

Abstract
Alcohol exposure during central nervous system (CNS) development can lead to fetal alcohol spectrum disorder (FASD). Human imaging studies have revealed significant white matter (WM) abnormalities linked to cognitive impairment in children with FASD, however the underlying mechanisms remain unknown. Here, we evaluated both the acute and long-term impacts of alcohol exposure on oligodendrocyte number and WM integrity in a third trimester-equivalent mouse model of FASD, in which mouse pups were exposed to alcohol during the first two weeks of postnatal development. Our results demonstrate a 58% decrease in the number of mature oligodendrocytes (OLs) and a 75% decrease in the number of proliferating oligodendrocyte progenitor cells (OPCs) within the corpus callosum of alcohol-exposed mice at postnatal day 16 (P16). Interestingly, neither mature OLs nor OPCs derived from the postnatal subventricular zone (SVZ) were numerically affected by alcohol exposure, indicating heterogeneity in susceptibility based on OL ontogenetic origin. Although mature OL and proliferating OPC numbers recovered by postnatal day 50 (P50), abnormalities in myelin protein expression and microstructure within the corpus callosum of alcohol-exposed subjects persisted, as assessed by western immunoblotting of myelin basic protein (MBP; decreased expression) and MRI diffusion tensor imaging (DTI; decreased fractional anisotropy). These results indicate that third trimester-equivalent alcohol exposure leads to an acute, albeit recoverable, decrease in OL lineage cell numbers, accompanied by enduring WM injury. Additionally, our finding of heterogeneity in alcohol susceptibility based on the developmental origin of OLs may have therapeutic implications in FASD and other disorders of WM development.
Keywords: Myelin Basic Protein, Diffusion Tensor Imaging, Corpus Callosum, Subventricular Zone, Oligodendrogenesis
Graphical Abstract
INTRODUCTION
Alcohol exposure during the development of the central nervous system (CNS) results in an array of behavioral and cognitive sequelae, described under fetal alcohol spectrum disorder (FASD) (Riley and McGee 2005). FASD represents a significant public health concern, as indicated in a recent report from the American Academy of Pediatrics estimating that as many as 2%-5% of first-grade students in the United States have the physical, behavioral, or learning impairments related to FASD (May et al. 2014; Williams et al. 2015). Despite this high prevalence, limited treatment options are available for affected individuals (Kodituwakku and Kodituwakku 2011).
White matter abnormalities have long been associated with prenatal ethanol (EtOH) exposure in humans, with notable alterations in the morphology of the corpus callosum ranging from severe agenesis to callosal thinning (Lebel et al. 2011; Norman et al. 2009). MRI techniques that utilize diffusion tensor imaging (DTI) have further revealed changes to the organization and microstructure of callosal white matter in individuals with FASD (Moore et al. 2014). Microstructural abnormalities as assessed by fractional anisotropy (FA) occur throughout the corpus callosum and are correlated with impaired cognition (Wozniak et al. 2009) and oculomotor control (Green et al. 2013; Paolozza et al. 2014). A recent MRI study relating improved cognitive function with increasing white matter volume in the corpus callosum of adolescent FASD subjects underscores the need to elucidate the cellular underpinnings of white matter injury and plasticity for improved therapeutic targeting (Gautam et al. 2014).
Oligodendrocytes (OLs) provide myelination and trophic support for maintenance of axonal integrity in CNS white matter (Funfschilling et al. 2012; Franklin and Gallo 2014) and may represent a target of EtOH toxicity (Guizzetti et al. 2014; Wilhelm and Guizzetti 2015). Genetic fate mapping studies in mice have demonstrated that OLs represent a heterogeneous population of cells with respect to developmental origin (Kessaris et al. 2006). Only recently have studies revealed a functional significance within different populations of OPCs in terms of myelination and response to demyelinating injury (Crawford et al. 2016; Tripathi et al. 2011; Xing et al. 2014). During embryonic development, oligodendrocyte progenitor cells (OPCs) populate the forebrain from distinct telencephalic germinal domains. At birth, the majority of forebrain OPCs and mature OLs are derived from the ventral telencephalic ventricular zone, however, by postnatal day 10 this population is almost entirely replaced by OPCs and OLs that originate from more dorsal regions of the embryonic ventricular zone (Kessaris et al. 2006; Takebayashi and Ikenaka 2015). Postnatally, OPCs continue to populate the forebrain from reservoirs of nestin+ progenitor cells within the postnatal subventricular zone (SVZ) of the lateral ventricles (Levison and Goldman 1993; Marshall et al. 2003; Luskin and McDermott 1994; Rowitch and Kriegstein 2010; Marshall et al. 2003), where they contribute to neural plasticity and repair (Dimou et al. 2008; Simon et al. 2011; Young et al. 2013; Burns et al. 2009). Changes in OL morphology (Dalitz et al. 2008), OL maturation and differentiation (Zoeller et al. 1994), and OL survival (Creeley et al. 2013) have been reported in third trimester-equivalent preclinical models of FASD, but nothing is known regarding EtOH’s impact on OL lineage cells derived from distinct telencephalic germinal zones (Guerri et al. 2001; Guizzetti et al. 2014).
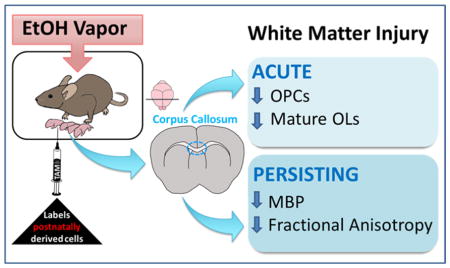
In humans, the third trimester of development represents a period of rapid oligodendrogenesis and myelination, which continues throughout the first few decades of postnatal life (Bartzokis et al. 2012; Gallo and Deneen 2014; Jakovcevski et al. 2009). In mice, oligodendrogenesis and myelination mostly occur during the first and second postnatal weeks (Dobbing and Sands 1979; Semple et al. 2014). The extraordinary metabolic demands required for the synthesis and maintenance of myelin sheaths render the process of myelination vulnerable to insult (Bartzokis 2004; Nave 2010). In the present study, we evaluated the acute and long-term impacts of EtOH exposure on OL number and white matter integrity within the corpus callosum using a third trimester-equivalent mouse model of FASD, in which mouse pups were exposed to EtOH during the first two weeks of postnatal development. Additionally, we employed a genetic Cre-loxP fate mapping approach to selectively identify OL lineage cells derived from nestin+ progenitors of the postnatal subventricular zone (SVZ), along with immunohistochemical detection of mature OLs and OPCs. Following early postnatal EtOH exposure, we demonstrated a marked decrease in the number of mature myelinating OLs and OPCs that is transient in nature and selective for non-SVZ-derived cells. Analysis of the vulnerable non-SVZ-derived OPC pool showed a reduction in their proliferative capacity. These early insults are coupled with enduring alterations in biochemical and microstructural indices of white matter integrity. In sum, our observations indicate heterogeneity in EtOH susceptibility among oligodendrocyte populations and demonstrate persistent alterations in the white matter integrity of the corpus callosum.
MATERIALS AND METHODS
Mice
All animal procedures were performed using Nestin-CreERT2: tdTomato reporter bitransgenic mice maintained on a C57Bl/J6 genetic background with homozygosity at both transgenes (Chow et al. 2015; Patzlaff et al. 2017). These mice harbor the tamoxifen-inducible CreERT2 fusion protein under control of the nestin promoter (Lagace et al. 2007) and a Rosa26-STOP-tdTomato reporter allele (Madisen et al. 2010). Tamoxifen administration to Nestin-CreERT2: tdTomato mice induces red fluorescence reporter expression in nestin+ cells and their downstream progeny (Chow et al. 2015; Patzlaff et al. 2017). All mice were maintained in a reverse 12-h dark/light cycle (lights off at 0800 hours), temperature and humidity controlled facility with food and water available ad libitum. All animal procedures were approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee per the NIH Animal Welfare Regulations and Public Health Service Policy on Humane Care and Use of Laboratory Animals (OLAW 2015).
Tamoxifen Administration
At postnatal day 2 (P2) all pups received a single intraperitoneal (i.p.) injection of tamoxifen at a dose of 33 mg/kg, dissolved in 10% EtOH/90% sunflower seed oil (Li et al. 2014). The tamoxifen (T5648), 200 proof EtOH (459836), and sunflower oil (S5007) were obtained from Sigma-Aldrich, St. Louis, MO. After the injection, the pups were placed back into their home cage with their original mother.
Alcohol Exposure
Nestin-CreERT2: tdTomato female mice of equal age (P80–P90) were bred with males of the same strain. Once pregnancy was determined by weight gain, females were separated into individual cages. Following tamoxifen administration to P2 pups, litters were randomly assigned to either EtOH or air control groups. From P3 to P15, cages with mothers and pups inside of the vapor inhalation chambers were exposed to either air or EtOH vapors from 1000–1400 hours daily (FIGURE 1) (Morton et al. 2014; Zamudio-Bulcock et al. 2014). Throughout the exposure, EtOH vapor levels were monitored with a Breathalyzer (Intoximeters, St. Louis, MO, 12-0050-00) and adjusted accordingly. During the exposure, the concentration of EtOH vapor was gradually increased from P3–P9 (4.9 ± 0.3 g/dl) to allow animals in EtOH vapor chambers to acclimate, and then kept steady from P10–P15 (7.1 ± 0.1 g/dl) (TABLE 1). At P4, P7, P11, and P14 one pup per time point was sacrificed immediately following the four-hour exposure period for blood EtOH concentrations (BECs), measured with an alcohol dehydrogenase-based assay as previously described (Galindo and Valenzuela 2006). Immediately following the exposure on P15, cages were removed from the vapor chambers and placed on the normal housing racks. Mice were euthanized on P16 or P50 for all subsequent analyses. At P22 mice were weaned and placed into group (2–3 animals per cage), sex-separate, standard housing. There was no effect of treatment on litter size (Air 5.8 ± 1.2 pups/litter, n = 5 litters; EtOH 5.7 ± 0.2 pups/litter, n = 7 litters; p = 0.9098). This study was restricted to analysis of male offspring, as previous work demonstrates sexual dimorphism in the white matter of rodents (Cerghet et al. 2009).
Figure 1.

Experimental Design: Tamoxifen (TAM) was administered to Nestin-CreERT2: tdTomato mice at P2, followed by vapor chamber exposure from P3–P15. The level of ethanol (EtOH) in the vapor chamber was gradually increased from P3 to P9, whereupon it remained steady until P15. Cohorts of mice were sacrificed (SAC) at P16 or P50 for all subsequent analyses.
Table 1.
Ethanol Concentrations1
| Age | Blood [EtOH], mg/dl | Vapor [EtOH] in chamber, g/dl |
|---|---|---|
| P3 | n/d | 3.8 ± 0.2 |
| P4 | 128.2 | 4.2 ± 0.4 |
| P5 | n/d | 4.6 ± 0.3 |
| P6 | n/d | 4.6 ± 0.4 |
| P7 | 160.0 | 5.3 ± 0.2 |
| P8 | n/d | 5.5 ± 0.5 |
| P9 | n/d | 6.4 ± 1.4 |
| P10 | n/d | 7.2 ± 0.6 |
| P11 | 185.6 | 6.9 ± 0.5 |
| P12 | n/d | 7.3 ± 0.4 |
| P13 | n/d | 7.1 ± 0.4 |
| P14 | 167.7 | 7.0 ± 0.6 |
| P15 | n/d | 7.1 ± 0.8 |
Corresponding pup blood ethanol concentration (mg/dl) and vapor ethanol concentration Breathalyzer measurements (g/dl) taken during four cohorts of EtOH exposures, presented as means ± SEM. BECs were not determined (n/d) for every time point.
Immunohistochemistry
At P16 or P50, mice were anesthetized by overdose with an i.p. injection of sodium pentobarbital (Fort Dodge Animal Health, Fort Dodge, IA, NDC 0856-0471-01) and transcardially perfused with phosphate-buffered solution (PBS) containing the following compounds from Sigma-Aldrich: 0.1% procaine hydrochloride (P9879) and 2 U/ml heparin (H3393), followed with perfusion of 4% paraformaldehyde (P6148) in 0.1M PBS. Brains were post-fixed overnight and stored in a 30% sucrose solution (Fisher Scientific, Hampton, NH, BP2201) in 0.1M PBS at 4°C. Cerebral hemispheres were sectioned at 30μm in the coronal plane using a freezing sliding knife microtome (Phoenix Equipment Inc., Rochester, NY, 14621) and stored in cryoprotective solution. Floating sections were processed for immunofluorescence as previously published (Li et al. 2010). Coronal sections containing the corpus callosum were immunolabeled for Olig2, a transcription factor expressed in all oligodendrocyte lineage cells. Olig2 was co-localized with CC1 antigen, Ki67 protein or PDGFRα to identify all mature myelinating oligodendrocytes, proliferating OPCs or all OPCs, respectively, using the following primary antibodies: rabbit anti-Olig2 (1:1,000; RRID:AB_570666; EMD Millipore, Darmstadt, Germany, AB9610), mouse anti-CC1 (1:400; RRID:AB_2057371; EMD Millipore, OP80), mouse anti-Ki67 (1:400; RRID:AB_393778; BD Biosciences, San Jose, CA, 550609) and rabbit anti-PDGFRα (1:350; RRID:AB_631064; Santa Cruz Biotechnology, Dallas, TX, sc-338). Antigen expression was detected using FITC- (715-096-150), or Cy5-conjugated (711-175-152) secondary antibodies (1:250; Jackson ImmunoResearch Laboratories, West Grove, PA). Tissue sections were counterstained with 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI; Thermo Fisher Scientific, Carlsbad, CA, D1306) and coverslipped using Fluoromount G (Electron Microscopy Sciences, Hatfield, PA, 17984).
Confocal Fluorescence Imaging
All z-stack projection images of immunolabeled cells in the corpus callosum were obtained using LASX acquisition software with a Leica DMi8 TCS SP8 confocal microscope (Wetzlar, Germany). Sequential imaging scans were performed at 10 and 63x. Gain parameters, zoom, pinhole size, step size, scan speed, and resolution were uniform across scans of equivalent magnification.
Stereology
OL lineage cell densities were estimated within serial coronal sections using the Optical Fractionator probe in Stereoinvestigator software (Microbrightfield Bioscience, Williston, VT) linked to an Olympus IX-81 DSU spinning disk confocal microscope and a 40× objective (Shinjuku, Tokyo, Japan). Region of interest (ROI) contours were manually outlined in each section using a 10× objective with reference to consistent anatomical structures. ROIs at the level of septal nuclei were analyzed between Bregma coordinates 1.18 to 0.02 mm. White matter ROI contours started from the dorsomedial corner of the lateral ventricle and were drawn laterally to encompass the corpus callosum to the distal tail of the SVZ. For estimates within the parenchymal grey matter, a 400 μm by 400 μm ROI was placed across layer IV of the somatosensory cortex and within the dorsal striatum. ROIs at the level of the dorsal hippocampus were analyzed between Bregma coordinates −1.34 to −2.30 mm and encompassed two distinct regions of the posterior corpus callosum: callosal crossing fibers (CF) which extended from the midline to the proximal boundary of the dorsal fornix, and callosal projection fibers (PF) that extended from the apex of the callosal arch to the boundary of the primary somatosensory cortex.
The optical dissector height was set at 18 μm, with a top and bottom guard zone height set at 2 μm. The counting frame size was equal to the grid size at x = 150 μm and y = 150 μm. Estimates of mature OLs, identified with Olig2+/CC1+ labeling (Bin et al. 2016), and OPCs, identified with Olig2+/Ki67+ and Olig2+/PDGFRα+ labeling, were calculated across 3–4 histological sections per mouse. To determine postnatally derived mature OLs and OPCs the number of Olig2+/CC1+, Olig2+/PDGFRα+, and Olig2+/Ki67+ cells co-localized with tdTomato fluorescence was estimated. ROI contour areas did not differ across treatment groups at P16 (p = 0.8929) or at P50 in either anterior (p = 0.7924) or posterior (CC p = 0.4227; LWM p = 0.1106) levels of the corpus callosum. The density of immunolabeled cells was calculated by dividing the cell estimates by the volume of the corresponding contours. At P16, n = 4 EtOH-exposed mice (across 4 litters) and n = 7 control air-exposed mice (across 4 litters) were used in stereological analyses. At P50 n = 4 EtOH-exposed mice (across 4 litters) and n = 6 control air-exposed mice (across 4 litters) were used. The data were subjected to t tests with Welch’s correction and expressed as means ± SEM.
Western Blots
At P50, a western blot analysis was used to analyze myelin basic protein (MBP) expression in the corpus callosum; n = 4 EtOH exposed mice across 4 separate litters, and n = 7 control air exposed mice across 4 separate litters. Following rapid decapitation, white matter from the midline of the corpus callosum was microdissected from 1 mm coronal slices. The microdissection was restricted to crossing fibers of the corpus callosum that extend from the apical arch of one hemisphere to the apical arch of the contralateral hemisphere, as previous work has demonstrated distinct patterns of gene expression across regions of the corpus callosum (Tagge et al. 2016). Tissue was collected in sucrose containing homogenization buffer, snap frozen on dry ice, and stored in −80°C. Cell lysis was achieved by means of mechanical disruption with a hand homogenizer (749540-0000, Kimble, Rockwood, TN) and sonication, homogenates were centrifuged at 4200 relative centrifugal force at 4°C for 10 minutes. The supernatant from each sample was removed for assay of the cytosolic fraction as previously described (Jantzie et al. 2014; Jantzie et al. 2016). Protein concentrations were determined using a Bradford protein assay. 4–12% Criterion-XT precast gels (Bio-Rad, Hercules, CA, 345-0124) were loaded with 30μg of protein per well. Following electrophoresis, protein was transferred to polyvinylidene fluoride membranes (PVDF; Bio-Rad, 1620177) that were blocked in milk/1xTris Buffered Saline with Tween 20 (TBS-T) and incubated in anti-MBP (1:1000; EMD Millipore, AB980) or anti-actin (1:5000; Sigma-Aldrich, A5441) at 4°C overnight as previously published (Jantzie et al. 2014; Jantzie et al. 2016). The following day, membranes were incubated in species appropriate HRP-conjugated secondary antibodies (1:5000; Thermo Scientific, Waltham MA, 62460) at room temperature for 1 hour, after which, membranes were washed, incubated with femto-west electrochemiluminescent substrate (ECL; Thermo Scientific, 37075), and developed using the chemiluminescent function of a GE-LAS 4000 Digital image reader (GE Healthcare Life Sciences, Chicago, IL). The bands of interest were quantified using GE ImageQuant software (GE Healthcare Life Sciences), standardized to the actin bands, of which levels were unchanged between treatment groups, and normalized to controls.
Magnetic Resonance Imaging
At P50, n = 4 EtOH exposed mice across 4 litters, and n = 5 air exposed mice across 4 litters were transcardially perfused with PBS containing 0.1% procaine and 2 U/ml heparin, followed with perfusion of 4% paraformaldehyde in PBS. Brains were post-fixed overnight and stored in 30% sucrose in PBS at 4°C. For scanning, brains were embedded in 2% agarose containing 3 mM sodium azide. Images were acquired, as previously described (Jantzie et al. 2015a; Robinson et al. 2016), on a Bruker 4.7T BioSpec 47/40 Ultra-Shielded Refrigerated nuclear MRI system equipped with a quadrature RF coil (72 mm I.D.) and a small-bore (12 cm I.D.) gradient set with a maximum gradient strength of 50 Gauss/cm (Bruker, Billerica, MA). T2 weighted structural images were acquired using the multi-slice multi-echo imaging sequence (TR = 3000.0 ms, TE = 12.0 ms), in which twelve sequential coronal 1mm slices were obtained. Diffusion tensor weighted images (DTI) were acquired using an echo-planar imaging sequence with 30 diffusion gradient directions (TR = 5000.0 ms, TE= 38 ms). Within Brunker’s Paravision 5.1 imaging software T2 structural maps, fractional anisotropy maps, and DTI directionally encoded color maps were generated for each animal. On processed DTI scans ROIs of the callosal crossing fibers (CF) and projection fibers (PF) in both hemispheres were analyzed across 2 sections. For each of the ROIs, fractional anisotropy (FA), axial diffusivity (AD, λ1), and radial diffusivity (RD, (λ2+ λ3)/2) were calculated.
Statistical Analysis
All analyses were performed blinded to treatment. Data were subjected to either unpaired parametric t test or 2-way ANOVA using GraphPad Prism 7.01 (La Jolla, CA). The data are expressed as means ± S.E.M., with p < 0.05 considered statistically significant. The statistical unit of determination (n) was defined as one pup.
RESULTS
Third trimester-equivalent ethanol exposure in Nestin-CreERT2:tdTomato mice
NestinCreERT2:tdTomato bitransgenic mice were used across all treatment groups to distinguish mature OLs and OPCs originating from the postnatal SVZ from those originating earlier in embryonic development. Reporter gene expression was selectively induced in postnatal nestin+ SVZ progenitors by tamoxifen administration at P2, as previously described (Petrik et al. 2013). Pups were then exposed to gradually increasing concentrations of EtOH within vapor chambers over the following 13 days from P3 to P15 (FIGURE 1). The mean concentration of EtOH within the vapor chambers over this period was 6.1 ± 0.3 g/dl (range = 3.8 – 7.3 g/dl), resulting in a mean pup blood EtOH concentration (BEC) of 160.4 ± 12.0 mg/dl (range = 128.2 – 185.6 mg/dl) (Table 1). The mean mouse pup BEC achieved in this study is twice the legal intoxication limit (80 mg/dl = 17.4 mM), which is consistent with BECs reported in some cases of human exposure during late pregnancy (Burd et al. 2012; Gohlke et al. 2008; Valenzuela et al. 2012). Maternal EtOH levels are expected to be approximately 10% of pup levels (Galindo and Valenzuela 2006). Tamoxifen-treated pups exposed only to air within the vapor chambers served as controls. We observed no postnatal mortality in air-control or EtOH exposed groups. Casual observations performed daily indicated no gross differences in maternal care between treatment groups. Analysis of pup weight at P2 and P15 showed no difference between air and EtOH treatment groups (Air, 6.3 ± 0.3 g/pup, n = 3 litters; EtOH 6.4 ± 0.3 g/pup, n = 4 litters; p = 0.7395). This is consistent with previous observations that mouse pups do not differ in their capacity to achieve normal body weight during third trimester-equivalent EtOH exposure in studies with levels comparable to those in this study (Karaçay et al. 2008; Pal and Alkana 1997; Wozniak et al. 2004).
Origin-dependent loss of oligodendrocyte lineage cells following ethanol
To assess both the acute and the long-term impact of early postnatal EtOH exposure on the number of oligodendrocyte lineage cells, we utilized a combination of immunofluorescence labeling and reporter gene expression to quantify mature OLs and OPCs at P16 or P50. The nuclear transcription factor Olig2 that is expressed throughout all stages of oligodendrocyte maturation, from proliferating OPCs to mature myelinating OLs (Back et al. 2007), was used to identify all oligodendrocyte lineage cells. Mature myelinating OLs were further identified by co-expression of Olig2 with CC1 (adenomatous polyposis coli, APC), a cytoplasmic protein highly expressed in mature myelinating OLs (Yuen et al. 2014). OPCs were identified by co-expression of Olig2 with PDGFRα, a receptor specific to OPCs (Nishiyama et al. 1996); whereas proliferating OPCs were identified by co-expression of Olig2 with Ki67, a nuclear cell proliferation marker (Gerdes et al. 1983). In addition to these markers, tdTomato (tdTom) reporter expression was used to distinguish mature OLs and OPCs originating from the postnatal SVZ from those originating earlier in embryonic development. For the purpose of this manuscript, we define all tdTom− oligodendrocytes as embryonically derived. However, this population may also include progeny of embryonic oligodendrocytes, as well as oligodendrocytes derived prior to tamoxifen administration at P2. Both embryonically-derived (tdTom−) and postnatal SVZ-derived (tdTom+) OL lineage cells were readily detectable throughout the septal region of the corpus callosum (FIGURE 2). In the corpus callosum of air-control mice, the tdTom+Olig2+ cells represented 45.3 ± 5.6% (n = 7) and 58.8 ± 7.8% (n = 6) of the total population of Olig2+ cells at P16 and P50, respectively, indicating a consistent level of tamoxifen-induced recombination across all mice with little variability in recombination efficiency.
Figure 2.

At P16 the maturational stage and cell origin of oligodendrocyte lineage cells in septal corpus callosum were determined using fluorescent labeling. (A) The distribution of tamoxifen induced fluorescence expression in postnatal SVZ-derived cells (tdTomato+, red) and immunolabeled mature oligodendrocytes (Olig2+CC1+, green) across the corpus callosum (scale bar = 100 μm) (B) High resolution merged and split channel images, scale bars = 10 μm, demonstrating colabeled cells within the corpus callosum (DAPI, blue; Olig2, white; and tdTomato, red). Mature oligodendrocytes (OLs) are identified by Olig2 and CC1 (green) coexpression. Vertical white arrows point to a DAPI+ Olig2+ CC1+ cell that is tdTomato positive, indicative of a postnatally derived mature OL. C) Proliferating OPCs are identified by Olig2 and Ki67 (green) coexpression. Horizontal white arrows point to a DAPI+ Olig2+ Ki67+ cell that is tdTomato negative, indicative of an embryonically derived proliferating OPC. (D) Oligodendrocyte progenitor cells (OPCs) are identified by Olig2 and PDGFRα (green) coexpression. Split channel images demonstrate an embryonically derived tdTomato- (white horizontal arrow) and a postnatally derived tdTomato+ (white vertical arrow) OPCs (Olig2+ PDGFRα +).
To determine the acute effects of EtOH exposure on oligodendrocyte lineage cells, we estimated mature OL and OPC densities within a specific ROI outlined at the level of the septal corpus callosum in histological sections. The ROI encompassed the lateral region of the septal corpus callosum, from the dorsomedial corner of the lateral ventricle to the distal tail of the SVZ, in order to capture the radial migration of postnatal SVZ-derived cells through this region (Kakita and Goldman 1999). EtOH exposure resulted in a significant 58% loss of embryonically-derived tdTom− mature OLs (16,138 ± 4,896 cells/mm3, n = 4) compared to air controls (38,162 ± 4,935 cells/mm3, n = 7, p = 0.0131) at P16 (FIGURE 3B), with no effect on the density of postnatal SVZ-derived tdTom+ mature OLs (p = 0.6066; FIGURE 3C). The total number of tdTom+ cells was unchanged between air and EtOH groups (p = 0.3235).
Figure 3.

EtOH exposure during white matter development results in acute loss of mature myelinating oligodendrocytes. (A) The corresponding regions of interest (ROIs) were drawn over DAPI fluorescence from the dorsomedial corner of the LV to the tail end of the SVZ to encompass the white matter of the septal corpus callosum, as shown, using the 4x objective for air- and EtOH-exposed animals. Markers recorded by an observer blinded to treatment were used to estimate the cellular density of tdTom+ and tdTom− mature OLs (OLs; Olig2+/CC1+). Representative marker maps of one coronal section from one air and one EtOH animal show the distribution of tdTom+ and tdTom− mature OLs. (B) At P16, EtOH exposure results in a significant reduction in the density of tdTom− mature OLs in septal corpus callosum (*p = 0.0131), (C) while the density of tdTom+ mature OLs derived from postnatal SVZ progenitors remains unchanged, (p = 0.6066; n = 4–7 animals per treatment group).
This acute loss of embryonically-derived mature OLs was accompanied by significant attenuation of OPC density. Exposure to EtOH elicited a 75% reduction in the density of tdTomato− proliferating OPCs (EtOH: 1,288 ± 565 cells/mm3, n = 4; air: 5,184 ± 1105 cells/mm3, p = 0.0130; FIGURE 4B), and had no effect on the density of tdTomato+ proliferating OPCs (p = 0.1696; FIGURE 4C), as assessed by Olig2+Ki67+ coexpression. The total OPC population, as assessed by Olig2+PDGFRα+ coexpression, was correspondingly affected. Embryonically-derived tdTom− OPCs were reduced by 60% in EtOH exposed animals (3,958 ± 879 cells/mm3, n = 4) compared to air-exposed controls (9,999 ± 1244 cells/mm3, n = 7, p = 0.0042; FIGURE 4D), whereas the density of postnatal SVZ-derived tdTom+ OPCs did not differ between EtOH and air treatment groups (p = 0.4825; FIGURE 4E).
Figure 4.

Developmental EtOH exposure results in an acute and origin dependent loss of oligodendrocyte progenitor cells. (A) ROIs encompassing septal corpus callosum pertaining to the adjacent marker maps illustrating the distribution of tdTom+ and tdTom− proliferating OPCs (Olig2+/Ki67+) in air and EtOH exposed animals. (B) At P16, stereological analysis of immunohistochemically processed sections containing the corpus callosum at the level of the septal nuclei revealed a significant reduction in the density of tdTom− proliferating OPCs in the corpus callosum following EtOH exposure (*p = 0.0130), (C) whereas the density of tdTom+ proliferating OPCs was unaffected (p = 0.1696; n = 4–7 animals per treatment group). (D) At P16, EtOH exposure diminished the density of the total tdTom− OPC (Olig2+ PDGFRα+) population (**p = 0.0042; n = 4–6 animals per treatment group). (E) No discrepancy in tdTom+ OPCs between Air and EtOH exposed animals was observed (p = 0.4825; n = 4–6 animals per treatment group).
To investigate whether the acute depletion in oligodendrocytes we observed in the corpus callosum was isolated to white matter, we estimated the density of tdTomato− and tdTomato+ mature OL in the cortex and striatum. Following EtOH exposure at P16, we observed a significant 46% decrease in embryonically-derived tdTom− mature OL density compared to air exposed controls in the cortex (air: 5,200 ± 541 cells/mm3, n = 5; EtOH: 2,812 ± 809 cells/mm3, n = 4, p = 0.0385) and striatum (air: 12,590 ± 1,038 cells/mm3, n = 5; EtOH: 6,802 ± 1,630 cells/mm3, n = 4, p = 0.0168) (FIGURE 5B). Consistent with our data from the corpus callosum, the density of tdTom+ mature OLs in the grey matter regions selected was not statistically different between air and EtOH groups (FIGURE 5C).
Figure 5.

The acute and origin dependent loss of mature oligodendrocytes following EtOH exposure extends to multiple grey matter structures and is not an isolated phenomena within the white matter of the corpus callosum A) ROIs within the cortex (ctx, orange) and striatum (str, yellow) were selected for stereological estimation of mature OL density. (B) At P16, the densities of tdTom− mature OLs within the ctx and str were significantly diminished in EtOH-exposed animals compared to air exposed controls, (*p = 0.0385 and *p = 0.0168, respectively), (B) whereas the densities of tdTom+ mature OLs did not statistically differ between treatment groups, (ctx, p = 0.8405; str, p = 0.5967; n = 4–5 animals per group).
To determine whether the decrease in OL lineage cell number persisted into adulthood, we quantified the OL lineage cells in the P50 cohort using the same quantitative analysis as for P16. We observed no effect of EtOH exposure on the number of tdTom− mature OLs or tdTom− proliferating OPCs at P50 (FIGURE 6), indicating a complete recovery in the number of embryonically-derived mature OLs and restored proliferative capacity. We also observed no effect of early postnatal EtOH exposure on the number of postnatal SVZ-derived tdTom+ OLs or of tdTom+ OPC proliferation at P50. Taken together, these data suggest an acute, origin-dependent loss of oligodendrocyte lineage cells that recovers by P50.
Figure 6.

Oligodendrocyte cell numbers recover from early deficits by adulthood (A) At P50, the densities of both tdTom− and tdTom+ mature OLs in the septal corpus callosum of EtOH-exposed animals were not different from the densities of air exposed controls, (p = 0.8767 and p = 0.8414, respectively). (B) Similarly, by P50 the densities of tdTom− and tdTom+ proliferating OPCs were not statistically different between EtOH and air exposed animals, (p = 0.0711 and p = 0.9480, respectively; n = 4–6 per treatment group).
Third trimester-equivalent ethanol exposure leads to persistent white matter injury
Cognitive and behavioral disabilities resulting from prenatal EtOH exposure endure into adulthood (Moore and Riley 2015). Recent imaging studies reveal abnormal white matter microstructure in adults with FASD (Li et al. 2009; Ma et al. 2005), and have linked cognitive deficits with structural abnormalities in white matter tracts of children and adolescents diagnosed with FASD (Treit et al. 2013). Although our initial findings indicated that the severe loss of oligodendrocyte lineage cells numerically recovered by P50, we next asked whether abnormalities in other indices of white mater integrity might persist at this later time point in our mouse model.
Myelin Basic Protein
MBP, an essential component of the myelin sheath of oligodendrocytes, exists as four isoforms that each contributes to myelin formation (Boggs 2006; Harauz et al. 2009). Western blotting of the microdissected crossing fibers of the corpus callosum (FIGURE 7A) was performed at P50 to examine the enduring effects of early postnatal EtOH exposure on MBP expression in adulthood. We found that MBP expression was reduced in the corpus callosum of EtOH-exposed animals compared to air controls across all four isoforms (p = 0.0005, 2-way ANOVA; FIGURE 7B–C). Of note, the 18-kD isoform, essential for the development and stability of CNS myelin (Harauz and Boggs 2013), was decreased by 45% in the corpus callosum of EtOH exposed animals compared to air controls (air: 100 ± 13, n = 7; EtOH 55 ± 8, n = 4, p = 0.0312, unpaired t test). These observations suggest that despite the numerical recovery of oligodendrocytes at P50, defects in myelin protein expression persist.
Figure 7.

EtOH exposure during peak white matter development results in diminished MBP expression in the midline of the corpus callosum at P50. (A) Tissue was isolated from the midline along the rostro-caudal axis of the corpus callosum (red), restricting the compositional analysis to the callosal crossing fibers. (B) Western blot at P50 shows the reduced expression of MBP in EtOH exposed animals. (C) EtOH exposure results in a significant decrease in MBP expression across the four isoforms (***p = 0.0005, n = 4–7 per group).
Diffusion Tensor Imaging
At P50, DTI was performed to advance our assessment of persisting white matter injury within regions of the corpus callosum following EtOH exposure. T2 structural maps and color coded diffusion maps were generated and the fractional anisotropy (FA), radial diffusivity (RD), and axial diffusivity (AD) were calculated for ROIs encompassing the callosal crossing fibers (CF) and projection fibers (PF) of the corpus callosum across 2 sections per animal (FIGURE 8A–B). FA reflects the presence of oriented structures generated by preferred diffusion orientations. Variance in cell type and cell density, fiber orientation and compaction, in conjunction with myelin integrity all affect the diffusion of water molecules within white matter structures. A decrease in FA has been characterized in paradigms of white matter injury due to oligodendrocyte ablation (Harsan et al. 2006) and correlated with the functional consequence of diminished compound action potential amplitude to reflect diminished myelin health and integrity (Wang et al, 2012). In our model, microstructural and diffusion abnormalities were indicated by diminished FA in the callosal crossing fibers of EtOH-exposed animals (0.342 ± 0.016, n = 4) compared to air-exposed controls (0.426 ± 0.023, n = 5, p = 0.0246) (FIGURE 8D). Within the projection fiber region, FA was increased in EtOH-exposed animals (0.294 ± 0.011), compared to controls (0.250 ± 0.005, p = 0.0047) (FIGURE 8E). Of note, our data show a trend toward decreased axial diffusivity in the callosal crossing and projection fibers of EtOH-exposed animals, although this did not reach statistical significance (TABLE 2).
Figure 8.

MRI analysis at P50 shows microstructural changes in the white matter of EtOH brains compared to air controls. (A) Corresponding anterior and posterior T2 structural maps and directionally colored diffusion maps (green = dorsoventral, red = mediolateral, blue = rostrocaudal) of air and EtOH exposed animals. (B) Per brain, two slices were selected for quantification. Representative ROIs encompassing the callosal crossing fibers (CF, red) and projection fibers (PF, green). (C) At P50, the fractional anisotropy (FA) of EtOH exposed animals is significantly decreased (*p = 0.0246) in the CF region, (D) and significantly increased in the PF white matter compared to air controls (**p = 0.0047, n = 4–5 per treatment group). (E) Oligodendrocyte lineage cell densities were estimated within the CF and PF regions of posterior corpus callosum at P50. The densities of OLs did not differ based on treatment (p = 0.5632), however, the densities of OL lineage cells were statistically different between regions (*p = 0.0175, n = 4 per treatment group, 2-way ANOVA).
Table 2.
Diffusion Tensor Imaging Measures in the Corpus Callosum2
| ROI | Treatment | ||
|---|---|---|---|
| Air (n = 5) | EtOH (n = 4) | ||
| Crossing Fibers | FA | 0.426 ± 0.023 * | 0.342 ± 0.016 * |
| AD, mm2/s | 6.957 ± 0.190 × 10−4 | 6.669 ± 0.955 × 10−4 | |
| RD, mm2/s | 3.759 ± 0.131 × 10−4 | 3.876 ± 0.195 × 10−4 | |
| Projection Fibers | FA | 0.250 ± 0.005 ** | 0.294 ± 0.011 ** |
| AD, mm2/s | 6.363 ± 0.065 × 10−4 | 6.298 ± 0.101 × 10−4 | |
| RD, mm2/s | 4.504 ± 0.059 × 10−4 ** | 4.231 ± 0.042 × 10−4 ** | |
Mean Fractional Anisotropy (FA), Axial Diffusion (AD), and Radial Diffusion (RD) measured in the corpus callosum and lateral white matter of EtOH- and air-exposed mice. A significant difference in FA between treatment groups was observed in the callosal crossing fibers (*p = 0.0487) and projection fibers (*p < 0.05; **p < 0.01; n = 4–5 animals per treatment group).
Oligodendrocyte lineage cell densities were estimated within equivalent ROIs in the posterior corpus callosum at P50. Consistent with the estimates of mature OL density in anterior corpus callosum regions, the density of mature OLs in the crossing fiber (CF) and projection fiber (PF) ROIs did not differ between EtOH or air treatments (2-way ANOVA, p = 0.5632). However, there was a significant effect of region on the density of OL lineage cells (p = 0.0175) (FIGURE 8F). This finding emphasizes the diversity of cellular environment within the spatially distinct regions of the corpus callosum white matter and directs our interpretation of DTI measurements. Together, these data indicate that third trimester-equivalent EtOH exposure in mouse results in persistent, regional changes in white matter diffusivity patterns that likely reflect complex microstructural abnormalities, similar to those previously described following other models of early postnatal myelin injury (Travis et al. 2015).
DISCUSSION
Using a third trimester-equivalent mouse model of EtOH exposure, we investigated the acute and long-term impact of early postnatal EtOH exposure on OL number and white matter integrity. Immediately following EtOH exposure, we observed a striking 58% reduction in the number of mature OLs within the corpus callosum. This acute loss of mature OL was accompanied by an even greater decrease in the number of proliferating OPCs (75%). Intriguingly, mature OLs and OPCs derived from the postnatal SVZ were resistant to EtOH toxicity compared to their embryonically derived counterparts, demonstrating differential susceptibility dependent on ontogenetic origin. Although recruitment of postnatal SVZ-derived OPCs can result in regeneration and repair in certain white matter pathologies (Aguirre and Gallo 2007), our study indicates that postnatal SVZ-derivatives do not boost their production to compensate for the loss of embryonically derived OPCs following EtOH exposure. Despite the complete recovery of OL lineage cell numbers by P50, our analysis revealed persisting myelin protein dysregulation and irregular microstructure in the corpus callosum of EtOH-exposed animals.
Our finding that developmental EtOH exposure resulted in decreased density of proliferating OPCs might suggest that the observed loss of in the total OPC population and mature OLs could be secondary to impaired OPC proliferation. Previous studies demonstrate that EtOH exposure inhibits the ability of neural progenitors and neural stem cells to initiate molecular programs leading to cellular differentiation and proliferation (Arzumnayan et al. 2009; Talens-Visconti et al. 2011). EtOH has also been shown to alter PDGRFα expression (Luo and Miller 1998), as well as Wnt signaling (Vangipuram and Lyman 2012), both important regulators of OPC proliferation and differentiation (Barres et al. 1992; Fruttiger et al. 1999; Yuen et al. 2014). OPCs differentiate into immature OLs only after multiple rounds of cell division (Barres et al. 1994; Durand and Raff 2000). Disruptions in oligodendrocyte lineage progression that manifests as stalled differentiation could deplete proliferating OPC pools, in addition to mature myelinating OLs (Back et al. 2002; Marinelli et al. 2016).
The acute loss of OLs following EtOH exposure could also be the result of excitotoxicity. Mouse pups in our intermittent model of EtOH exposure experienced daily EtOH withdrawal following each 4 hour exposure period. Previous studies using mouse models of postnatal EtOH exposure in which pup BECs reach similarly moderate or higher levels show evidence of acute withdrawal occurring between periods of EtOH administration (Karaçay et al. 2008; Morton et al. 2014; Pal and Alkana 1997). EtOH withdrawal is known to cause glutamate release and NMDA-dependent excitotoxicity (Hoffman et al. 1989; Hughes et al. 1998; Karl et al. 1995; Thomas and Riley 1998; Thomas et al. 1997). OL lineage cells are particularly vulnerable to acute excitotoxicity in a maturation-dependent manner (Butts et al. 2008; Follett et al. 2000; Jantzie et al. 2015b; Marinelli et al. 2016; Matute et al. 2006; Talos et al. 2006a; Talos et al. 2006b). Acute EtOH exposure can lead to apoptosis of oligodendrocyte lineage cells, as shown in recent studies demonstrating the particular susceptibility of early myelinating OLs to EtOH-induced apoptosis following intravenous administration of high doses of EtOH in macaque monkeys during the third trimester (BECs ranging from 300–400 mg/dl) (Creeley et al. 2013).
Our findings also demonstrate heterogeneity in the susceptibility of oligodendrocyte lineage cells to EtOH toxicity depending upon ontogenetic origin. OPCs are known to be generated from distinct ventral and dorsal germinal domains within the developing CNS. Recent evidence indicates that these different populations mount distinct responses to demyelination in adulthood and display distinct vulnerabilities to age-associated functional decline (Crawford et al. 2016). Our study also suggest a functional significance related to ontogenetic diversity within the oligodendrocyte populations by demonstrating differential susceptibilities of embryonically and postnatally derived oligodendrocyte lineage cells to acute EtOH exposure during early postnatal development. During the period of our EtOH exposure, the corpus callosum undergoes considerable cellular transformation, with ongoing replacement at P0–P10 of OPCs derived from the Nkx2.1+ ventral embryonic ventricular zone by OPCs derived from more dorsal Gsh2+ and Emx1+ embryonic derivatives (Kessaris et al. 2006; Richardson et al. 2006). Additionally, there is concurrent migration of OPCs from the postnatal SVZ during this early postnatal period (Levison and Goldman 1993). Further fate mapping studies that follow these spatially and temporally distinct waves of OPC and mature OL production will be important in determining whether EtOH may selectively impact the timing or nature of this developmental transition. Considering that EtOH exposure is known to impact migration of glial progenitors during development (Clarren et al. 1978; Guizzetti et al. 2014), an interesting possibility is that EtOH impairs or delays the migration OPCs during this transitional period between P0–P10. Overall, mounting evidence suggests that OLs are far more diverse than previously assumed (Azim et al. 2016; Srubek Tomassy and Fossati 2014; Dimou and Gallo 2015), not only in terms of ontogenetic origin, but also in terms of myelination, and response to injury (Barradas et al. 1998; Crawford et al. 2016; Dionne et al. 2016; Nishiyama et al. 2014; Spassky et al. 2000; Tomassy et al. 2014; Tripathi et al. 2011; Xing et al. 2014).
Although we found that OL numbers recovered by early adulthood (P50), we observed persistent changes to myelin gene expression and white matter microstructural organization. Myelin compaction is driven primarily by MBP which binds to the two opposing cytosolic membranes of myelin drawing them tightly together (Roach et al. 1983). Previous studies have found diminished levels of myelin protein expression immediately following EtOH exposure (Ozer et al. 2000; Zoeller et al. 1994) and in oligodendrocyte primary cultures exposed to EtOH (Chiappelli et al. 1991), but neither of these studies assessed the long-term impact of developmental EtOH exposure on the level of MBP expression later in life. Here, we report significantly diminished levels of MBP in corpus callosum of adult mice exposed to EtOH. Future studies will be required to determine whether this decrease is associated with altered myelination or conduction properties of myelinated axons within the corpus callosum.
Several imaging studies describe microstructural abnormalities within the corpus callosum of adolescents and adults with FASD (Bookstein et al. 2002b; Lebel et al. 2011; Treit et al. 2013). Our DTI data show significantly decreased FA in the corpus callosum of EtOH-exposed mice at P50, consistent with previous reports in human FASD subjects (Wozniak et al. 2009). The parameters of DTI provide a measure related to the structure and integrity of white matter tissue (Basser and Pierpaoli 1996; Hüppi and Dubois 2006). Our data demonstrating decreased FA in the corpus callosum indicates the disruption of white matter microstructural integrity. It will be important for future studies to assess the ultrastructure of the crossing fiber and projection fiber myelin to further examine microstructural integrity in our exposure paradigm.
It is important to note that only male mice were assessed in the present study to avoid variability associated with sex-specific differences in white matter development and EtOH toxicity. Sex-separate analysis of neurophysiological data due to the variance in vulnerability between males and females to the constellation of teratogenic effects induced by perinatal EtOH is increasingly recognized as important (Paolozza et al. 2014; Tesche et al. 2015). Sexual dimorphism of oligodendrocytes and myelin gene expression in the corpus callosum has been consistently observed in the brains of normally developing rodents (Cerghet et al. 2009), but the sex-specific effects of prenatal EtOH exposure on oligodendrocyte cells has not been comprehensively explored (Wilhelm and Guizzetti 2015). Recent research supports the hypothesis that sex-specific effects in white matter regions exist in models of FASD. For instance, studies have demonstrated sex-specific effects of EtOH on spatial and motor learning (Goodlett and Peterson 1995; Reekes et al. 2016), a oligodendrogenesis dependent task (Xiao et al. 2016), and sex-specific deficits in eye movement have been detected in FASD subjects (Paolozza et al. 2015), potentially resulting from optic nerve hypoplasia following developmental EtOH exposure, characterized by abnormal myelin integrity (Parson et al. 1995; Pinazo-Duran et al. 1997). Although beyond the scope of the current investigation, it will be important in future studies to determine the impact of sex as a biological variable in our mouse model,
In summary, individuals exposed to EtOH during CNS development experience a wide range of challenges throughout their lives. The corpus callosum has been consistently implicated in the abnormal FASD brain. Studies in adults with FASD suggest these individuals experience a higher incidence of adult-onset neuropsychiatric disorders (Famy et al. 1998; Streissguth et al. 2004), which many be attributable to persisting white matter abnormalities in the corpus callosum (Bartzokis 2012; Bookstein et al. 2002a; Bookstein et al. 2002b; Fields 2008). Understanding the mechanisms underlying these persisting white matter deficits following developmental EtOH exposure will be essential for designing new therapeutic approaches. Our finding that the ontogenetic origin of OPCs determines their susceptibility to developmental EtOH exposure has important implications for therapeutic targeting. In addition, our findings of persistent deficits in myelin protein expression and abnormal white matter microstructural integrity suggest that long-term disruption of oligodendrocyte function and early, transient OPC loss may be functionally linked.
MAIN POINTS.
Oligodendrocyte susceptibility to early postnatal EtOH exposure indicates heterogeneity based on ontogenetic origin.
Despite the recovery of acute oligodendrocyte losses by adulthood, MBP dysregulation and WM microstructural abnormalities persist.
Acknowledgments
We thank Dr. Yirong Yang at the UNM School of Medicine’s BRaIN Imaging Center for his MRI expertise, and the staff of the UNM Animal Resource Facility for excellent animal care and assistance. Images of fluorescent labeling in this paper were generated at the University of New Mexico & Cancer Center Fluorescence Microscopy Shared Resource, funded as detailed on: http://hsc.unm.edu/crtc/microscopy/acknowledgement.shtml.
References
- Aguirre A, Gallo V. Reduced EGFR signaling in progenitor cells of the adult subventricular zone attenuates oligodendrogenesis after demyelination. Neuron Glia Biol. 2007;3:209–20. doi: 10.1017/S1740925X08000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzumnayan A, Anni H, Rubin R, Rubin E. Effects of ethanol on mouse embryonic stem cells. Alcohol Clin Exp Res. 2009;33:2172–9. doi: 10.1111/j.1530-0277.2009.01057.x. [DOI] [PubMed] [Google Scholar]
- Azim K, Berninger B, Raineteau O. Mosaic subventricular origins of forebrain oligodendroglia. Frontiers in Neuroscience. 2016:10. doi: 10.3389/fnins.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol. 2002;61:197–211. doi: 10.1093/jnen/61.2.197. [DOI] [PubMed] [Google Scholar]
- Back SA, Riddle A, McClure MM. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke. 2007;38:724–30. doi: 10.1161/01.STR.0000254729.27386.05. [DOI] [PubMed] [Google Scholar]
- Barradas PC, Gomes SS, Cavalcante LA. Heterogeneous patterns of oligodendroglial differentiation in the forebrain of the opossum Didelphis marsupialis. J Neurocytol. 1998;27:15–25. doi: 10.1023/a:1006930818708. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply 49–62. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Heydari P, Couvrette A, Lee GJ, Kalashyan G, et al. Multimodal magnetic resonance imaging assessment of white matter aging trajectories over the lifespan of healthy individuals. Biol Psychiatry. 2012;72:1026–34. doi: 10.1016/j.biopsych.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Neuroglialpharmacology: myelination as a shared mechanism of action of psychotropic treatments. Neuropharmacology. 2012;62:2137–53. doi: 10.1016/j.neuropharm.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–19. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Bin JM, Harris SN, Kennedy TE. The oligodendrocyte-specific antibody ‘CC1’ binds Quaking 7. J Neurochem. 2016;139:181–186. doi: 10.1111/jnc.13745. [DOI] [PubMed] [Google Scholar]
- Boggs JM. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci. 2006;63(17):1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookstein FL, Sampson PD, Connor PD, Streissguth AP. Midline corpus callosum is a neuroanatomical focus of fetal alcohol damage. Anat Rec. 2002a;269:162–74. doi: 10.1002/ar.10110. [DOI] [PubMed] [Google Scholar]
- Bookstein FL, Streissguth AP, Sampson PD, Connor PD, Barr HM. Corpus callosum shape and neuropsychological deficits in adult males with heavy fetal alcohol exposure. Neuroimage. 2002b;15:233–51. doi: 10.1006/nimg.2001.0977. [DOI] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd L, Blair J, Dropps K. Prenatal alcohol exposure, blood alcohol concentrations and alcohol elimination rates for the mother, fetus and newborn. J Perinatol. 2012;32:652–9. doi: 10.1038/jp.2012.57. [DOI] [PubMed] [Google Scholar]
- Burns KA, Murphy B, Danzer SC, Kuan C-Y. Developmental and post-injury cortical gliogenesis: a genetic fate-mapping study with Nestin-CreER mice. Glia. 2008;57(10):1115–1129. doi: 10.1002/glia.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts BD, Houde C, Mehmet H. Maturation-dependent sensitivity of oligodendrocyte lineage cells to apoptosis: implications for normal development and disease. Cell Death Differ. 2008;15:1178–86. doi: 10.1038/cdd.2008.70. [DOI] [PubMed] [Google Scholar]
- Caprariello AV, Batt CE, Zippe I, Romito-DiGiacomo RR, Karl M, Miller RH. Apoptosis of Oligodendrocytes during Early Development Delays Myelination and Impairs Subsequent Responses to Demyelination. J Neurosci. 2015;35:14031–41. doi: 10.1523/JNEUROSCI.1706-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerghet M, Skoff RP, Swamydas M, Bessert D. Sexual dimorphism in the white matter of rodents. J Neurol Sci. 2009;286:76–80. doi: 10.1016/j.jns.2009.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappelli F, Taylor AN, Espinosa de los Monteros A, de Vellis J. Fetal alcohol delays the developmental expression of myelin basic protein and transferrin in rat primary oligodendrocyte cultures. Int J Dev Neurosci. 1991;9:67–75. doi: 10.1016/0736-5748(91)90074-v. [DOI] [PubMed] [Google Scholar]
- Chow CL, Guo W, Trivedi P, Zhao X, Gubbels SP. Characterization of a Unique Cell Population Marked by Transgene Expression in the Adult Cochlea on Nestin-CreERT2/tdTomato-Reporter Mice. J Comp Neurol. 2015;523(10):1474–1487. doi: 10.1002/cne.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarren SK, Alvord EC, Sumi SM, Streissguth AP, Smith DW. Brain malformations related to prenatal exposure to ethanol. The Journal of pediatrics. 1978;92:64–67. doi: 10.1016/s0022-3476(78)80072-9. [DOI] [PubMed] [Google Scholar]
- Crawford AH, Tripathi RB, Richardson WD, Franklin RJ. Developmental Origin of Oligodendrocyte Lineage Cells Determines Response to Demyelination and Susceptibility to Age-Associated Functional Decline. Cell Rep. 2016;15(4):761–773. doi: 10.1016/j.celrep.2016.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creeley CE, Dikranian KT, Johnson SA, Farber NB, Olney JW. Alcohol-induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathol Commun. 2013;1:23. doi: 10.1186/2051-5960-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalitz P, Cock M, Harding R, Rees S. Injurious effects of acute ethanol exposure during late gestation on developing white matter in fetal sheep. Int J Dev Neurosci. 2008;26:391–9. doi: 10.1016/j.ijdevneu.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Dimou L, Gallo V. NG2-glia and their functions in the central nervous system. Glia. 2015;63(8):1429–1451. doi: 10.1002/glia.22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne N, Dib S, Finsen B, Denarier E, Kuhlmann T, Drouin R, Kokoeva M, Hudson TJ, Siminovitch K, Friedman HC, Peterson AC. Functional organization of an Mbp enhancer exposes striking transcriptional regulatory diversity within myelinating glia. Glia. 2016;64(1):175–194. doi: 10.1002/glia.22923. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Human Development. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Dumas L, Heitz-Marchaland C, Fouquet S, Suter U, Livet J, Moreau-Fauvarque C, Chédotal A. Multicolor analysis of oligodendrocyte morphology, interactions, and development with Brainbow. Glia. 2015;63(4):699–717. doi: 10.1002/glia.22779. [DOI] [PubMed] [Google Scholar]
- Durand B, Raff M. A cell-intrinsic timer that operates during oligodendrocyte development. Bioessays. 2000;22:64–71. doi: 10.1002/(SICI)1521-1878(200001)22:1<64::AID-BIES11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of Oligodendrocyte Differentiation and Myelination. Science. 2010;330(6005):779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Famy C, Streissguth AP, Unis AS. Mental illness in adults with fetal alcohol syndrome or fetal alcohol effects. Am J Psychiatry. 1998;155:552–4. doi: 10.1176/ajp.155.4.552. [DOI] [PubMed] [Google Scholar]
- Fields RD. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 2008;31:361–70. doi: 10.1016/j.tins.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–41. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM, Gallo V. The translational biology of remyelination: past, present, and future. GLIA. 2014;62(11):1905–1915. doi: 10.1002/glia.22622. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo R, Valenzuela CF. Immature hippocampal neuronal networks do not develop tolerance to the excitatory actions of ethanol. Alcohol. 2006;40:111–8. doi: 10.1016/j.alcohol.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Deneen B. Glial development: the crossroads of regeneration and repair in the CNS. Neuron. 2014;83:283–308. doi: 10.1016/j.neuron.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P, Nunez SC, Narr KL, Kan EC, Sowell ER. Effects of prenatal alcohol exposure on the development of white matter volume and change in executive function. Neuroimage Clin. 2014;5:19–27. doi: 10.1016/j.nicl.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 1983;31:13–20. doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- Gohlke JM, Griffith WC, Faustman EM. Computational models of ethanol-induced neurodevelopmental toxicity across species: Implications for risk assessment. Birth Defects Res B Dev Reprod Toxicol. 2008;83:1–11. doi: 10.1002/bdrb.20137. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Peterson SD. Sex differences in vulnerability to developmental spatial learning deficits induced by limited binge alcohol exposure in neonatal rats. Neurobiology of Learning and Memory. 1995;64(3):265–275. doi: 10.1006/nlme.1995.0009. [DOI] [PubMed] [Google Scholar]
- Green CR, Lebel C, Rasmussen C, Beaulieu C, Reynolds JN. Diffusion tensor imaging correlates of saccadic reaction time in children with fetal alcohol spectrum disorder. Alcohol Clin Exp Res. 2013;37:1499–507. doi: 10.1111/acer.12132. [DOI] [PubMed] [Google Scholar]
- Green PP, McKnight-Eily LR, Tan CH, Mejia R, Denny CH. Vital Signs: Alcohol-Exposed Pregnancies - United States, 2011–2013. MMWR Morb Mortal Wkly Rep. 2016;65:91–7. doi: 10.15585/mmwr.mm6504a6. [DOI] [PubMed] [Google Scholar]
- Guerri C, Pascual M, Renau-Piqueras J. Glia and fetal alcohol syndrome. Neurotoxicology. 2001;22:593–9. doi: 10.1016/s0161-813x(01)00037-7. [DOI] [PubMed] [Google Scholar]
- Guizzetti M, Zhang X, Goeke C, Gavin DP. Glia and neurodevelopment: focus on fetal alcohol spectrum disorders. Front Pediatr. 2014;2:123. doi: 10.3389/fped.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, Boggs JM. Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J Neurochem. 2013;125:334–61. doi: 10.1111/jnc.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, Ladizhansky V, Boggs JM. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. doi: 10.1021/bi901005f. [DOI] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Steibel J, Parizel N, de Sousa PL, Boehm N, Grucker D, Ghandour MS. Brain dysmyelination and recovery assessment by noninvasive in vivo diffusion tensor magnetic resonance imaging. J Neurosci Res. 2006;83(3):392–402. doi: 10.1002/jnr.20742. [DOI] [PubMed] [Google Scholar]
- Hoffman PL, Rabe CS, Moses F, Tabakoff B. N-methyl-D-aspartate receptors and ethanol: inhibition of calcium flux and cyclic GMP production. J Neurochem. 1989;52:1937–40. doi: 10.1111/j.1471-4159.1989.tb07280.x. [DOI] [PubMed] [Google Scholar]
- Hughes PD, Kim YN, Randall PK, Leslie SW. Effect of prenatal ethanol exposure on the developmental profile of the NMDA receptor subunits in rat forebrain and hippocampus. Alcohol Clin Exp Res. 1998;22:1255–61. [PubMed] [Google Scholar]
- Hüppi PS, Dubois J. Diffusion tensor imaging of brain development. Seminars in Fetal and Neonatal Medicine. 2006;11:489–497. doi: 10.1016/j.siny.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Temporally-controlled site-specific mutagenesis in the basal layerof the epidermis: comparison of the recombinase activity of thetamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. NucleicAcids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N. Oligodendrocyte Development and the Onset of Myelination in the Human Fetal Brain. Frontiers in Neuroanatomy. 2009;3:5. doi: 10.3389/neuro.05.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Corbett CJ, Berglass J, Firl DJ, Flores J, Mannix R, Robinson S. Complex pattern of interaction between in utero hypoxia-ischemia and intra-amniotic inflammation disrupts brain development and motor function. J Neuroinflammation. 2014;11:131. doi: 10.1186/1742-2094-11-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Getsy PM, Denson JL, Firl DJ, Maxwell JR, Rogers DA, Wilson CG, Robinson S. Prenatal Hypoxia-Ischemia Induces Abnormalities in CA3 Microstructure, Potassium Chloride Co-Transporter 2 Expression and Inhibitory Tone. Front Cell Neurosci. 2015a;9:347. doi: 10.3389/fncel.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Talos DM, Jackson MC, Park HK, Graham DA, Lechpammer M, Folkerth RD, Volpe JJ, Jensen FE. Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb Cortex. 2015b;25:482–95. doi: 10.1093/cercor/bht246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Winer JL, Corbett CJ, Robinson S. Erythropoietin Modulates Cerebral and Serum Degradation Products from Excess Calpain Activation following Prenatal Hypoxia-Ischemia. Dev Neurosci. 2016;38:15–26. doi: 10.1159/000441024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakita A, Goldman JE. Patterns and dynamics of SVZ cell migration in the postnatal forebrain: monitoring living progenitors in slice preparations. Neuron. 1999;23:461–72. doi: 10.1016/s0896-6273(00)80800-4. [DOI] [PubMed] [Google Scholar]
- Karaçay B, Li S, Bonthius DJ. Maturation-Dependent Alcohol Resistance in the Developing Mouse: Cerebellar Neuronal Loss and Gene Expression During Alcohol-Vulnerable and -Resistant Periods. Alcoholism: Clinical and Experimental Research. 2008;32:1439–1450. doi: 10.1111/j.1530-0277.2008.00720.x. [DOI] [PubMed] [Google Scholar]
- Karl PI, Kwun R, Slonim A, Fisher SE. Ethanol elevates fetal serum glutamate levels in the rat. Alcohol Clin Exp Res. 1995;19:177–81. doi: 10.1111/j.1530-0277.1995.tb01488.x. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–9. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodituwakku PW, Kodituwakku EL. From research to practice: an integrative framework for the development of interventions for children with fetal alcohol spectrum disorders. Neuropsychol Rev. 2011;21:204–23. doi: 10.1007/s11065-011-9170-1. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Whitman MC, Noonan MA, Ables JL, DeCarolis NA, Arguello AA, Donovan MH, Fischer SJ, Farnbauch LA, Beech RD, et al. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27:12623–9. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Roussotte F, Sowell ER. Imaging the Impact of Prenatal Alcohol Exposure on the Structure of the Developing Human Brain. Neuropsychology Review. 2011;21:102–118. doi: 10.1007/s11065-011-9163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–12. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Li L, Candelario KM, Thomas K, Wang R, Wright K, Messier A, Cunningham LA. Hypoxia inducible factor-1alpha (HIF-1alpha) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J Neurosci. 2014;34:16713–9. doi: 10.1523/JNEUROSCI.4590-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Coles CD, Lynch ME, Hu X. Voxelwise and skeleton-based region of interest analysis of fetal alcohol syndrome and fetal alcohol spectrum disorders in young adults. Hum Brain Mapp. 2009;30:3265–74. doi: 10.1002/hbm.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Harms KM, Ventura PB, Lagace DC, Eisch AJ, Cunningham LA. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia. 2010;58:1610–9. doi: 10.1002/glia.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Miller MW. Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Research Reviews. 1998;27:157–167. doi: 10.1016/s0165-0173(98)00009-5. [DOI] [PubMed] [Google Scholar]
- Luskin MB, McDermott K. Divergent lineages for oligodendrocytes and astrocytes originating in the neonatal forebrain subventricular zone. Glia. 1994;11:211–26. doi: 10.1002/glia.440110302. [DOI] [PubMed] [Google Scholar]
- Ma X, Coles CD, Lynch ME, Laconte SM, Zurkiya O, Wang D, Hu X. Evaluation of corpus callosum anisotropy in young adults with fetal alcohol syndrome according to diffusion tensor imaging. Alcohol Clin Exp Res. 2005;29:1214–22. doi: 10.1097/01.alc.0000171934.22755.6d. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli C, Bertalot T, Zusso M, Skaper SD, Giusti P. Systematic Review of Pharmacological Properties of the Oligodendrocyte Lineage. Frontiers in Cellular Neuroscience. 2016;10:27. doi: 10.3389/fncel.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CA, Suzuki SO, Goldman JE. Gliogenic and neurogenic progenitors of the subventricular zone: who are they, where did they come from, and where are they going? Glia. 2003;43(1):52–61. doi: 10.1002/glia.10213. [DOI] [PubMed] [Google Scholar]
- Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–24. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- May PA, Baete A, Russo J, Elliott AJ, Blankenship J, Kalberg WO, Buckley D, Brooks M, Hasken J, Abdul-Rahman O, et al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics. 2014;134:855–66. doi: 10.1542/peds.2013-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EM, Migliorini R, Infante MA, Riley EP. Fetal Alcohol Spectrum Disorders: Recent Neuroimaging Findings. Curr Dev Disord Rep. 2014;1:161–172. doi: 10.1007/s40474-014-0020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EM, Riley EP. What Happens When Children with Fetal Alcohol Spectrum Disorders Become Adults? Curr Dev Disord Rep. 2015;2:219–227. doi: 10.1007/s40474-015-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton RA, Diaz MR, Topper LA, Valenzuela CF. Construction of vapor chambers used to expose mice to alcohol during the equivalent of all three trimesters of human development. J Vis Exp. 2014 doi: 10.3791/51839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010;468:244–52. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 Proteoglycan and PDGF α-Receptor on O2A Progenitor Cells in the Developing Rat Brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Zhu X, Suzuki R. NG2 cells (polydendrocytes) in brain physiology and repair. Frontiers in Neuroscience. 2014:8. doi: 10.3389/fnins.2014.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman AL, Crocker N, Mattson SN, Riley EP. Neuroimaging and Fetal Alcohol Spectrum Disorders. Developmental disabilities research reviews. 2009;15:209–217. doi: 10.1002/ddrr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLAW; Health NIo, editor. Public Health Serive Policy on Humane Care and Use of Laboratory Animals. Bethesda, MD: 2015. [Google Scholar]
- Ozer E, Sarioglu S, Güre A. Effects of prenatal ethanol exposure on neuronal migration, neuronogenesis and brain myelination in the mice brain. Clinical neuropathology. 2000;19:21–25. [PubMed] [Google Scholar]
- Pal N, Alkana RL. Use of inhalation to study the effect of ethanol and ethanol dependence on neonatal mouse development without maternal separation: a preliminary study. Life Sci. 1997;61:1269–81. doi: 10.1016/s0024-3205(97)00672-3. [DOI] [PubMed] [Google Scholar]
- Paolozza A, Munn R, Munoz DP, Reynolds JN. Eye movements reveal sexually dimorphic deficits in children with fetal alochol spectrum disorder. Front Neurosci. 2015;9:76. doi: 10.3389/fnins.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolozza A, Treit S, Beaulieu C, Reynolds JN. Response inhibition deficits in children with Fetal Alcohol Spectrum Disorder: Relationship between diffusion tensor imaging of the corpus callosum and eye movement control. NeuroImage : Clinical. 2014;5:53–61. doi: 10.1016/j.nicl.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parson SH, Dhillon B, Findlater GS, Kaufman MH. Optic nerve hypoplasia in the fetal alcohol spectrum: a mouse model. J Anat. 1995;186(Pt 2):313–320. [PMC free article] [PubMed] [Google Scholar]
- Patzlaff NE, Nemec KM, Malone SG, Li Y, Zhao X. Fragile X related protein 1 (FXR1P) regulates proliferation of adult neural stem cells. Hum Mol Genet. 2017 doi: 10.1093/hmg/ddx034. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrik D, Yun S, Latchney SE, Kamrudin S, LeBlanc JA, Bibb JA, Eisch AJ. Early Postnatal In Vivo Gliogenesis From Nestin-Lineage Progenitors Requires Cdk5. PLoS ONE. 2013;8(8):e72819. doi: 10.1371/journal.pone.0072819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinazo-Duran MD, Renau-Piqueras J, Guerri C, Strömland K. Optic nerve hypoplasia in fetal alcohol syndrome: an update. Eur J Ophthalmol. 1997;7(3):262–70. doi: 10.1177/112067219700700311. [DOI] [PubMed] [Google Scholar]
- Reekes TH, Vinyard HT, III, Echols W, Eubank AJ, Bouldin MD, Murray WH, Brewer S, Brown BT, Willis HL, Tabrani Z, Favero CB, Clabough EB. Moderate chronic fetal alcohol exposure causes a motor learning deficit in adult outbred Swiss-Webster mice [version 1; referees: 1 approved, 1 approved with reservations] F1000Research. 2016;5:1896. [Google Scholar]
- Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–18. doi: 10.1038/nrn1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230:357–65. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Roach A, Boylan K, Horvath S, Prusiner SB, Hood LE. Characterization of cloned cDNA representing rat myelin basic protein: absence of expression in brain of shiverer mutant mice. Cell. 1983;34:799–806. doi: 10.1016/0092-8674(83)90536-6. [DOI] [PubMed] [Google Scholar]
- Robinson S, Winer JL, Berkner J, Chan LA, Denson JL, Maxwell JR, Yang Y, Sillerud LO, Tasker RC, Meehan WP., III Imaging and serum biomarkers reflecting the functional efficacy of extended erythropoietin treatment in rats following infantile traumatic brain injury. Journal of Neurosurgery: Pediatrics. 2016:1–17. doi: 10.3171/2015.10.PEDS15554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2014;0:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Götz M, Dimou L. Progenitors in the adult cerebral cortex: Cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59(6):869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- Smith GST, Paez PM, Spreuer V, Campagnoni CW, Boggs JM, Campagnoni AT, Harauz G. Classical 18.5-and 21.5-kDa isoforms of myelin basic protein inhibit calcium influx into oligodendroglial cells, in contrast to golli isoforms. Journal of Neuroscience Research. 2011;89:467–480. doi: 10.1002/jnr.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Olivier C, Perez-Villegas E, Goujet-Zalc C, Martinez S, Thomas JL, Zalc B. Single or multiple oligodendroglial lineages: A controversy. Glia. 2000;29(2):143–148. [PubMed] [Google Scholar]
- Streissguth AP, Bookstein FL, Barr HM, Sampson PD, O’Malley K, Young JK. Risk factors for adverse life outcomes in fetal alcohol syndrome and fetal alcohol effects. J Dev Behav Pediatr. 2004;25:228–38. doi: 10.1097/00004703-200408000-00002. [DOI] [PubMed] [Google Scholar]
- Tagge I, O’Connor A, Chaudhary P, Pollaro J, Berlow Y, Chalupsky M, Bourdette D, Woltjer R, Johnson M, Ronney W. Spatio-temporal patterns of demyelination and remyelination in the cuprizone mouse model. PloS one. 2016;11(4):e0152480. doi: 10.1371/journal.pone.0152480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talens-Visconti R, Sanchez-Vera I, Kostic J, Perez-Arago MA, Erceg S, Stojkovic M, Guerri C. Neural differentiation from human embryonic stem cells as a tool to study early brain development and the neuroteratogenic effects of ethanol. Stem Cells Dev. 2011;20:327–39. doi: 10.1089/scd.2010.0037. [DOI] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol. 2006a;497:42–60. doi: 10.1002/cne.20972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006b;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesche CD, Kodituwakku PW, Garcia CM, Houck JM. Sex-related differences in auditory processing in adolescents with fetal alcohol spectrum disorder: A magnetoencephalographic study. NeuroImage: Clinical. 2015;7:571–587. doi: 10.1016/j.nicl.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Riley EP. Fetal alcohol syndrome: does alcohol withdrawal play a role? Alcohol Health Res World. 1998;22:47–53. [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Weinert SP, Sharif S, Riley EP. MK-801 administration during ethanol withdrawal in neonatal rat pups attenuates ethanol-induced behavioral deficits. Alcohol Clin Exp Res. 1997;21:1218–25. [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, Arlotta P. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science. 2014;344:319–24. doi: 10.1126/science.1249766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassy GS, Fossati V. How big is the myelinating orchestra? Cellular diversity within the oligodendrocyte lineage: facts and hypotheses. Frontiers in Cellular Neuroscience. 2014:8. doi: 10.3389/fncel.2014.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B. Oligodendorycte death results in immune-mediated CNS demyelination. Nat Neurosci. 2016;19(1):65–74. doi: 10.1038/nn.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis KE, Adams JN, Ben-Shachar M, Feldman HM. Decreased and Increased Anisotropy along Major Cerebral White Matter Tracts in Preterm Children and Adolescents. PLoS ONE. 2015;10:e0142860. doi: 10.1371/journal.pone.0142860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treit S, Lebel C, Baugh L, Rasmussen C, Andrew G, Beaulieu C. Longitudinal MRI reveals altered trajectory of brain development during childhood and adolescence in fetal alcohol spectrum disorders. The Journal of Neuroscience. 2013;33:10098–10109. doi: 10.1523/JNEUROSCI.5004-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Clarke LE, Burzomato V, Kessaris N, Anderson PN, Attwell D, Richardson WD. Dorsally and ventrally dervied oligodendrocytes have similar electrical properties but myelinate preferred tracts. J Neurosci. 2011;31(18):6809–6819. doi: 10.1523/JNEUROSCI.6474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela CF, Morton RA, Diaz MR, Topper L. Does moderate drinking harm the fetal brain? Insights from animal models. Trends in Neurosciences. 2012;35:284–292. doi: 10.1016/j.tins.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangipuram SD, Lyman WD. Ethanol Affects Differentiation-Related Pathways and Suppresses Wnt Signaling Protein Expression in Human Neural Stem Cells. Alcoholism: Clinical and Experimental Research. 2012;36:788–797. doi: 10.1111/j.1530-0277.2011.01682.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Vlkolinksky R, Xie M, Obenaus A, Song SK. Diffusion tensor imaging detected optic nerve injury correlates with decreased compound action potentials after murine retinal ischemia. Invest Ophthalmol Vis Sci. 2012;53(1):136–142. doi: 10.1167/iovs.11-7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm CJ, Guizzetti M. Fetal Alcohol Spectrum Disorders: an overview from the glia perspective. Frontiers in Integrative Neuroscience. 2015:9. doi: 10.3389/fnint.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JF, Smith VC Committee On Substance A. Fetal Alcohol Spectrum Disorders. Pediatrics. 2015;136:e1395–406. doi: 10.1542/peds.2015-3113. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–14. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE, Nelson ML, Chang PN, Lim KO. Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: an extension of previous diffusion tensor imaging findings. Alcoholism, clinical and experimental research. 2009;33:1825–35. doi: 10.1111/j.1530-0277.2009.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Alexander Sinclair-Wilson, Wright JL, Fudge AD, Emery B, Li H, Richardson WD. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nature Neuroscience. 2016;19:1210–1217. doi: 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing YL, Roth PT, Stratton JAS, Chuang BHA, Danne J, Ellis SL, Ng SW, Kilpatrick TJ, Merson TD. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J Neurosci. 2014;34(42):14128–14146. doi: 10.1523/JNEUROSCI.3491-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77(5):873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 2014;158:383–396. doi: 10.1016/j.cell.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio-Bulcock PA, Morton RA, Valenzuela CF. Third trimester-equivalent ethanol exposure does not alter complex spikes and climbing fiber long-term depression in cerebellar Purkinje neurons from juvenile rats. Alcohol Clin Exp Res. 2014;38:1293–300. doi: 10.1111/acer.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeller RT, Butnariu OV, Fletcher DL, Riley EP. Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin-associated glycoprotein in cerebellum. Alcohol Clin Exp Res. 1994;18:909–16. doi: 10.1111/j.1530-0277.1994.tb00059.x. [DOI] [PubMed] [Google Scholar]