these results suggest that ML1 plays an essential role in TV exocytosis.
Fig. 4. ML1 is necessary and sufficient to trigger TV exocytosis.

(A) Anti-HK-α immunocytochemistry with phalloidin-labeled F-actin. Cells were treated with ML-SA5 (10 μM) or histamine (50 μM+ 10 μM IBMX) in the presence or absence of ML-SI3 (20 μM) or ML-SI4 (10 μM) for 30 min at 37 °C. Scale bar, 10 μm. Blue and white arrows indicate basolateral and apical membranes, respectively. (B) VAC total surface area, an index of TV exocytosis associated with gastric acid secretion, under various treatment conditions (average of 3–6 z-plane sections/cell). (C) ML-SI4 (10 μM) effects on constitutive VAC formation in ML1PC parietal cells. Scale bar, 10 μm. (D) ML-SI effects on VAC total surface area. Bar graphs (B, D) show means ± SEM from ≥3 experiments. ***P < 0.001 one-way ANOVA, Bonferroni’s post-hoc analysis. (E) Summary of the percentages of large TVs as determined from the G2 gate (> 1 μm) of FSC/SSC dot plots. Mean ± SEMs from 4 experiments were shown. *P < 0.05, **P < 0.01; Student’s t-test.
ML1 mediated Ca2+ release increases homotypic fusion of TVs in vitro
Tubulovesicles may fuse with each other upon secretagogue stimulation (Duman et al., 2002; Forte and Zhu, 2010). We investigated the roles of ML1 and Ca2+ in this process using isolated TVs with flow cytometry to determine TV size. Large-sized TVs (>1 μm), which presumably resulted from homotypic fusion of small ones, were increased by more than 20-fold upon addition of external Ca2+ to isolated TVs (Fig. 4E and S4I–S4N). Notably, application of ML-SA5 also increased the size and granularity of TVs, and this effect was inhibited by ML-SI4 (Fig. 4E and S4I–S4N). Hence, ML1 activation may readily increase membrane fusion between TVs.
ML1 promotes polarized trafficking of TVs to apical, but not basolateral, membranes
Consistent with the hypothesis that histamine induces trafficking and fusion of TVs towards apical canalicular membranes (Forte and Zhu, 2010), our immunofluorescence analysis of cultured parietal cells revealed that the α subunit of H+/K+-ATPase accumulated in VAC membranes, but was absent from basolateral membranes (Fig. 4A, 4C and S4G). In ML1PC cells, ML1 proteins also accumulated in VAC membranes, in addition to the presumed lysosomal and tubulovesicular localizations (Fig. 4C and S4G). To further probe TV trafficking, we developed a patch-clamp method to record directly from VAC membranes isolated from ML1PC cells or histamine-stimulated WT cells (whole-VAC recording; Fig. 5A). Native VAC membranes are comprised of both apical membranes and TV membranes (Nakada et al., 2012) (Fig. 5A). Conversely, the plasma membranes of cultured parietal cells consist exclusively of basolateral membranes, which can be subjected to whole-cell recording (Fig. 5A). In the whole-VAC patch configuration, inwardly rectifying ML1-like currents were observed in histamine-treated WT cells (basal IML1 = 33 ± 11 pA/pF, n = 7) and ML1PC cells (Fig. 5B, 5C, 5F), and these currents were potentiated by ML-SA5 (149 ± 45 pA/pF, n = 7; Fig. 5B, 5C, 5F). Substantial IML1 was detected even in VAC-cytosolic-side-out patches (Fig. S5). Whole-cell IML1 was not detected in histamine-stimulated WT cells (1 ± 0.5 pA/pF, n = 8) or ML1PC cells (Fig. 5D, 5E, 5F and S5). These results implicate ML1 in polarized trafficking and exocytosis of TVs towards apical membranes.
Fig. 5. ML1 promotes polarized TVs trafficking towards apical membranes.

(A) Patch-clamp recording from basolateral (standard whole-cell recording) or apical (lyse cell to expose VACs before whole-VAC recording) membranes. Note that the extracellular side of the apical membrane is facing the VAC lumen. (B, C) Representative whole-VAC (apical membrane) recordings of ML1-like currents in histamine-stimulated WT (B) and ML1PC (C) parietal cells. (D, E) Representative whole-cell (basolateral membrane) detection of IML1 in WT and ML1PC cells. (F) Group averages of data from experiments shown in panels B–E (n = 3–5 patches/condition). Bar graphs show means ± SEM from ≥3 experiments. *P < 0.05, **P < 0.01, ***P < 0.001 one-way ANOVA, Bonferroni’s post-hoc analysis.
Parietal cell-specific expression of ML1 restores gastric acid secretion in ML1 KO mice
To further investigate the role of ML1 in gastric acid secretion, ML1 KO (also carrying loxP alleles) were crossed with ML1PC mice, resulting in Cre-dependent expression of ML1 restricted to parietal cells (Fig. S6A–S6I). In these ML1 KO: ML1PC mice, whole-endolysosome ML1 currents, TV Ca2+ release, histamine-induced acid secretion, and normal plasma gastrin levels (Fig. 2P) were largely restored (NIPR = 0.075 ± 0.019 ΔpH/min, n = 13; Fig. S6B–S6H). Notably, ML1 KO: ML1PC mice had a relatively normal stomach size (Fig. S6I), suggesting that ML1 deficiency in parietal cells underlies the abnormal stomach anatomy seen in ML1 KO mice.
Histamine-induced TV exocytosis and acid secretion require cAMP/protein kinase A (PKA) and Ca2+ signaling
Histamine-dependent activation of parietal H2 receptors activates cAMP-dependent PKA to phosphorylate multiple target proteins required for TV exocytosis (Forte and Zhu, 2010). Consistent with previous studies (Forte and Zhu, 2010), robust NIPR was induced in WT parietal cells treated with the cAMP-elevating agent forskolin or the membrane-permeable cAMP analogue 8-Br-cAMP (Fig. 6A, 6B). Conversely, histamine-stimulated NIPR in WT parietal cells was blunted by the PKA inhibitor H89 (20 μM; Fig. 6D, 6E). Hence, cAMP/PKA signaling was found to be necessary for histamine-induced acid secretion and TV exocytosis in WT cells. On the other hand, ML-SA or ML1PC-induced NIPR was not affected by PKA inhibition (Fig. S6J and S6K). Likewise, PKA inhibitors blocked histamine-induced, but not ML-SA5-induced, VAC formation in cultured parietal cells (Fig. 6F, 6G and S6M). Treatment of WT parietal cells with a membrane-permeable Ca2+ chelator (BAPTA-AM) also blocked NIPR (Fig. 6C, 6E) as well as histamine-induced VAC formation (Fig. 6F, 6G). In contrast, removal of external Ca2+ did not affect NIPR (Fig. S6L). These results demonstrate that Ca2+ release from an intracellular store is required for histamine-induced acid secretion and TV exocytosis.
Fig. 6. Histamine evokes ML1-mediated TV Ca2+ release through cAMP/PKA signaling.
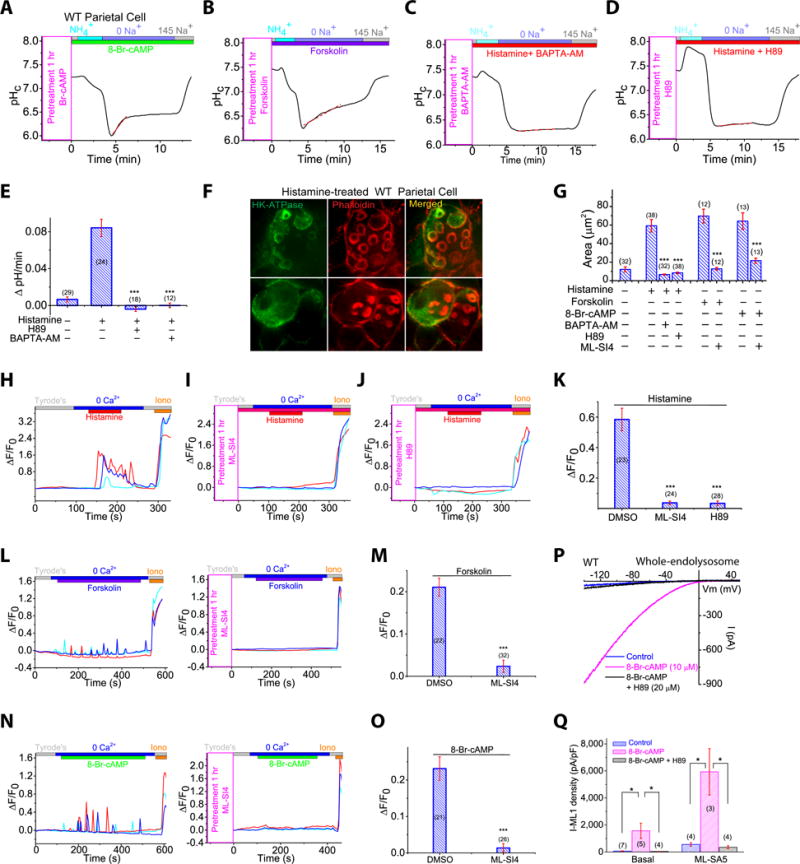
(A, B) Effects of cAMP (A) and forskolin (B) on NIPR in WT parietal cells. (CE) PKA inhibitor H89 (20 μM) (C) or Ca2+-chelator BAPTA-AM (20 μM) (D) block histamine-stimulated NIPR. Data were summarized in (E). (F) H89 and BAPTA-AM effects on VAC formation induced by histamine (50 μM + 10 μM IBMX). Scale bar, 10 μm. (G) Forskolin (50 μM) or 8-Br-cAMP (20 μM) stimulate VAC formation, measured as total surface area. (H–K) Histamine (50 μM) induced TV Ca2+ release in GCaMP3-ML1-expressing ML1PC cells (See also Movie S4), was blocked by ML-SI4 (10 μM) (I) or H89 (20 μM) (J); experiment averages in (K). (L, M) ML-SI4 (10 μM) blocked forskolin (50 μM)-induced Ca2+ oscillations in ML1PC parietal cells (see also Movie S5). (N and O) ML-SI4 (10 μM) blocked 8-Br-cAMP (20 μM)-induced Ca2+ oscillations in ML1PC cells (See also Movie S6). (P) Whole-endolysosomal IML1 in WT parietal cells with and without 8-Br-cAMP (20 μM) and H89 (20 μM). (Q) Mean current densities under treatments in P. Mean ± SEMs from ≥3 experiments are shown. *P < 0.05, **P < 0.01, ***P < 0.001 one-way ANOVA, Bonferroni’s post-hoc analysis (E) or Student’s t-test (K, M, O, Q).
ML1 mediates histamine-induced PKA-dependent Ca2+ release from TV stores
The Ca2+- and cAMP/PKA-dependence of TV exocytosis and acid secretion suggests that ML1, as a TV Ca2+ release channel, is regulated, directly or indirectly, by cAMP/PKA signaling. Indeed, 8-Br-cAMP and forskolin-induced TV exocytosis, as measured by VAC formation, was inhibited in the presence of ML-SI4 (Fig. 6G and S6N). To test the hypothesis that histamine-initiated signaling promotes TV Ca2+ release through ML1 to facilitate gastric acid secretion (Fig. 7), we measured Ca2+ release in GCaMP3-ML1-expressing ML1PC cells using time-lapse confocal imaging at 37 °C. Bath application of histamine produced robust Ca2+ release, as well as small Ca2+ oscillations (Fig. 6H, 6K and Movie S4). GPN pretreatment did not affect the responses (Fig. 6O), suggesting that they were mediated by TV stores, rather than lysosomal stores. At room temperature (22–24 °C), using conventional Fura-2-based Ca2+ imaging, we detected small Ca2+ increases in a subset of WT cells after histamine treatment (Fig. S6P). Much larger Fura-2 Ca2+ increases were seen consistently in ML1PC cells (Fig. S6Q).
Fig. 7. Signaling pathways that mediates histamine-stimulated ML1-dependent TV exocytosis and acid secretion.
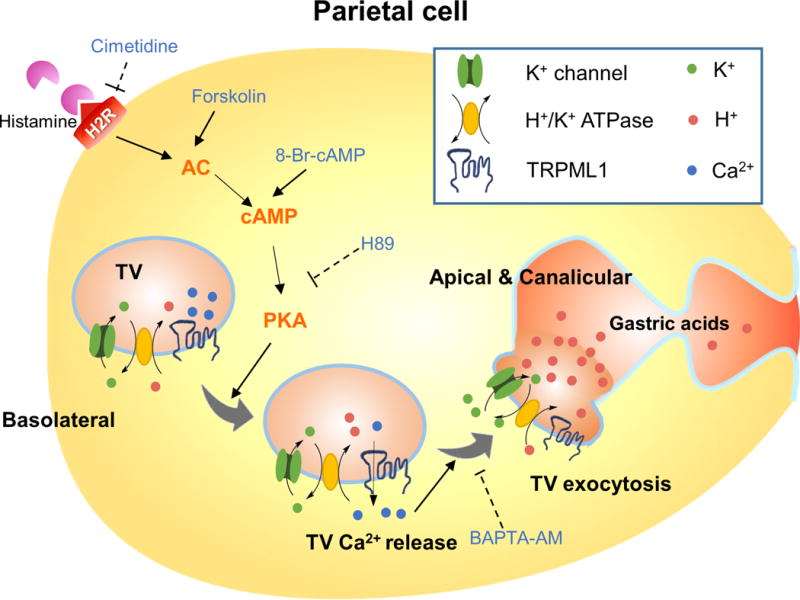
Activation of type 2 histamine receptor (H2Rs) in parietal cells by histamine induces cAMP-dependent PKA activation. Localized on TVs, ML1 may mediate a PKA-sensitive Ca2+ release pathway to trigger Ca2+-dependent fusion of TVs with each other and with apical membranes. Histamine-induced exocytosis of H+/K+-ATPase-enriched TVs toward lumen-facing apical membranes in a cAMP/PKA-dependent manner.
Histamine-induced TV Ca2+ release in ML1PC parietal cells was blocked by ML-SI4 or PKA inhibition (Fig. 6I–6K). Interestingly, forskolin (Movie S5) and 8-Br-cAMP (Movie S6) induced spontaneous Ca2+ oscillations in a zero-Ca2+ external solution, and these spontaneous oscillations were largely diminished in the presence of ML-SI4 (10 μM; Fig. 6L–6O). Together, these results suggest that histamine induces ML1-mediated TV Ca2+ release through cAMP and PKA signaling.
Reactive oxygen species and phosphatidylinositol 3,5-bisphosphate are endogenous agonists of ML1 (Dong et al., 2010; Zhang et al., 2016). Treatment with the antioxidant NAC or the PIKfyve inhibitor apilimod (to attenuate phosphatidylinositol 3,5-bisphosphate production) did not affect histamine-induced VAC formation in WT parietal cells (Fig. S6T and S6U). In contrast, in WT cells that were pretreated with 8-Br-cAMP or histamine, basal and ML-SA-activated whole-endolysosome ML1 currents (IML1) were increased significantly (Fig. 6P, 6Q and S6R, S6S). These increases were not seen in the presence of PKA inhibitors (Fig. 6P, 6Q). These results indicate that IML1 in partial cells is upregulated potently by PKA signaling.
Discussion
In the present study, we used pharmacological and genetic manipulation methods to discern missing links between histamine and acid secretion in gastric parietal cells. Our results indicate that ML1 channels expressed on parietal-cell TVs provide a PKA-sensitive Ca2+ release conduit that triggers Ca2+-dependent fusion of TVs with each other and with apically-directed canalicular membranes, resulting in apical localization of the H+/K+-ATPase that pumps H+ into the gastric lumen (Fig. 7). The putative Ca2+-dependence of TV fusion had been controversial because conventional Ca2+ imaging studies had not shown consistent increases in response to histamine treatment, as was demonstrated finally in the current study. Using organelle-targeted Ca2+ probes and super-resolution imaging, we showed unequivocally here that TV trafficking and exocytosis are dependent on Ca2+ release from TVs mediated by ML1 proteins, which are in turn regulated by histamine-stimulated PKA signaling. Our demonstration of ML1 expression in TVs has provided an answer to a long-standing puzzle as to why the achlorhydric phenotype is only seen in ML-IV, but not any other LSDs (Chandra et al., 2011; Xu and Ren, 2015).
It is likely that additional regulators of this ML1 pathway remain to be discovered. Indeed, a number of TV-localized channels and transporters, including K+ channels, have been reported to regulate acid secretion (Forte and Zhu, 2010; Lambrecht et al., 2005). Some of them may affect TV membrane potential and thus ML1-mediated Ca2+ release, which is strongly voltage-dependent (Xu and Ren, 2015). The Ca2+ effector associated with TV fusion is not yet known. However, Ca2+-sensing proteins, such as apoptosis-linked gene 2 and Syt-VII proteins, have been shown to link ML1-mediated lysosomal Ca2+ release with retrograde transport and membrane fusion of lysosomes (Li et al., 2016), making them candidates for this role in TVs. The pathway elucidated in the current study provides rapid, bidirectional regulation of gastric acid secretion. Given the well-established roles of gastric acids in stomach homeostasis and gastric biology (Forte and Zhu, 2010; Hersey and Sachs, 1995), our identification of ML1 as the first known TV Ca2+ channel may lead to the development of new therapeutic approaches to treat acid-related gastric diseases.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Haoxing Xu (haoxingx@umich.edu).
Experimental Model and Subject Details
Mice
ML1 KO mice were developed in a B6:129 mixed genetic background and their phenotype has been characterized as described elsewhere (Venugopal et al., 2007). The GCaMP3-ML1 ROSA-lSl mice were generated in a C57BL/6J genetic background, as described in Fig. S2A and S2B and crossed with an ATP4B Cre line (Syder et al., 2004) to generate the ML1 ROSA-lSl:ATP4B Cre (ML1PC) mice, in which the GCaMP3-ML1 transgene is selectively expressed in parietal cells. Prior to most experiments, mice were fasted overnight with free access to water. Both sexes of littermate or age-matched mice were used under an approved animal protocol (#6577) following the Institutional Animal Care Guidelines at the University of Michigan.
Gastric gland isolation
Gastric glands were isolated as described previously (Pasham et al., 2013). Briefly, mice were fasted overnight with free access to tap water. After the animals were euthanized, their stomachs were isolated and sliced longitudinally to separate the forestomach, antrum, and corpus regions. The corpus tissues were sliced into small sections and digested in collagenase at 37 °C for 1 h. The digested samples were then filtered through 40 μm filters and pelleted by light centrifugation at the speed of 50 ×g for 8 min. Isolated gastric glands were plated on Matigel (BD Biosciences)-coated cover slips and incubated at 37 °C in a culture medium (Medium A, pH 7.4) containing: DMEM, 20 mM HEPES, 1 mM glutamine supplement, 10 mM glucose, 0.2% BSA, 50 ng/ml EGF, 5% selenite-insulin-transferrin liquid medium, 50 U/ml penicillin/streptomycin, 200 μg/ml gentamycin, and 50 μg/ml novobiacin.
Primary parietal cell isolation and culture
After mucosal digestion of isolated glands, supernatants were pelleted by centrifugation at 200 ×g, washed three times with HEPES-MEM, and re-suspended in Medium A. Approximately 70% of the total gastric cells suspended in Medium A were parietal cells. The cells were plated onto Matrigel-coated 18-mm round coverslips or 35-mm dishes and incubated at 37 °C.
Method Details
NIPR measurement
In isolated glands and cultured parietal cells, NIPR (see Fig. S1O) was measured with a pH-sensitive BCECF dye (Pasham et al., 2013). Briefly, glands and cells were loaded with 10 μM BCECF-AM (Thermo-Fisher, USA) for 15 min at 37 °C. Acid loading was achieved by a brief application of NH4Cl (20 mM). NIPR was triggered upon Na+ removal (0 Na+). A linear fit, within 5 min of reaching peak acidity, was used to determine the rate of pH recovery (ΔpH/min) during re-alkalization. BCECF fluorescence was recorded by an EasyRatio Pro system (PTI) at 440-nm and 490-nm wavelengths. The fluorescence ratio (F490/F440) was used to calculate cytoplasmic pH values based on a standard calibration curve constructed for a range of pH 5 to 8.5 with K+-H+ ionophore nigericin (Pasham et al, 2013). The standard 145 Na+ solution contained (in mM): 125 NaCl, 3 KCl, 1 CaCl2, 1.2 MgSO4, 5 glucose, and 32.2 HEPES (pH 7.4). The 0 Na+ solution contained (in mM): 125 NMDG-Cl, 3 KCl, 1 CaCl2, 1.2 MgSO4, 5 glucose, 32.2 HEPES (pH 7.4). For the NH4Cl application, 10 mM NMDG-Cl was replaced with 20 mM NH4Cl. The high K+ solution used for calibration contained (in mM): 105 KCl, 1 CaCl2, 1.2 MgSO4, 32.2 HEPES, 10 mannitol, and 10 μg/ml nigericin (pH 7.0).
[14C] aminopyrine uptake assay
Aminopyrine accumulates in the acidic lumen of isolated glands (Mettler et al, 2007). Briefly, 1-ml gland samples in HEPES-MEM medium (saturated with 100% oxygen) were mixed with various pharmacological regents and 10 μl [14C] -aminopyrine (115 mCi/mmol; 5 × 103 cpm) in test tubes. The samples were incubated in a shaking water bath at 37 °C for 30 min. Pelleted samples were dried out overnight and then dissolved in 125 μl NaOH (1 M) at 80 °C for 20 min. Afterwards, 125 μl HCl (1 M) was added to neutralize the sample. Aliquots (200 μl) of suspended pellets were mixed with scintillation fluid (500 μl) for analysis in a Beckman LS3801 counter. The remaining sample (50 μl) was used to determine the protein content for normalization.
Whole stomach acid measurement
Overnight-fasted mice were anesthetized with a mixture of xylazine and ketamine. A non-survival surgery was then performed. Briefly, 1–2 cm midline laparotomy incision was made on the abdomen. A small incision was made in the duodenum through which PE-50 tubing was inserted and secured with a ligature around the pylorus. At the beginning of each experiment, each stomach was rinsed several times with normal saline solution and then filled with 400 μl of the same saline solution via the tubing. Saline (400 μl) was injected through the fistula and collected every 10 min. Histamine (1 mg/kg, Sigma) was administered via IP injection. ML-SAs and ML-SIs were administered into the stomach via the PE tubing. Acid output (in μmol H+) was determined by titration.
Gastrin measurement
Animals were fasted overnight with free access to tap water. About 150 μl of blood specimens were collected into EDTA-coated tubes. ML-SA5 (100 μM) was administered by oral gavage and histamine (1 mg/kg) was administered intraperitoneally. Total plasma gastrin concentrations were determined with a gastrin enzyme-linked immunoassay kit (Raybiotech, USA) according to manufacturer’s instruction.
Transmission Electron Microscopy
Mouse tissue sections were immersed in a mixture of 2.5% glutaraldehyde and 4% formaldehyde in phosphate-buffered saline (PBS, pH 7.2) for 4 h at 4 °C. The samples were postfixed in 1% osmium tetroxide for 1 h, followed by 0.1 M cacodylate buffer for 1 h at room temperature. The fixed samples were stained with 2% uranyl acetate in maleate buffer for 1 h in the dark. The samples were dehydrated with ethanol and embedded with Epon (Electron Microscopy Sciences). After the polymerization of Epon, ultrathin sections (60–80 nm) were prepared with an LKB MK III Ultratome. Samples were stained with 2% uranyl, contrasted with lead citrate, and examined with a Philips CM10 transmission electron microscope. An anti-GFP antibody from Invitrogen (Cat No: A-6455) was used for immuoelectron microscopic analysis ((Jabs et al., 2005)).
Immuno-electron microscopy
Ultrathin sections (60–80 nm) were etched in 10% H2O2 and 5% sodium metaperiodate for 10 min. After several washes and blocking with 1% BSA in PBS, the sections were incubated overnight with rabbit anti-GFP antibody (Invitrogen (Cat No: A-6455)) at 4 °C. After several washes, the sections were then incubated with anti-rabbit IgG secondary antibody conjugated with 10-nm colloidal gold particles (Sigma, USA) for 2–3 h at room temperature. The sections were washed and contrasted with uranyl acetate and then examined with a Philips CM10 and Joel 1400 plus transmission electron microscope.
Biochemistry
Standard western blotting analyses were performed (Zhang et al, 2016). The lysis buffer contained 1% NP-40, 0.25% Na-deoxycholate, 1 mM NaF, 150 mM NaCl, 1 mM Na3VO4, 0.5 mM CaCl2, and 50 mM Tris-HCl (pH 7.4). For the immunoprecipitation experiments, after a 10-s sonication, whole-cell lysates were centrifuged at 14,000 ×g for 10 min. Supernatants were incubated with primary antibodies at 4 °C for 1 h, followed by overnight incubation with Protein A/G plus-agarose. Western blot analyses were performed with rabbit anti-GFP (1:200, Life technologies, USA), anti-HK-α, and anti-HK-β (1: 1,000) antibodies.
Immunohistochemistry
Mouse stomachs were sliced longitudinally from the antrum to the forestomach. Tissues were fixed in 4% paraformaldehyde in PBS, from which paraffin-embedded 5-μm-thick sections were prepared. The tissue sections were permeabilized with 0.1% Triton X-100 plus 1% BSA for 10 min, followed by blocking with 1% BSA for 1 h. The sections were incubated with various antibodies, including mouse anti-HK-α or β (1: 1,000; MBL international) and mouse anti-Lamp-1 (1:200; DSHB). The sections were then incubated with anti-mouse IgG secondary antibodies conjugated with Alexa Flour 488, 546, or 647 (Life Technology) for 2 h. F-actin rich apical vacuole immunolabeling was performed with phalloidin-TRITC (1:1000; ECM Bioscience); ML1PC parietal cells were labeled with anti-GFP antibodies (1:200; Life technologies, USA). The sections were mounted with Fluoromount-G (Southern Biotechnology Associates, Inc.). Whole-gland immunostaining was performed using freshly isolated glands from overnight starved WT and ML1PC mice (Zavros et al, 2008). Drug-treated glands were allowed to settle down by gravity, and then fixed using 4% PFA in PBS. The glands were permeabilized with 0.3% Triton X-100 plus 1% BSA for 10 min prior to immunostaining procedures. All representative images represent experiments repeated at least 3 times each. Quantification were performed on randomly selected images.
Cellular fractionation
Subcellular membrane fractions were obtained from corpus glands as described previously (He et al., 2011; Wang et al., 2012). Briefly, tissues were homogenized in PBS supplemented with 10 mM sucrose and 1 mM EDTA (pH 7.2) in a Potter-Elvehjem homogenizer. The homogenates were then subjected to sequential centrifugation steps to produce pellets 1–3 (P1–3): P1 (3,200 ×g for 10 min, the plasma membrane fraction), P2 (20,000 ×g for 10 min, the lysosome fraction), and P3 (100,000 ×g for 1 h, the light microsomal membrane fraction consisting mostly TV-derived membranes).
Immunoisolation of TVs
The P3 pellets were re-suspended in a sucrose buffer that contained 300 mM sucrose (10 mM HEPES, pH 7.4), and fractionated using 20%, 27% and 33% of sucrose gradients (Calhoun and Goldenring, 1997; Lapierre et al., 2007). Enriched TVs, solubilized in 500 μl of PBS containing protease and phosphatase inhibitors, were then pulled down by protein G beads coated with an anti-H+-K+-ATPase-α antibody and IgG at 4°C for 1 h.
Confocal imaging
Time-lapse imaging was conducted on an Olympus spinning-disk confocal microscope equipped with a temperature controller. To detect Ca2+ release from lysosomes and TVs, we used GCaMP3-ML1-expressing ML1PC parietal cells. GCaMP3, a single-wavelength genetically-encoded Ca2+ indicator was engineered using GFP and calmodulin (Tian et al., 2009), was fused directly to the cytoplasmic N-terminus of ML1 (Fig. S3F; detailed characterization see ref. (Shen et al., 2012)). Upon Ca2+ release from TV stores, GCaMP3-ML1 fluorescence increased, which was monitored at an excitation wavelength of 480 nm (F480) by an EasyRatio Pro imaging system (PTI). Cells were bathed in Tyrode’s solution containing 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM Glucose, and 20 mM HEPES (pH 7.4). TV Ca2+ release was monitored in a zero Ca2+ solution containing 145 mM NaCl, 5 mM KCl, 3 mM MgCl2, 10 mM glucose, 1 mM EGTA, and 20 mM HEPES (pH 7.4). Free Ca2+ concentration was estimated by Maxchelator software (http://maxchelator.stanford.edu/). Images were analyzed with MetaMorph Advanced Imaging acquisition software v.7.7.8.0 (Molecular Devices) and Image J (NIH).
STED microscopy
STED microscopy was performed using the STED module of a Leica TCS SP8 STED 100X microscope (Leica Microsystems) on paraffin-embedded tissue sections. Hybrid detectors were used to detect signals at a certain time gate after a laser excitation pulse with a pixel size of ∼20 nm. The images were further de-convolved with the Huygens Professional Software (Scientific Volume Imaging).
Fura-2 Ca2+ imaging
Cultured parietal cells were loaded with Fura-2-AM (3 μM) in the culture medium at 37 °C for 60 min. Fluorescence was recorded by an EasyRatio Pro system (PTI) at two different wavelengths (340 nm and 380 nm) and the Fura-2 ratio (F340 /F380) was used to determine changes in intracellular [Ca2+].
Whole-endolysosome electrophysiology
Isolated enlarged endolysosomes were subjected to whole-endolysosomal electrophysiology by a modified patch-clamp method (Dong et al., 2010; Wang et al., 2012). Briefly, cells were treated with 1 μM vacuolin-1 overnight to increase selectively the size of late endosomes and lysosomes (Cerny et al., 2004). Enlarged vacuoles were released into the dish by mechanical disruption of the cell membrane with a finetip glass electrode. Unless otherwise indicated, vacuoles were bathed continuously in an internal (cytoplasmic) solution containing 140 mM K+-gluconate, 4 mM NaCl, 1 mM EGTA, 2 mM Na2-ATP, 2 mM MgCl2, 0.39 mM CaCl2, 0.1 mM GTP, and 10 mM HEPES (pH adjusted to 7.2 with KOH; free [Ca 2+]i ∼ 100 nM calculated by MaxChelator software). The pipette (luminal) solution contained 145 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM MES, and 10 mM glucose (pH adjusted to 4.6 with NaOH). The whole-endolysosome configuration was achieved as described previously (Wang et al., 2012). After formation of a gigaseal between the patch pipette and an enlarged endolysosome, voltage steps of several hundred millivolts with a millisecond duration were applied to break into the vacuolar membrane (Wang et al., 2012). All bath solutions were applied via a fast perfusion system that produced a complete solution exchange within a few seconds. Data were collected via an Axopatch 2A patch clamp amplifier, Digidata 1440, and processed in pClamp 10.0 software (Axon Instruments). Whole-endolysosome currents were digitized at 10 kHz and filtered at 2 kHz. All experiments were conducted at room temperature (21–23 °C) and all recordings were analyzed in pCLAMP10 (Axon Instruments) and Origin 8.0 (OriginLab).
Whole-VAC electrophysiology
Whole-VAC patch-clamp was performed on apical vacuoles isolated from parietal cells, as illustrated in Fig. 5A. Briefly, cultured parietal cells were treated with 50–100 μM histamine + 20 μM IBMX for 30–40 min to induce VAC formation in WT parietal cells. ML1PC parietal cells contained large VACs. Large VACs were released into the dish by mechanical disruption of the cell membrane with a fine-tip glass electrode. Subsequently, whole-VAC and cytoplasmic-side-out recordings were performed in a manner similar to that of endolysosomal recordings (Wang et al., 2012).
VAC surface area measurement
The apical membrane vacuole surface area during resting and stimulated states was measured for all VACs that were visible in multiple (3–6) Z-cross sections. Data were analyzed in ImageJ (NIH). Quantifications of were done by researchers blind to the experimental groups.
Flow-cytometry-based TV fusion assay
This was performed using enriched light microsomal TV fractions (P3) as prepared from ML1PC mice parietal cells as described above. P3 fractions were re-suspended in the fusion buffer (140 mM KCl, 20 mM HEPES, 1 mM MgCl2, pH 7.4) with the addition of 50 μM EGTA (free [Ca2+] is estimated to be 20–50 nM). The sizes of TVs were determined in a Flow Cytometer (Synergy, iCyt). Using a side-scatter (SSC) threshold of 300 arbitrary units the lower sensitivity of the instrument was established and the SSC and forward scatter (FSC) voltages were set. A (G2) gate was set to include TVs of 1–2 μm in diameter, and a threshold gate (G1) was set based on a densely-populated region. The data were presented as the percentages of the changes in the FSC-SSC area.
Quantification and Statistical Analysis
A minimum of 3 independent repeats were performed for each experiment. No statistical methods were used to predetermine sample size. Data are presented as means ± standard errors of the mean (SEMs). Statistical comparisons were performed with one-way analyses of variance (ANOVAs) with Bonferroni’s post-hoc analysis or Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001). P values < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
ML1 antagonists inhibit gastric acid secretion in parietal cells.
Transgenic expression of ML1 in parietal cells causes constitutive acid secretion.
ML1 is a Ca2+ release channel in the tubulovesicle.
Histamine-PKA signaling activates ML1-dependent exocytosis of tubulovesicles.
Acknowledgments
This work was supported by NIH project grants (R01-NS062792 and R01-AR060837 to H.X; P01-DK06041 to J.M. and L.C.S.) and Molecular Biology Core support from the Michigan Gastrointestinal Research Center (P30-DK34933). We are grateful to Dr. Loren Looger (Janelia Farm HHMI) for the GCaMP3 construct, Dr. Susan Slaugenhaupt (Massachusetts General Hospital) for the ML1 KO mice, Dr. Andrea Todesco for providing the ATP4B-Cre mice, Dr. Isabel Martinez for the help on in vivo experiments, Arthur Tessier for the help on C14-aminipyrine experiment, Ms. Felichi Arines for the assistance, and Dr. Richard Hume for comments on the manuscript. We appreciate the encouragement and helpful comments from other members of the Xu laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions:
N.S. and H.X. designed the study; N.S., M.G., X.Z., J.Y., M.B., T.K., and S.S. performed the laboratory experiments; R.C., Q.G., W.W., S.P., X.H., M.F., G.K., S.S., M.S., L.S., J.M., and J. L. M. contributed the reagents; N.S., M.G., X.Z., N.R., L.S., J. L. M., X.Z and H.X. analyzed and interpreted the data; N.S. and H.X. wrote the paper with inputs and final approval from all authors.
References
- Berg TO, Stromhaug E, Lovdal T, Seglen O, Berg T. Use of glycyl-L-phenylalanine 2-naphthylamide, a lysosome-disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. The Biochemical journal. 1994;300(Pt 1):229–236. doi: 10.1042/bj3000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun BC, Goldenring JR. Two Rab proteins, vesicle-associated membrane protein 2 (VAMP-2) and secretory carrier membrane proteins (SCAMPs), are present on immunoisolated parietal cell tubulovesicles. The Biochemical journal. 1997;325(Pt 2):559–564. doi: 10.1042/bj3250559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004;5:883–888. doi: 10.1038/sj.embor.7400243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra M, Zhou H, Li Q, Muallem S, Hofmann SL, Soyombo AA. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology. 2011;140:857–867. doi: 10.1053/j.gastro.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew CS, Brown MR. Release of intracellular Ca2+ and elevation of inositol trisphosphate by secretagogues in parietal and chief cells isolated from rabbit gastric mucosa. Biochimica et biophysica acta. 1986;888:116–125. doi: 10.1016/0167-4889(86)90077-7. [DOI] [PubMed] [Google Scholar]
- Chew CS, Hersey SJ, Sachs G, Berglindh T. Histamine responsiveness of isolated gastric glands. The American journal of physiology. 1980;238:G312–320. doi: 10.1152/ajpgi.1980.238.4.G312. [DOI] [PubMed] [Google Scholar]
- Courtois-Coutry N, Roush D, Rajendran V, McCarthy JB, Geibel J, Kashgarian M, Caplan MJ. A tyrosine-based signal targets H/K-ATPase to a regulated compartment and is required for the cessation of gastric acid secretion. Cell. 1997;90:501–510. doi: 10.1016/s0092-8674(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, et al. PI (3, 5) P (2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nature communications. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman JG, Singh G, Lee GY, Machen TE, Forte JG. Ca(2+) and Mg(2+)/ATP independently trigger homotypic membrane fusion in gastric secretory membranes. Traffic. 2002;3:203–217. doi: 10.1034/j.1600-0854.2002.030306.x. [DOI] [PubMed] [Google Scholar]
- Forte JG, Zhu L. Apical recycling of the gastric parietal cell H, K-ATPase. Annual review of physiology. 2010;72:273–296. doi: 10.1146/annurev-physiol-021909-135744. [DOI] [PubMed] [Google Scholar]
- Forte TM, Machen TE, Forte JG. Ultrastructural changes in oxyntic cells associated with secretory function: a membrane-recycling hypothesis. Gastroenterology. 1977;73:941–955. [PubMed] [Google Scholar]
- He W, Liu W, Chew CS, Baker SS, Baker RD, Forte JG, Zhu L. Acid secretion-associated translocation of KCNJ15 in gastric parietal cells. American journal of physiology Gastrointestinal and liver physiology. 2011;301:G591–600. doi: 10.1152/ajpgi.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersey SJ, Sachs G. Gastric acid secretion. Physiological reviews. 1995;75:155–189. doi: 10.1152/physrev.1995.75.1.155. [DOI] [PubMed] [Google Scholar]
- Jabs R, Pivneva T, Huttmann K, Wyczynski A, Nolte C, Kettenmann H, Steinhauser C. Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. Journal of cell science. 2005;118:3791–3803. doi: 10.1242/jcs.02515. [DOI] [PubMed] [Google Scholar]
- Lambrecht NW, Yakubov I, Scott D, Sachs G. Identification of the K efflux channel coupled to the gastric H-K-ATPase during acid secretion. Physiological genomics. 2005;21:81–91. doi: 10.1152/physiolgenomics.00212.2004. [DOI] [PubMed] [Google Scholar]
- Lapierre LA, Avant KM, Caldwell CM, Ham AJ, Hill S, Williams JA, Smolka AJ, Goldenring JR. Characterization of immunoisolated human gastric parietal cells tubulovesicles: identification of regulators of apical recycling. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1249–1262. doi: 10.1152/ajpgi.00505.2006. [DOI] [PubMed] [Google Scholar]
- Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, Gao Q, Cheng X, Xu H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nature cell biology. 2016;18:404–417. doi: 10.1038/ncb3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinowska DH, Sachs G, Cuppoletti J. Gastric H+ secretion: histamine (cAMP-mediated) activation of protein phosphorylation. Biochimica et biophysica acta. 1988;972:95–109. doi: 10.1016/0167-4889(88)90106-1. [DOI] [PubMed] [Google Scholar]
- Mettler SE, Ghayouri S, Christensen GP, Forte JG. Modulatory role of phosphoinositide 3-kinase in gastric acid secretion. American journal of physiology Gastrointestinal and liver physiology. 2007;293:G532–543. doi: 10.1152/ajpgi.00138.2007. [DOI] [PubMed] [Google Scholar]
- Nakada SL, Crothers JM, Jr, Machen TE, Forte JG. Apical vacuole formation by gastric parietal cells in primary culture: effect of low extracellular Ca2+ American journal of physiology Cell physiology. 2012;303:C1301–1311. doi: 10.1152/ajpcell.00244.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu PA, Reenstra WW, Machen TE. Intracellular Ca requirements for stimulus-secretion coupling in parietal cell. The American journal of physiology. 1989;256:C241–251. doi: 10.1152/ajpcell.1989.256.2.C241. [DOI] [PubMed] [Google Scholar]
- Pasham V, Rotte A, Mia S, Alesutan I, Chatterjee S, Hosseinzadeh Z, Bhandaru M, Noegel AA, Lang F. Annexin 7 in the regulation of gastric acid secretion. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;32:1643–1654. doi: 10.1159/000356600. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Dwyer NK, Lubensky IA, Tsokos M, Sutliff VE, Latimer JS, Frei KP, Brady RO, Barton NW, Blanchette-Mackie EJ, et al. Constitutive achlorhydria in mucolipidosis type IV. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1207–1212. doi: 10.1073/pnas.95.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nature communications. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JM, Munson K, Vagin O, Sachs G. The gastric HK-ATPase: structure, function, and inhibition. Pflugers Archiv : European journal of physiology. 2009;457:609–622. doi: 10.1007/s00424-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda J, Zhu L, Okamoto CT, Karvar S. Rab27b localizes to the tubulovesicle membranes of gastric parietal cells and regulates acid secretion. Gastroenterology. 2011;140:868–878. doi: 10.1053/j.gastro.2010.09.044. [DOI] [PubMed] [Google Scholar]
- Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V, Gordon JI. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4471–4476. doi: 10.1073/pnas.0307983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn P, Zorec R, Rettig J, Keating DJ. Exocytosis in non-neuronal cells. Journal of neurochemistry. 2016 doi: 10.1111/jnc.13602. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nature methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau WL, McGuigan JE. Relations between serum gastrin levels and rates of gastric hydrochloric acid secretion. The New England journal of medicine. 1971;284:408–412. doi: 10.1056/NEJM197102252840803. [DOI] [PubMed] [Google Scholar]
- Tsunoda Y, Takeda H, Otaki T, Asaka M, Nakagaki I, Sasaki S. Intracellular Ca2+ shift and signal transduction from the tubulovesicular portion of gastric parietal cells during gastrin stimulation or Ca2+ ionophore treatment: comparison between luminescent and fluorescent probes, and electron probe X-ray microanalyzer. Biochemistry and cell biology = Biochimie et biologie cellulaire. 1988;66:279–287. doi: 10.1139/o88-037. [DOI] [PubMed] [Google Scholar]
- Venugopal B, Browning MF, Curcio-Morelli C, Varro A, Michaud N, Nanthakumar N, Walkley SU, Pickel J, Slaugenhaupt SA. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. American journal of human genetics. 2007;81:1070–1083. doi: 10.1086/521954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E1373–1381. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang X, Dong XP, Samie M, Li X, Cheng X, Goschka A, Shen D, Zhou Y, Harlow J, et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell. 2012;151:372–383. doi: 10.1016/j.cell.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Martinoia E, Szabo I. Organellar channels and transporters. Cell calcium. 2015;58:1–10. doi: 10.1016/j.ceca.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D. Lysosomal physiology. Annual review of physiology. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Forte JG. Cell biology of acid secretion by the parietal cell. Annual review of physiology. 2003;65:103–131. doi: 10.1146/annurev.physiol.65.072302.114200. [DOI] [PubMed] [Google Scholar]
- Zavros Y, Orr MA, Xiao C, Malinowska DH. Sonic hedgehog is associated with H+-K+-ATPase-containing membranes in gastric parietal cells and secreted with histamine stimulation. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G99–G111. doi: 10.1152/ajpgi.00389.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nature communications. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Hou N, Sun Y, Teng Y, Yang X. Atp4b promoter directs the expression of Cre recombinase in gastric parietal cells of transgenic mice. Journal of genetics and genomics = Yi chuan xue bao. 2010;37:647–652. doi: 10.1016/S1673-8527(09)60083-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.