

Abstract
Background
The genetic variation underlying many heritable forms of cardiovascular disease is incompletely understood, even in patients with strong family history or early age at onset.
Methods and Results
We used whole exome sequencing to detect pathogenic variants in 55 patients with suspected monogenic forms of cardiovascular disease. Diagnostic analysis of established disease genes identified pathogenic variants in 21.8% of cases, and variants of uncertain significance in 34.5% of cases. Three patients harbored heterozygous nonsense or splice-site variants in the nucleoporin genes NUP37, NUP43, and NUP188, which have not been implicated previously in cardiac disease. We also identified a heterozygous splice site variant in the nuclear envelope gene SYNE1 in a child with severe dilated cardiomyopathy that underwent transplant, as well as in his affected father. To confirm a cardiovascular role for these candidate genes in vivo, we used morpholinos to reduce SYNE1, NUP37, and NUP43 gene expression in zebrafish. Morphant embryos displayed cardiac abnormalities, including pericardial edema and heart failure. Furthermore, lymphoblasts from the patient carrying a SYNE1 splice-site variant displayed changes in nuclear morphology and protein localization that are consistent with disruption of the nuclear envelope.
Conclusions
These data expand the repertoire of pathogenic variants associated with cardiovascular disease, and validate the diagnostic and research utility of whole exome sequencing. We identify NUP37, NUP43, and NUP188 as novel candidate genes for cardiovascular disease, and suggest that dysfunction of the nuclear envelope may be an under-recognized component of inherited cardiac disease in some cases.
Keywords: gene mutation, nucleoporin, cardiomyopathy, atrial fibrillation arrhythmia
Introduction
Familial cardiac disease accounts for significant morbidity and mortality worldwide.1 Many monogenic heritable forms of cardiomyopathy, ion channelopathy, and aortic aneurysm have been documented clinically. A genetic etiology has been identified for many cardiovascular disease (CVD) cases previously deemed idiopathic, and multiple genes are now clearly associated with monogenic forms of CVD. Despite this, a significant percentage of putatively familial cases lack a molecular diagnosis, 2 indicating that many genetic contributions to Mendelian forms of cardiovascular disease remain undiscovered.
One of the most readily recognized genes associated with monogenic forms of cardiac disease is LMNA, which encodes the intermediate filament proteins lamins A and C, with lamin A being a major component of the nuclear envelope (NE). Several LMNA mutations have been identified in patients displaying a particularly aggressive form of dilated cardiomyopathy (DCM) and conduction system defects, 3,4 and mutations in the lamin A binding protein, emerin, cause Emery Dreifuss Muscular Dystrophy 1 with cardiac conduction defects and atrial arrhythmia.5, 6 Mutations in additional NE genes have also been proposed as causes of various cardiac disease phenotypes, including the presenilin genes PSEN1 and PSEN2, associated with DCM, and TMEM43, associated with Emery-Dreifuss muscular dystrophy 7 and arrhythmogenic right ventricular dysplasia.7-10 Despite these associations, relatively little is known about the 80 or more proteins that reside in the NE regarding a contribution to familial CVD, particularly in comparison to other gene classes, such as genes encoding sarcomere proteins, or ion channels.
The recognition that alterations in NE genes can lead to human disease, including CVD, has created interest in understanding the full repertoire of NE gene mutations seen in humans.11 Whole exome sequencing (WES) can detect coding variants throughout the genome, and thus offers an opportunity to survey the contribution of numerous genes in suspected monogenic forms of CVD. Accordingly, we screened 55 CVD patients for pathogenic variants via WES, and identified a molecular diagnosis for over 20% of our cohort. WES also identified candidate mutations in novel disease genes that encode components of the NE. In this report, we describe four putative truncating variants in the NE genes NUP37, NUP43, NUP188 and SYNE1 found in patients with severe but distinct cardiovascular conditions.
Methods
Patient enrollment
Individuals provided informed consent and were enrolled in the North Carolina Clinical Genomic Evaluation by Next Generation Exome Sequencing (NCGENES) study by a certified genetic counselor. Reported variants were Sanger confirmed in UNC's College of American Pathologists (CAP) and Clinical Laboratory Improvement Amendments (CLIA)- certified clinical molecular genetics laboratory. Reports and clinical interpretation were provided to patients by a board certified medical geneticist and genetic counselor.
Whole exome sequencing
With UNC Institutional Review Board approval, genomic DNA was isolated from whole blood in the UNC Biospecimen Processing Facility. Sample quality and quantity were measured using an Agilent Bioanalyzer or Tape Station. Libraries were prepared using the Agilent SureSelect All Exon V4 kit and paired-end sequenced on a HiSeq2500 at the UNC High Throughput Sequencing Facility at an average depth of 50×, and achieving at least 8× coverage for 98% of all CVD diagnostic genes. Mapping (hg19), alignment, and variant calling were done according to the Broad Institute's best practices using BWA and GATK.12,13 Variants were computationally annotated and filtered using an in-house bioinformatics pipeline based upon genomic position, minor allele frequency (MAF) within the 1000 Genomes database and Exome Aggregation Consortium database (n=61,486 exomes), and predicted protein effect. 14
Diagnostic analysis
Patient WES data was filtered on all gene lists relevant to their phenotype. Diagnostic lists were generated for cardiomyopathy, arrhythmia, and thoracic aortic aneurysm. The diagnostic gene lists were curated from commercial CVD gene panels, Online Mendelian Inheritance in Man (OMIM) entries, and PubMed searches (see Table S1), and variants were annotated with respect to conservation, protein functional domains, and ConDel pathogenicity predictions.15 Variants were classified according to ACMG guidelines.16 Case-level results were defined as “positive,” “negative,” or “uncertain” depending on the degree of phenotypic overlap between the patient and the variant(s)/condition identified 48. Sanger fill-in for hypertrophic cardiomyopathy cases was performed on two regions poorly covered by WES: exons 9 and 14 in MYH7, and exon 8 of MYBPC3.
For the research sweep on negative cases, python scripts were used to make SQL database queries to select rare (MAF < 0.001) frame-shift, splice-site, or nonsense variants throughout the whole exome. Evaluation of Panther cellular compartment classifications (release 20150430) in the AmiGO database revealed ontology for genes on our CVD diagnostic lists. Candidate NE variants were also Sanger verified. SYNE1
(F: TCCTTGAATAACGCTTTGTCCA / R: ACTTAGATGTGGGTAATTCATGCT), NUP37 (F: AGTTTTCTTATTTGTTGTTCCTACCT / R: GCCACAACATTCTTCTTTCCC), NUP43 (F: CCACATGTTCACAATCTTTTGCC / R: TCACCTGCTTTTCCTGAAGACT), and NUP188 (F: ACTCTCCTTAGAACAAAAATGGGGA / R: GCATGTTGAGGGCCACATTC).
Cell culture, Immunofluorescence, and Microscopy
With informed consent and approval from the UNC IRB, follow-up blood samples were obtained from NCG_00024 and his father. Peripheral blood lymphocytes were transformed with Epstein Barr virus by the UNC Tissue Culture Facility. Control cells used were GM12765 from Coriell human cell repository. 6000 cells / well from passage three or earlier were plated onto NuNc / LabTekII CC-2 coated chamber slides (Sigma), adhered for five days, fixed with 4% paraformaldehyde for 30 minutes, and stained in the chamber slides. Primary antibodies used were: rabbit anti-nesprin1 (Sigma; 1:250) and goat anti-laminA (Millipore; 1:250). Cellular actin was visualized by Alexafluor-conjugated phalloidin (Life Technologies). Alexafluor secondary antibodies were used at 1:2000. Slides were cover slipped with Prolong Gold containing DAPI and imaged on a Zeiss AxioImager M2. Samples were blinded and nuclei from two separate experiments were scored. Abnormal shapes were defined as micronuclei, blebbed, or irregular nuclei. Normal morphology was defined as a smooth spherical shape. The difference in the number of abnormal cells between control and patient cells was evaluated using Fisher's t-test.
Expression analysis
RNA from murine heart and zebrafish embryos was isolated using Trizol or RNeasy mini kit (Qiagen). RNA quality was checked on an Agilent Bioanalyzer, and reverse transcribed using QuantiTect Reverse Transcription Kit (Qiagen) or Superscript III supermix (Invitrogen). 1μl of 1:20 cDNA was used to amplify transcripts using Ssofast Evagreen kit (BioRad). Reactions were run in triplicate, and relative expression levels were compared to ACTB or GAPDH. A two-way ANOVA was done to compare gene expression and morpholinos, followed by a post-hoc Fisher's t-test for individual morpholinos.
Animal husbandry
Adult wild type ICR mice and wild type and Tg (myl7:GFP)twu26 zebrafish were maintained at UNC-CH animal facilities. Embryos were produced and recovered by natural spawning or timed matings (mice) in accordance with IACUC protocols. Zebrafish embryos were maintained at 28.5° in system water and treated from 24 hours post fertilization (hpf) with 1-phenyl 2-thoiurea (PTU) to prevent pigmentation.
Morpholinos
Splice-blocking antisense morpholino oligos (MO's) against syne1b, nup37, and nup43 were designed by and purchased from GeneTools, Inc. Sequences (5′-3′): zsyne1b MO-I, e44i44, CCTGGAAATCAAACTTACCTGTAGT; zsyne1b MO-II, e38i38, GCTCTGAAGATGAAGCGTACCTTGA; znup43 MO-I, e7i7, GCAGCGAAATCATTGCTTACTCTGT; znup43 MO-II, e4i4, ATGCGCCACAAAACACTTACCAATA; znup37 MO-I, e4i4, AAAAAGAGAGCTACCTTCACATCAC; znup37 MO-II, e3i3, ACACAAGTTCAAAACTATACCTGA; Standard Control MO, CCTCTTACCTCAGTTACAATTTATA. Nup37 and nup43 MO were used at 7ng, and syne1b MO was used at 8ng. Zebrafish embryos at the 1-2 cell stage were injected with 1 nL of morpholino or 350 pg RNA in water buffered with 5 mM HEPES. For RNA-rescue experiments, pCS2 clones containing sequence-verified Nup37 and Nup43 open reading frames were obtained from the Harvard plasmid repository (HsCD00324272; HsCD00339012). Full length mRNA was transcribed using mMessage machine (Ambion), purified, and analyzed with the Tape Station.
Zebrafish phenotyping
Unfertilized and dead embryos were removed from dishes within 12 hpf, and again at 24 hpf. For phenotyping, 3dpf embryos were placed 1 per well and scored under a brightfield microscope. The significance of the difference in percentage of animals with pericardial edema under different experimental conditions at 72hpf was evaluated first using a Chi-square analysis to compare all groups, followed by a two-tailed Fisher's t-test at alpha < 0.05 with a bonferroni correction of 0.007. Heart chamber abnormality was evaluated in Tg animals by examining GFP-labeled heart chambers at 100×. Any tail curvature was noted. Embryos that died between 24hpf and 72hpf were counted as dead. To assess heart rate, 15-second videos were recorded at 30 frames per second on unanesthetized 3dpf embryos using a Leica M205C fluorescence stereoscope and Leica Application Suite software. Heartbeats were manually scored by a blinded observer and multiplied by four to calculate heart rate in beats per minute.
Results
Exome data was obtained from 55 patients enrolled in the North Carolina Clinical Genomic Evaluation by Next-Generation Exome Sequencing (NCGENES) study. Individuals were referred by a cardiologist with hallmarks of a Mendelian cardio-genetic disease, including severity, age of onset, or positive family history. The cohort reflects the diversity typically encountered clinically; most patients were enrolled for cardiomyopathy or long QT, and patients with thoracic aortic aneurysm (TAA) or severe mitral valve prolapse (MVP), were also enrolled.
Diagnostic Analysis
To investigate WES diagnostic yield of this relatively unselected cohort, we first filtered for variants present in genes associated with the patient's phenotype (Table S1). These diagnostic lists include genes encoding sarcomeric proteins (38 genes; 36.2% of all genes on the diagnostic lists) and ion channels (17 genes; 16.2% of all genes), molecular classes that are well recognized in CVD etiology. Interestingly, we noted that a substantial number of the other genes on these lists encoded proteins that reside in the NE (7 genes; 6.7%). NE genes were present on all three CVD phenotype gene lists: cardiomyopathy (LMNA, EMD, PSEN1, PSEN2, TMEM43, TMPO), arrhythmia (TMEM43, EMD), and thoracic aortic aneurysm (SMAD3).
WES variants from all gene lists relevant to the patient's phenotype were examined. 12 out of 55 (21.8%) patients harbored a known or likely pathogenic variant in an established CVD gene, and an additional 19 out of 55 patients (34.5%) had an uncertain result (Figure 1A). A total of 37 variants were reported to patients, and 16 of these had been previously seen in the primary literature or ClinVar (see Table S2). Individuals with arrhythmia were most likely to get a positive result whereas only 1 out of 11 (12.5%) patients with aneurysms or valvular conditions received a positive result. Cardiomyopathy cases had the greatest number of VUS, although VUS were encountered in all phenotypic classes (Figure 1B).
Figure 1.
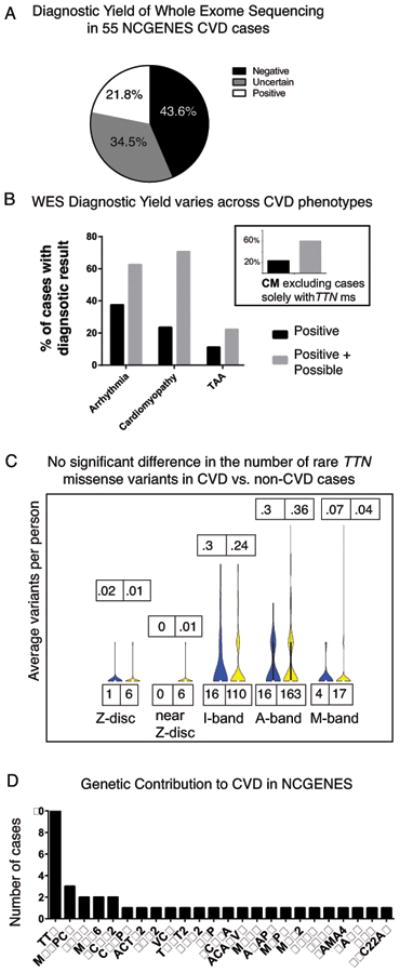
A. WES diagnostic yield in a clinically diverse set of 55 CVD patients. B. Diagnostic yield differs among CVD phenotypic classes. Grey bars: VUS. Inset shows yield minus cases solely with TTN ms variants. C. The distribution of rare TTN missense variants does not differ significantly between CVD and non-CVD NCGENES participants. For each gene region, the number of rare variants (MAF < 0.002) in cases (blue) and controls (yellow) is shown on the bottom. Top numbers show average number of variants in cases then controls for each region of amino acids (NM_001267550.1): Z-band (1-831), Near Z-band (832-2169), I-band (2170-15655), A-band (15656-33588), M-band (33589-35992). D. Genes in which variants were reported back to NCGENES CVD participants. The number of cases in which variants in each gene were reported is shown on the y-axis.
In agreement with others, we found that TTN truncating variants comprise the bulk of mutations for DCM patients.17 Due to the gene's large size, TTN variants are routinely encountered in WES. Because many rare missense variants in TTN could not be excluded as potentially pathogenic, TTN contributed a large number of VUS for our cardiomyopathy cohort. We compared the distribution of all TTN MS variants (MAF < 0.002 in ExAC) in our entire NCGENES cohort, and found no significant difference in the distribution of TTN MS variants that were rare or had higher CADD scores (an in silico prediction of deleteriousness) in DCM cases compared to 451 controls (NCGENES participants without a cardiovascular phenotype, Figure 1C, Table S3).
While TTN variants were reported in 10 of 55 CVD cases, other genes contributed pathogenic variants to only one or two cases. Two individuals with hypertrophic cardiomyopathy (HCM) carried distinct pathogenic variants in MYH6, and two HCM cases harbored the same rare pathogenic missense variant in MYBPC3 (Table S2). All other genes only explained one case (Figure 1D).
Research Analysis
A significant number of cases were negative in our diagnostic analysis (43.6%, Figure 1A). To identify novel candidate disease-causing variants in these cases, we broadened our analysis to the entire captured exome. Examination of all rare (MAF < 0.001) nonsense, splice-site, and frame-shifting variants identified four candidate mutations in NE genes. (Figure 2). Interestingly, three of these predicted loss of function (pLOF) variants were in genes encoding nucleoporins (NUPs), proteins that comprise the nuclear pore complexes (NPC). NPCs are multiprotein structures that facilitate nucleocytoplasmic shuttling of mRNAs and proteins, provide structural integrity to the NE, bind other NE components such as lamin A, and may also interact with chromatin.18-21
Figure 2. Sanger confirmation of variants in NUP37, NUP43, NUP188, and SYNE1.
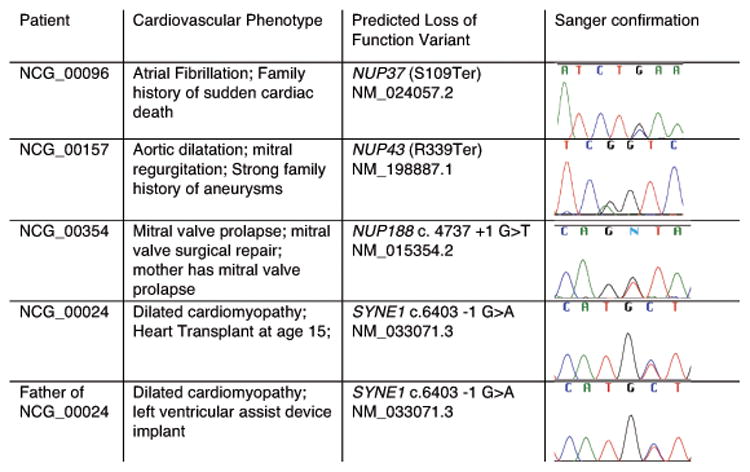
Heterozygous nonsense variants were identified in the nucleoporins NUP37 and NUP43, and a heterozygous consensus splice donor variant was identified in NUP188 (Figure 2, 3A). While each individual in whom these variants were identified was enrolled for an apparently autosomal dominant form of CVD, each had a distinct phenotype. Candidate variants were Sanger confirmed. Thus, we identified three different pLOF variants in nucleoporins in CVD patients with severe, but distinct cardiovascular phenotypes.
Figure 3.
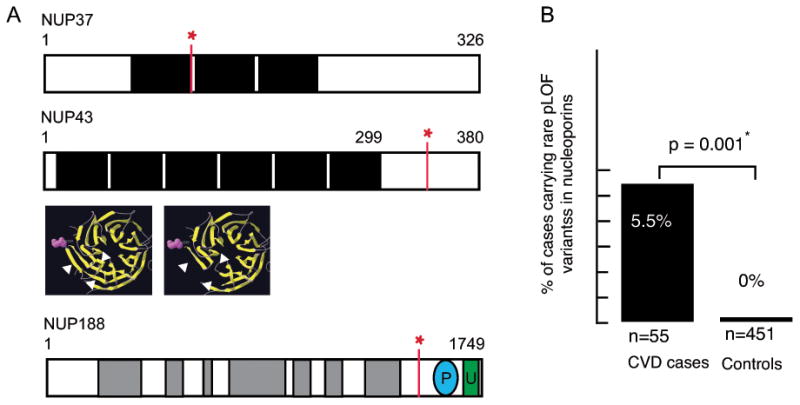
A. Schematic of the three nucleoporin variants found in CVD patients. The nonsense variant in NUP37 is located just 3′ to the first WD domain, shaded black. The nonsense variant in NUP43 may produce a protein missing several C-terminal beta sheets (arrows; model shown in yellow). The location of the NUP188 splice site variant is shown in relation to armadillo repeats (grey) and known phosphorylation and ubiquitination sites. B. Three out of 55 CVD patients carried a rare (MAF < 0.001) truncating variant in a nucleoporin gene, vs. none in 451 patients without CVD. P = 0.001, Fisher's exact t-test.
We also identified a splice acceptor variant in the NE gene SYNE1 in a 17-year old male with DCM that underwent heart transplant at age 15. His father also has DCM and has a left ventricular assist device implanted. SYNE1 has previously been implicated in cardiomyocyte function and cardiomyopathy in mice, as well as a myopathy with variable cardiac and skeletal muscle involvement, Emery-Dreifuss muscular dystrophy 4 (EDMD4, OMIM 612998). 22-25 Both of these individuals were followed clinically for years by a cardiologist and were not noted to have any muscle weakness. The ability to perform cascade testing for these cases was limited; however, the affected father of the subject with severe DCM was available for follow-up analysis. Targeted sequencing showed that the affected father also carries the SYNE1 c.6403-1 splice variant (Figure 2).
In vitro and in vivo evaluation of candidate disease-associated NE genes
The NUP37, NUP188, and SYNE1 variants are not present in the ExAC database of 61,486 exomes, and the NUP43 (R339Ter) variant is rare (MAF= 0.00002), indicating these variants cannot be considered benign polymorphisms based upon population frequency. Moreover, we found that while three out of 55 CVD patients carried a truncating variant of MAF < 0.001, none were found in NCGENES participants enrolled for non-cardiovascular conditions (n=451, p =0.001, Figure 3B). These data show that rare pLOF variants in nucleoporins are enriched in patients with CVD.
Very little functional data exists on the nucleoporins we identified, and their expression in heart has not previously been described. To address this, we generated cDNA from adult as well as embryonic day 11 (E11.0) mouse heart, and screened for expression of our candidate genes. All genes were expressed in both E11.0 and adult mouse heart, and expression levels vary between stage of development and adult heart compartment (Figure 4A). Expression of NUP37 was 7-fold higher in atrium vs. ventricle, which was notable given the atrial fibrillation phenotype seen in this patient (Figure 4B). These data are in agreement with gene expression data found in the Genotype-Tissue Expression database (GTEx; http://www.gtexportal.org; accessed July 8, 2016) showing that these genes are expressed in human heart.
Figure 4.
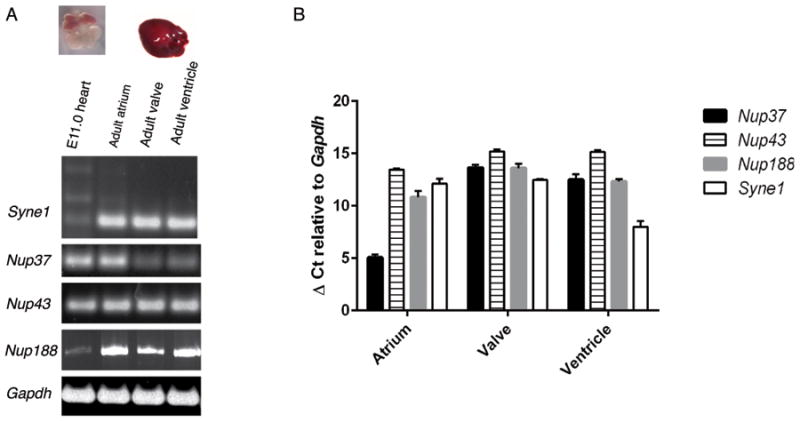
A. Syne1, Nup37, Nup43, and Nup188 transcripts were detected in embryonic day 11.0 mouse heart (lane 1), as well as adult mouse atrium (lane 2), valve (lane 3), and ventricle (lane 4). B. Relative expression levels of Nup37, Nup43, Nup188 and Syne1 in adult mouse heart normalized to expression of Gapdh. Data are represented as average of triplicate reactions, with standard deviation error bars.
Mice missing a portion of the SYNE1 gene display abnormal nuclear shape, mislocalization of NE components, and cardiomyopathy.23 Similar changes have been shown in cells from a patient with cardiomyopathy carrying a SYNE1 missense variant 24. To determine whether our WES-identified SYNE1 splice site variant was associated with cytological changes in NE morphology, we analyzed patient lymphoblasts. Nesprin1 protein in control cells was present on the nuclear surface and extended into the cytoplasm (Figure 5A). In the DCM patient, nesprin1 staining was detected in the NE in a pattern similar to control cells, although the staining appeared more confined to the nucleus, and less homogenous, with large nuclear regions devoid of nesprin1 staining (Figure 5B). SYNE1-encoded nesprin protein binds to lamin A, a marker of the inner NE, and alterations in lamin A are known to cause DCM, so we examined expression of lamin A protein in patient cells. In contrast to the spherical perinuclear staining in control cells, patient nuclei demonstrated immunoreactive puncta within nuclear blebs and bulges (Figure 5C, D). Patient cells also had a globular pattern of actin in contrast to the well-organized linear arrays of actin typically seen in control cells (Figure 5E, F), consistent with a previously reported role for SYNE1 in proper actin binding, formation and localization.25-27 The number of patient cells with an abnormal nuclear morphology was significantly increased compared to control cells (Figure 5G). Thus, examination of cells from the patient carrying a splice-site variant in SYNE1 is consistent with morphological alterations in the nuclear envelope.
Figure 5.
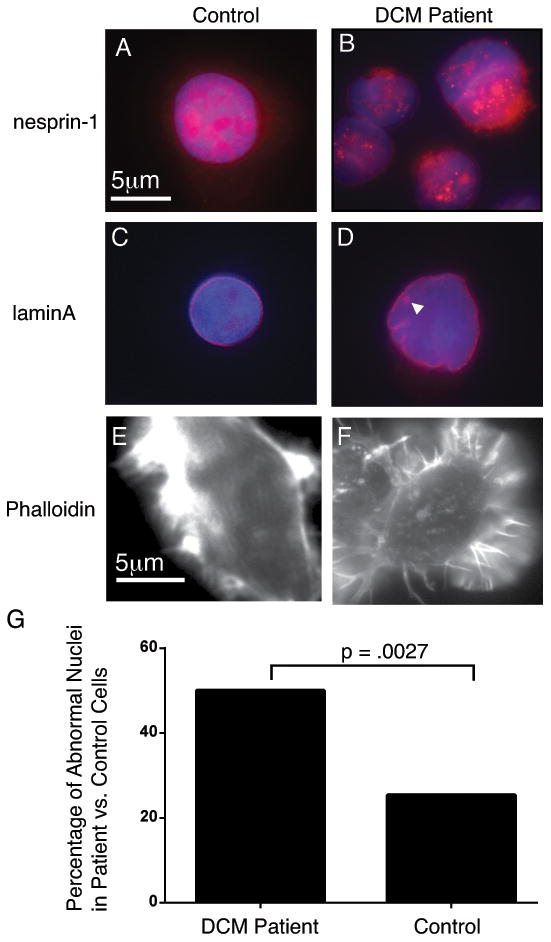
Cells from the patient carrying a SYNE1 splice site variant display nuclear morphology consistent with NE disruption. A. Control cells exhibit nesprin 1 staining throughout the nucleus and into the cytoplasm. B. Cells from the DCM patient express nesprin-1, although staining appears less consistent, with some nuclear regions appearing unstained. C. Lamin A staining in control cells defines a spherical perinuclear ring. D. Lamin A staining in DCM patient cells with irregular nuclei. Note the ring of lamin A staining seen inside the nucleus (arrowhead). E. Phalloidin staining in control cells show linear arrays of actin. F. The actin in DCM patient cells appears less organized and more globular than control cells. G. Percentage of cells with abnormal nuclei is significantly higher in cells from the DCM patient with the SYNE1 (50/94) variant vs. control cells (25/63).
Functional evaluation of candidate genes
The phenotypes of the patients carrying pLOF variants in NE genes suggested that mutations in these genes might preferentially affect cardiovascular development or function, and knockdown of one of the candidate genes, nup188, has previously been shown to result in heart looping defects in Xenopus.28 We therefore investigated the functional role of SYNE1, NUP37, and NUP43 in vivo through morpholino knockdown in zebrafish.
Because zebrafish embryos rely on diffusion to circulate oxygen throughout their bodies for the first several days of life, it is possible to study mutations which severely impact cardiovascular development and circulation, and which ultimately lead to heart failure. Accordingly, zebrafish have become a model organism of choice for investigating the function of candidate CVD genes.29, 30
We confirmed the presence of zebrafish orthologues of human SYNE1, NUP37, and NUP43, and all are expressed in zebrafish heart during development. Expression of nup37 and nup43 is stable from day 2 to day 3, while expression of syne1b increases approximately 12-fold from day 2 to day 3, when phenotypic scoring was done and animals were sacrificed (data not shown).
Zebrafish embryos injected with 4nM standard control morpholino (MO), which does not bind any physiological target, were indistinguishable from un-injected clutch mates (Figure 6A). Injection of an antisense (MO) that disrupts splicing of the normal nup37 transcript resulted in reduced nup37 expression at 3 dpf, as well as severe pericardial edema, a characteristic manifestation of zebrafish heart failure, along with reduced blood flow and apparent arrhythmia in some animals (Figure 6B; Video S1). Injection of the nup37 MO into Tg (myl7:EGFP) embryos,31 which have fluorescently labeled cardiac myocytes, demonstrated abnormalities of the heart chambers in several animals, including small or stretched atrium or malpositioned chambers that had not looped properly (Figure S1). A second, independent nup37 morpholino designed against a separate splicing boundary yielded similar results (Figure S1). Co-injection of full-length human NUP37 mRNA rescued the morphant phenotype to a significant extent (Figure 6G).
Figure 6.
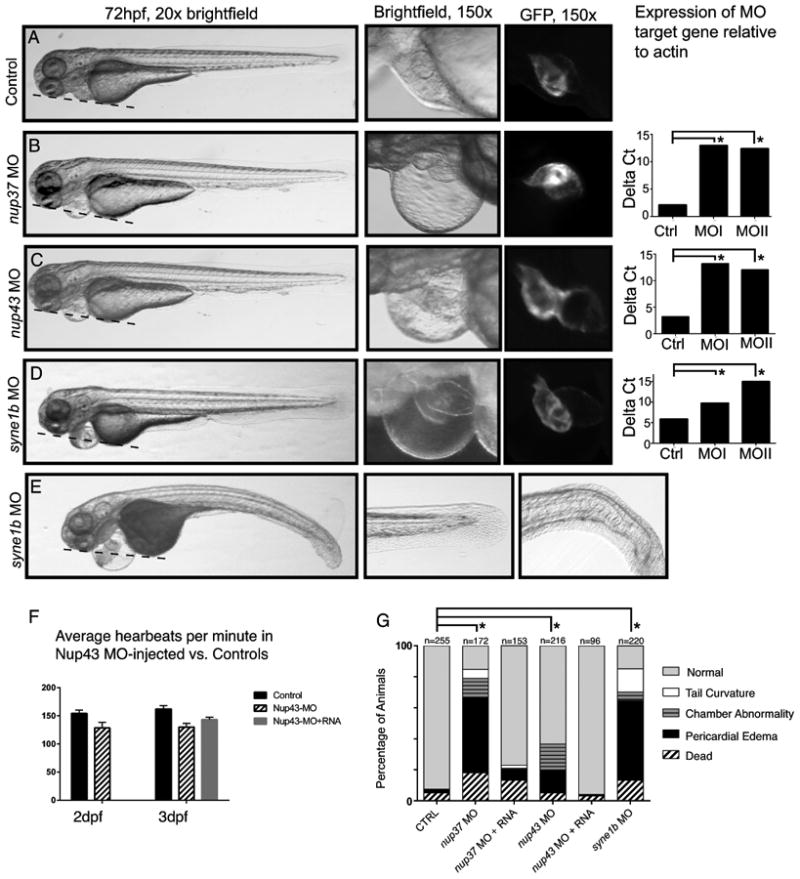
Representative images from embryos injected with control MO (A), nup37 MO (B), nup43 MO (C), and syne1b MO (D). 20× (left) and 150× (middle) brightfield or fluorescence images of in Tg embryos. Dashed lines denote plane across the head and yolk sac. In wild type animals, the pericardial chamber is in the same plane, while edema in morphant animals causes the pericardial chamber to swell significantly outside that plane. Right-most panels show qPCR results from control (Ctrl) and MO-injected animals harvested at 3dpf. Asterisks denote statistically significant differences in gene expression using two-way ANOVA analysis and follow-up t-test. E. Representative images of the tail phenotypes seen in some syne1b MO-injected zebrafish. F. The heart rate of nup43 morphants is slower than control zebrafish at 2dpf and 3dpf, and this phenotype is significantly rescued in nup43 MO + nup43 mRNA-injected animals. G. Summary of morphant phenotypes. The asterisk designates significant difference, p < 0.0001 in the percentage of animals with pericardial edema.
Previously described mutations associated with TAA result in pericardial edema when evaluated in zebrafish.32 Nup43 morphants also developed the expected pericardial edema, although the proportion was less than in nup37 MO-injected animals (Figure 6C, G). Fifteen percent of nup43 morphants displayed pericardial edema or heart chambers that were in a linear orientation, and appeared to have not fully looped properly (Figure 6C); this phenotype was ameliorated in embryos injected with nup43 MO plus human NUP43 RNA (Figure 6G). At 3dpf, the heart rate of many nup43 morphants appeared slower than control-injected animals. We quantified the apparent difference in heart rate by optical recording of Tg animals injected with either nup43 MO, control MO, or nup43 MO followed by 350 pg NUP43 mRNA. nup43 morphants had a statistically significant slower heart rate than control animals at both 2 and 3dpf. Co-injection of full-length human NUP43 mRNA significantly ameliorated the bradyarrythmia in nup43-MO injected embryos (Figure 6F). Thus, reduced expression of nup37 and nup43 results in cardiovascular defects without any apparent effects on the rest of the body.
The majority of embryos injected with a morpholino targeting syne1b developed severe pericardial edema (Figure 6 D,E,G). A small proportion of morphants displayed a curved tail phenotype (Figure 6E, Figure S2), consistent with SYNE1 being implicated in the variable skeletal and cardiac myopathy EDMD4. Heart morphology appeared normal in many syne1b morphants with pericardial edema, while some morphants showed small or stretched heart chambers in severely affected animals (Figure S2). Many syne1b morphants with pericardial edema had pooled blood and reduced or absent blood flow (Video S2). Rescue with full-length SYNE1 mRNA was not feasible given the length of its 27kb open reading frame; a second, independent morpholino designed against a separate splicing boundary yielded similar results (Figure S2). These results indicate that aberrant expression of nuclear envelope genes can lead to abnormal cardiac development and heart failure in zebrafish.
Discussion
Diagnostic Analysis
Our results demonstrate that WES has both diagnostic and research utility for a diversity of suspected monogenic forms of cardiovascular disease. WES identified a pathogenic variant in 21.8% of CVD cases, with diagnostic yield varying among phenotypic classes. Due to lack of uniform coverage for all exons, it is possible that causative variants were missed in this analysis, although most genes showed good coverage across all individuals (Table S4). We found a substantial number of novel likely pathogenic variants, and our annotations of these variants in well-phenotyped individuals will aid the clinical interpretation of these variants by future groups. Truncating A-band mutations in TTN are a significant cause of cardiomyopathy, and thus, TTN variants must be evaluated for any DCM patient undergoing clinical WES. Our analysis found no increase in rare or deleterious TTN MS variants in any region of the gene, in CVD cases compared with non-CVD cases. These results support a growing body of evidence suggesting caution is warranted in interpreting rare TTN missense variants as pathogenic in cases of autosomal dominant cardiomyopathy. 33,34
While cardiomyopathy and arrhythmia cases had a relatively high yield, the yield for TAA and MVP cases was much lower, likely due to a smaller number of known disease genes for these phenotypes. We felt strongly that diagnostic results should only be reported in genes with a strong gene-disease association; our yield is thus more akin to running a targeted gene panel. A major advantage with the use of WES as a diagnostic tool, as opposed to using a gene panel approach, is that WES allows further investigation of negative cases.
The yield of “uncertain” results in this study is consistent with the experience of practicing clinicians; while such results do not change clinical management, they are a valid type of result routinely returned by clinical laboratories and might be useful if an extended pedigree were available for co-segregation studies to allow them to be classified further 49.
Nucleoporins and Cardiac Disease
Unexpectedly, three cases in our cohort carried rare pLOF variants in nucleoporin genes not previously associated with CVD, despite truncating variants in these genes being absent in our broader NCGENES cohort, or exceedingly rare in the ExAC database. The sum of all pLOF variants reported in ExAC for NUP37 for example, is less than 1 in 10,000, indicating that truncating variants in this gene are quite rare. This is the second report to our knowledge of a nucleoporin associated with familial CVD. Mutations in NUP155 have been implicated in familial atrial fibrillation and sudden death, 35 the same phenotype and family history present in NCG_00096 who carries the NUP37 (R339Ter) variant. NUP37, NUP43, and NUP188 are, like NUP155, scaffold nucleoporins, whose expression is important both for NPC assembly as well as transport function.19
As NUPs have been shown to play a number of different mechanistic roles beyond maintaining structural integrity of the NE, including shuttling of specific cargo molecules36, 37 and interacting with chromatin to regulate gene expression, 20 the exact mechanisms by which the NUP variants could lead to a cardiovascular phenotype remain unclear. Truncating variants producing nonsense-mediated decay would be expected to lead to haploinsufficiency, a mechanism that is supported by morpholino knock-down experiments. Decreased nup188 expression in Xenopus embryos results in heart looping defects, a phenotypic finding we also noted in a subset of our nup37 and nup43 morphants. Alternatively, several of the truncating variants could create proteins missing functional domains, which could result in loss-of-function or possibly dominant negative effects. While our zebrafish data indicate a potential role for nucleoporins in cardiovascular development, these proteins are likely to function in adult heart as well, and ultimately, it is difficult to correlate phenotypes seen in fish with phenotypes one would see in a human.38
Nesprin in Cardiomyopathy
The SYNE1-encoded protein nesprin-1 is part of the linker of the nucleus to the cytoskeleton (LINC) complex. Multiple isoforms have been reported for nesprin-1, and at least one heart-specific regulatory element has been described.26 The NE-anchored nesprin-1 protein responds to mechanical force initiated at the cell surface through its interaction with cytoplasmic actin, and thereby affects downstream molecular changes in the nucleus.39 Studies in mice demonstrate that nesprin-1 coordinates the proper biomechanical gene response in cardiomyocytes and contributes to the cytoskeletal defects in LMNA-deficient cardiomyopathy; heterozygous nesprin mice display cardiac defects (M. Pucklewartz, personal communication 40), and SYNE1 variants have been identified in at least two families with DCM, with or without skeletal muscle involvement. 24,47 Physical disruption of normal LINC complex interactions is therefore a plausible biological mechanism underlying the cardiomyopathy in our patient and father.
The cardiovascular system, which undergoes constantly changing force and pressure, may be particularly vulnerable to alterations in NE components, including nuclear pores and LINC proteins.41-44 The phenotype of each patient in our cohort carrying a candidate mutation in these NE genes is distinct, suggesting a spectrum of possible cardiovascular phenotypes dependent upon the NE gene affected. Nevertheless, the WES results suggest that human mutations in this gene class may be particularly important for cardiovascular disease, a conclusion supported by our statistical analysis in non-CVD NCGENES patients, expression analysis in murine heart, and in vitro cell studies, as well as our zebrafish studies, where morphant animals developed cardiovascular phenotypes. Together, our results support the idea that a healthy, intact nuclear envelope is important for normal heart function and expand the repertoire of genetic variants in NE genes that are associated with cardio-genetic disease.
Few NE genes have been sufficiently studied to establish a strong gene-disease association. We find the association of inner nuclear membrane (INM) NE genes is stronger than that of outer nuclear membrane (ONM) NE genes. The INM genes LMNA, EMD, SMAD3, and TMEM43 genes are more established as causative according to more rigorous criteria45, and numerous reports have upheld a role for these NE genes in CVD. While multiple lines of evidence are converging to support a role for NE genes in CVD, many of these should still be considered candidate genes until further evidence is generated (Figure 7). The NE and nuclear transport have emerged as potential therapeutic targets for modulating genetic programs associated with disease.46 Identifying individuals with mutations in NE genes that could benefit from these approaches is therefore important. Future work will further delineate the spectrum of phenotypes associated with mutations in NE genes and investigate the mechanism by which NE genes, and nucleoporins specifically, contribute to CVD in humans.
Figure 7.
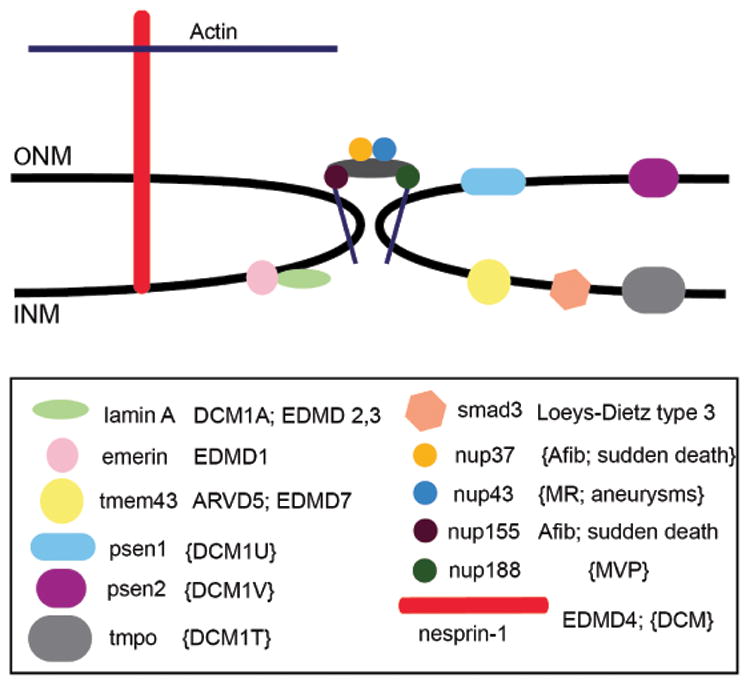
NE genes associated with familial CVD are schematically depicted within the inner nuclear membrane (INM), outer nuclear membrane (ONM), or nuclear pore complex. Established gene-disease associations are unbracketed; candidate associations are shown in brackets.
Supplementary Material
Clinical Perspective.
Although monogenic cases of various forms of cardiac disease have been suspected clinically for some time, the genes that contribute to pathogenesis in those cases that are “negative” via traditional genetic testing have often eluded us. Although one nucleoporin, NUP155, has been reported to cause familial atrial fibrillation, nucleoporins as a whole, are not well-recognized as playing a substantial role in cardiovascular disease. We observed that truncating variants in nucleoporins were enriched in patients with cardiovascular disease, compared to over 400 controls without cardiovascular disease, and provide functional evidence in zebrafish that reduction of gene expression for two nucleoporins, NUP37 and NUP43, results in a cardiac phenotype. We also provide functional evidence that SYNE1 splice variants can be associated with cardiovascular disease in the absence of skeletal muscle disease, in both humans and zebrafish. Given the importance of LMNA in Mendelian cases of cardiovascular disease, our results highlight the overall importance of nuclear envelope genes as an entire physiological group, beyond just LMNA, that could be considered, along with other important gene groups, such as sarcomere proteins, or ion channels in relation to cardiovascular traits. . The techniques used – whole exome sequencing followed by a research analysis of negative individuals, along with functional follow-up, provides an additional framework for use in cardiovascular genetics.
Acknowledgments
We thank the family members that participated in this study.
Sources of Funding: National Human Genome Research Institute award U01HG006487 to J. P. Evans, J. S. Berg, K. C. Wilhelmsen, K. Weck, and G.T. Haskell; NIH K08HL096836 and a grant from the UAI Research Foundation to B. Jensen; UNC TraCS Institute UL1RR025747 and 550KR61305 to J.S. Berg.
Footnotes
Disclosures: None
References
- 1.Ganesh SK, Arnett DK, Assimes TL, Assimes TL, Basson CT, Chakravarti A, et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: update: a scientific statement from the American Heart Association. Circulation. 2013;128:2813–2851. doi: 10.1161/01.cir.0000437913.98912.1d. [DOI] [PubMed] [Google Scholar]
- 2.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 4.Lu JT, Muchir A, Nagy PL, Worman HJ. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4:562–568. doi: 10.1242/dmm.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 6.Brown CA, Scharner J, Felice K, Meriggioli MN, Tarnopolsky M, Bower M, et al. Novel and recurrent EMD mutations in patients with Emery-Dreifuss muscular dystrophy, identify exon 2 as a mutation hot spot. J Hum Genet. 2011;56:589–594. doi: 10.1038/jhg.2011.65. [DOI] [PubMed] [Google Scholar]
- 7.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor MRG, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, et al. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 9.Merner ND, Hodgkinson KA, Haywood AFM, Connors S, French VM, Drenckhahn JD, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2005;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69:1005–1013. doi: 10.1002/ana.22338. [DOI] [PubMed] [Google Scholar]
- 11.Worman HJ. Components of the nuclear envelope and their role in human disease. Novartis Found Symp. 2005;264:35–42. discussion 42–50, 227–30. [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reilly J, Stanley A, McGee J, Owen P, Schmitt C, Wilhelmsen K. MaPSeq, A Service-Oriented Architecture for Genomics Research within an Academic Biomedical Research Institution. Informatics. 2015;(2):20–30. [Google Scholar]
- 14.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;16:601–608. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014 doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- 18.Ibarra A, Hetzer MW. Nuclear pore proteins and the control of genome functions. Genes Dev. 2015;29:337–349. doi: 10.1101/gad.256495.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loïodice I, Alves A, Rabut G, Van Overbeek M, Ellenberg J, Sibarita JB, et al. The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell. 2004;15:3333–3344. doi: 10.1091/mbc.E03-12-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D'Angelo MA, Hetzer MW. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 2008;18:456–466. doi: 10.1016/j.tcb.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ptak C, Aitchison JD, Wozniak RW. The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr Opin Cell Biol. 2014;28C:46–53. doi: 10.1016/j.ceb.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee I, Zhang J, Moore-Morris T, Pfeiffer E, Buchholz KS, Liu A, et al. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 2014;10:e1004114. doi: 10.1371/journal.pgen.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puckelwartz MJ, Kessler E, Zhang Y, Hodzic D, Randles KN, Morris G, et al. Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Hum Mol Genet. 2009;18:607–620. doi: 10.1093/hmg/ddn386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–2833. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG. The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics. 2002;80:473–481. [PubMed] [Google Scholar]
- 26.Rajgor D, Mellad JA, Autore F, Zhang Q, Shanahan CM. Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One. 2012;7:e40098. doi: 10.1371/journal.pone.0040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellad JA, Warren DT, Shanahan CM. Nesprins LINC the nucleus and cytoskeleton. Curr Opin Cell Biol. 2011;23:47–54. doi: 10.1016/j.ceb.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci USA. 2011;108:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res. 2011;91:279–288. doi: 10.1093/cvr/cvr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih YH, Zhang Y, Ding Y, Ross CA, Li H, Olson TM, et al. Cardiac transcriptome and dilated cardiomyopathy genes in zebrafish. Circ Cardiovasc Genet. 2015;8:261–269. doi: 10.1161/CIRCGENETICS.114.000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- 32.Guo D, Gong L, Regalado ES, Santos-Cortez RL, Zhao R, Cai B, et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet. 2015;96:170–177. doi: 10.1016/j.ajhg.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, et al. Role of Titin Missense Variants in Dilated Cardiomyopathy. J Am Heart Assoc. 2014;4:e002645. doi: 10.1161/JAHA.115.002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–1027. doi: 10.1016/j.cell.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 36.Terry LJ, Wente SR. Nuclear mRNA export requires specific FG nucleoporins for translocation through the nuclear pore complex. J Cell Biol. 2007;178:1121–1132. doi: 10.1083/jcb.200704174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Xu L. Specific nucleoporin requirement for Smad nuclear translocation. Mol Cell Biol. 2010;30:4022–4034. doi: 10.1128/MCB.00124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarazón E, Rivera M, Roselló-Lletí E, Molina-Navarro MM, Sánchez-Lázaro IJ, España F, Montero JA, et al. Heart failure induces significant changes in nuclear pore complex of human cardiomyocytes. PLoS One. 2012;7:e48957. doi: 10.1371/journal.pone.0048957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, Garcia-Mata R, Burridge K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol. 2014;16:376–381. doi: 10.1038/ncb2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikolova-Krstevski V, Leimena C, Xiao XH, Kesteven S, Tan JC, Yeo LS, et al. Nesprin-1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C-deficient cardiomyopathy. J Mol Cell Cardiol. 2011;50:479–486. doi: 10.1016/j.yjmcc.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Stroud MJ, Banerjee I, Veevers J, Chen J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res. 2014;114:538–548. doi: 10.1161/CIRCRESAHA.114.301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dahl KN, Ribeiro AJS, Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ Res. 2008;102:1307–1318. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol. 2010;2:a000760. doi: 10.1101/cshperspect.a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meinke P, Nguyen TD, Wehnert MS. The LINC complex and human disease. Biochem Soc Trans. 2011;39:1693–1697. doi: 10.1042/BST20110658. [DOI] [PubMed] [Google Scholar]
- 45.MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faustino RS, Nelson TJ, Terzic A, Perez-Terzic C. Nuclear transport: target for therapy. Clin Pharmacol Ther. 2007;81:880–886. doi: 10.1038/sj.clpt.6100141. [DOI] [PubMed] [Google Scholar]
- 47.Pucklewartz MJ, Kessler EJ, Kim G, Dewitt MM, Zhang Y, Earley JU, et al. Nesprin-1 mutations in human and murine cardiomyopathy. J Mol Cell Cardiol. 2010;48:600–608. doi: 10.1016/j.yjmcc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strande NT, Berg JS. Defining the Clinical Value of a Genomic Diagnosis in the Era of Next-Generation Sequencing. Annu Rev Genomics Hum Genet. 2016;17:303–332. doi: 10.1146/annurev-genom-083115-022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, et al. Genetic Testing for Dilated Cardiomyopathy in Clinical Practice. J Card Fail. 2012;18:296–303. doi: 10.1016/j.cardfail.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.