

Abstract
In pathway‐targeted cancer drug therapies, the relatively rapid emergence of drug‐tolerant persisters (DTPs) substantially limits the overall therapeutic benefit. However, little is known about the roles of DTPs in drug resistance. In this study, we investigated the features of epidermal growth factor receptor–tyrosine kinase inhibitor‐induced DTPs and explored a new treatment strategy to overcome the emergence of these DTPs. We used two EGFR‐mutated lung adenocarcinoma cell lines, PC9 and II‐18. They were treated with 2 μM gefitinib for 6, 12, or 24 days or 6 months. We analyzed the mRNA expression of the stem cell‐related markers by quantitative RT‐PCR and the expression of the cellular senescence‐associated proteins. Then we sorted DTPs according to the expression pattern of CD133 and analyzed the features of sorted cells. Finally, we tried to ablate DTPs by glucose metabolism targeting therapies and a stem‐like cell targeting drug, withaferin A. Drug‐tolerant persisters were composed of at least two types of cells, one with the properties of cancer stem‐like cells (CSCs) and the other with the properties of therapy‐induced senescent (TIS) cells. The CD133high cell population had CSC properties and the CD133low cell population had TIS properties. The CD133low cell population containing TIS cells showed a senescence‐associated secretory phenotype that supported the emergence of the CD133high cell population containing CSCs. Glucose metabolism inhibitors effectively eliminated the CD133low cell population. Withaferin A effectively eliminated the CD133high cell population. The combination of phloretin and withaferin A effectively suppressed gefitinib‐resistant tumor growth.
Keywords: Cell interaction, drug resistance, EGFR‐TKI, senescent cells, stem‐like cells
Abbreviations
- 2DG
2‐deoxy‐D‐glucose
- Akt
protein kinase B
- AMPK
adenosine monophosphate‐activated protein kinase
- CSC
cancer stem‐like cell
- DTP
drug‐tolerant persister
- EGFR
epidermal growth factor receptor
- HDAC
histone demethylase
- IGF‐1R
insulin‐like growth factor 1 receptor
- mTOR
mammalian target of rapamycin
- NSCLC
non‐small‐cell lung cancer
- PI3K
phosphatidylinositol 3‐kinase
- qRT‐PCR
quantitative RT‐PCR
- Rb
retinoblastoma protein
- SASP
senescence‐associated secretory phenotype
- TIS
therapy‐induced senescence
- TKI
tyrosine kinase inhibitor
- WFA
withaferin A
Epidermal growth factor receptor–TKIs such as gefitinib and erlotinib are first‐line treatments for advanced NSCLC harboring EGFR‐activating mutations, and have been reported to improve the clinical outcome and quality of life of patients with EGFR‐mutated NSCLC.1, 2, 3, 4 However, the relatively rapid emergence of resistance to EGFR‐TKIs substantially limits the overall therapeutic benefit.5, 6 A secondary EGFR T790M mutation is the most common type of acquired resistance to EGFR‐TKIs7 and c‐MET amplification, PIK3CA mutations, BRAF mutations and small‐cell lung cancer transformation are also associated with acquired resistance to EGFR‐TKIs.8, 9, 10 However, the mechanisms responsible for approximately 30% of cases of acquired resistance to EGFR‐TKIs are still unknown.
Recent studies have revealed novel non‐mutational mechanisms of drug resistance. For example, a small population of CSCs is intrinsically more refractory to the effects of a variety of anticancer drugs, possibly through enhanced drug efflux.11 Cancer stem‐like cells are defined as cells within a tumor that possesses the capacity to self‐renew and to generate the heterogeneous lineages of cancer cells that comprise the tumor.12 Pre‐existing or de novo emerging CSCs can survive anticancer drug treatment, continue sustained growth, and result in the emergence of drug‐resistant subclones.13 However, the possible roles of CSCs in acquired resistance to EGFR‐TKIs are still unknown. Effective treatment is not available for acquired resistance to EGFR‐TKIs except for third‐generation EGFR‐TKIs targeting the EGFR T790M mutation.14 It is therefore necessary to analyze the transition state of EGFR‐TKI resistance and to eliminate drug‐resistant clones in the transition phase.
Sharma et al.15 reported that treatment with EGFR‐TKIs leads to the emergence of a small subpopulation of drug‐tolerant cells, named DTPs, in the transition phase, during which EGFR‐TKIs show acute and dramatic antitumor activity in NSCLC patients harboring EGFR‐activating mutations.16 The EGFR‐TKI‐induced DTPs show >100‐fold reduced drug sensitivity and maintain viability through engagement of IGF‐1R signaling and an altered chromatin state that requires the HDAC RBP2/KDM5A/Jarid1A.15 Although an HDAC inhibitor and an IGF‐1R inhibitor did prevent the emergence of DTPs in vitro, the combination of an HDAC inhibitor (vorinostat) and erlotinib did not show a meaningful clinical benefit in EGFR‐mutated NSCLC patients.17 In addition, a phase III trial (ADVIGO 1018) that examined the effects of figitumumab, a fully human anti‐IGF‐1R G2 mAb, in combination with erlotinib as a second/third‐line treatment in previously treated NSCLC patients, failed and was discontinued.18
A major contributor to EGFR‐TKI treatment failure is the formation and emergence of EGFR‐TKI‐induced DTPs. Conventional anticancer drug treatments are of minor benefit to overcome and prevent EGFR‐TKI‐induced DTPs. In this context, we investigated the features of EGFR‐TKI‐induced DTPs and explored a new treatment strategy to overcome the emergence of these DTPs.
Materials and Methods
Cells lines and reagents
Human EGFR‐mutant PC9 (exon19del E746‐A750; Sigma‐Aldrich, St. Louis, MO, USA), HCC827 (exon19del E746‐A750; ATCC, Manassas, VA, USA) and II‐18 (L858R; RIKEN Cell Bank, Riken, Tsukuba, Japan) cells were purchased. All of the cell lines were cultured in RPMI‐1640 media supplemented with 10% FBS and 1% penicillin–streptomycin (Wako, Osaka, Japan), and were grown in a humidified incubator with 5% CO2 at 37°C. Gefitinib (Selleck Chemicals, Houston, TX, USA), cisplatin (Nichi‐Iko, Toyama, Japan), docetaxel (Sanofi, Paris, France), pemetrexed (Eli Lilly, Indianapolis, IN, USA), WFA (Wako), 2‐deoxy‐D‐glucose (Sigma‐Aldrich), and phloretin (Sigma‐Aldrich) were purchased.
RNA extraction and qRT‐PCR
Total RNA was extracted from cell lines as previously described.19 Single‐stranded cDNA was synthesized using SuperScript III First‐Strand reagent kits (Life Technologies, Waltham, MA, USA). Real‐time PCR was performed using a Thermal Cycler Dice Real Time System II (Takara) with primers purchased from Life Technologies (Table S1). Amplifications were carried out in triplicate with SYBR Premix Ex Taq II (Takara, Kusatsu, Japan), according to the manufacturer's instructions. Target mRNA levels were normalized against GAPDH, and the relative mRNA expression levels were calculated. The experiments were carried out in triplicate.
Western blot analysis
The detailed protocol for Western blotting has been described previously.19 The indicating antibodies to the following proteins were used in this study: β‐actin (#4967), EGFR (D38B1) (#4267), phosphor‐EGFR (Tyr1068) (#2234), phospho‐Rb (Ser780) (#9307), p21 Waf1/Cip1 (12D1) (#2947), p27 Kip1 (D69C12) (#3686), phosphor‐Histone H2A.X (Ser139/Tyr142) (#5438), AMPKα (23A3) (#2603), and phospho‐AMPKα (Thr172) (#2535) were purchased from Cell Signaling Technology (Danvers, MA, USA).
In vitro sphere‐forming assay
To study stem‐like cell properties, a sphere‐forming assay was undertaken as described previously.20 Sorted or drug‐treated viable PC9 cells were plated in ultra‐low attachment 6‐well plates (Corning, Corning, NY, USA) at a density of 10 000 cells/mL in serum‐free DMEM/F12 medium (Life Technologies) supplemented with 20 ng/mL epidermal growth factor (Sigma), 10 ng/mL basic fibroblast growth factor (Sigma‐Aldrich), 5 μg/mL insulin (Sigma‐Aldrich), 1× B27 supplement (Life Technologies), and 0.4% BSA (Sigma‐Aldrich). The cells were cultured under 5% CO2 at 37°C for 1 week.
Cell surface marker analysis using flow cytometry
Cells (1 × 105) were resuspended in 100 μL Hanks’ balanced salt solution supplemented with 2% BSA were incubated with 5 μL polyclonal mouse anti‐human CD133‐FITC conjugated antibody (1:20; Miltenyi Biotec, Bergisch Gladbach, Germany) and were analyzed using a BD FACSAria III (BD Biosciences, Franklin Lakes, NJ, USA). In this study, we sorted DTPs into CD133high and CD133low cell fractions based on CD133 expression level using the FACSAria III cell sorter.
Tumorigenicity in nude mice
Female BALB/c nude mice (6 weeks old) were purchased from SLC (Shizuoka, Japan). Mice were inoculated s.c. in the flank with 5.0 × 104 cells/100 μL suspension of parental PC9 cells and CD133high‐ and CD133low‐sorted PC9 DTPs. This study was approved by the Institutional Animal Care and Use Committee (permit number: 150108) and was carried out according to the Animal Experimentation Regulations of Kobe University (Kobe, Japan).
In vivo tumor growth inhibition assay
Suspensions of PC9 cells (5 × 106) were injected s.c. into the backs of female BALB/c nude mice (6 weeks old) (SLC). The length (x) and width (y) of the tumor masses were measured twice a week, and tumor volume (TV) was calculated using the formula TV = (x × y2) / 2. Relative tumor volume on day n was calculated using the formula: relative tumor volume = TVn / TV0, where TVn is the tumor volume on day n and TV0 is the tumor volume on day 0. When TV reached 100 mm3, mice were divided into five groups consisting of four mice per group (day 0) and were then allocated into one vehicle group and four gefitinib (6.25 mg/kg daily) groups. When the TV re‐reached 100 mm3 after it was transiently reduced by gefitinib, the four gefitinib groups were re‐allocated as follows: (i) a gefitinib (6.25 mg/kg daily) treatment group;21 (ii) gefitinib (6.25 mg/kg daily) and phloretin (10 mg/kg three times per week) combination treatment group;22 (iii) a gefitinib (6.25 mg/kg daily) and WFA (2 mg/kg three times per week) combination treatment group;23 and (iv) gefitinib (6.25 mg/kg daily), phloretin (10 mg/kg three times per week), and WFA (2 mg/kg three times per week) combination treatment group. Gefitinib was dissolved in DMSO and in glucose solution for the in vivo study. Phloretin and WFA were dissolved in methanol and in glucose solution for the in vivo study.
Results
Treatment with EGFR‐TKI induces TIS cells and CSC marker expression
Drug‐tolerant persisters were generated within a few days of exposure of PC9 and II‐18 cells to a concentration (2 μM) of an EGFR‐TKI (gefitinib) that was higher than the IC50 value. Their drug‐tolerant phenotype was confirmed using flow cytometric and pathological analyses as described by Sharma et al.15 (data not shown). Because the genotoxic stress induced by anticancer drugs, including gefitinib, is known to induce cellular senescence (TIS),23 we investigated the characteristics of these gefitinib‐induced DTPs, focusing on cellular senescence. β‐Galactosidase staining revealed the induction of senescence in PC9 cells following gefitinib treatment (Fig. 1a). Similarly, Western blot analyses showed time‐course‐dependent upregulation of senescence‐associated proteins (pRb, p27, p21, and γH2AX) (Fig. 1b). Cellular senescence is considered to be a state of stable exit from the cell cycle and senescent cells are assumed to be permanently arrested.24 However, response to gefitinib treatment in EGFR‐mutated lung cancer is often short‐lived and has failed to result in cures.5 Hence, the acquisition of resistance and recurrence to gefitinib treatment is thought to result from a feature of DTPs other than the cellular senescence of cells. We therefore further explored whether DTPs have CSC properties. The upregulation of stem cell‐related markers such as Oct3/4, Nanog, sox2, myc, CD133, CD44, ALDH1A1, ABCB, and ABCG following gefitinib treatment of PC9 and II‐18 cells was confirmed using qRT‐PCR (Fig. 1c).20 These data suggested that gefitinib induced DTPs that have both cellular senescent and CSC properties.
Figure 1.
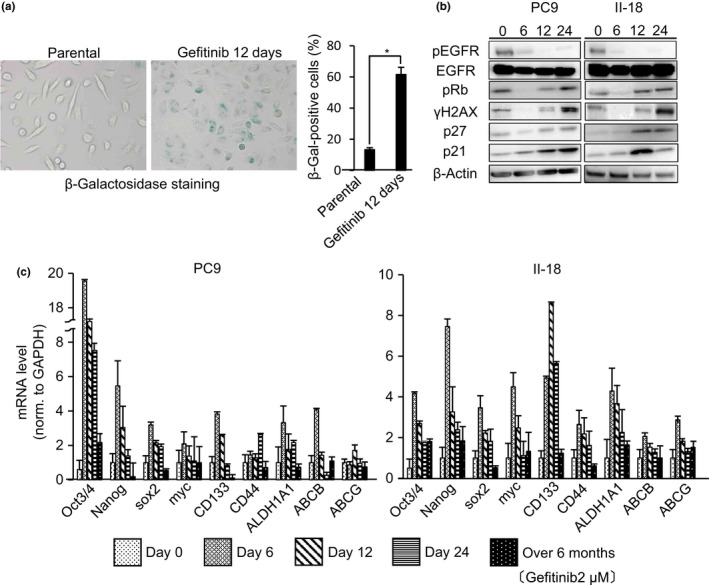
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor treatment induces cellular senescence‐associated protein production and stem cell marker expression in EGFR‐mutated non‐small‐cell lung cancer cell lines PC9 and II‐18. (a) PC9 cells were treated with gefitinib (2 μM) for 12 days, stained with β‐galactosidase (β‐Gal), and quantified. Several fields were counted and data are the means ± SD of 300 cells counted over three independent experiments (right panel). *P < 0.001. (b) PC9 and II‐18 cells were treated with gefitinib (2 μM) for the times indicated on day 0, 6, 12, and 24, following which the cells were lysed and immunoblotted as indicated. Cropped gels are shown for greater clarity. (c) PC9 and II‐18 cells were treated with gefitinib (2 μM) for the indicated times. Cellular mRNA expression levels of stem cell‐related markers were analyzed by quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
Gefitinib treatment increases the fraction of CD133high cells in gefitinib‐induced DTPs
Next we determined if it was possible to discriminate senescent cells from CSCs of gefitinib‐induced DTPs. Recent studies have indicated that NSCLC contains cells that express the glycoprotein prominin‐1 (CD133), which is a CSC marker that is essential for tumor cell propagation and metastasis.25 We therefore examined the expression of CD133 in gefitinib‐induced DTPs. We exposed PC9 cells to gefitinib (2 μM) and followed the CD133high cell population using flow cytometric analysis. We removed apoptotic PC9 cells (17%–31% of total cells) before flow cytometric analysis because such cells lose the CD133 membrane marker and could therefore bias the analysis. A significant increase in the CD133high cell population was observed 12 days after gefitinib treatment (Fig. 2). We could separate DTPs into two cell fractions based on CD133 expression level using the FACSAria III cell sorter.
Figure 2.
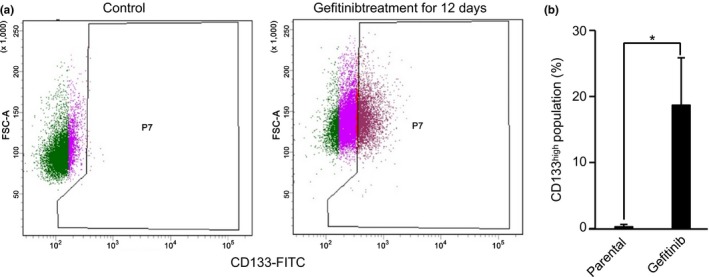
Epidermal growth factor receptor–tyrosine kinase inhibitor treatment induces CD133high cells in drug‐tolerant persisters. (a) PC9 cells treated with gefitinib (2 μM) for 12 days were stained with CD133 and analyzed using flow cytometry. Gating was used to identify CD133high cells. (b) CD133high cells were quantified using flow cytometry. Results are the means ± SD of three independent experiments. *P < 0.001.
CD133high cell population has CSC properties and CD133low cell population has senescent cell properties
To investigate the differences between CD133high and CD133low cell populations, we first carried out Western blotting for senescence‐associated proteins (Figs 3a,S1). The expression of these proteins was increased in the CD133low cell population compared to the CD133high population. The expression of stem cell‐related markers was then evaluated using qRT‐PCR (Figs 3b, S2) and relatively higher expression of these markers was observed in the CD133high cell population compared to the CD133low cell population.
Figure 3.
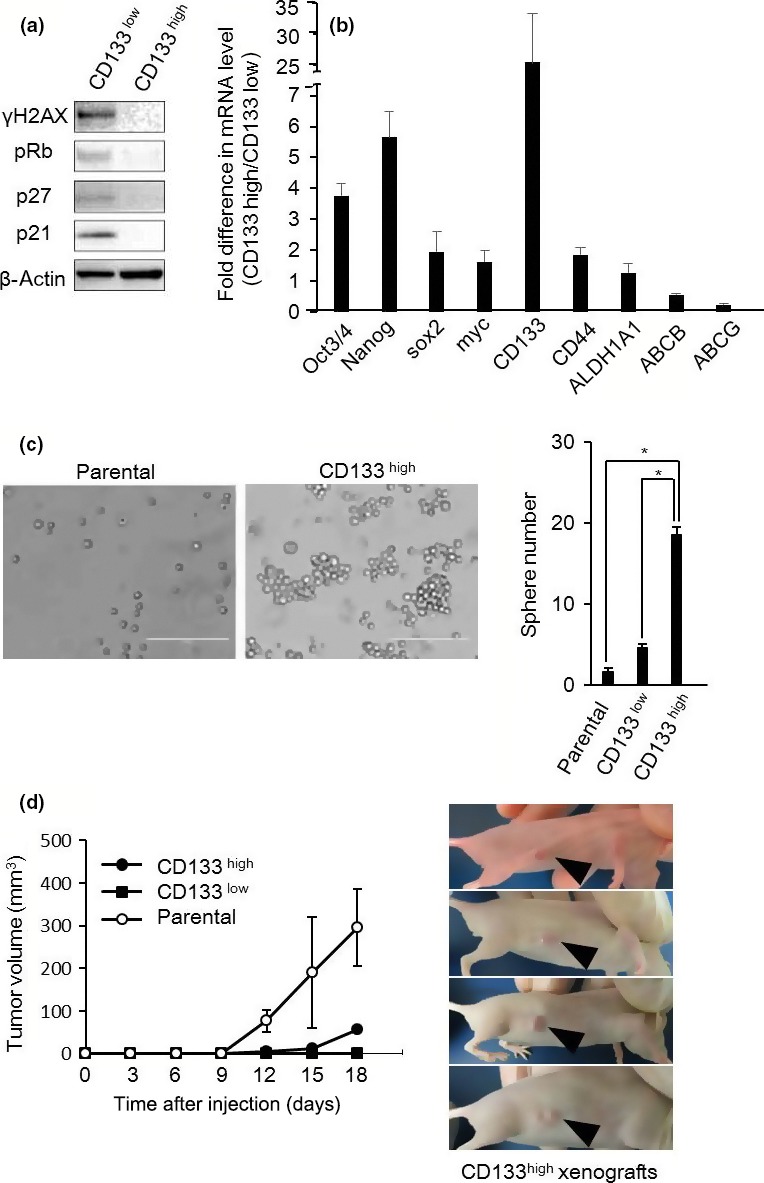
Comparison of the features of CD133high and CD133low non‐small‐cell lung cancer cells. (a) CD133high and CD133low cells were sorted from PC9 drug‐tolerant persisters 12 days after gefitinib (2 μM) treatment. Whole‐cell lysates were prepared and analyzed by Western blotting as indicated. (b) mRNA expressions of stem cell‐related markers in CD133high and CD133low cells sorted from PC9 drug‐tolerant persisters 12 days after gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Relative expression levels were obtained by dividing the mRNA level in CD133high cells by that in CD133low cells. Results are the means ± SD of three independent experiments. (c) Sphere‐forming assay of parental, CD133high, and CD133low cells. Left, phase contrast images. Scale bar = 50 μm. Right, quantification of sphere number. CD133high cells showed an enhanced capacity for self‐renewal compared with parental and CD133low cells. Results are the means ± SD of at least three independent experiments. *P < 0.01. (d) Xenograft tumor volume was measured on the indicated days following parental, CD133high, and CD133low cell injection (left panel). Results are means ± SD (four mice). Images show the CD133high tumors of four mice 18 days after injection (right panel).
We next undertook an in vitro sphere forming assay (Fig. 3c) to explore CSC properties of the CD133high cell population. Compared to the parental and the CD133low cell population, the CD133high cell population gradually formed significantly more sphere colonies of various sizes and irregular shapes after approximately 1 week of culture. Furthermore, we injected the CD133high cell population s.c. into BALB/c nude mice. After approximately 10 days, the injected CD133high cells started to gradually form tumors (Fig. 3d). These results indicated that the CD133high cell population possessed strong tumorigenicity. Collectively, these data suggested that the CD133low cell population has senescent cell properties and the CD133high cell population has CSC properties.
Senescence‐associated secretory phenotype of CD133low cell population drives the emergence of CD133high cell population
We then investigated the relationship between the CD133high and the CD133low cell populations. We first undertook in vitro assays that focused on the assay of soluble factors from the CD133low cell population. Recent increasing evidence indicates that senescent cells actively communicate with neighboring cells through a plethora of secretory factors including inflammatory cytokines, chemokines, and growth factors. This senescence‐related phenotype is called the SASP.26, 27 We evaluated the SASP of gefitinib‐treated PC9 and II‐18 cells using qRT‐PCR (Fig. 4a). Gefitinib‐induced SASP was enhanced in a time‐course‐dependent manner. Interestingly, the CD133low cell population containing senescent cells showed a stronger SASP than the CD133high cell population (Fig. 4b). To understand how the SASP of senescent cells might contribute to an increase in the proportion of CSCs (the CD133high cell population), we compared the effect of incubation of PC9 cells with gefitinib plus the conditioned medium of gefitinib‐treated (12 days) cells with incubation of PC9 cells with gefitinib plus standard, non‐conditioned medium. When PC9 cells were cultured in the conditioned medium plus gefitinib, there was a greater induction of the CD133high cell population than when the PC9 cells were cultured in the standard, non‐conditioned medium containing gefitinib (Fig. 4c). In contrast, neutralizing anti‐bodies against some SASP effectively suppressed the induction of the CD133high cell population Fig. (S3). These results suggest that the SASP regulates an increase in the proportion of cancer stem cells. Rapamycin, an mTOR inhibitor and autophagy inducer, evokes cellular senescence but represses the SASP.28 We investigated whether rapamycin could suppress the SASP induced by gefitinib treatment. Quantitative RT‐PCR analyses of the SASP showed that the addition of rapamycin suppressed the gefitinib‐induced SASP (Fig. 4d). Furthermore, flow cytometric analysis indicated that the addition of rapamycin suppressed the gefitinib‐induced increase in the proportion of the CD133high cell population (Fig. 4e). These data suggested that the gefitinib‐induced senescent cells (the CD133low cell population) contributed to an increase in the proportion of CSCs (CD133high cell population) through the SASP.
Figure 4.
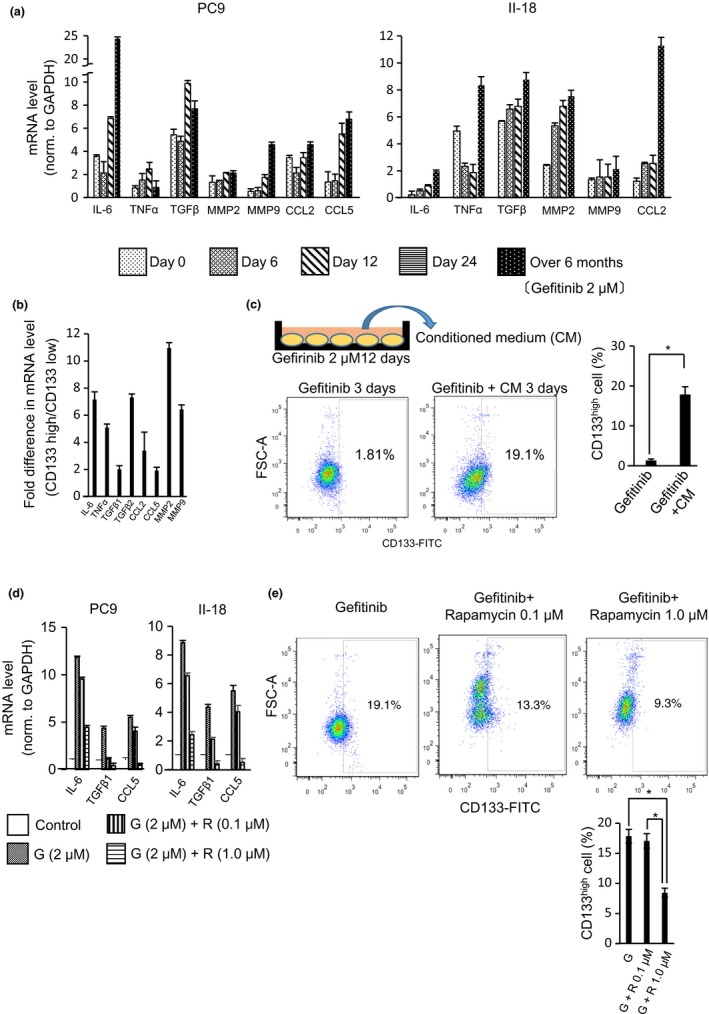
Senescence‐associated secretory phenotype (SASP) induced by epidermal growth factor receptor–tyrosine kinase inhibitor treatment contributes to the emergence of CD133high cells in drug‐tolerant persisters. (a) PC9 and II‐18 cells were treated with gefitinib (2 μM) over the indicated time. An SASP was determined by analysis of the cellular mRNA levels of the indicated proteins using quantitative (q)RT‐PCR. Results are means ± SD of three independent experiments. (b) mRNA expressions of SASP‐associated proteins in CD133low and CD133high cells sorted from PC9 drug‐tolerant persisters 12 days after gefitinib (2 μM) treatment was analyzed using qRT‐PCR. Relative expression levels were obtained by dividing the mRNA level in CD133low cells by that in CD133high cells and results are the means ± SD of three independent experiments. (c) PC9 cells were cultured either in medium containing gefitinib (2 μM) or in the conditioned medium (obtained from PC9 cells treated with gefitinib [2 μM] for 12 days). Cells were stained with anti‐CD133 antibody after 3 days of culture and were analyzed using flow cytometry. Results are the means ± SD of at least three independent experiments. *P < 0.01 (d) mRNA expressions of SASP‐associated proteins in PC9 cells (control) and PC9 cells treated with gefitinib (G) or with gefitinib combined with rapamycin (R) were analyzed using qRT‐PCR. Results are the means ± SD of three independent experiments. (e) PC9 cells that were treated for 7 days with gefitinib (G, 2 μM), or with gefitinib combined with 0.1 μM or 1.0 μM rapamycin (R), were stained for CD133. CD133high cells were quantified using flow cytometry. Results are the means ± SD of three independent experiments. *P < 0.05.
Glucose metabolism‐targeting therapies and CSC targeting drug, WFA, are new therapeutic strategies for gefitinib‐induced DTPs
We determined whether conventional anticancer drugs including cisplatin and pemetrexed were effective against gefitinib‐induced DTPs. We generated gefitinib‐induced DTPs by exposing PC9 cells to gefitinib (2 μM) for 7 days. The DTPs were examined for their response to conventional anticancer drugs by using a growth inhibition assay (Fig. 5a). Conventional anticancer drugs were not effective against these DTPs. Recently, therapy of induced senescent cells has been reported to be dependent on senescence‐related metabolic reprogramming, which is composed of enhanced glycolysis and a hypermetabolic phenotype.24 We therefore investigated whether gefitinib‐induced senescent cells were dependent on glucose metabolism. Compared to the CD133high cell population, the CD133low cell population showed higher phosphorylation reflecting activation of AMPK (Figs 5b,S4) and qRT‐PCR analysis showed upregulation of the glucose transporters Glut1 and Glut3, and the glycolytic enzyme hexokinase 2 (Hk2) in the CD133low cell population (Fig. 5c). These results suggested that the CD133low cell population that contained senescent cells was more dependent on glucose metabolism than the CD133high cell population. Thus, glucose metabolism targeting therapies and WFA, which are novel therapeutic agents for CSCs,29 are promising strategies for combating gefitinib‐induced DTPs. To evaluate their efficacy, we assayed the effect of glucose metabolism‐targeting therapies including 2DG and phloretin, and WFA on gefitinib‐induced DTPs (Fig. 5a). Interestingly, these drugs had an antitumor activity against the parent cells as well as the DTPs. To assess the efficacy of phloretin and WFA in a separate in vivo model, we treated mice bearing xenografts of PC9 cells. To generate gefitinib‐induced DTPs in this in vivo model, we first treated PC9 cell xenograft models with gefitinib (6.25 mg/kg daily) and, after the tumors had reprogressed on gefitinib treatment, we added phloretin, WFA, or their combination, to gefitinib. These additional drugs had a dramatic inhibitory effect on progressed tumor progress (Fig. 5d). Thus, glucose metabolism‐targeting drugs and the CSC targeting drug, WFA, are promising drugs to overcome gefitinib‐induced DTPs and to prevent cancer recurrence.
Figure 5.
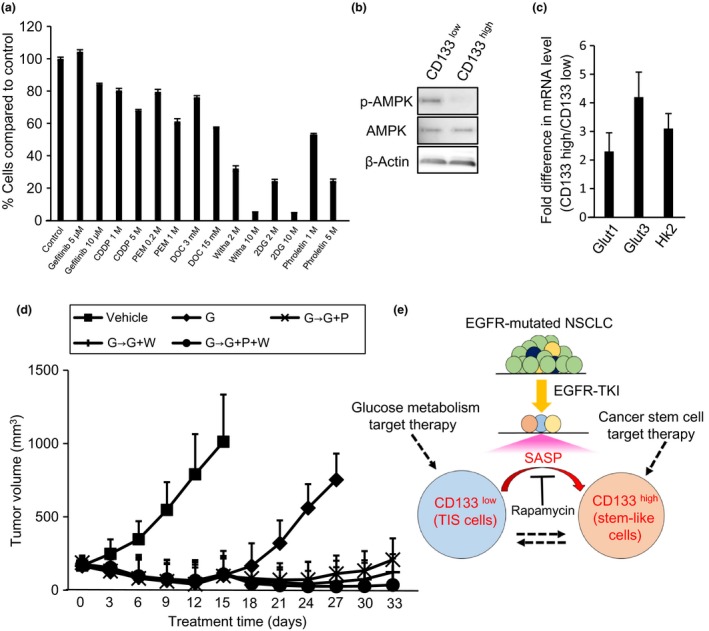
Glucose metabolism targeting treatment and withaferin A (Witha) treatment are effective in vivo for treatment of gefitinib‐induced drug‐tolerant persisters (DTPs). (a) DTPs were induced by exposing PC9 cells to gefitinib (2 μM) for 7 days. DTPs were treated with DMSO (control), gefitinib, conventional drugs (cisplatin [CDDP], pemetrexed [PEM], or docetaxel [DOC]), stem cell‐targeting with a, or glucose metabolism‐targeting drugs (2‐deoxy‐D‐glucose [2DG] and phloretin) with the indicated concentrations of each drug for 3 days. After 3 days of treatment, the percentage of DTPs (relative to control) was measured. Results are the means ± SD of three independent experiments. (b,c) CD133high and CD133low cells were sorted from PC9 DTPs 12 days after gefitinib (2 μM) treatment. (b) Whole‐cell lysates were prepared and analyzed by Western blotting as indicated. (c) mRNA expression of glucose transporters (GLUT1 and GLUT3) and the glycolytic enzyme hexokinase2 (HK2) was analyzed using qRT‐PCR. Relative expression levels were obtained by dividing the expression level in CD133high cells by that in CD133low cells and the results are the means ± SD of three independent experiments. (d) Gefitinib‐treated (6.25 mg/kg daily) PC‐9 bearing mice were divided into four groups of four mice each. After treatment was maintained for 3 days, gefitinib (6.25 mg/kg daily) was continued in one group (◆), one group was switched to gefitinib (6.25 mg/kg daily) combined with phloretin (10 mg/kg three times per week) (X), one group was switched to gefitinib (6.25 mg/kg daily) combined with Witha (2 mg/kg three times per week) (+), and the other was switched to gefitinib (6.25 mg/kg daily) combined with phloretin (10 mg/kg three times per week) and Witha (2 mg/kg three times per week) (●). Tumor volume was determined on the indicated days of treatment. Bars indicate SEM. (e) Hypothetical model showing the DTP‐targeting therapy strategy of this study.
Discussion
In this study, we analyzed the features of gefitinib‐induced DTPs. Our analysis suggested that gefitinib‐induced DTPs are composed of at least two types of cells: one has the properties of CSCs, and the other has the properties of TIS cells. The FACS analysis of CD133 (Prominin‐1) expression, a marker for CSCs,25 divided the DTPs into CD133high and CD133low cell populations. The CD133high cell population has CSC properties and the CD133low cell population has senescent cell properties. Numerous studies have found that high levels of CD133 expression confers chemotherapy resistance and cancer stemness on various types of cancer cells.30, 31, 32, 33 In glioblastoma, CD133 expression induces chemoresistance by increasing expression of MDR1 through the PI3K‐Akt signal pathway.34 A high level of expression of CD133 therefore suggested that the CD133high cell population in gefitinib‐induced DTPs possesses cancer stem‐like properties and chemoresistance.
Whether DTPs emerge de novo during gefitinib treatment or exist in the parental cell population before treatment is an intriguing and significant question. Recent studies have reported that NSCLC cells before treatment contained CD133 high expressing population with strong self‐renewal and multipotent differentiation capacities.35, 36 The ClonTracer barcode system37 tracking of more than 1 million individual cells in cultured cancer models revealed that chemoresistant clones are present before treatment. Gefitinib treatment abolishes chemosensitive parental clones. However, CD133‐expressing parental chemoresistant clones would survive this treatment and proliferate, resulting in the emergence of CD133‐expressing DTPs. The CD133low cell population would be induced in response to gefitinib‐induced genotoxic stress. In the current study (Fig. 1b), senescence‐associated protein expression was identified in parental cells before treatment but initially disappeared after gefitinib treatment. These proteins were re‐expressed on day 12 after gefitinib treatment. The senescence‐associated protein bands observed by Western blotting before treatment (day 0) imply oncogene‐induced senescence, which is characterized as a proliferative arrest elicited by oncogenic RAS and RAF through induction of p16Ink4a.38, 39 However, the senescence‐associated protein bands that emerged after treatment imply TIS, which is thought to be an irreversible cell cycle arrest induced by anticancer drug treatment, including gefitinib, through genotoxic stress of the cancer cells.24 Based on the above, it was considered that parental oncogene‐induced senescence cells abolished by gefitinib treatment and that chemoresistant TIS cells emerged de novo.
The TIS CD133low cell population showed an SASP (Fig. 4a,b). Recently, increasing evidence from sophisticated mouse models indicates that senescent cells actively communicate with neighboring cells through an SASP.26, 27 Our analysis showed that the CD133low cell population has higher potential for secreting various inflammatory cytokines and chemokines including interleukin‐6, transforming growth factor‐β, tumor necrosis factor‐α than the CD133high population. Some studies have found that these pro‐inflammatory cytokines and survival signals, such as transforming growth factor‐β and interleukin‐6 can induce functional changes in some cell populations that not only increase their drug resistance but also contribute to the preservation of stemness in certain conditions.40, 41 To analyze the correlation between the SASP of the CD133low cell population after gefitinib treatment and the increase in the proportion of the CD133high cell population, we used rapamycin, which effectively suppressed the SASP.28 Addition of rapamycin to gefitinib treatment effectively suppressed the TIS‐induced SASP and simultaneously inhibited the increase in the proportion of the CD133high cell population. These results suggested that the CD133low cell population forms a CSC niche that produces an SASP, and that blocking the SASP with rapamycin is an effective strategy for prevention of gefitinib induced DTP emergence, although we need to examine the adequacy of SASP carefully because the CD133high cell population started to increase before the upregulation of SASP on day 6 (Figs 1c,4a). In support of this possibility, the combination of BGT226, a novel PI3K/mTOR dual inhibitor and gefitinib showed supra‐additive growth inhibitory effects on EGFR‐mutated NSCLC cells in vitro and in vivo.42 Furthermore, mTOR inhibitors temsirolimus and everolimus overcome hepatocyte growth factor‐dependent resistance to EGFR‐TKIs in EGFR‐mutated NSCLC cells.43 Although recent clinical studies of mTOR inhibitors combined with EGFR‐TKIs have failed to show clinical benefit in unselected NSCLC patients,44, 45, 46 mTOR inhibitor and EGFR‐TKI combination therapy should be re‐evaluated in EGFR‐mutated NSCLC patients after acquired resistance to EGFR‐TKIs.
To investigate other anticancer drugs that might show promise for overcoming DTPs, and that might act as an alternative to mTOR inhibitors, we examined the sensitivity of gefitinib‐induced DTPs to various types of drugs, including conventional chemotherapeutic agents (cisplatin, pemetrexed, and docetaxel) and EGFR‐TKI. However, the DTPs showed significant resistance to conventional chemotherapeutic agents and gefitinib (Fig. 5a). The recent IMPRESS trial47 is the only randomized phase III trial to compare continuation of gefitinib in combination with conventional cytotoxic chemotherapy versus chemotherapy alone in patients with advanced EGFR‐mutation‐positive NSCLC with resistance to first‐line gefitinib. This trial showed that the additional conventional cytotoxic chemotherapy does not contribute to improvement in progression‐free survival. This result means that the combination of existing chemotherapeutic agents is not a promising strategy for overcoming gefitinib‐induced DTPs.
We therefore considered new strategies to eliminate gefitinib‐induced DTPs. Anticancer chemotherapy has recently been reported to induce not only TIS but also senescence‐related metabolic reprogramming in cancer cells.24 Therapy‐induced senescence resulted in increased glycolytic activity in cancer cells and enforced the Warburg effect. The enhanced glycolytic activity induces insensitivity to TKIs used for chronic myeloid leukemia.48 Our analysis showed that TIS cells of the CD133low cell population increased expression levels of the glucose transporters Glut1 and Glut3, and the glycolytic enzyme hexokinase 2 (Hk2), and enhanced AMPK activity compared to the CD133high cell population (Figs 5b,c,S4,(S5). The CD133low cell population was susceptible to a glucose transporter inhibitor (phloretin) and a glycolytic blocking agent (2DG) in vitro or phloretin in vivo. To our knowledge, there is no report that 2DG has been used in vivo analyses. Further functional in vitro analysis is necessary to link between glucose metabolism and the resistance to TKIs. The CSC‐containing CD133high cell population was also resistant to conventional chemotherapeutic agents. In order to overcome this treatment limitation, novel therapeutic strategies to eliminate CSCs have been developed.49, 50, 51 Withaferin A, a bioactive compound isolated from the plant Withania somnifera, is a potent inhibitor of CSCs.29 To the best of our knowledge, we are the first to show that WFA has a promising antitumor effect on lung cancer cells both in vitro and in vivo, and that WFA is a potent therapeutic agent to overcome gefitinib‐induced DTPs. Unfortunately, we could not culture CD133low cell and CD133high cell populations separately (data not shown). After isolation of the CD133high cell population from gefitinib‐induced DTPs, we attempted to culture the CD133high cell population alone. However, after a few days of culture, a CD133low cell population emerged in the culture dish. Addition of some cytokines in the supernatant failed to maintain the CD133high cell population. These results meant that we could not evaluate the drug effects of phloretin, 2DG, and WFA on individual CD133low and CD133high cell populations.
Based on our results, we conclude that gefitinib‐induced DTPs are composed of TIS cells containing a CD133low cell population and CSCs containing a CD133high cell population. Glucose metabolism‐targeting therapeutic agents have effective antitumor activity against the CD133low cell population; the CSC‐targeting agent, WFA, has effective antitumor activity against the CD133high cell population, as summarized in Figure 5(e). Although these glucose‐targeting therapeutic agents and WFA are not available in a clinical setting at present, they are potent agents for overcoming gefitinib‐induced DTPs and are worth evaluating in prospective clinical trials. The combination of these agents and gefitinib will be a promising therapeutic strategy to overcome EGFR‐TKI resistance.
Disclosure Statement
Y. Nishimura received research grants from Eli Lilly Japan, AstraZeneca, and Novartis Pharma. The other authors have no conflict of interest.
Supporting information
Fig. S1. Cellular senescence‐associated proteins production from parental cells and drug‐tolerant persisters. Whole‐cell lysates were prepared and analyzed by Western blotting as indicated.
{kind=link}
Fig. S2. The CD133high cell population showed relatively higher expression of stem cell‐related markers compared to the CD133low cell population. mRNA expressions of stem cell‐related markers in parental, drug‐tolerant persister (DTP), CD133high, and CD133low cells sorted from PC9 cells 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
{kind=link}
Fig. S3. Neutralizing antibodies against the senescence‐associated secretory phenotype suppressed the induction of the CD133high cell population. PC9 cells were cultured either in medium or in medium containing gefitinib (2 μM) for 12 days. Drug‐tolerant persisters were incubated with gefitinib (2 μM), gefitinib plus anti‐transforming growth factor‐β (TGF‐β) antibody (1 mg/mL; R&D Systems, Minneapolis, MN, USA), gefitinib plus anti interleukin (IL)‐6 antibody (1 mg/mL; R&D Systems), or gefitinib plus anti‐CCL5 antibody (1 mg/mL; R&D Systems) for the last 3 days. The cells were stained with anti‐CD133 antibody after 3 days of culture and were analyzed using flow cytometry. The results are the means ± SD of at least three independent experiments. The CD133high cell population showed relatively higher expression of stem cell‐related markers compared to the CD133low cell population. The mRNA expressions of stem cell‐related markers in parental, drug‐tolerant persisters, and CD133high and CD133low cells sorted from PC9 cells 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
{kind=link}
Fig. S4. Adenosine monophosphate‐activated protein kinase (AMPK) activity was enhanced in drug‐tolerant persisters compared to parental cells. Whole‐cell lysates were prepared and analyzed by Western blotting as indicated.
{kind=link}
Fig. S5. Drug‐tolerant persisters showed relatively higher expression of glucose transporters (GLUT1 and GLUT3) and the glycolytic enzyme hexokinase2 (HK2) compared to the parental cells. The mRNA expressions of GLUT1, GLUT3, and HK2 in parental cells and drug‐tolerant persisters 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
{kind=link}
Table S1. Primer sequences used for RT‐PCR.
Acknowledgments
The authors thank Dr. Keisuke Nishimura for kindly supporting the flow cytometry techniques, Dr. Koji Yamamoto for technical assistance in taking images for immunochemical staining, and Dr. Michiru Nishita for technical assistance in Western blot analyses. This work was supported by research grants from the Japan Society for the Promotion of Science (KAKENHI no. 25860643 to D. Tamura), a Kobe University School of Medicine Alumni Association (Shinryokukai General Incorporated Association) grant (to D. Tamura), and the Medical Research Fund of Hyogo Medical Association (to K. Kunimasa).
Cancer Sci 108 (2017) 1368–1377
Funding Information
Eli Lilly Japan; AstraZeneca; Novartis Pharma; the Japan Society for the Promotion of Science; Kobe University School of Medicine Alumni Association Shinryokukai General Incorporated Association; Hyogo Medical Association.
References
- 1. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–8. [DOI] [PubMed] [Google Scholar]
- 2. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomized phase 3 trial. Lancet Oncol 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 3. Zhou C, Wu YL, Chen G et al Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol 2011; 12: 735–42. [DOI] [PubMed] [Google Scholar]
- 4. Keedy VL, Temin S, Somerfield MR et al American Society of Clinical Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR) mutation testing for patients with advanced non‐small‐cell lung cancer considering first‐line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011; 29: 2121–7. [DOI] [PubMed] [Google Scholar]
- 5. Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol 2013; 31: 3987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumors: learning from lung cancer. Nat Rev Clin Oncol 2014; 11: 473–81. [DOI] [PubMed] [Google Scholar]
- 7. Arcila ME, Oxnard GR, Nafa K et al Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid‐based assay. Clin Cancer Res 2011; 17: 1169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bar J, Onn A. Overcoming molecular mechanisms of resistance to first‐generation epidermal growth factor receptor tyrosine kinase inhibitors. Clin Lung Cancer 2012; 13: 267–79. [DOI] [PubMed] [Google Scholar]
- 9. Hammerman PS, Jänne PA, Johnson BE. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small cell lung cancer. Clin Cancer Res 2009; 15: 7502–9. [DOI] [PubMed] [Google Scholar]
- 10. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor‐resistant disease. J Clin Oncol 2013; 31: 1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trumpp A, Wiestler OD. Mechanisms of disease: cancer stem cells–targeting the evil twin. Nat Clin Pract Oncol 2008; 5: 337–47. [DOI] [PubMed] [Google Scholar]
- 12. Clarke MF, Dick JE, Dirks PB et al Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66: 9339–44. [DOI] [PubMed] [Google Scholar]
- 13. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501: 328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Politi K, Ayeni D, Lynch T. The next wave of EGFR tyrosine kinase inhibitors enter the clinic. Cancer Cell 2015; 27: 751–3. [DOI] [PubMed] [Google Scholar]
- 15. Sharma SV, Lee DY, Li B et al A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell 2010; 141: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gazdar AF. Activating and resistance mutations of EGFR in non‐small‐cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009; 28: S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reguart N, Rosell R, Cardenal F et al Phase I/II trial of vorinostat (SAHA) and erlotinib for non‐small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 2014; 84: 161–7. [DOI] [PubMed] [Google Scholar]
- 18. Basu B, Olmos D, de Bono JS. Targeting IGF‐1R: throwing out the baby with the bathwater? Br J Cancer 2011; 104: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatakeyama Y, Kobayashi K, Nagano T et al Synergistic effects of pemetrexed and amrubicin in non‐small cell lung cancer cell lines: potential for combination therapy. Cancer Lett 2014; 343: 74–9. [DOI] [PubMed] [Google Scholar]
- 20. Shien K, Toyooka S, Yamamoto H et al Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell‐like properties in cancer cells. Cancer Res 2013; 73: 3051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taguchi F, Koh Y, Koizumi F, Tamura T, Saijo N, Nishio K. Anticancer effects of ZD6474, a VEGF receptor tyrosine kinase inhibitor, in gefitinib (“Iressa”)‐sensitive and resistant xenograft models. Cancer Sci 2004; 95: 984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu CH, Ho YS, Tsai CY et al In vitro and in vivo study of phloretin‐induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int J Cancer 2009; 124: 2210–19. [DOI] [PubMed] [Google Scholar]
- 23. Kakar SS, Ratajczak MZ, Powell KS et al Withaferin a alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PLoS ONE 2014; 9: e107596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dörr JR, Yu Y, Milanovic M et al Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013; 19: 421–5. [DOI] [PubMed] [Google Scholar]
- 25. Bertolini G, Roz L, Perego P et al Highly tumorigenic lung cancer CD133+ cells display stem‐like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A 2009; 106: 16281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pérez‐Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer 2014; 14: 547–58. [DOI] [PubMed] [Google Scholar]
- 27. Kuilman T, Peeper DS. Senescence‐messaging secretome: SMS‐ing cellular stress. Nat Rev Cancer 2009; 9: 81–94. [DOI] [PubMed] [Google Scholar]
- 28. Laberge RM, Sun Y, Orjalo AV et al MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015; 17: 1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishi M, Akutsu H, Kudoh A et al Induced cancer stem‐like cells as a model for biological screening and discovery of agents targeting phenotypic traits of cancer stem cell. Oncotarget 2014; 5: 8665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamura K, Aoyagi M, Ando N et al Expansion of CD133‐positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J Neurosurg 2013; 119: 1145–55. [DOI] [PubMed] [Google Scholar]
- 31. Liu G, Yuan X, Zeng Z et al Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Won C, Kim BH, Hee Yi E et al STAT3‐mediated CD133 upregulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015; 62: 1160–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song W, Li Q, Wang L, Huang W. Wang L FoxO1‐negative cells are cancer stem‐like cells in pancreatic ductal adenocarcinoma. Sci Rep 2015; 5: 10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xi G, Hayes E, Lewis R et al CD133 and DNA‐PK regulate MDR1 via the PI3K‐ or Akt‐NF‐κB pathway in multidrug‐resistant glioblastoma cells in vitro. Oncogene 2016; 35: 241–50. [DOI] [PubMed] [Google Scholar]
- 35. Eramo A, Lotti F, Sette G et al Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 2008; 15: 504–14. [DOI] [PubMed] [Google Scholar]
- 36. Zhang DG, Jiang AG, Lu HY, Zhang LX, Gao XY. Isolation, cultivation and identification of human lung adenocarcinoma stem cells. Oncol Lett 2015; 9: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V et al Studying clonal dynamics in response to cancer therapy using high‐complexity barcoding. Nat Med 2015; 21: 440–8. [DOI] [PubMed] [Google Scholar]
- 38. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 39. Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 1998; 12: 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yao Z, Fenoglio S, Gao DC et al TGF‐beta IL‐6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 2010; 107: 15535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim SM, Kwon OJ, Hong YK et al Activation of IL‐6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non‐small cell lung cancer with T790M resistance mutation. Mol Cancer Ther 2012; 11: 2254–64. [DOI] [PubMed] [Google Scholar]
- 42. Katanasaka Y, Kodera Y, Yunokawa M, Kitamura Y, Tamura T, Koizumi F. Synergistic anti‐tumor effects of a novel phosphatidyl inositol‐3 kinase/mammalian target of rapamycin dual inhibitor BGT226 and gefitinib in non‐small cell lung cancer cell lines. Cancer Lett 2014; 347: 196–203. [DOI] [PubMed] [Google Scholar]
- 43. Ishikawa D, Takeuchi S, Nakagawa T et al mTOR inhibitors control the growth of EGFR mutant lung cancer even after acquiring resistance by HGF. PLoS ONE 2013; 8: e62104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Price KA, Azzoli CG, Krug LM et al Phase II trial of gefitinib and everolimus in advanced non‐small cell lung cancer. J Thorac Oncol 2010; 5: 1623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soria JC, Bennouna J, Leighl N et al Phase II study of everolimus plus erlotinib in previously treated patients with advanced non‐small cell lung cancer. Ann Oncol 2014; 25: 409–15. [DOI] [PubMed] [Google Scholar]
- 46. Papadimitrakopoulou VA, Soria JC, Jappe A, Jehl V, Klimovsky J, Johnson BE. Everolimus and erlotinib as second‐ or third‐line therapy in patients with advanced non‐small‐cell lung cancer. J Thorac Oncol 2012; 7: 1594–601. [DOI] [PubMed] [Google Scholar]
- 47. Soria JC, Wu YL, Nakagawa K et al Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR‐mutation‐positive non‐small‐cell lung cancer after progression on first‐line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol 2015; 16: 990–8. [DOI] [PubMed] [Google Scholar]
- 48. Rovida E, Peppicelli S, Bono S et al The metabolically‐modulated stem ecll niche: a dynamic scenario regulating cancer cell phenotype and resistance to therapy. Cell Cycle 2014; 13: 3169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khan IN, Al‐Karim S, Bora RS, Chaudhary AG, Saini KS. Cancer stem cells: a challenging paradigm for designing targeted drug therapies. Drug Discov Today 2015; 20: 1205–16. [DOI] [PubMed] [Google Scholar]
- 50. Scarpa ES, Ninfali P. Phytochemicals as innovative therapeutic tools against cancer stem cells. Int J Mol Sci 2015; 16: 15727–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia P, Xu XY. PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application. Am J Cancer Res 2015; 5: 1602–9. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cellular senescence‐associated proteins production from parental cells and drug‐tolerant persisters. Whole‐cell lysates were prepared and analyzed by Western blotting as indicated.
Fig. S2. The CD133high cell population showed relatively higher expression of stem cell‐related markers compared to the CD133low cell population. mRNA expressions of stem cell‐related markers in parental, drug‐tolerant persister (DTP), CD133high, and CD133low cells sorted from PC9 cells 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
Fig. S3. Neutralizing antibodies against the senescence‐associated secretory phenotype suppressed the induction of the CD133high cell population. PC9 cells were cultured either in medium or in medium containing gefitinib (2 μM) for 12 days. Drug‐tolerant persisters were incubated with gefitinib (2 μM), gefitinib plus anti‐transforming growth factor‐β (TGF‐β) antibody (1 mg/mL; R&D Systems, Minneapolis, MN, USA), gefitinib plus anti interleukin (IL)‐6 antibody (1 mg/mL; R&D Systems), or gefitinib plus anti‐CCL5 antibody (1 mg/mL; R&D Systems) for the last 3 days. The cells were stained with anti‐CD133 antibody after 3 days of culture and were analyzed using flow cytometry. The results are the means ± SD of at least three independent experiments. The CD133high cell population showed relatively higher expression of stem cell‐related markers compared to the CD133low cell population. The mRNA expressions of stem cell‐related markers in parental, drug‐tolerant persisters, and CD133high and CD133low cells sorted from PC9 cells 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
Fig. S4. Adenosine monophosphate‐activated protein kinase (AMPK) activity was enhanced in drug‐tolerant persisters compared to parental cells. Whole‐cell lysates were prepared and analyzed by Western blotting as indicated.
Fig. S5. Drug‐tolerant persisters showed relatively higher expression of glucose transporters (GLUT1 and GLUT3) and the glycolytic enzyme hexokinase2 (HK2) compared to the parental cells. The mRNA expressions of GLUT1, GLUT3, and HK2 in parental cells and drug‐tolerant persisters 12 days after medium or gefitinib (2 μM) treatment was analyzed using quantitative RT‐PCR. Results are the means ± SD of three independent experiments.
Table S1. Primer sequences used for RT‐PCR.