

SUMMARY
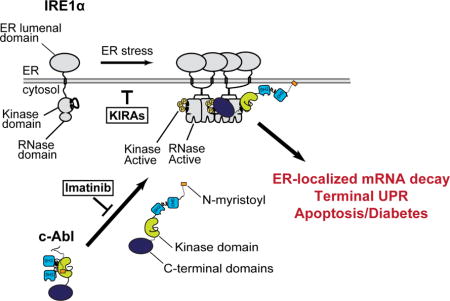
In cells experiencing unrelieved endoplasmic reticulum (ER) stress, the ER transmembrane kinase/endoribonuclease (RNase)—IRE1α—endonucleolytically degrades ER-localized mRNAs to promote apoptosis. Here we find that the ABL family of tyrosine kinases rheostatically enhances IRE1α’s enzymatic activities, thereby potentiating ER stress-induced apoptosis. During ER stress, cytosolic ABL kinases localize to the ER membrane, where they bind, scaffold, and hyperactivate IRE1α’s RNase. Imatinib—an anti-cancer tyrosine kinase inhibitor—antagonizes the ABL-IRE1α interaction, blunts IRE1α RNase hyperactivity, reduces pancreatic β-cell apoptosis, and reverses type 1 diabetes (T1D) in the non-obese diabetic (NOD) mouse model. A mono-selective kinase inhibitor that allosterically attenuates IRE1α’s RNase—KIRA8—also efficaciously reverses established diabetes in NOD mice by sparing β-cells and preserving their physiological function. Our data support a model wherein ER-stressed β-cells contribute to their own demise during T1D pathogenesis and implicate the ABL-IRE1α axis as a drug target for the treatment of an autoimmune disease.
eTOC Blurb
Morita et al. show that the non-receptor ABL tyrosine kinases enhance the enzymatic activities of the ER transmembrane kinase/endoribonuclease, IRE1a, thereby potentiating ER stress-induced apoptosis. Targeting the ABL-IRE1 pathway with Imatinib and selective KIRA kinase inhibitors reverses autoimmune diabetes in mice.
INTRODUCTION
Diverse perturbations compromise folding and structural maturation of secretory proteins in the endoplasmic reticulum (ER). If uncorrected, such “ER stress” promotes cell degeneration and apoptosis. ER stress activates unfolded protein response (UPR) signaling pathways that determine cell fate. Remediable ER stress activates adaptive (‘A’)-UPR outputs that favor cell survival. But under irremediably high, chronic ER stress, these adaptive measures wane, as alternate terminal (‘T’)-UPR outputs trigger apoptosis.
High/chronic ER stress promotes numerous diseases of premature cell loss (Oakes and Papa, 2015). For example, pancreatic islet β-cells, responsible for synthesizing and secreting sufficient quantities of insulin to maintain glucose homeostasis, commonly experience high ER stress and secretory exhaustion (Scheuner and Kaufman, 2008). Peripheral insulin resistance further elevates β-cell insulin secretory demand during development of type 2 diabetes (T2D)(Back and Kaufman, 2012). Insulin gene mutations cause encoded proinsulin to become structurally arrested in the β-cell ER, and various UPR gene deletions debilitate insulin production by β-cells. Dysregulated UPR signaling promotes β-cell autonomous apoptosis in these diverse diabetes syndromes (Ozcan et al., 2004; Papa, 2012).
Type 1 diabetes (T1D) is triggered by immune dysregulation and autoreactive T cell responses against β-cells. However, the autoreactivity does not inevitably result in direct β-cell destruction (fratricide) but will also induce β-cells to autonomously undergo apoptosis (suicide) during disease progression (Atkinson et al., 2011; Bottazzo, 1986). Both human and mouse studies have suggested that at the time of T1D diagnosis as much as 30–40% of β-cells remain and are functionally unresponsive but can recover following removal of stress, suggesting that a window of opportunity may exist for therapies that prevent further β-cell deterioration and restore β-cell function (Alanentalo et al., 2010; Krogvold et al., 2015).
The non-obese diabetic ‘NOD’ mouse develops β-cell failure subsequent to innate immune and T-cell islet infiltration (as in humans with T1D)(Anderson and Bluestone, 2005). We found that treating NOD mice with the anti-cancer drug, imatinib, both prevents and reverses diabetes, inducing prolonged remission (Louvet et al., 2008). This remarkable efficacy in the NOD has prompted a phase II clinical trial to repurpose imatinib for new-onset T1D. However, a full understanding of the underlying mechanism of imatinib’s efficacy has remained unclear. Enigmatically, imatinib shows minimal effects on T cell effector function and trafficking in the NOD. Insulitis scores, CD4+/CD8+ ratios in spleen and pancreatic lymph nodes, and regulatory T cell function remained unchanged, supporting the notion that imatinib’s anti-diabetic effect is not simply due to immune modulation (Louvet et al., 2008). Recently, investigators reported high ER stress signaling in autoimmune-targeted β-cells of the NOD (Engin et al., 2013; Tersey et al., 2012), which prompted us to inquire whether imatinib may instead protect β-cells in the NOD by modulating the UPR.
The ER transmembrane kinase/endoribonuclease (RNase), IRE1α, determines cell fate based on ER stress severity. Under ER stress, IRE1α monomers in the ER membrane undergo trans-autophosphorylation and RNase activation, thereby initiating frame-shift splicing of the mRNA encoding XBP1 transcription factor to trigger adaptive UPR transcriptional programs (Yoshida et al., 2001). If ER stress remains unrelieved, IRE1α organizes into high-order, oligomeric complexes as its autophosphorylation and RNase activation state rise further, thereby causing endonucleolytic degradation of many ER-localized mRNAs and apoptosis (Han et al., 2009a). Despite this mechanistic understanding, the exact components of the IRE1α complex remain largely unresolved.
Here we find that imatinib’s anti-diabetogenic effects in the NOD derive from ameliorating pro-apoptotic terminal UPR signaling in β-cells through an unexpected link between IRE1α and the non-receptor ABL tyrosine kinases, which play diverse, intracellular signaling functions but have not previously been characterized as UPR components. We find that this ABL-IRE1α axis functions upstream in the UPR to potentiate apoptosis during ER stress, and that imatinib reduces apoptosis by attenuating a stimulatory interaction of ABL with IRE1α. These findings predicted that direct inhibition of IRE1α should prove anti-diabetogenic in the NOD. Enabled with newly optimized compounds called ‘KIRA’s (kinase-inhibitory RNase attenuators) that inhibit IRE1α kinase/RNase activity, we found that a mono-selective KIRA induces near-complete reversal of established diabetes in the NOD model.
RESULTS
Imatinib blunts the terminal UPR and apoptosis
High ER stress signaling has been reported in autoimmune-targeted NOD islets (Engin et al., 2013; Tersey et al., 2012). To characterize signature A- and T-UPR events in NOD islets, we examined IRE1α activation state prior to diabetes onset. IRE1α became progressively more induced and activation-loop autophosphorylated in NOD islets at 10 and 12 weeks of age, coinciding with increasing pre-diabetic insulitis (Figure 1A)(Anderson and Bluestone, 2005). Islets from 12 week-old NOD mice show elevated thioredoxin-interacting protein (TXNIP), a key T-UPR mediator induced by IRE1α hyperactivation, that activates the NLRP3 inflammasome to promote islet inflammation and β-cell death (Figure 1A)(Lerner et al., 2012). TXNIP mRNA progressively rose, and proinsulin–encoding, Ins1 and Ins2, mRNAs, which we identified as IRE1α RNase substrates (Han et al., 2009a), progressively decayed in pre-diabetic NOD islets compared to age-matched immune-deficient NSG controls, which neither develop immune infiltrates nor β-cell dysfunction (Figure 1B). During the pre-diabetic window of rising T-UPR outputs, A-UPR mediators, including spliced XBP1 mRNA, and mRNAs encoding the ER chaperone BiP and cytoprotective MANF (Lindahl et al., 2014), waned (Figure 1C). By the time of overt disease, insulin mRNAs declined further, while TXNIP mRNA continued to rise (Figure 1D). Thus, NOD islets morph from an A-to a T-UPR signature after development of insulitis, and before progression to frank diabetes (Figure 1E).
Figure 1. A T-UPR signature in NOD islets precedes diabetes onset and is attenuated by imatinib.
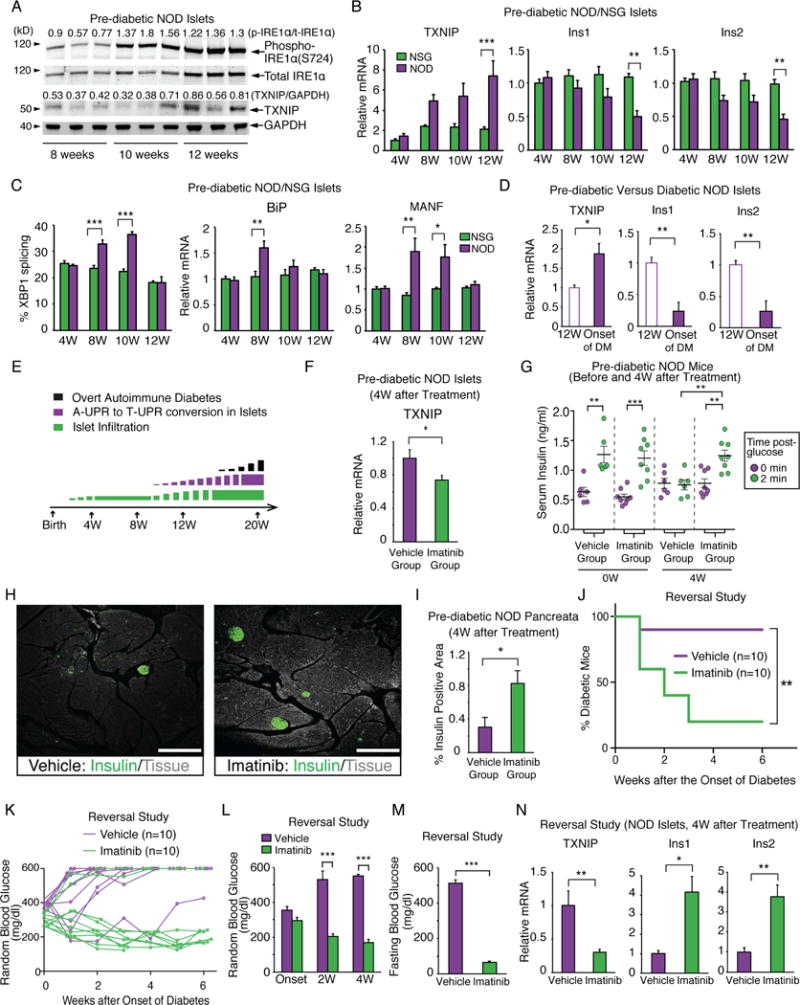
(A) Immunoblots, with signal intensity ratios, for phospho-(S724) and total IRE1α and TXNIP in islets from NOD mice at indicated ages. Each lane is from an individual mouse. (B) qPCR of relative TXNIP, Ins1 and Ins2 mRNA levels in NOD (n=5) and control NSG (n=5) islets. (C) % spliced XBP1 mRNA and qPCR of relative BiP and MANF mRNA levels from (B). (D) Relative Ins1, Ins2 and TXNIP mRNA levels in 12-wk-old pre-diabetic (n=3) and new-onset diabetic NOD mice (n=5) islets. (E) Temporality of insulitis, adaptive (A-) to terminal (T-) UPR conversion, and overt diabetes (range 16–30 weeks) in NOD mice. (F) Relative TXNIP mRNA levels in islets from pre-diabetic NOD mice treated with imatinib or vehicle for 4 weeks. (G) First-phase insulin response in pre-diabetic NOD mice before and after 4 weeks of imatinib (n=8) or vehicle (n=6). Each symbol denotes an individual mouse. (H and I) Immunofluorescence of insulin and % insulin-positive area in pancreata from NOD mice treated with imatinib or vehicle for 4 weeks. 12–16 sections per group were analyzed. 3–4 mice per group. (H) Representative images of insulin (green) and tissue sectional area (grey). Scale bar, 200 μm. Imatinib was started at 10 weeks of age for (F–I). (J) % diabetic NOD mice treated upon disease onset (blood glucose >250 mg/dl) for 4 weeks with imatinib (n=10) or vehicle (n=10). Log-rank test used for statistical analysis. (K) Individual blood glucose (BG) levels for (J). (L and M) Random (L) and 17 hr-fasting (M) BGs of mice treated for 4 weeks with imatinib (n=6) or vehicle (n=4) at disease onset. (N) qPCR of relative TXNIP, Ins1 and Ins2 mRNA levels in islets of mice in (L and M). Two-way ANOVA followed by post-hoc Tukey’s test used for statistical analysis (B, C, G, and L). Bars; mean±SEM. n; number of mice. P-values: * < 0.05, ** < 0.01, *** < 0.001. See also Figure S1 and Table S3.
As imatinib can prevent and reverse diabetes in the NOD (Louvet et al., 2008), we hypothesized that its efficacy may derive in part from attenuating the T-UPR in islets. Daily oral dosing of pre-diabetic NOD mice with imatinib for 4 weeks significantly decreased islet TXNIP mRNA, preserved first-phase serum insulin, and doubled insulin positive area in pancreata, without changing body weight (Figures 1F–I, S1A). Newly-diabetic NODs started on daily imatinib showed 80% reversal within three weeks (Figure 1J). Random and fasting blood glucose levels were significantly lower in the imatinib group within two and four weeks, respectively, while their islets showed attenuated TXNIP and preserved insulin mRNAs (Figure 1K–N).
While imatinib blunts the T-UPR in NOD islets, whether it confers a direct, immune-independent, cytoprotective effect on β-cells was unclear. To address this, we asked whether immune-deficient NSG mice islets are protected by imatinib from toxic doses of tunicamycin (Tm), a protein glycosylation inhibitor that causes apoptosis (Ghosh et al., 2014). In Tm-exposed NSG islets, imatinib blocks elevations in spliced XBP1 and TXNIP mRNAs and declines in insulin mRNAs, with similar results in human islets (Figures 2A,B,S1B). In NSG islets, Tm-promoted decreases in insulin content and glucose-stimulated insulin secretion (GSIS) are prevented by imatinib (Figure 2C,D). In the absence of ER stress agents, imatinib did not change TXNIP and Ins1/2 mRNA, and insulin secretion (Fig. S1C–E). Also, imatinib reduces Tm-promoted TXNIP induction in C57BL/6 islets (Figure 2E), showing that its effects are not strain-specific. Finally, imatinib decreases Tm-promoted apoptosis in NSG islets (Figure 2F,G).
Figure 2. Imatinib reduces ER stress-induced T-UPR endpoints and β-cell death.
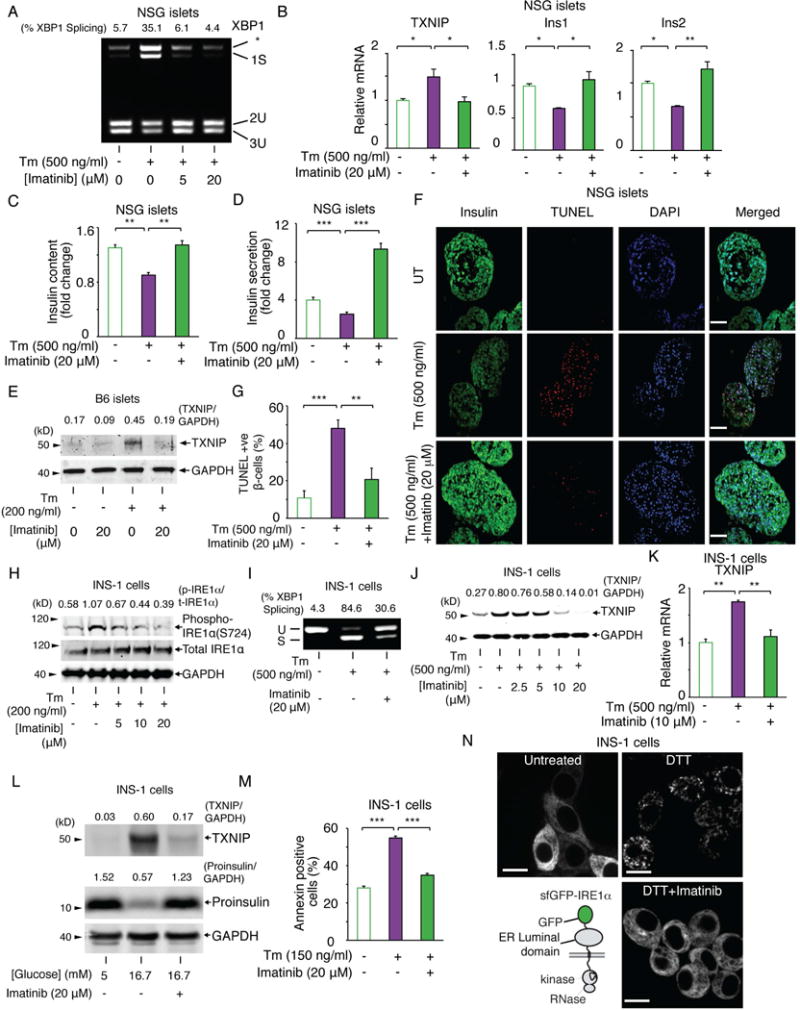
(A) PstI-digested XBP1 cDNA amplicons from NSG islets treated for 4 hr with indicated [imatinib], then ± Tm for 4 hr. (B) qPCR of relative mRNAs from NSG islets treated for 4 hr ± imatinib, then ± Tm for 4 hr. (C) Cellular insulin protein content in NSG islets treated ± imatinib for 2 hr, then ± Tm for 16 hr. (D) Glucose-stimulated insulin secretion in NSG islets treated ± imatinib for 2 hr, then ± Tm for 16 hr. For (C and D), [Glucose] was 2.5 mM or 28 mM for 30 min. (C and D) Data shown as ratio of insulin levels at 28 mM over 2.5 mM glucose. (E) TXNIP immunoblots from C57BL/6 islets treated ± imatinib for 2 hr, then ± Tm for 16 hr. (F) Immunofluorescence of NSG islets treated ± imatinib for 2 hr, then ± Tm for 16 hr; DAPI (blue), Insulin (green), and TUNEL (red). Scale bar, 50 μm. (G) Quantified TUNEL-positive β-cells normalized to DAPI-positive cells in (F). (H) Immunoblots for indicated proteins in INS-1 cells treated ± Tm and indicated [imatinib] for 8 hr. (I) XBP1 cDNA amplicons from INS-1 cells pretreated for 4 hr ± imatinib, then ± Tm for 4 hr. (J) TXNIP immunoblots from INS-1 cells co-treated with ± Tm and indicated [imatinib] for 24 hr. (K) qPCR for TXNIP mRNA in INS-1 cells treated for 2 hr with imatinib, then Tm for 4 hr. (L) TXNIP and proinsulin immunoblots from INS-1 cells treated with indicated [glucose] ± imatinib for 72 hr. (M) Annexin V staining of INS-1 cells co-treated with Tm and ± imatinib for 72 hr. (N) sfGFP-IRE1α reporter (left bottom). Images of INS-1 expressed sfGFP-IRE1α treated ± imatinib for 1 hr, then ± 10 mM DTT for 1 hr. Scale bar, 10 μm. Bars; mean±SEM. Three independent biological samples were used for qPCR, insulin content, insulin secretion, immunofluorescence experiments, and Annexin V staining. P-values: * < 0.05, ** < 0.01, *** < 0.001. See also Figure S1.
We next used a β-cell derived insulinoma line, INS-1, which can be genetically manipulated, to study imatinib’s mechanistic effects. In INS-1 cells, Tm-promoted IRE1α autophosphorylation, XBP1 mRNA splicing and TXNIP mRNA/protein induction are all inhibited by imatinib (Figure 2H–K). Nilotinib, an equally selective but more potent inhibitor than imatinib, reduced Tm-promoted XBP1 splicing (Figure S1F,G), TXNIP mRNA induction (Figure S1H), and apoptosis at lower concentrations than imatinib (Figure 2M, S1I). Imatinib’s salutary effects extend broadly to other ER stress regimes, including SERCA pump inhibition by thapsigargin (Tg) and anterograde trafficking blockage by brefeldin A (Figure S1J,K). Glucotoxicity-promoted insulin mRNA decay (Lipson et al., 2006), TXNIP induction, and proinsulin depletion are all also prevented by imatinib (Figures 2L, S1L). As in islets, imatinib significantly reduces ER stress-induced INS-1 cell apoptosis (Figure 2M).
Suggesting that it inhibits the upstream-most UPR signaling step of IRE1α oligomerization, imatinib blocks ER membrane focal aggregation of a superfolder green fluorescent protein (sf)GFP-IRE1α reporter during DTT-induced ER stress (Figure 2N)(Ghosh et al., 2014). But while imatinib inhibits IRE1α in cells, it does not directly inhibit either the kinase or RNase activities of a recombinant IRE1α* mini-protein (Figure S1M–P). Therefore, imatinib’s inhibitory effects in cells appear to occur through a different target than IRE1α.
c-Abl drives the T-UPR through IRE1α
As imatinib was optimized for BCR-Abl (Capdeville et al., 2002), we next monitored the activation state of its non-oncogenic counterpart, c-Abl, in NOD islets. Like IRE1α, c-Abl becomes progressively more induced and activation-loop phosphorylated in pre-diabetic NOD islets (Figures 1A, 3A). Also, in INS-1 cells, high glucose triggered acute c-Abl mRNA induction and c-Abl activation, which was abrogated by imatinib (Figures 3B,C, S2A,B). c-Abl staining was evident in β-cells, but not in infiltrating immune cells based on lack of co-localization with CD45 and DAPI (Figure S2C–T), with higher protein and mRNA levels in NOD compared to NSG islets (Figure S3A,B). Other imatinib targets PDGFRα, PDGFRβ, and c-KIT, were not detectable in β-cells (Figure S3C). In islets, c-Abl co-localizes with synaptophysin in neuroendocrine cells, and with glucagon in α-cells, but is undetectable in cells expressing pancreatic polypeptide or somatostatin (Figure S2C–T). Together, these data suggested that imatinib’s efficacy in the NOD may derive through targeting β-cell-expressed c-Abl.
Figure 3. ABL family tyrosine kinases are necessary and sufficient for driving T-UPR-mediated apoptosis through IRE1α.
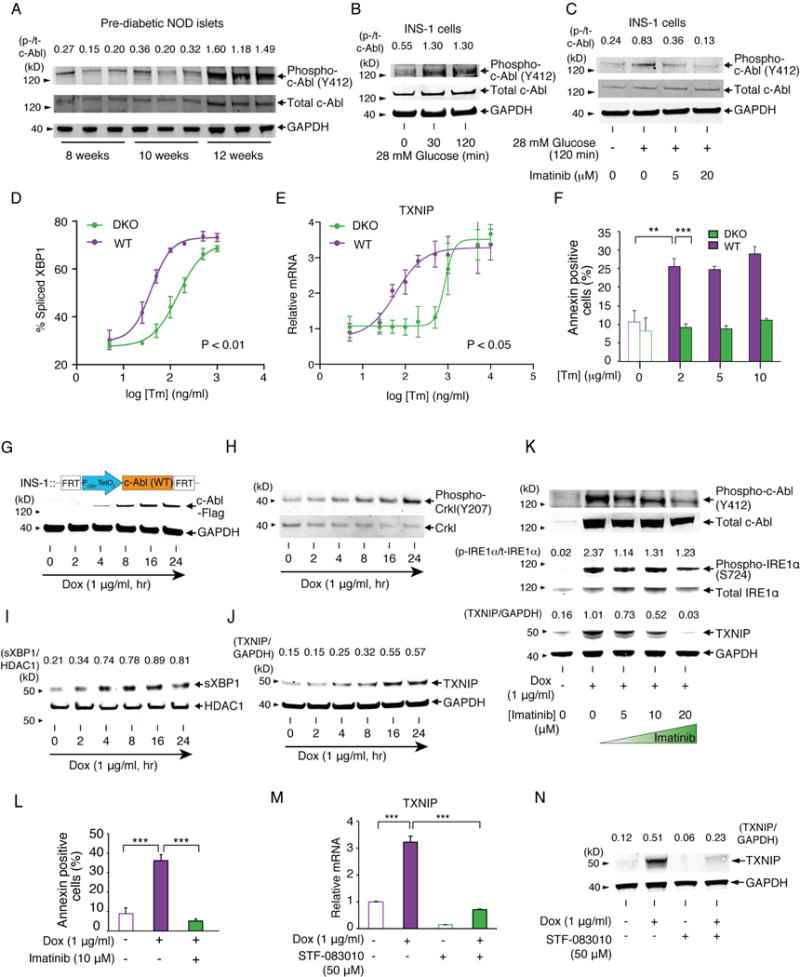
(A) Phospho-(Y412) and total c-Abl immunoblots from islets in Figure 1A with phospho/total c-Abl signal intensity ratios. The same GAPDH blot from Figure 1A is again shown for protein normalization. (B) Phospho- and total c-Abl immunoblots from INS-1 cells incubated in 5 mM glucose for 17 hr, then 28 mM glucose for indicated times. (C) Phospho- and total c-Abl immunoblots for INS-1 cells incubated in 5 mM glucose for 17 hr, then indicated [imatinib] for 2 hr, then ± 28 mM glucose for 2 hr. (D and E) % spliced XBP1 (quantified from PstI-digested XBP1 cDNA amplicons) (D) and qPCR of relative TXNIP mRNAs normalized to no Tm (E) from c-Abl/Arg DKO or WT MEFs treated with indicated [Tm] for 3 hr. Statistical analysis; two-way ANOVA (D and E). (F) Annexin V staining of c-Abl/Arg DKO or WT MEFs treated with indicated [Tm] for 24 hr. Statistical analysis; two-way ANOVA followed by post-hoc Tukey’s test. (G-J) Immunoblots for indicated proteins in INS-1 stably cells expressing WT c-Abl under Dox for indicated times. Nuclear protein extract used for (I). (K) Immunoblots for indicated proteins in INS-1 cells expressing WT c-Abl ± Dox, co-treated with indicated [imatinb] for 72 hr. (L) Annexin V staining of INS-1 cells expressing WT c-Abl ± Dox, co-treated with ± imatinib for 96 hr. (M) qPCR of relative TXNIP mRNA in INS-1 cells expressing WT c-Abl under Dox, co-treated ± STF-083010 for 48 hr. (N) Immunoblots for TXNIP in INS-1 cells expressing WT c-Abl under Dox, co-treated ± STF-083010 for 48 hr. Bars; mean±SEM. Three independent biological samples were used for XBP1 splicing, qPCR, and Annexin V staining. P-values: ** < 0.01, *** < 0.001. See also Figure S2, S3, and S4.
The ABL tyrosine kinase family comprises c-Abl (ABL1), and Arg (ABL2), which display redundant and unique functions (Wang, 2014). To study the UPR roles of c-Abl and Arg, we subjected Abl/Arg (−/−) double-knockout (DKO) mouse embryonic fibroblasts (MEFs) to ER stress. XBP1 mRNA splicing, dose-dependently induced by Tm, is significantly blunted in Abl/Arg DKO MEFs (Figure 3D, S4A); c-Abl and Arg appear to play redundant roles because c-Abl (−/−) single knockout MEFs show no XBP1 splicing defect (Figure S4B). Under ER stress, Abl/Arg DKO MEFs show crippled TXNIP mRNA induction and apoptosis (Figures 3E,F, S4C).
Given the necessity of c-Abl/Arg in promoting the T-UPR, we next tested their sufficiency in isogenic INS-1 and T-REx293 lines that stably overexpress c-Abl or Arg under doxycycline (Dox). In T-REx293 cells, induction of c-Abl, capable of phosphorylation of endogenous Crkl, causes spontaneous autophosphorylation of endogenous IRE1α—without ER stress—and rapidly triggers XBP1 mRNA splicing and nuclear accumulation of XBP1s transcription factor (Figure S4D–H). Similarly, c-Abl induction in INS-1 cells leads to nuclear XBP1s enrichment and TXNIP induction, while imatinib inhibits IRE1α autophosphorylation, TXNIP elevation, and apoptosis (Figure 3G–L).
Overexpression of Arg is also sufficient to induce TXNIP (Figure S4I). In contrast, overexpression of other imatinib targets, PDGFRα and c-kit, did not induce T-UPR events (Figure S4J,K). c-Abl and Arg did not affect PERK, another UPR sensor kinase, based on unchanged phosphorylation of its substrate eiF2α in Abl/Arg DKO MEFs under ER stress, or in INS-1 cells overexpressing c-Abl (Figure S4L,M). The IRE1α RNase inhibitor—STF-083010—(Papandreou et al., 2011) blocks c-Abl-induced TXNIP mRNA and protein elevation (Figure 3M,N), further supporting that c-Abl-driven T-UPR signaling proceeds through IRE1α RNase activation.
c-Abl binds and activates IRE1α
Tyrosine phosphorylation has not previously been reported to play a role in the UPR, despite the necessity and sufficiency of ABL kinases for stimulating IRE1α. Therefore, the necessity of c-Abl’s tyrosine kinase activity was tested in an INS-1 line conditionally expressing a kinase-dead mutant, (K290R). Surprisingly, K290R c-Abl triggers nuclear accumulation of XBP1s and TXNIP induction to similar levels as WT c-Abl (Figure 4A–D). Remarkably, imatinib reduces K290R c-Abl-induced TXNIP protein elevation (Figure 4E), like WT c-Abl (Figure 3K).
Figure 4. Co-localization of c-Abl with IRE1α at the ER membrane drives the T-UPR through a scaffolding effect.
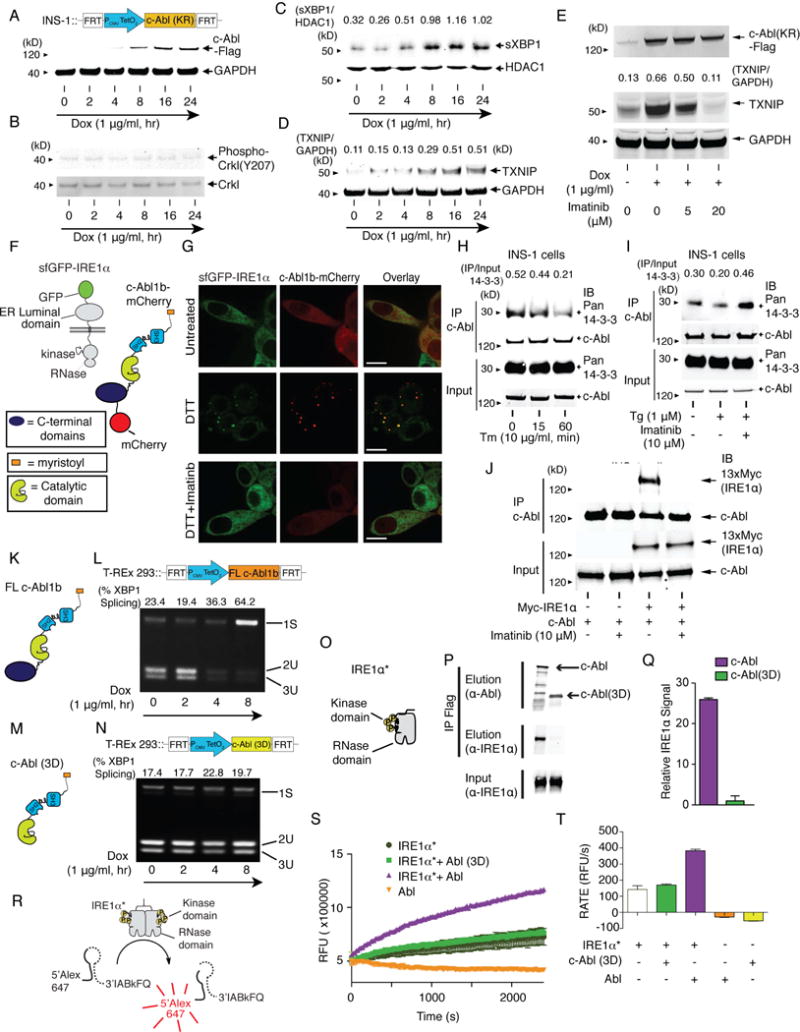
(A–D) Immunoblots for indicated proteins in INS-1 cells stably expressing K290R c-Abl under Dox for indicated times. Nuclear protein extract used for (C). (E) Immunoblots for indicated proteins in INS-1 cells expressing K290R c-Abl ± Dox, co-treated with indicated [imatinib] for 48 hr. (F) c-Abl1b-mCherry and sfGFP-IRE1α reporters. (G) Confocal micrographs of INS-1 cells expressing sfGFP-IRE1α and transiently expressing full-length c-Abl1b-mCherry treated ± imatinib for 1 hr, then 10 mM DTT for 1 hr. Scale bar, 10 μm. (H) Immunoprecipitation (IP) of c-Abl, then immunoblot for pan 14-3-3 and c-Abl from INS-1 cells treated with Tm for indicated times. (I) IP of c-Abl, then immunoblot for pan 14-3-3 and c-Abl from INS-1 cells treated ± imatinib for 2 hr, then Tg for 2 hr. (J) IP of c-Abl, then immunoblot for Myc-IRE1α and c-Abl from INS-1 cells expressing WT c-Abl ± transient expression of a 13×-Myc tagged-IRE1α ± imatinib for 48 hr. (K and M) Full-length (FL) c-Abl1b and c-Abl(3D), lacking residues C-terminal to the kinase. (L and N) PstI-digested XBP1 cDNA amplicons from T-REx 293 cells expressing FL c-Abl1b (L) or c-Abl(3D) (N) under Dox for indicated times. (O) IRE1α*, a recombinant mini-protein containing cytosolic kinase and RNase domains. (P) IP of IRE1α* with Flag-tagged FL c-Abl1b or c-Abl(3D) transiently expressed in T-REx 293. (Q) Quantification of purified IRE1α* (normalized to eluted c-Abl levels) IP-ed in (P). Mean±SEM. Triplicated. (R) XBP1 mini-substrate. (S) RNase activity of IRE1α* (200 ng) ± Abl(3D) (50 ng) or ± c-Abl (50 ng) by real time fluorescence. Mean±SEM. Triplicated. (T) Rate of XBP1 mini-substrate cleavage by IRE1α* ± Abl(3D) or ± c-Abl. Mean±SEM. Triplicated. See also Figure S5.
These results suggest that c-Abl phosphotransfer-independently stimulates IRE1α, and that imatinib counters this effect apart from kinase catalytic inhibition. We therefore reasoned that under ER stress c-Abl may co-localize with IRE1α to scaffold and stimulate IRE1α’s activity, with imatinib countering this interaction. To test this, we performed live cell imaging with sfGFP-IRE1α and a c-Abl-mCherry fusion. In INS-1 cells, c-Abl-mCherry is diffusely localized, but with significant ER enrichment, consistent with previous reports (Figure 4F,G)(Qi and Mochly-Rosen, 2008). Under ER stress, c-Abl-mCherry co-localizes with sfGFP-IRE1α in ER punctate foci, while imatinib co-treatment prevents c-Abl-mCherry focal re-localization, as with sfGFP-IRE1α (Figure 2N, 4G).
We next tested whether c-Abl’s association with cytosolic 14-3-3 proteins that modulate localization of c-Abl and other kinases is disrupted under ER stress, as during oxidative stress (Nihira et al., 2008; Yoshida et al., 2005). Indeed, ER stress causes rapid dissociation of the c-Abl/14-3-3 complex, which imatinib prevents (Figures 4H,I, S5A). These data further support a model of c-Abl re-localizing from the cytosol to an IRE1α complex at the ER membrane under stress. Also, c-Abl co-immunoprecipitates (co-IPs) either endogenous or transgenic IRE1α, which imatinib abrogates (Figures 4J, S5B,C).
The N-terminal myristoyl group of c-Abl is necessary for directing c-Abl to IRE1α because a non-myristoylated splice variant—Abl1a—cannot rescue XBP1 splicing or TXNIP mRNA elevation when reconstituted in Abl/Arg DKO MEFs, unlike N-terminally myristoylated, Abl1b (Figure S5D–G). We then asked if the N-terminal SH2, SH3, and kinase domains of c-Abl are sufficient for IRE1α stimulation by testing an N-terminal truncation–Abl(3D). XBP1 splicing is abrogated in cells expressing Abl(3D) (Figure 4K–N, S5F,H), showing that c-Abl’s C-terminal domains are also required for IRE1α activation. Also, unlike full-length c-Abl, Abl(3D) cannot co-IP recombinant IRE1α* (Figure 4O–Q). Finally, to determine if c-Abl and IRE1α directly interact, we conducted biochemical experiments using recombinant c-Abl (Figure S5I) and IRE1α*. Purified, full-length c-Abl, immobilized on beads with an ATP-competitive inhibitor, co-IPs IRE1α* (Figure S5J). Finally, purified c-Abl or c-Abl K290R can—with equal potency—directly stimulate IRE1α*’s RNase catalytic activity in vitro, whereas Abl(3D) cannot (Figure 4R–T, S5K–M).
Thus, under ER stress, c-Abl scaffolds and stimulates IRE1α at the ER membrane independent of its phosphotransfer activity, but requires both its N-myristoyl group and domains C-terminal to its kinase. Thus, we predicted that forcibly directing c-Abl into IRE1α foci should hyperactivate IRE1α without ER stress. To test this, we used GNF-2 (Figure 5A), an inhibitor that interacts with the myristate-binding pocket in c-Abl’s catalytic domain, thereby displacing c-Abl’s N-terminal myristoyl group and enhancing ER localization (Choi et al., 2009). Consistent with previous studies, we further find that GNF-2 promotes spontaneous formation of ER foci containing both c-Abl-mCherry and sfGFP-IRE1α, correlating with diminished c-Abl/14-3-3 interaction, without ER stress (Figure 5B,C). In INS-1 cells, GNF-2 reduces Crkl phosphorylation, while increasing IRE1α autophosphorylation, XBP1 splicing, TXNIP mRNA/protein induction, Ins1 mRNA decay, and apoptosis (Figure 5D–K); GNF-2 does not increase XBP1 splicing in Abl/Arg DKO MEFs or PERK-mediated eiF2α phosphorylation (Figure S6A,B). In summary, although GNF-2 allosterically inhibits c-Abl’s phosphotransfer activity, its ability to promote c-Abl co-localization with IRE1α suffices to hyperactivate IRE1α’s RNase, which can be inhibited with STF-083010 (Figure S6C).
Figure 5. Allosteric c-Abl inhibitor, GNF-2, forces c-Abl to co-localize with IRE1α to promote apoptosis in the absence of ER stress.
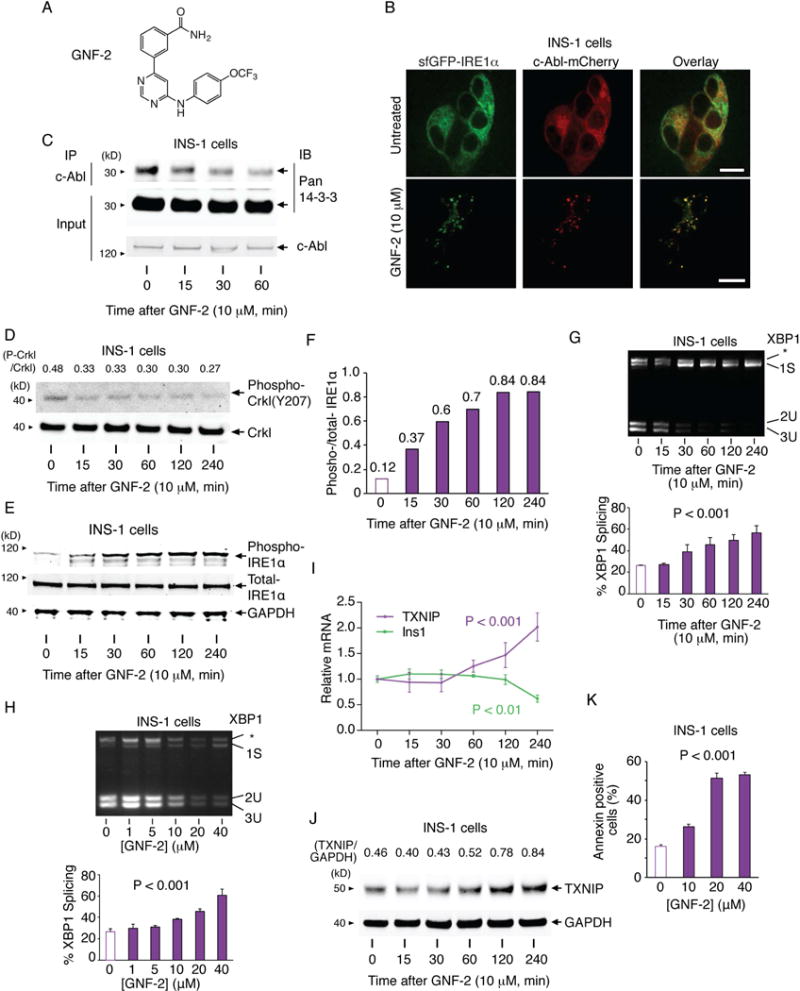
(A) Structure of GNF-2. (B) Confocal micrographs of INS-1 cells stably expressing sfGFP-IRE1α and transiently expressing c-Abl1b-mCherry, ± GNF-2 for 1 hr. Scale bar, 10 m. (C) IP of c-Abl with blotting for pan 14-3-3 and c-Abl from INS-1 cells treated with GNF-2 for indicated times. (D) Immunoblots for indicated proteins in INS-1 cells treated with GNF-2 for indicated times. (E and F) Immunoblots for phospho (S724) and total IRE1α in INS-1 cells treated with GNF-2 for indicated times. (F) Relative signal intensity for phospho/total IRE1α. (G and H) PstI-digested XBP1 cDNA amplicons from INS-1 cells treated with GNF-2 for indicated times (G) or indicated [GNF-2] for 1 hr (H). Quantified % spliced XBP1 (bottom). (I) qPCR of relative indicated mRNAs in INS-1 cells treated with GNF-2 for indicated times. (J) TXNIP immunoblots from INS-1 cells treated with GNF-2 for indicated times. (K) Annexin V staining of INS-1 cells treated with indicated [GNF-2] for 48 hr. Statistical analysis; one-way ANOVA with post-hoc test for trend (G-I and K). Bars; mean±SEM. Three independent biological samples were used for XBP1 splicing, qPCR, and Annexin V staining. See also Figure S6.
IRE1α inhibition spares β-cells to reverse diabetes in the NOD mouse
By investigating the mechanism of imatinib’s efficacy in the NOD, we identified an ABL-IRE1α signaling axis, leading to a final prediction: if IRE1α hyperactivity promotes β-cell death in the NOD, then direct IRE1α inhibition should be anti-diabetogenic. To test this, we treated pre-diabetic NOD females with KIRA6, an ATP-competitive ligand that allosterically inhibits IRE1α’s RNase by breaking oligomers. Using an intraperitoneal (i.p.) dosing regime that reduces diabetes in the Akita mouse— which expresses an oxidative-folding defective proinsulin mutant, Ins2(C96Y), that triggers autonomous β-cell apoptosis (Ghosh et al., 2014; Lerner et al., 2012)—we noted significant reduction of TXNIP and recovery of Ins1/Ins2, BiP, and MANF mRNAs within one week of treatment in 10 week-old NODs (Figure S6D–H). After a 6 week KIRA6 treatment of 8 week old pre-diabetic NODs, preserved first-phase insulin response—and significantly greater pancreatic insulin protein staining—was evident in the KIRA6 group compared to vehicles (Figure S6I–K). Diabetes reversal was attempted by dosing KIRA6 upon disease onset. Random blood glucoses stabilized in the KIRA6 cohort, whereas hyperglycemia continued to rise in controls, without significant weight differences between groups (Figure S6L,M). By 4 weeks, significant differences in the percentage of diabetic mice in the two cohorts became apparent, and KIRA6-treated mice showed preserved fasting insulin (Figure S6N,O).
Despite these encouraging results, KIRA6 inhibits c-Abl at micromolar concentrations (Figure S7A), raising the possibility that KIRA6’s anti-diabetogenic effects proceed partially through ABL inhibition. But recently a mono-selective IRE1α inhibitor–compound 18 (Figure 6A)–that possesses all the properties of a KIRA, and will be referred to as KIRA8 henceforth, was described (Harrington et al., 2015). Specifically, KIRA8 blocks IRE1α* oligomerization, and potently inhibits IRE1α* RNase activity against XBP1 and Ins2 RNAs (Figure 6B–E). KIRA8 more potently reduces IRE1α-driven apoptosis in INS-1 cells than KIRA6 (Figure 6F) and also reverses XBP1 splicing promoted by GNF-2 (Figure 6G). We confirmed KIRA8’s mono-selectivity for IRE1α in vitro, noting that it even has minimal inhibition on IRE1α’s closely related paralog, IRE1β (Wang et al., 1998)(Figure S7B, Table S1); other kinases operating in the UPR, including PERK, c-Abl, and Arg are not inhibited by KIRA8.
Figure 6. Mono-selective KIRA8 spares β-cells and reverses established diabetes in NOD mice.
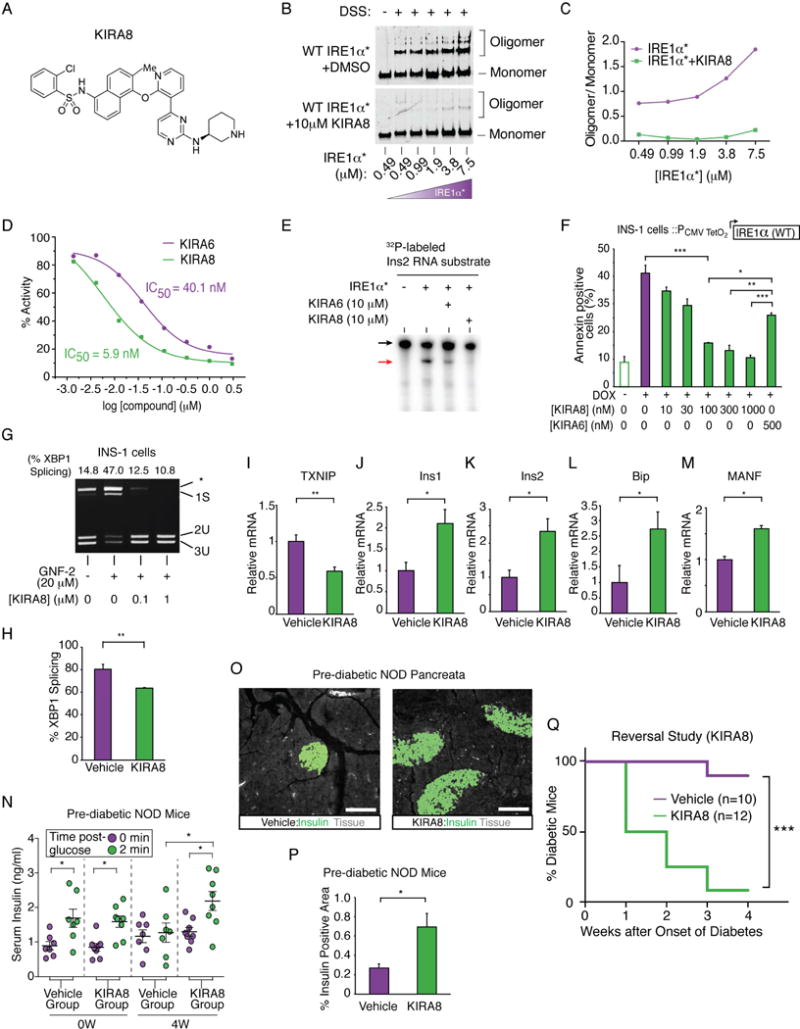
(A) Structure of KIRA8. (B) Immunoblots of DSS-crosslinked varying [IRE1α*] ± KIRA8. (C) Quantified oligomer/monomer ratio. (D) % IRE1α* RNase activity at indicated [compound]. IC50: KIRA6, 40.1 nM; KIRA8, 5.9 nM. (E) Cleavage of α32P-labeled mouse Ins2 RNA by IRE1α* ± KIRA6 or KIRA8. (F) Annexin V staining of INS-1 cells expressing IRE1α under Dox, co-treated with indicated [compound] for 72 hr. Three independent biological samples were used. (G) PstI-digested XBP1 cDNA amplicons from INS-1 cells treated with indicated [KIRA8] for 2 hr, then GNF-2 for 1 hr. (H-M) Indicated mRNAs from islets of NOD females injected with 50 mg/kg KIRA8 (n=4) or vehicle (n=3) i.p., daily, starting at 10 wks age for 1 wk. (N) First-phase insulin response in pre-diabetic NOD mice before and after 4 weeks of KIRA8 (n=8) or vehicle (n=7), daily, starting at 10 weeks of age. (O and P) Immunofluorescence of insulin and quantified insulin-positive area in pancreata from NOD mice treated daily with KIRA8 (n=5) or vehicle (n=4) for 6 weeks starting at 10 weeks of age. (O) Representative images of insulin stain (green) and tissue sectional area (grey). Scale bar, 100 μm. (Q) % diabetic mice in KIRA8- (n=12) and vehicle-treated (n=10) groups; KIRA8 or vehicle was initiated at disease onset. Statistical analysis; log-rank test. Bars; mean±SEM. n; number of mice. P-values: * < 0.05, ** < 0.01, *** < 0.001. See also Figure S7, Table S2 and S3.
KIRA8 was first tested in vivo in 3-week-old male pre-diabetic Akita mice by i.p. daily dosing of 50 mg/kg (Table S2), whereupon significant reduction of hyperglycemia became apparent over several weeks (Figure S7C). This encouraging anti-diabetogenic efficacy of KIRA8 in the focal Akita model motivated us to test it in the complex NOD. Showing target engagement, one week treatment of pre-diabetic NODs with KIRA8 led to significant reductions in islet XBP1 splicing and TXNIP mRNAs, and preserved Ins1/Ins2, BiP and MANF mRNAs (Figures 6H–M); no significant effects were apparent in T-UPR endpoints in B6 mice treated similarly with KIRA8 (Figure S7D–F). Upon longer treatment durations in 10-week-old prediabetic NODs, KIRA8 preserved first-phase insulin responses by 4 weeks, and by 6 weeks significantly increased—by three-fold—the levels of pancreatic insulin positive area (Figure 6N–P). Although a trend towards an increased proportion of islets without insulitis was noted in KIRA8-treated mice, overall insulitis severity was not significantly reduced relative to vehicle (Figure S7G).
Finally, we conducted an NOD diabetes reversal study, starting KIRA8 daily upon disease onset. Random blood glucose rapidly decreased in the KIRA8 cohort relative to controls, with no weight differences seen (Figure S7H–J). By four weeks, statistically significant differences in the percentages of diabetic mice became apparent, with a remarkable >90% diabetes reversal rate in the KIRA8 cohort (Figure 6Q).
DISCUSSION
Role of the ABL-IRE1α axis in amplifying the UPR
The UPR is canonically viewed as a network of signaling cascades emanating from three ER transmembrane sensors—IRE1α, PERK, and ATF6—activated by unfolded proteins in the ER. However, it is becoming apparent that many cytosolic proteins characterized as components of other signaling pathways interact with UPR sensors to regulate their outputs, constituting a complex and dynamically-regulated “UPRosome” (Hetz and Glimcher, 2009). For example, protein-tyrosine phosphatase 1B, BAX inhibitor-1, ASK1, RACK1, and nonmuscle myosin IIB tune IRE1α outputs to affect cell fate (Gu et al., 2004; He et al., 2012; Lisbona et al., 2009; Nishitoh et al., 2002; Qiu et al., 2010). Here we found that ABL kinases are also integral UPR components that rheostatically regulate IRE1α.
Under high/chronic ER stress, a scaffolding interaction between ABL and IRE1α at the ER membrane increases IRE1α’s enzymatic activities to critical levels, potentiating the T-UPR (Figure 7). This ABL-IRE1α link was found by delineating the mechanism of how the anti-cancer drug—Gleevec (imatinib)—prevents and reverses diabetes in the NOD mouse, then confirmed and validated in multiple, reductionist, cell-based systems. Based on these studies, c-Abl and Arg play necessary, overlapping roles in amplifying IRE1α’s response to ER stress. Furthermore, forced activation of either ABL kinase is sufficient to drive IRE1α autophosphorylation, XBP1 mRNA splicing, ER-localized mRNA decay, induction of TXNIP, and apoptosis. All these outputs are curtailed by imatinib.
Figure 7. Model of the ABL-IRE1α axis.
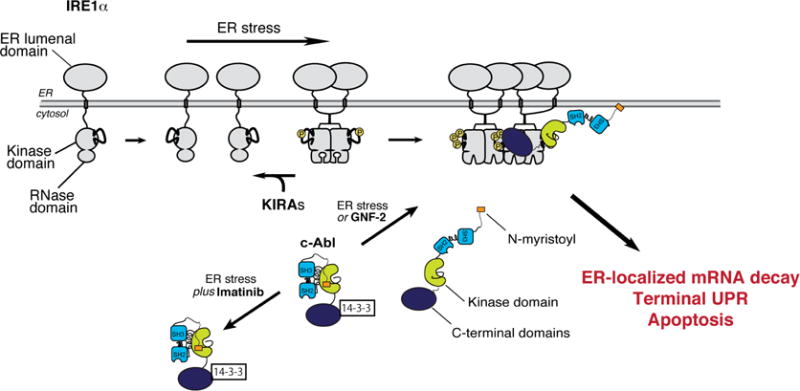
Under ER stress, c-Abl, dissociates from cytosolic 14-3-3 proteins to co-localize with IRE1α at the ER membrane, thus driving high-order oligomerization, ER-localized mRNA decay, T-UPR induction, and apoptosis. Imatinib prevents c-Abl’s re-localization to blunt the T-UPR. GNF-2 forces c-Abl localization to IRE1α, without ER stress, activating the T-UPR. Under ER stress or GNF-2, KIRAs block IRE1α hyperactivation and the T-UPR.
While c-Abl is best understood as the kinase component of the oncoprotein, BCR-Abl, WT ABL kinases play both pro-survival and pro-death roles (Wang, 2000, 2014). For example, c-Abl is a major driver of apoptosis during oxidative stress and DNA damage (Yoshida et al., 2005). Multi-domain ABL kinases interact with diverse proteins and shuttle between various organelles, playing numerous biological roles (Wang, 2014). During UV-induced DNA damage, c-Abl rapidly mobilizes to the nucleus, interacting with DNA through its C-terminal DNA-binding domain. Under oxidative stress, c-Abl promotes apoptosis by localizing to mitochondria and the nucleus (Nihira et al., 2008). Thus, c-Abl’s ability to co-localize with IRE1α at the ER membrane under ER stress, as its interactions with 14-3-3 proteins become disrupted, is consistent with ABL re-localization from a cytosolic pool to specific organelles serving as a general mechanism for diversified, contextual signaling.
Two inhibitors of ABL’s phosphotransfer activity–imatinib and GNF-2–cause divergent UPR signaling based on differential effects on ABL ER localization (Figure 7). Under ER stress, imatinib blocks ABL/IRE1α co-localization at the ER, preserving ABL/14-3-3 protein interactions and blunting the UPR. Imatinib’s UPR-inhibitory effects do not rely on inhibition of ABL’s phosphotransfer activity, which appears to be dispensable based on kinase-dead c-Abl’s ability to drive IRE1α activation. That GNF-2 can directly activate IRE1α—without ER stress—by disengaging kinase-inhibited endogenous ABL from 14-3-3 proteins and into IRE1α−foci further demonstrates the dispensability of ABL’s phosphotransfer activity. This UPR scaffolding role is consistent with c-ABL playing phosphotransfer-independent functions in other signaling pathways (Cong and Goff, 1999; Galan-Moya et al., 2008; Rauch et al., 2011; Theis and Roemer, 1998).
ABL scaffolding provides gain control over IRE1α’s RNase, which triggers ER-localized mRNA decay and apoptosis when hyperactivated. Amplification of IRE1α RNase activity by ABL under ER stress, or through GNF-2 without ER stress, can be defeated through direct–STF-083010–or allosteric–KIRA8–RNase inhibition. Thus, despite c-Abl playing a mitochondrial role late during ER stress (Ito et al., 2001), its rheostatic control over IRE1α is essential to adjusting an early set point that potentiates apoptosis. Amplification of IRE1α’s enzymatic activities by ABL, thereby sensitizing cells to apoptosis, was demonstrated in engineered cell lines and in endogenous systems with pharmacological agents. Finally, direct enhancement of IRE1α’s RNase activity by ABL was confirmed in reconstitution experiments with purified proteins, including a kinase-dead mutant.
In sum, these results support a growing view that IRE1α’s cell fate outputs are not solely dependent on autonomous signaling events relayed linearly in direct proportion to conditions within the ER, but that other non-canonical UPR components can tune IRE1α activity. This opens up the potential to drug the UPR and its outputs through other targets.
UPR modulation in autoimmune-initiated β-cell degeneration
Mono-selective IRE1α inhibitor—KIRA8—enabled us to precisely show that attenuating IRE1α pharmacologically in the Akita mouse preserves glycemic control in this monogenetic, fulminant diabetes model driven by autonomous β-cell apoptosis. This result motivated us to test whether KIRA8 would also demonstrate anti-diabetogenic effects in the complex NOD model, which like the Akita shows islet IRE1α hyperactivation in the period leading up to frank diabetes (Lerner et al., 2012). At dosing regimes in the NOD that led to partial inhibition of IRE1α (i.e., attenuated, but nonetheless preserved, XBP1 mRNA splicing in islets), declines in BiP, MANF, and insulin mRNAs (and insulin protein), were reversed, even as reductions in TXNIP mRNA were observed. Concomitant with T-UPR blunting, β-cell functional mass was preserved in diabetes prevention studies. In the more stringent endpoint of reversal of established diabetes, KIRA8 showed >90% efficacy in NOD mice within three weeks.
Therefore, pharmacologically attenuating IRE1α activity in murine diabetes models is phenotypically distinct from removal of the Ire1α gene in the β-cell, which leads to translational blocks and β-cell hypoplasia (Hassler et al., 2015). Also, β-cell conditional knockout of the Xbp1 gene compensatorily activates IRE1α, confounding interpretations of the natural role of the IRE1α-XBP1 arm in β-cell physiology (Lee et al., 2011). Such compensatory, dysregulated UPR effects may be general as Perk deletion, likewise, hyperactivates IRE1α in β-cells, which suffer early apoptosis, leading to postnatal diabetes (Harding and Ron, 2002). Therefore, selective UPR kinase inhibitors are powerful tools for deconvoluting the role of parallel UPR signaling in diverse in vivo models of ER stress cell degeneration.
Analogously, whole body genetic knockouts of Abl1 and Abl2 are embryonic lethal, but pharmacological inhibition of ABL kinases reveals cell-sparing effects. And despite the essential function of ABL kinases in development, human patients with CML treated with imatinib (or nilotinib) can tolerate chronic ABL inhibition. Indeed, a multitude of mounting reports document paradoxical cell-sparing effects of tyrosine kinase inhibitory drugs originally developed against cancers (Fountas et al., 2015; Hagerkvist et al., 2007; Han et al., 2009b; Paniagua et al., 2006). Clinical studies and case reports show improved glycemic control and reversal of diabetes in humans treated with imatinib, sunitinib, or dasatanib (Fountas et al., 2015). While the cell-sparing mechanism of action of these drugs and their relevant kinase targets is not generally understood, here the cytoprotective basis of one member, imatinib, is clarified. Importantly, our results provide mechanistic rationale to support the clinical use of imatinib for the treatment of new-onset T1D, currently being tested in a Phase II clinical trial (NCT01781975). Similarly, other FDA-approved TKIs may conceivably be repurposed for cell-degenerative indications after clarifying their underlying mechanism of action.
Lastly, our results implicate ER stress-induced β-cell degeneration centrally in the pathogenesis of T1D (as in other diabetic syndromes), although the disease is initiated by autoimmune attack of pancreatic islets. It is tempting to speculate that temporary revival of β-cell function in human T1D patients experiencing a “honeymoon period” shortly after starting insulin may result from ER stress/T-UPR reduction in islets as β-cell secretory workload becomes reduced. Thus, our results support an evolving notion that targeting the significant percentage of potentially salvageable β-cells in early periods of autoimmune attack is a promising therapeutic strategy in T1D (Krogvold et al., 2015). The ability, therefore, to blunt, from either node, the ABL-IRE1α axis and curtail premature degeneration of ER stressed β-cells in autoimmune diabetes raises the promise of further optimizing related small molecules into drugs to treat T1D, and perhaps other diseases driven by unchecked ER stress.
EXPERIMENTAL PROCEDURES
Please see supplemental information.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by Lead Contact, Feroz R. Papa (frpapa@medicine.ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Studies
Female NOD and NOD.SCID.IL2Rγ-null (NSG) mice were obtained from Taconic and Jackson Laboratories, respectively. Male C57BL/6 “Akita” Ins2 WT/C96Y (Ins2+/Akita) mice were obtained from Jackson Laboratories. Akita mice were genotyped by following the instruction of Jackson Laboratories. Glucose levels were measured from tail snips obtained between 9:00 and 11:00 AM using a LifeScan glucose meter (OneTouch Ultra) twice per week. Diabetic mice were defined as having blood glucose levels in the range of 250 to 400 mg/dL. All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. Animals were kept in a specific pathogen-free animal facility on a 12 hr light-dark cycle at an ambient temperature of 21°C. They were given free access to water and food.
Tissue Culture
INS-1 cells (rat insulinoma cell line) were grown in RPMI, 10% fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, 2 mM glutamine, 50 μM β-mercaptoethanol. SV40-transformed MEFs were maintained in Dulbecco’s Modified Eagle Media (DMEM) containing 5mM glucose. The DMEM was supplemented with 10% heat-inactivated fetal bovine serum (FBS, J.R. Scientific), 100U penicillin, and 100U streptomycin. Flag-tagged, full length mouse type IV c-Abl (WT), mouse type IV c-Abl (K290R), mouse PDGFRα, mouse c-KIT, mouse Arg, human type 1b full-length c-Abl (1b), and deletion forms of human c-Abl(3D) were subcloned into pcDNA5/FRT/TO plasmid (Invitrogen) by PCR. INS-1/FRT/TO or T-REx 293/FRT/TO cells were grown in the above media with 10 μg/ml blasticidin. Cells were then grown in 200 μg/ml zeocin, cotransfected with each 1 μg pcDNA5/FRT/TO constructs and 1 μg FLP recombinase constructs pOG44 (Invitrogen) using Lipofectamine LTX (Invitrogen). After 4 hr, cells were switched to zeocin-free media, trypsinized 48 hr later, and then plated in media containing hygromycin (150 μg/ml), which was replaced every 3 days until colonies appeared.
Ex vivo Islet Studies
Non-diabetic human islets were obtained from Prodo Labs (Irvine, CA) and cultured in Prodo Islet Medium (PIM from Prodo Labs). Islets were pretreated with or without imatinib for 2 hr followed by treatment with Tm for 16 hr. For mouse islet experiments, extracted islets were cultured in RPMI + 10% FBS with 500 ng/ml Tm with or without Imatinib (20 μM) or left untreated for 16 hr. Approximately 50 islets were cultured for each condition in triplicate.
METHOD DETAILS
Imatinib Treatments
Imatinib mesylate (Gleevec) tablets were purchased from Novartis, ground and suspended in peanut oil or methylcellulose to a concentration of 5 mg/ml. Mice were treated daily by gavage with a single dose of 1.5 mg/mouse (0.3 ml). Control mice received 0.3 ml of vehicle only. 10-week-old euglycemic NOD mice were used for pre-diabetic studies. Mice that succumbed to complications attributed to overt diabetes during the experiments were excluded from the final analysis. In reversal studies, imatinib or vehicle treatment immediately commenced at the onset of diabetes and continued for 4 weeks. The imatinib- or vehicle-treated groups were randomly selected. For reversal studies, mice were treated by gavage daily with 1.5 mg of imatinib or methycellulose for 6 weeks.
Intra-Peritoneal Glucose Tolerance Tests and First-phase Insulin Response
Mice were fasted for 17 hr before intraperitoneal injection with glucose (1.5 g/kg in saline). Blood samples (25 ml) were collected from the tail vein immediately before the injection and 2 minutes afterwards to determine the first-phase insulin response, and separated to serum. Serum insulin levels were measured using mouse ultra-sensitive insulin-ELISA (Mercodia).
Histological Staining of Pancreatic Tissues and Insulitis Score
Paraffin-embedded and frozen sections prepared from human and mouse pancreata were analyzed using standard immunofluorescence staining procedures. Sections were subsequently washed and incubated with the species-appropriate, Alexa-conjugated secondary antibodies (Invitrogen). Sections were stained with DAPI for 5 minutes to stain nuclei and mounted with FluorSave reagent (Calbiochem). The insulin positive area in pancreatic sections was highlighted and quantified with Metamorph by a blinded operator, and was expressed as a percentage of the total pancreatic area. 12–16 sections per group were analyzed. For insulitis score, multiple 10 μm sections were stained with hematoxylin and eosin and scored blindly for severity of insulitis. The insulitis was considered peri-insulitis when lymphocytes were found surrounding, but not infiltrating the architecture of the islets; moderate insulitis if less than half of the islet architecture was infiltrated with lymphocytes; and severe insulitis if more than half of the islet architecture was infiltrated with lymphocytes. Score; 0=no insulitis; 1=peri insulitis; 2=moderate insulitis; 3=severe insulitis.
Western Blots and Antibodies
For protein analysis, cells were lysed in M-PER buffer (ThermoScientific) plus complete EDTA-free protease inhibitor (Roche) and phosphatase inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined using BCA Protein Assay (Thermo). Western blots were performed using 4–12% Bis-Tris (NuPage), 3–8% Tris-Acetate (NuPage), or Phos-tag (Wako Pure Chemical Industries) precast gels on Invitrogen XCell SureLock Mini-Cell modules. Gels were run using MES, Tris-Acetate, or Tris-Glycine buffer and transferred onto nitrocellulose transfer membrane using an XCell II Blot Module. Antibody binding was detected with near-infrared-dye-conjugated secondary antibodies (Li-Cor) on the LI-COR Odyssey scanner. Blocking, antibody incubation, and washing were done in TBS with 0.05% Tween-20 (v/v) and 0.5–5% (w/v) non-fat dry milk. Antibody-binding was detected with near-infrared-dye-conjugated secondary antibodies (Licor) on the LI-COR Odyssey scanner and quantified by densitometry using ImageJ (U. S. National Institutes of Health). GAPDH and HDAC1 were used as a loading control. Please note that the immunoblot membrane of Figure 1A—used to detect IRE1α species—phospho- and total-was stripped and reprobed for detection of Abl species— phosphor- and total- for Figure 3A. Thus, the GAPDH blot used for normalization is common to both figures, and has been shown in Figures 1A and 3A.
RNA Isolation, Quantitative Real-time PCR, and Primers
RNA was isolated from whole cells using either QIAGEN RNeasy Mini kits or Trizol (Invitrogen). For standard mRNA detection, generally 1 mg total RNA was reverse transcribed using the QuantiTect Reverse Transcription Kit (QIAGEN). For qPCR, we used SYBR green (QIAGEN) and StepOnePlus Real-Time PCR System (Applied Biosystems). Thermal cycles were: 5 minutes at 95 °C, 40 cycles of 15 s at 95 °C, 30 s at 60 °C. Gene expression levels were normalized to Beta Actin or Hprt1. Primers used for qPCR were as follows: Rat TXNIP: 5′-CTGATGGAGGCACAGTGAGA-3′ and 5′-CTCGGGTGGAGTGCTTAGAG-3′; Rat Ins1: 5′-GTCCTCTGGGAGCCCAAG-3′ and 5′- ACAGAGCCTCCACCAGG-3′; Rat c-Abl: 5′-CTCCTTGACTGACCCAGAGC-3′ and 5′-GCACCGACATCAGCTACAGA-3′; Rat Beta Actin: 5′-GCAAATGCTTCTAGGCGGAC-3′ and 5′-AAGAAAGGGTGTAAAACGCAGC-3′; Human Insulin: 5′-GCCTTTGTGAACCAACACCTG-3′ and 5′-GTTGCAGTAGTTCTCCAGCTG-3′; Human TXNIP: 5′-CCTCTGGGAACATCCTTCAA-3′ and 5′-GGGGTATTGACATCCACCAG-3′; Human Beta Actin: 5′-AGAGCTACGAGCTGCCTGAC-3′ and 5′-AGCACTGTGTTGGCGTACAG-3′; Mouse Hprt1: 5′-CTCATGGACTGATTATGGACAGGAC-3′ and 5′- GCAGGTCAGCAAAGAACTTATAGCC-3′; Mouse BiP: 5′-TTCAGCCAATTATCAGCAAACTCT-3′ and 5′- TTTTCTGATGTATCCTCTTCACCAGT-3′; Mouse MANF: 5′- AGGTCCACTGTGCTCAGGTC-3′ and 5′-CCACCATATCCCTGTGGAAA-3′; Mouse TXNIP: 5′-TCAAGGGCCCCTGGGAACATC-3′ and 5′-GACACTGGTGCCATTAAGTCAG-3′; Mouse Ins1: 5′-ACCCACCCAGGCTTTTGTC-3′ and 5′-TCCCCACACACCAGGTAGAGA-3′; Mouse Ins2: 5′-TGGCTTCTTCTACACACCCATGT-3′ and 5′-AGCTCCAGTTGTGCCACTTGT-3′; Mouse c-Abl: 5′-GCAGTTCCTTCCGAGAGATG-3′ and 5′-GAGAACGGAAGCCTGAGTTG-3′
XBP-1 mRNA Splicing
RNA was isolated from whole cells or tissues and reverse transcribed as above to obtain total cDNA. Then, XBP-1 primers were used to amplify an XBP-1 amplicon spanning the 26 nt intron from the cDNA samples in a regular 3-step PCR. Thermal cycles were: 5 min at 95 °C, 30 cycles of 30 s at 95 °C, 30 s at 60 °C, and 1 minute at 72 °C, followed by 72 °C for 15 minutes, and hold at 4 °C. Primers used for XBP-1 mRNA splicing were as follows: sense primer XBP1.3S (5′-AAACAGAGTAGCACAGACTGC-3′) and antisense primer XBP1.2AS (5′-GGATCTCTAAGACTAGAGGCTTGGTG-3′). PCR fragments were then digested by PstI, resolved on 3% agarose gels, stained with EtBr and quantified by densitometry using ImageJ (U. S. National Institutes of Health).
Glucose-Stimulated Insulin Secretion (GSIS) assay
Freshly isolated islets were cultured in RPMI-1640 with 10% FCS, 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, and 11.1 mM glucose with or without Tm (500 ng/ml) for 16 hr before the GSIS assay. Imatinib (20 μM) was added 2 hr before treating with Tm. In the GSIS assay, islets were preincubated in HEPES-buffered Krebs-Ringer bicarbonate solution (KRBH) (10 mM HEPES [pH 7.4], 129 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 5 mM NaHCO3, and 0.1% BSA) containing 2.5 mM glucose for 30 min at 37°C. Fifty islets per condition were incubated with either 2.5 mM or 28 mM glucose in KRBH at 37°C for 30 minutes. Collected media were analyzed by anti-insulin ELISA (EMD Millipore) and insulin levels were normalized to total protein amount.
Flow Cytometry
For assaying apoptosis by Annexin V staining, cells were plated in 12-well plates overnight. Cells were then treated with either various ER stress agents or Dox for the indicated times. On the day of analysis, cells were trypsinized and washed in PBS and resuspended in Annexin V binding buffer with Annexin V FITC (FITC Annexin V Apoptosis Detection Kit I, BD PharMingen). Flow cytometry was performed on a Becton Dickinson LSRII flow cytometer.
Islet Staining
After islet extraction, islets were spun, washed once with PBS, and fixed for 30 minutes with 4% PFA. After fixation, islets were washed twice with PBS, followed by a wash in 100% ethanol. After removal of all ethanol, 100 μL of prewarmed Histogel (Thermo Scientific) was added to the eppendorf tube and placed at 4°C to solidify before paraffin embedding and 5 μm sectioning of the islets. Islets were stained with TUNEL using ApopTag® Red In Situ Apoptosis Detection Kit (Millipore) according to the manufacturer’s instructions. Islets were also co-stained with guinea pig anti-insulin (Zymed), DAPI (Sigma-Aldrich), and goat anti-guinea pig secondary (Rockland) before mounting onto slides with VectaShield (Vector Laboratories). At least 10 islets and > 500 beta-cell nuclei were counted per group (in triplicate). Cells were considered TUNEL positive if staining was present and co-localized with DAPI staining, indicating nuclear localization.
Superfolder GFP-IRE1α Construction and Microscopy
The first 27 amino acids of mouse IRE1α containing the signal peptide were cloned just before the first ATG of sfGFP lacking the stop codon, and the remaining IRE1α sequence (WT) was cloned in-frame after the sfGFP in a pcDNA5/FRT/TO mammalian expression plasmid. INS-1-FRT/TO cells were transfected to generate stable cell lines expressing the above constructs. INS1-sfGFP-IRE1α cells were inducted with 1 ng/ml Dox (a sub-apoptotic dose sufficient for imaging the reporter) for 24 hr. Live cells were imaged (Nikon Eclipse Ti-Yokogawa CSU22 spinning disk confocal) with Apo 60×/1.49 oil objective and a 491nm excitation/525-50 emission filter. Composite figures were prepared using ImageJ (NIH).
Expression and Purification of Abl(3D)
The first 538 amino acids of human cAbl1b (Abl(3D)) followed by a 6× His (HisTag) sequence was cloned in a pET-28a vector. The plasmid for Abl(3D), YopH (pCDFDuet-1 vector), and GroEL (pGRO7 vector) were co-transformed into BL21 cells and plated onto triple selective (kanamycin, streptomycin, and chloramphenicol) LB agar plates and grown overnight at 37 °C. A single colony from the plate was grown in an overnight culture of 50 mls of Terrific Broth + kanamycin/streptomycin/chloramphenicol overnight at 37°C. A large 2L culture was then inoculated with the overnight culture and grown to an OD600 of 1.2 at 37 °C (approximately 5–6 hr). Culture was then cooled to 18°C for one hr while shaking. Expression was induced with 0.2 mM IPTG for 16 hr (overnight) at 18°C and pelleted. Pellet was re-suspended in chilled lysis buffer (50mM HEPES pH 8.0, 20 mM Imidazole, 300 mM NaCl, 0.1% Triton-X, and 1 mM PMSF). Lyse cells by sonication and clear the lysate by spinning for 30–45 minutes at 10,000× rcf. Decant the clarified lysate and add 1 ml 50% Ni-NTA slurry and rotate at 4°C for 1 hr. Spin down to pellet the resin and aspirate off the supernatant. Wash resin with 3× resin volume of cold lysis buffer. Abl(3D) was then eluted in 7 mls of 50 mM HEPES pH 8.0, 300 mM Imidazole, 300 mM NaCl, 0.1% Triton-X, and 1 mM PMSF. Eluted Abl(3D) was dialyzed overnight into 50 mM Tris pH 8.0, 150 mM NaCl, 5% Glycerol, and 1 mM DTT. FPLC buffers A (50 mM Tris pH 8.0, 1 mM DTT, 5% Glycerol) and B (50 mM Tris pH 8.0, 15 M NaCl, 1 mM DTT, 5% Glycerol) were prepared and filtered. Abl(3D) was eluted on an anion exchange column (QFF HiTrap; GE Healthcare) with a 0–35% B gradient over 15 column volumes at a flow rate of 2 mls per minute. Fractions with UV absorbance were analyzed by SDS-PAGE. Pure fractions were combined, concentrated, and stored for later use.
Expression and Purification of cAbl1b K290R
cAbl1b K290R was cloned using a c-Abl1b FLAG pcDNA5/FRT/TO template and the following QuikChange primers: Forward: 5′-GTGGCCGTGAGGACCTTGAAGGAGGACACCATG–3′, Reverse: 5′-GTCCTCACGGCCACCGTCAGGCTGTATTTCTTCC–3′. The plasmid (10 μg) was transfected into HEK293T cells that had been plated onto 15 cm dishes at 3×105 cells/ml and allowed to grow for 16 hr. The cells were grown for another 16 hr in media containing plasmid and transfection reagent (Turbofectin). Cells were then washed twice with ice cold PBS and lysed in 1 ml of chilled lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton-X). Anti-Flag M2 Magnetic Resin (Sigma Aldrich) (60 μL in a 50% slurry) was added to an eppendorf tube and diluted with 500 μL of TBS and aspirated to wash the beads, this step was repeated twice. Lysate was then added to the beads and rotated at 4 °C for 2 hr. After two hr the supernatant was removed and the beads were washed three times with TBS. To elute cAbl1b K290R, 100 μL of 1 μg/μL of 3×FLAG peptide in TBS was added to beads and rotated for one hr at 4 °C. After one hr the supernatant was collected and it’s purity and concentration was determined via SDS-PAGE.
In vitro IRE1α * Protein Preparation, Kinase, RNase and Crosslinking Assays
A construct containing the cytosolic kinase and RNase domains of human IRE1α (residues 469-977, IRE1α*) was expressed in SF9 insect cells using the Bac-to-Bac baculovirus expression system (Invitrogen) with a 6-His-tag at the N-terminus, and purified with a Ni-NTA (Qiagen) column. To determine the effect of compounds on IRE1α* kinase activity, imatinib or nilotinib (20 μM and 6.7 μM) was incubated with IRE1α* in cleavage buffer (20 mM HEPES at pH 7.5, 0.05% Triton X-100 (v/v), 50 mM potassium chloride, 1 mM magnesium chloride, 1 mM DTT) for 30 min, followed by incubation with 10 μCi [γ-32P] ATP (3,000 Ci mmol−1, PerkinElmer) at 23 °C for 3 hr. Samples were then spotted onto phosphocellulose paper and washed in triplicate with 0.5% phosphoric acid and autoradiographed. Percent activity was quantified relative to DMSO treated IRE1α*. The RNase assay for the endpoint readings of IRE1α* was performed by using 5′FAM-3′BHQ-labeled XBP1 single stem-loop mini-substrate (5′FAM- CUGAGUCCGCAGCACUCAG-3′BHQ, from Dharmacon). For the endpoint readings of IRE1α* RNase activity, 0.1 mg/ml IRE1α* was incubated with 3 mM XBP1 mini-substrate for 20 min. Reaction mixtures were subsequently resolved by urea 15% PAGE. For the kinetic assay with c-Abl constructs, 200 ng IRE1α* and 50 ng c-Abl or Abl(3D) were incubated at room temperature in buffer A (20mM HEPES pH 7.5, 50mM K2OAc, 1mM MgOAc, 0.05% Triton X-100) with 200 nM ATP. For the kinetic assay with c-Abl1b WT and K290R contracts, 230nM IRE1α* and varying concentrations of c-Abl1b K290R (520 nM, 260 nM, 130 nM, 65 nM, and 32.5 nM) or cAbl1b WT were incubated at room temperature in buffer A. After 20 minutes, 2 μL of the above mixture was added into a Corning 384 well plate. XBP1 mini-substrate (5′-Alex647-CAUGUCCGCAGCGCAUG-IowaBlack-FQ-3′; IDT) was added to the wells to a final concentration of 2 μM and a final well volume of 30 μL. Fluorescence was detected on a Perkin Elmer Envision Microplate Reader at excitation and emission wavelengths of 650 nm and 665 nm. Reaction process was monitored real time in 15- or 20-second intervals for 40 minutes. Rates were determined by plotting the linear range of fluorescent curves using GraphPad Prism analysis software. To determine the effect of compounds using the RNase kinetic assay, 10 nM IRE1α* were incubated with 3 mM XBP1 mini-substrate under varying concentrations of compounds for one hr. Fluorescence signal was monitored in 20-second intervals on SpectraMax M5 (Molecular Devices) and the reaction velocity (V0) was defined as Δ(Fluorescence Intensity) × (min−1) to calculate % activity. Internally 32P-labeled mouse Insulin2 (Ins2) RNA was also used as a substrate. For the crosslinking experiments, IRE1α* was crosslinked with 250 mM disuccinimidyl suberate (DSS) with DMSO or 10 mM KIRA8 for one hr.
In vitro IRE1α* Immunoprecipitations
T-REx 293 cells were plated onto 10-cm2 dishes 24 hr before transfection with c-Abl-flag or c-Abl(3D)-flag DNA (X-tremegene HP protocol). After 24 hr, cells were trypsinized, washed with PBS, and lysed in 500 μl RIPA buffer (50 mM Tris pH 7.8, 150 mM NaCl, 1mM EDTA, 1% Igepal CA-630, 1× Roche PhosSTOP phosphatase inhibitor cocktail, 1× Pierce Protease inhibitor cocktail, 1mM PMSF) for 30 minutes on ice. Lysates were cleared for 20 minutes at 14,000 g and 10 μL anti-flag magnetic beads (Sigma-Aldrich) were added to each cleared lysate. Immunoprecipitation was performed for 2 hr at 4°C. Immediately following immunoprecipitation, resin was washed three times with 200 μL RIPA buffer, followed by three 200 μL TBS washes, and a one hr incubation with 100 nM IRE1α* in TBS at 4°C. Resin was washed three times with TBS. Bound Abl and associated IRE1α* were eluted with 100 μL 0.18 mg/ml 3× Flag peptide in TBS for 15 minutes at room temperature. Fractions were analyzed by SDS-PAGE and probed with flag (Cell Signaling) and IRE1α (Santa Cruz) antibodies following immunoblotting.
c-Abl-mCherry Constructs and Lentivirus Production
Full-length Flag-tagged human c-Abl1b was used as a template to create deletion forms of c-Abl(3D) by PCR and Gibson Assembly (NEB). Flag-tagged human c-Abl (1b, 1a, and 3D) constructs were subcloned into pcDNA5 (Invitrogen), and then into pLVX-mCherry-N1 vector (Clontech) for lentivirus expression. For lentivirus production, T-REx 293 packaging cells were cotransfected at 70% confluence by calcium phosphate method with 5 μg lentivirus vectors, 3 μg psPAX2 vector, and 2 μg pCMV-VSV-G vector. After 48 hr, the viral supernatant was collected and filtered. Cells were incubated overnight with the viral supernatant. Subsequently, puromycin was used for selection.
Synthesis of Dasatinib-amine and Dasatinib-amine Precursor
5-Thiazolecarboxamide, 2-[[6-[4-(2-aminoethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl] amino]-N-(2-chloro-6-methylphenyl)
One equivalent of commericially available Dasatinib-amine precursor (2-((6-(4-(14-amino-4-oxo-6,9,12-trioxa-3-azatetradecyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)-N-(2-chloro-6 methylphenyl)thiazole-5-carboxamide (CAS#: 302964-08-5) was added to five equivalents of 1((-2-N-Boc-amino)ethyl)piperazine (CAS#: 140447-78-5) and three equivalents of DIEA in 0.1 M 1,4-dioxane. The reaction was sealed in a microwave vial and placed in an organic synthesis microwave for one hr at 110°C. The crude mixture was then dissolved in 5 ml of dichloromethane and 2 ml of trifluoroacetic acid are stirred at room temperature for two hr. Solvent was removed under vaccum and 5-thiazolecarboxamide, 2-[[6-[4-(2-aminoethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl] amino]-N-(2-chloro-6-methylphenyl) was purified by flash column chromatography using DCM:Methanol (2% NH4OH).
2-[[6-[4-(14-amino-4-oxo-6,9,12-trioxa-3-azatetradec-1-yl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-N-(2-chloro-6-methylphenyl)-5-Thiazolecarboxamide (Dasatinib-amine, CAS#: 1622947-07-2)
A mixture of 5-Thiazolecarboxamide, 2-[[6-[4-(2-aminoethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-N-(2-chloro-6-methylphenyl) (1 eq.) and 2,2-Dimethyl-4-oxo-3,8,11,14-tetraoxa-5-azahexadecan-16-oic acid (1.2 eq.) was added to EDCI (1.5 eq.), HOAt (1.5 eq.), DIEA (3 eq.) in 0.1 M DMF and stirred at room temperature overnight. The mixture was diluted in EtOAc and the organic layer extracted in a separatory funnel. Ethyl acetate was removed under vacuum from the organic layer. Crude was re-dissolved in a 1:4 mixture of DCM and TFA and stirred at room temperature for 2 hr. Solvent was removed under vacuum and the crude dissolved in a 1:1 mixture of water and acetonitrile and purified using reverse-phase chromatography (HPLC). 2,2-Dimethyl-4-oxo-3,8,11,14-tetraoxa-5-azahexadecan-16-oic acid (1.2 eq.) was added to EDCI (1.5 eq.), HOAt (1.5 eq.), DIEA (3 eq.) in 0.1 M DMF and stirred at room temperature overnight. The mixture was diluted in EtOAc and the organic layer extracted in a separatory funnel. Ethyl acetate was removed under vacuum from the organic layer. Crude was re-dissolved in a 1:4 mixture of DCM and TFA and stirred at room temperature for 2 hr. Solvent was removed under vacuum and the crude dissolved in a 1:1 mixture of water and acetonitrile and purified using reverse-phase HPLC. Rf = 0.5 (CHCl3 containing 13% v/v MeOH and 2% v/v NH3 in MeOH). ESI-MS: m/z = 676.9 [M+H]+ (consistent with previously described characterization (Fischer et al., 2011)).
Preparation of Dasatinib Resin
NHS-activated Sepharose™ 4 Fast Flow resin in a 50% slurry with isopropanol was pipetted into a Poly Prep® Chromatography Column (BioRad). Isopropanol was drained from the resin and the resin was washed twice with 2× resin volumes of ice cold 50% DMF:Ethanol (1:1). Resin was drained and 1× resin volume of DMF:Ethanol (1:1) was added. 2-[[6-[4-(14-amino-4-oxo-6,9,12-trioxa-3-azatetradec-1-yl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-N-(2-chloro-6-methylphenyl)-5-Thiazolecarboxamide (Dasatinibamine, CAS#: 1622947-07-2) was added to the resin to a concentration of 15 mM. The resin mixture was adjusted to a pH of 8.0 with DIEA and allowed to rotate overnight at room temperature. Resin was then drained and two resin volumes of quenching mixture (0.5 M Ethanolamine, 0.5 M NaCl pH 8.5) was added and allowed to rotate overnight at room temperature. Resin was drained and washed three times with 3× resin volumes of 0.1M Tris-HCl at pH 8.5 and then washed three times with three resin volumes 0.1 M Acetate, 0.5 M NaCl at pH 5.0. Resin was drained and stored at 4°C in 20% Ethanol as a 50% slurry.
In Vitro Pull Down Assay with Dasatinib Resin
Dasatinib resin (40 μL) was added to a microcentrifuge tube; centrifuged and aspirated. Resin was washed with 100 μL pull down buffer (50 mM Tris pH 7.5, 100mM NaCl, 0.5 mg/ml BSA, 1 mM DTT) three times and aspirated. IRE1α* (500 nM) with and without c-Abl (Carnebio; 250 nM) in pull down buffer (120 μL) was added to washed resin and allowed to rotate for 1.5 hr at room temperature. After incubation, resin was centrifuged and aspirated. Resin was subsequently washed with 120 μL pull down buffer, centrifuged and aspirated. Immobilized proteins were then eluted with 120 μL 1× SDS and boiled. Samples were resolved via SDS-PAGE gel and visualized via Western Blot using a total IRE1α antibody (#3294; Cell Signaling) and an SH2-domain specific c-Abl antibody (sc-56887; Santa Cruz Biotechnology). Antibody-binding was detected with near-infrared-dye-conjugated secondary antibodies (Licor) on the LI-COR Odyssey scanner.
Kinome
KIRA8 was tested at a single dose at least in duplicate at a concentration of 1 μM against 365 kinases by Reaction Biology. Percent enzymatic activity was determined relative to DMSO-treated kinases. Curve fits were performed for KIRA8 when the remaining enzymatic activity was less than 65%.
In vitro c-Abl Tyrosine Kinase Activity
Titrations of inhibitors (3-fold serial dilutions starting at 20 μM, eight data points) were assayed in a black 384-well plate (Corning, 3573) in buffer containing 50 nM Abl(3D), 75 mM HEPES (pH 7.5), 155 mM MgCl2, 3.75 mM EGTA, 1 mM Na3VO4, 150 mM NaCl, 0.2 mg/ml BSA, 100 mM ATP, and 20 μM Abl pyrene substrate (Ac-AEAIYAA(dap-pyrene)-LA-NH2). The final volume of each assay well was 30 μL and the total DMSO concentration was 4% per well. The enzymatic reaction was initiated with Abl pyrene substrate after one hr incubation at room temperature of Abl(3D), inhibitor, and ATP. The reaction proceeded for two hr after which the plate was read on a Perkin Elmer EnVision fluorimeter (Ex 340, Em 405).
KIRA6 Treatments
KIRA6 1-(4-(8-amino-3-tert-butylimidazo[1,5-a]pyrazin-1-yl)naphthalen-1-yl)-3-(3-(trifluoromethyl)phenyl)urea (KIRA6). A mixture of I1 (60.0 mg, 0.120 mmol), A (66 mg, 0.15 mmol), tetrakis(triphenylphosphine)palladium (5 mg, 4 μmol) and sodium carbonate (928 mg, 0.27 mmol) was dissolved in a 3:1 mixture of DME/water (0.5 ml). The mixture was heated overnight at 85°C. The crude mixture was cooled to room temperature, diluted in a mixture of acetonitrile/water and purified by reverse phase chromatography (HPLC) to obtain 53 mg of KIRA6. TLC (CH2Cl2:MeOH, 95:5 v/v): Rf = 0.4; 1H NMR (300 MHz, MeOD): δ 8.26 (m, 1H), 8.08–7.99 (m, 2H), 7.90–7.86 (m, 1H), 7.83–7.79 (m, 1H), 7.69–7.52 (m, 5H), 7.35 (d, J = 7.4 Hz, 1H), 6.98 (m, 1H), 1.65 (s, 9H); 13C NMR (126 MHz, MeOD): δ 154.8, 140.2, 135.7, 133.0, 132.4, 131.7, 131.6, 131.0, 130.7, 129.4, 128.8, 128.6, 128.5, 127.0, 126.6, 125.9, 125.4, 123.2, 121.9, 120.1, 118.7, 115.0, 114.4, 110.1, 33.6, 27.3; ESI-MS (m/z): [M]+ calculated for C28H25F3N6O [M+H]+: 519.2; found 519.4. The purity of KIRA6 was determined with two analytical RP-HPLC methods, using a Varian Microsorb-MV 100-5 C18 column (4.6 mm × 150 mm), and eluted with either H2O/CH3CN or H2O/MeOH gradient solvent systems (+0.05% TFA) run over 30 min. Products were detected by UV at 254 nm. KIRA6 was found to be >95% pure in both solvent systems. Female NOD mice were randomized to KIRA6 or vehicle groups, and injected i.p. with a 2 mg/ml solution of KIRA6 (5 mg/kg) or vehicle (3% ethanol: 7% Tween-80: 90% saline) twice a day (b.i.d.). 8- or 10-week-old euglycemic NOD mice were used for pre-diabetic studies. In reversal studies, KIRA6 or vehicle treatment immediately commenced at the onset of diabetes. The first 6 enrolled mice in each group were treated for 8 weeks. Blood glucose and body weight were monitored weekly. At 6 weeks, 2 (vehicle) and 1 (KIRA6) mice with blood glucose levels greater than 600 mg/dL were euthanized due to complications of hyperglycemia. As some mice were euthanized due to complications attributed to overt diabetes, subsequent KIRA6 trials were conducted for no more than 4 weeks, and blood glucose was monitored weekly. Mice that were euthanized due to complication attributed to overt diabetes were excluded from the final analysis.
Synthesis of KIRA8
(S)-2-chloro-N-(6-methyl-5-((3-(2-(piperidin-3-ylamino)pyrimidin-4-yl)pyridin-2-yl)oxy)naphthalen-1-yl)benzenesulfonamide (KIRA8)
A solution of (S)-tert-butyl 3-((4-(2-((5-(2-chlorophenylsulfonamido)-2-methylnaphthalen-1-yl)oxy)pyridin-3-yl)pyrimidin-2-yl)amino)piperidine-1-carboxylate (4.36 g, 6.22 mmol) in DCM (15 ml) was treated with HCl in dioxane (3.5 M, 4 ml) and stirred at 50°C for one hr. The solution was then concentrated, and the remaining solid was triturated with Et2O. Drying over P2O5 in a vacuum desiccator gave (S)-2-chloro-N-(6-methyl-5-((3-(2-(piperidin-3-ylamino)pyrimidin-4-yl)pyridin-2-yl)oxy)naphthalen-1-yl)benzenesulfonamide as an off-color and free-flowing solid (3.56 g, 90%). Product was determined to be 97.4% pure by reverse phase analytical chromatography (HPLC). ESI-MS: m/z = 601.3 [M+H]+ (consistent with previously described characterization. Compound #18 in (Harrington et al., 2015)).
1H-NMR of KIRA8
KIRA8 Treatments
Female NOD mice were randomized to KIRA8 or vehicle groups, and injected i.p. with KIRA8 (50 mg/kg) or vehicle (3% ethanol: 7% Tween-80: 90% saline) once a day. 8- or 10-week-old euglycemic NOD mice were used for pre-diabetic studies. Mice that were euthanized due to complication attributed to overt diabetes were excluded from the final analysis. In reversal studies, KIRA8 or vehicle treatment immediately commenced at the onset of diabetes and continued for 6 weeks. Blood glucose and body weight were monitored weekly. Male Ins2+/Akita mice were injected i.p. with KIRA8 or vehicle.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism version 6.00. Student’s t test or one-way analysis of variance (ANOVA) followed by post-hoc Tukey’s test were applied to determine statistical difference between two groups or between more than two groups, respectively, unless otherwise noted. The difference of the percent diabetic mice between the treatment and vehicle group in the reversal study was estimated using the Kaplan–Meier method, as indicated in the figure legends, and compared using log-rank test between groups, setting the endpoint as reversal of diabetes. Use of one-way analysis of variance followed by post-hoc test for trend and two-way analysis of variance to determine statistical significance are indicated in the figure legends. The number of mice were selected based on our previous observations in performing similar assays in the NOD mouse model (Louvet et al., 2008; Villalta et al., 2013). Animal number (n) and experimental repeats are indicated in figures and figure legends. Data are presented as mean ± SEM. P<0.05 was considered significant throughout the study. P-values: * < 0.05; ** < 0.01; *** < 0.001; N.S., non-significant.
Supplementary Material
Highlights.
During endoplasmic reticulum (ER) stress, ABL kinases localize to the ER membrane
At the ER, ABL scaffolds IRE1α to hyperactivate the unfolded protein response (UPR)
Imatinib blunts the UPR and apoptosis by maintaining ABL in a 14-3-3 cytosolic pool
Direct targeting of IRE1α, using mono-selective KIRA8, reverses autoimmune diabetes
Acknowledgments
This work was supported by National Institutes of Health (NIH): RO1DK080955 (F.R.P), RO1GM086858 (D. J. M.), RO1AI046643 (J.A.B.), RO1DK100623 (D.J.M. and F.R.P), P30 DK063720 (F.R.P. and J.A.B.) and T32GM008268 (A.C.R.), Burroughs Wellcome Fund (F.R.P.); Juvenile Diabetes Research Foundation JDRF # 17-2013-513 (J.A.B. and F.R.P.), #3-PDF-2015-80-A-N (S.M.), #2-SRA-2016-234-S-N (F.R.P); Sumitomo Life Social Welfare Sciences Foundation (S.M.); Lundbeck Foundation Clinical Research Fellowship Program (I.H.P.); Brehm Coalition (J.A.B.); Breakthrough Therapeutics Initiative from Leona M. and Harry B. Helmsley Charitable Trust (F.R.P.). We thank J.Y. Wang for providing MEFs, Vinh Nguyen for islet isolation, Anthony Hernandez for technical assistance, Dorothy Fuentes for animal husbandry. We thank members of the Bluestone, Maly, and Papa labs, and Art Weiss, Scott Oakes, Dean Sheppard and Mark Anderson for helpful suggestions, edits, and comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: All authors conceived and designed the experiments. S.M., S.A.V., A.C.R., H.F., W.R., I.T.H., M.M., R.G., L.W., K.C., and R.M. performed the experiments. All authors analyzed the data. F.R.P., J.A.B, and D.J.M wrote the paper.
Author Information: Correspondence and requests for materials should be addressed to D.J.M., J.A.B. or F.R.P. Conflict of interest disclosure: D.J.M, B.J.B, and F.R.P. are founders and equity holders of OptiKira, LLC, a UPR-ophthalmology biotech company.
SUPPLEMENTAL INFORMATION
Document S1. Figures S1–S7, Tables S2 and S3.
Table S1. Percent cross reactivity of KIRA8 against 365 different kinases. Related to Figure S7.
References
- Alanentalo T, Hornblad A, Mayans S, Karin Nilsson A, Sharpe J, Larefalk A, Ahlgren U, Holmberg D. Quantification and three-dimensional imaging of the insulitis-induced destruction of beta-cells in murine type 1 diabetes. Diabetes. 2010;59:1756–1764. doi: 10.2337/db09-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, et al. How does type 1 diabetes develop?: the notion of homicide or beta-cell suicide revisited. Diabetes. 2011;60:1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottazzo GF. Lawrence lecture. Death of a beta cell: homicide or suicide? Diabet Med. 1986;3:119–130. doi: 10.1111/j.1464-5491.1986.tb00722.x. [DOI] [PubMed] [Google Scholar]
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, Gray NS. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J Biol Chem. 2009;284:29005–29014. doi: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong F, Goff SP. c-Abl-induced apoptosis, but not cell cycle arrest, requires mitogen-activated protein kinase kinase 6 activation. Proc Natl Acad Sci U S A. 1999;96:13819–13824. doi: 10.1073/pnas.96.24.13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D, Hotamisligil GS. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Science translational medicine. 2013;5:211ra156. doi: 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JJ, Dalhoff C, Schrey AK, Graebner OY, Michaelis S, Andrich K, Glinski M, Kroll F, Sefkow M, Dreger M, et al. Dasatinib, imatinib and staurosporine capture compounds - Complementary tools for the profiling of kinases by Capture Compound Mass Spectrometry (CCMS) J Proteomics. 2011;75:160–168. doi: 10.1016/j.jprot.2011.05.035. [DOI] [PubMed] [Google Scholar]
- Fountas A, Diamantopoulos LN, Tsatsoulis A. Tyrosine Kinase Inhibitors and Diabetes: A Novel Treatment Paradigm? Trends Endocrinol Metab. 2015;26:643–656. doi: 10.1016/j.tem.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Galan-Moya EM, Hernandez-Losa J, Aceves Luquero CI, de la Cruz-Morcillo MA, Ramirez-Castillejo C, Callejas-Valera JL, Arriaga A, Aranburo AF, Ramon y Cajal S, Silvio Gutkind J, et al. c-Abl activates p38 MAPK independently of its tyrosine kinase activity: Implications in cisplatin-based therapy. Int J Cancer. 2008;122:289–297. doi: 10.1002/ijc.23063. [DOI] [PubMed] [Google Scholar]
- Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Nguyen DT, Stuible M, Dube N, Tremblay ML, Chevet E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279:49689–49693. doi: 10.1074/jbc.C400261200. [DOI] [PubMed] [Google Scholar]
- Hagerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007;21:618–628. doi: 10.1096/fj.06-6910com. [DOI] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009a;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Chung KW, Cheon HG, Rhee SD, Yoon CH, Lee MK, Kim KW, Lee MS. Imatinib mesylate reduces endoplasmic reticulum stress and induces remission of diabetes in db/db mice. Diabetes. 2009b;58:329–336. doi: 10.2337/db08-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(Suppl 3):S455–461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, Dellamaggiore K, Kendall R, Beckmann H, Jaeckel P, et al. Unfolded Protein Response in Cancer: IRE1alpha Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS medicinal chemistry letters. 2015;6:68–72. doi: 10.1021/ml500315b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassler JR, Scheuner DL, Wang S, Han J, Kodali VK, Li P, Nguyen J, George JS, Davis C, Wu SP, et al. The IRE1alpha/XBP1s Pathway Is Essential for the Glucose Response and Protection of beta Cells. PLoS Biol. 2015;13:e1002277. doi: 10.1371/journal.pbio.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Beatty A, Han X, Ji Y, Ma X, Adelstein RS, Yates JR, 3rd, Kemphues K, Qi L. Nonmuscle myosin IIB links cytoskeleton to IRE1alpha signaling during ER stress. Developmental cell. 2012;23:1141–1152. doi: 10.1016/j.devcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Pandey P, Mishra N, Kumar S, Narula N, Kharbanda S, Saxena S, Kufe D. Targeting of the c-Abl tyrosine kinase to mitochondria in endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2001;21:6233–6242. doi: 10.1128/MCB.21.18.6233-6242.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogvold L, Skog O, Sundstrom G, Edwin B, Buanes T, Hanssen KF, Ludvigsson J, Grabherr M, Korsgren O, Dahl-Jorgensen K. Function of Isolated Pancreatic Islets From Patients at Onset of Type 1 Diabetes: Insulin Secretion Can Be Restored After Some Days in a Nondiabetogenic Environment In Vitro: Results From the DiViD Study. Diabetes. 2015;64:2506–2512. doi: 10.2337/db14-1911. [DOI] [PubMed] [Google Scholar]
- Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci U S A. 2011;108:8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. IRE1alpha Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl M, Danilova T, Palm E, Lindholm P, Voikar V, Hakonen E, Ustinov J, Andressoo JO, Harvey BK, Otonkoski T, et al. MANF is indispensable for the proliferation and survival of pancreatic beta cells. Cell reports. 2014;7:366–375. doi: 10.1016/j.celrep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell. 2009;33:679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, Bluestone JA. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008;105:18895–18900. doi: 10.1073/pnas.0810246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihira K, Taira N, Miki Y, Yoshida K. TTK/Mps1 controls nuclear targeting of c-Abl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene. 2008;27:7285–7295. doi: 10.1038/onc.2008.334. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annual review of pathology. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Paniagua RT, Sharpe O, Ho PP, Chan SM, Chang A, Higgins JP, Tomooka BH, Thomas FM, Song JJ, Goodman SB, et al. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest. 2006;116:2633–2642. doi: 10.1172/JCI28546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa FR. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb Perspect Med. 2012;2:a007666. doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Mochly-Rosen D. The PKCdelta -Abl complex communicates ER stress to the mitochondria - an essential step in subsequent apoptosis. J Cell Sci. 2008;121:804–813. doi: 10.1242/jcs.024653. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Mao T, Zhang Y, Shao M, You J, Ding Q, Chen Y, Wu D, Xie D, Lin X, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells. Sci Signal. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J, Volinsky N, Romano D, Kolch W. The secret life of kinases: functions beyond catalysis. Cell Commun Signal. 2011;9:23. doi: 10.1186/1478-811X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, Mirmira RG. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis S, Roemer K. c-Abl tyrosine kinase can mediate tumor cell apoptosis independently of the Rb and p53 tumor suppressors. Oncogene. 1998;17:557–564. doi: 10.1038/sj.onc.1201973. [DOI] [PubMed] [Google Scholar]
- Villalta SA, Lang J, Kubeck S, Kabre B, Szot GL, Calderon B, Wasserfall C, Atkinson MA, Brekken RA, Pullen N, et al. Inhibition of VEGFR-2 reverses type 1 diabetes in NOD mice by abrogating insulitis and restoring islet function. Diabetes. 2013;62:2870–2878. doi: 10.2337/db12-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19:5643–5650. doi: 10.1038/sj.onc.1203878. [DOI] [PubMed] [Google Scholar]
- Wang JY. The capable ABL: what is its biological function? Mol Cell Biol. 2014;34:1188–1197. doi: 10.1128/MCB.01454-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.