

Abstract
Hepatitis B virus X protein plays a crucial role in the pathogenesis of hepatocellular carcinoma. We previously showed that the tumor suppressor ARID2 inhibits hepatoma cell cycle progression and tumor growth. Here, we evaluated whether hepatitis B virus X protein was involved in the modulation of ARID2 expression and hepatocarcinogenesis associated with hepatitis B virus infection. ARID2 expression was downregulated in HBV‐replicative hepatoma cells, HBV transgenic mice, and HBV‐related clinical HCC tissues. The expression levels of HBx were negatively associated with those of ARID2 in hepatocellular carcinoma tissues. Furthermore, HBx suppressed ARID2 at transcriptional level. Mechanistically, the promoter region of ARID2 gene inhibited by HBx was located at nt‐1040/nt‐601 and contained potential ATOH1 binding elements. In addition, ectopic expression of ATOH1 or mutation of ATOH1 binding sites within ARID2 promoter partially abolished HBx‐triggered ARID2 transcriptional repression. Functionally, ARID2 abrogated HBx‐enhanced migration and proliferation of hepatoma cells, whereas depletion of ATOH1 enhanced tumorigenecity of HCC cells. Therefore, our findings suggested that deregulation of ARID2 by HBx through ATOH1 may be involved in HBV‐related hepatocellular carcinoma development.
Keywords: ARID2, ATOH1, hepatitis B virus infection, hepatitis B virus X protein, hepatocellular carcinoma
Chronic infection by hepatitis B virus (HBV) is a major cause of hepatocellular carcinoma (HCC), a highly prevalent malignant disease worldwide.1, 2 The X protein of HBV (HBx) plays a crucial role in hepatocellular carcinogenesis.3 As a multifunctional regulator, HBx modulates cellular processes, such as cell cycle progression, apoptosis, cell proliferation, and migration.4, 5, 6 HBx acts as a transactivator by interacting with transcriptional factors, thus regulating host gene expression.7, 8 Several studies have reported that a wide variety of cellular genes, promoters/enhancers, and transcriptional factors, such as nuclear factor kappaB (NF‐κB), activator protein (AP)‐1, activating transcription factor (ATF)/cAMP, and cAMP response element binding protein (CREB), are involved in HBx‐induced hepatocarcinogenesis.9 However, the underlying mechanisms through which HBx regulates gene transcription to induce hepatocarcinogenesis remain largely unknown.
AT‐rich DNA interaction (ARID) 2, a chromatin remodeling gene, belongs to the switch/sucrose nonfermenting (SWI/SNF) chromatin‐remodeling complex.10 ARID2 contains a conservative N‐terminal ARID region, followed by three LLxxLL motifs and two conservative C‐terminal C2H2 Zn‐finger motifs, which can bind directly to DNA or interact with proteins.11 Recently, ARID2 was identified as a tumor suppressor and found to be frequently mutated in various cancers, such as HCC,12, 13, 14, 15 melanoma,16 nonsmall lung carcinoma,17 and gastric adenomas.18 Recent studies have revealed that loss of ARID2 expression owing to inactivating mutations promotes hepatoma cell proliferation19, 20 and may be associated with tumor recurrence.21 Adenomatous polyposis coli (APC) and ARID2 inactivating mutations have recently been reported to be involved in the early stages of gastric carcinogenesis.18 Similarly, somatic mutations in components of the SWI/SNF complex, such as ARID1A, ARID2, alpha thalassemia/mental retardation syndrome X‐linked (ATRX), and polybromo‐1 (PBRM1), are found in esophageal squamous cell carcinoma, and these genetic alterations are induced at an early stage in patients with esophageal squamous cell carcinogenesis.22 Despite the prevalence of genetic mutations in ARID2, functional role of ARID2 in HCC, particularly HBV‐related HCC, remains largely undefined.
In this study, we explored the possible role of HBx protein in the modulation of ARID2 expression and in hepatoma cell proliferation and motility. Interestingly, we found that HBx decreased ARID2 protein expression and enhanced hepatoma tumorigenesis through modulation of the transcription factor atonal BHLH transcription factor 1 (ATOH1). Our results suggested that HBx induced the suppression of the tumor suppressor ARID2 and may contribute to HCC tumorigenesis.
Materials and Methods
Patient samples
Hepatitis B virus‐related HCC tissues and paired adjacent nontumorous tissues were obtained from patients who underwent surgery for HCC at the Second Affiliated Hospital of Chongqing Medical University. This study was approved by the Institutional Ethical Review Board of Chongqing Medical University, and informed consent was obtained from participating patients. Each specimen was frozen immediately after surgery and stored in liquid nitrogen for later use.
HBV‐transgenic mice
Hepatitis B virus‐transgenic (HBV‐Tg) mice were kindly provided by Professor Ning‐shao Xia from the School of Public Health, Xiamen University.23 All mice were maintained under specific pathogen‐free conditions in the laboratory animal center of Chongqing University. The experiments were carried out with approval of the Chongqing Medical University Animal Care and Use Committee.
Cell culture
The human hepatoma cell line HepG2 was obtained from the American Type Culture Collection (ATCC, VA, USA). SK‐Hep1, Huh7, SMMC‐7721, and normal human liver cells (LO2) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Hepatoma cells stably expressing HBx (LO2‐HBx, SK‐Hep1‐HBx, and Huh7‐HBx cells) were preserved in our laboratory.24 Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Rockville, MD, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in an atmosphere containing 5% CO2.
Adenoviruses and reporter plasmids
Recombinant adenoviruses AdHBx and AdARID2 were constructed as previously described.20, 24 The full‐length cDNA of ATOH1 (coding sequence of NM_005172) was subcloned from the pPCR‐Script‐ATOH1 plasmid (GeneCopoeia #FL30148) and inserted into the shuttle vector pAdTrack‐TO4 or His‐tagged vector pSEB‐3His vector. HBx (genotype C) was cloned into the Flag‐tagged pSEB‐3Flag vector (from Dr. T‐C He, University of Chicago, IL, USA). The adenoviral recombinant pAdATOH1 was generated using the AdEasy system, and an analogous adenovirus expressing only green fluorescent protein (GFP; AdGFP) was used as control.
The wild‐type (WT) human ARID2 promoter reporter and sequential deletion constructs were generated by cloning of the polymerase chain reaction (PCR) fragments of the ARID2 promoter into the pGL3‐Basic vector (Promega, Madison, WI, USA; #E1751). Primer sequences are listed in Table S1.
RNAi lentivirus production
The small double‐strand hairpin shRNA for ATOH1 were designed and inserted into the HpaI/XhoI sites of pLL3.7 lentivirus vector (kindly provided by Prof. Bing Sun from the Institute Pasteur of Shanghai, Chinese Academy of Sciences). Lentivirus were prepared as reported previously25 and was used to infect SK‐Hep1 cells in the presence of 5 μg/mL polybrene. Inhibition of ATOH1 was verified by western blot analysis. Oligonucleotides targeting ATOH1 were listed in Table S1.
Dual‐luciferase assay
Huh7 cells were plated into 25‐cm2 cell culture flasks and transfected with 3 μg ARID2 promoter reporter constructs along with 300 ng pRL‐TK (an internal control; Promega) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. At 24 h after transfection, cells were seeded into 24‐well plates (Life Sciences, Tewksbury, MA, USA). At 8 h after replating, cells were infected with AdGFP or AdHBx. Cells were harvested 36 h postinfection and subjected to Dual‐Luciferase Reporter Assays (Promega). All experiments were performed at least three times, and results are expressed as means ± standard deviations (SDs).
RNA extraction, reverse transcription (RT)‐PCR, and real‐time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega). Quantification of target genes was performed by SYBR Green qPCR on a CFX Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA). All primer sequences are listed in the Table S1. Relative expression was calculated as a ratio of the expression of the specific transcript to that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH).
Western blot analysis
Whole protein lysates of cells or liver tissues were extracted using cell lysis buffer (Beyotime Biotechnology, Jiangsu, China) containing 1 mM phenylmethylsulfonyl fluoride (Beyotime). Protein concentrations were measured using a BCA protein assay kit (Beyotime). Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) on 10% gels and electrotransferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The immunoblots were probed with the indicated antibodies targeting ARID2 (Abcam, Cambridge, UK; Ab113283), ATOH1 (Thermo Fisher, Waltham, MA, USA; PA5‐29392), E‐cadherin (Bioworld, St. Louis Park, MN 55416, USA; BS1098), and HBx (kindly provided by Pro. Ning‐shao Xia of Xiamen University). Secondary antibodies coupled to horseradish peroxidase were purchased from Abcam. Proteins bands were visualized with a Super Signal West Pico Chemiluminescent Substrate Kit (Millipore). GAPDH expression was used as the normalization control. The immunoblots were quantified by densitometric analysis using ImageJ software.
Immunohistochemistry
Liver tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin according to standard procedures. Serial tissue sections were incubated with anti‐ARID2 (Abcam), anti‐HBx (kindly provided by Professor Ning‐shao Xia of Xiamen University) or anti‐ATOH1 (Thermo Fisher) separately. Subsequently, the slides were incubated with secondary anti‐rabbit or anti‐mouse IgG (ZSGB‐BIO, Beijing, China) and visualized using 3,3′‐diaminobenzidine (ZSGB‐BIO).
Cell migration assay
Cell migration was measured using transwell‐24 units with polycarbonate filters (BD, San Jose, CA, USA). Cells were infected with AdGFP or AdARID2. For knockdown assay, cells were infected with lentiviruses carrying ATOH1 shRNA or control shRNA. At 24 h after infection, cells were suspended in 200 μL serum‐free medium and added at 6 × 103 cells/well to the upper chamber. DMEM (800 μL) containing 10% FBS was added to the lower chamber. Cells were incubated at 37°C in an atmosphere containing 5% CO2 for 12 h, fixed with 4% formaldehyde, and stained with crystal violet for 3 min. The number of migrated cells was counted in five fields (200×) on each membrane, and the average number per field was calculated.
Cell proliferation assay
Cells were transfected with pAdTrack‐ARID2 plasmid or vector control for 48 h in 6‐well plates. To examine the cell proliferation in ATOH1‐depleted hepatoma cells, SK‐Hep1 cells were infected with lentiviruses carrying ATOH1 shRNA or control shRNA. Cell proliferation was assayed by detecting the incorporation of 5‐ethynyl‐2′‐deoxyuridine (EdU) into DNA during DNA synthesis with an EdU Cell Proliferation Assay Kit (Ribobio, Guangzhou, China). According to the manufacturer's protocol, cells were incubated with 50 μM EdU for 2 h, followed by fixation, permeabilization, and EdU staining. The cell nuclei were stained with DAPI (Sigma, St. Louis, MO, USA) at a concentration of 1 μg/mL for 2 min. The proportion of cells containing EdU was determined using fluorescence microscopy. The percentage of EdU‐positive cells was quantified from five random fields (200×) in three wells.
Co‐immunoprecipitation (IP) assay
Sk‐Hep1 cells were cotransfected with pSEB‐3Flag‐HBx or vector control along with pSEB‐3His‐ATOH1. At 48 h post‐transfection, cells were resuspended in 600 μL IP lysis buffer (Beyotime) and precleared with protein G agarose beads (Millipore) for 1 h. Supernatants were incubated with anti‐His (ABM‐0008; Zhongding Biotechnology Company, Nanjing, China) or anti‐Flag antibodies (F1804; Sigma) overnight at 4°C, followed by a 2 h incubation with protein G agarose beads. Immune complexes were resolved by SDS‐PAGE on 12% gels, transferred to PVDF membranes, and then subjected to immunoblot analysis using the indicated antibodies.
Chromatin IP (ChIP)
SK‐Hep1 cells were infected with AdHBx or AdGFP for 36 h. The chromatin was crosslinked by treating the cells with 4% formaldehyde for 10 min at 37°C. Sonicated cell lysates were subjected to immunoprecipitation using 5 μg anti‐HBx or anti‐ATOH1 antibodies (normal IgG served as a control). Following elution, DNA was extracted and subjected to PCR analysis. Isolated chromatin immunoprecipitated by E2F1 antibody was amplified with ARID2 primers and CCND1 (cyclin D1) primers used us positive control.20 For ChIP‐qPCR, the immunoprecipitated DNA was quantified by real‐time PCR. Enrichment of ATOH1 at the examined regions of the ARID2 promoter was quantified relative to the input control.
Statistical analysis
All values are presented as means ± standard deviations (SDs). Statistical significance was determined using analysis of variance (anova) followed by the least significant differences (LSD) test for multiple comparisons, and Student's t‐tests were used to compare two groups. Differences with P‐values of less than 0.05 were considered statistically significant.
Results
HBx downregulated ARID2 expression in human hepatoma cells
Previously, we and others reported that ARID2 deficiency is prevalent in human HCC, suggesting that ARID2 deletion may play an important role in the pathogenesis of HCC.15, 20 However, the relationship between HBV infection and ARID2 expression in hepatoma cell lines is unclear. We first examined the levels of ARID2 in an HBV‐expressing hepatoma cell line. We found that ARID2 expression levels were significantly decreased in HepG2.2.15 cells stably expressing HBV DNA (Fig. 1a). Next, we examined ARID2 expression in liver tissues of HBV transgenic mice. The data revealed that ARID2 was markedly downregulated when compared with that in WT littermate mice (Fig. 1b). To determine whether viral‐encoded protein was responsible for ARID2 repression in hepatoma cells, HepG2 cells were transiently transfected with the HBV replicative plasmid pCH‐9 (HBV) or plasmid encoding different HBV viral proteins, including pCDNA‐HBs, pCDNA‐HBc, and pCDNA‐HBx. Compared with the vector control, both HBV and HBx protein markedly downregulated ARID2 expression, whereas HBc protein and HBs protein had little effect on modulation of ARID2 expression (Fig. 1c). Furthermore, when cells were transduced with the HBV replicative plasmid containing the stop mutation of HBx (HBx‐Stop), ARID2 expression was enhanced compared with that of WT HBV‐transfected cells. We further confirmed that X‐Stop‐induced ARID2 restoration was partially abolished by overexpression of HBx in HepG2 cells (Fig. 1d), indicating that HBx played a vital role in HBV‐induced ARID2 repression. Moreover, HBx‐induced inhibition of ARID2 expression was further confirmed in both HBx‐transient and stable‐transduced hepatoma cells (Fig. 1a,e,f). Collectively, these data indicated that HBV replication, particularly HBx protein, suppressed ARID2 expression in hepatoma cells.
Figure 1.
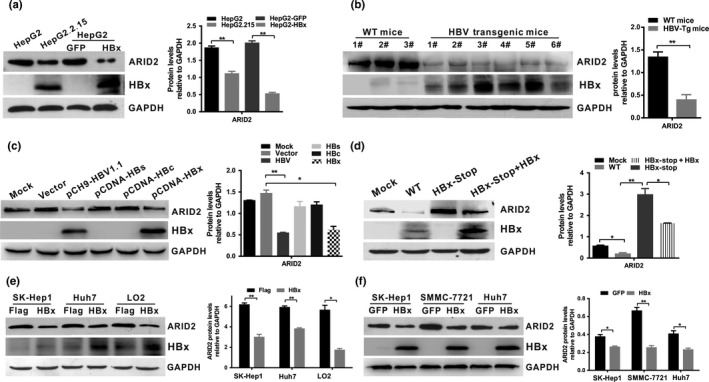
HBx downregulated ARID2 expression in hepatoma cells. (a) ARID2 and HBx protein was detected by western blot analysis in HepG2.2.15 cells and HepG2 cells infected with adenoviruses expressing HBx or GFP control (n = 3, **P < 0.01). (b) ARID2 and HBx protein expression in HBV‐transgenic mice and WT littermates (WT mice; **P < 0.01). (c) HepG2 cells were transfected with pCH‐9(HBV1.1), pCDNA‐HBs, pCDNA‐HBc, pCDNA‐HBx, or vector plasmid. ARID2 and HBx protein was detected using western blotting (n = 3, *P < 0.05; **P < 0.01). (d) HepG2 cells were transiently transfected with full‐length HBV (WT‐HBV), stop‐mutant HBx (HBx‐stop), or stop‐mutant HBx plasmids plus adenovirus encoding HBx. ARID2 and HBx expression was determined by western blot analysis (n = 3, * P < 0.05; **P < 0.01). (e,f) Western blot analysis of ARID2 and HBx expression in Sk‐Hep1/Sk‐Hep1‐HBx, Huh7/Huh7‐HBx, LO2/LO2‐HBx, and HBx‐transduced hepatoma cells. All data were acquired from three independent experiments, and representative results are shown. Integrated density was quantitatively analyzed using ImageJ software. *P < 0.05; **P < 0.01, Student's t‐test.
HBx was negatively correlated with ARID2 expression in HCC tissues
To further investigate the potential effects of HBx on ARID2 expression in HCC tissues, we evaluated HBx and ARID2 expression in paired tissues obtained from 24 patients with HBV‐HCC. Immunohistochemical staining showed that positive rate of ARID2 was significantly lower (7/24, 30%) in tumor tissues when compared to paired adjacent non‐tumor tissues (P < 0.05, Fig. S3c), whereas there was no significant difference in HBx staining intensity between HCC and pericarcinous tissue (Fig. S3c). Moreover, weak staining of ARID2 was coincident with higher expression of HBx in the same tissues, and vice versa (P < 0.05, Fig. 2a,b).
Figure 2.
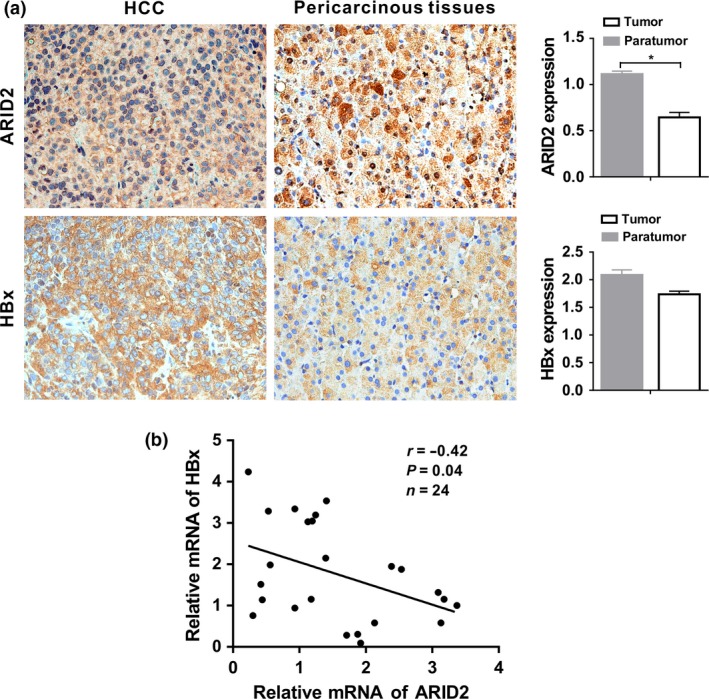
HBx was negatively correlated with ARID2 expression in HCC tissues. (a) Representative immunohistochemical staining of HBx and ARID2 in serial tissue slices from 24 paired HBV‐related HCC tissues and adjacent nontumorous tissues. Immunostaining intensity was assessed using Image‐Pro Plus 6.0 software. *P < 0.05; magnification: 400×. (b) Correlation analysis of ARID2 and HBx mRNA expression in 24 paired HCC tissues (*P < 0.05, r = −0.42, Pearson's correlation).
The correlations of the expression levels of ARID2 with HBx were further analyzed in patients with HCC. Strikingly, the expression levels of HBx were negatively correlated with those of ARID2 in HCC tissues (P < 0.05; Fig. 2b). Therefore, these data indicated that ARID2 was downregulated in HCC tissues and was negatively associated with HBx expression.
HBx inhibited ARID2 expression at the transcriptional level
To determine whether ARID2 expression was repressed by HBx at the transcriptional level, we constructed the human ARID2 promoter reporter pGL3‐ARID2 (position ‐1040 to +101) and serial 5′‐end deletion mutations of ARID2, including pGL3‐ARID2‐P1(position ‐780 to +101), pGL3‐ARID2‐P2 (position ‐601 to +101), pGL3‐ARID2‐P3 (position ‐365 to +101), and pGL3‐ARID2‐P4 (position ‐154 to +101; Fig. 3a,b). The reporter activity of pGL3‐ARID2 increased approximately 15‐fold when compared with that of the vector plasmid (P < 0.05). Ectopic expression of HBx significantly inhibited ARID2 luciferase signaling in Huh7 cells, as compared with that of the vector control (P < 0.05; Fig. 3a). Moreover, HBx was found to inhibit ARID2 promoter activity in a concentration‐dependent manner (Fig. 3a). Next, we sought to further clarify the ARID2 promoter region that was targeted by HBx. Our data showed that HBx repressed the luciferase activities of pGL3‐ARID2‐P (58%) and pGL3‐ARID2‐P1 (54%), but not that of other deletion reporters (Fig. 3b). Thus, the 5′‐flanking region located at ‐1040 to ‐601 bp upstream of the human ARID2 gene transcription start site may be mainly responsible for transcriptional repression by HBx. Next, we examined whether HBx bound directly to this promoter region of the ARID2 gene. ChIP assays showed that HBx could not directly interact with the ARID2 promoter (Fig. 3c), suggesting that HBx may inhibit ARID2 transcription through an indirect mechanism.
Figure 3.

HBx inhibited ARID2 mRNA expression at the transcriptional level. (a) Luciferase activity of the human ARID2 promoter construct pGL3‐ARID2 in Huh7 cells. Huh7 cells were transfected with pGL3‐ARID2 (position ‐1040 to +101) for 24 h and then infected with AdGFP control or AdHBx. At 36 h postinfection, cells were harvested for luciferase assays. Data are presented as the mean (±SD) relative luciferase activity compared with the activity of the pGL3‐Basic control sample. Three independent experiments were performed. *P < 0.05; **P < 0.01 by Student's t‐test. (b) Luciferase assays of human ARID2 promoter constructs with the wild‐type sequence (‐1040/+101 nt) or the indicated serial deletion mutations in AdHBx‐ or AdGFP‐infected Huh7 cells (n = 3, *P < 0.05). (c) ChIP assays of cell extracts from Huh7 cells infected with AdHBx or AdGFP. Huh7 cells were infected with AdHBx or AdGFP control. At 48 h post‐infection, cell lysates were collected, and ChIP analysis was performed using control IgG or anti‐HBx antibodies. Transcriptional factor E2F1 recruited on ARID2 and CCND1 promoter were used as ChIP positive control.
HBx‐mediated ATOH1 suppression contributed to downregulation of ARID2
Previous studies have indicated that HBx can regulate other transcription factors to bind to promoters or enhancers and regulate gene transcription.8 To identify the transcription factors that mediate HBx‐induced ARID2 inhibition, we predicted the transcription factor binding sites in the ‐1040 to ‐601 nt promoter region of the ARID2 gene using the JASPAR database (http://jaspar.genereg.net/). We found that there were several potential binding elements in this promoter region, including those for ATOH1, MZF1, E2F6, and BRCA1 (Fig. 4a). We then screened six transcription factors that may be involved in HBx‐induced ARID2 suppression. Our results indicated that HBx inhibited ATOH1 expression both at the mRNA and protein level (Fig. 4b,c; Fig. S1). We then tested whether rescue of ATOH1 expression restored HBx‐triggered ARID2 transcriptional repression. We found that the promoter activity of ARID2 was largely restored by overexpression of ATOH1 relative to the control (P < 0.05; Fig. 5a). Similarly, we found that ARID2 expression was restored by ATOH1 ectopic expression in HBx‐stable hepatoma cells (Fig. 5b). ChIP assays demonstrated that HBx blocked the recruitment of ATOH1 to the ARID2 promoter (Fig. 5c). Moreover, silencing of ATOH1 markedly inhibited ARID2 expression both at the mRNA and protein level (Fig. 5d,e). These results indicated that ATOH1 plays an important role in transcriptional regulation of ARID2.
Figure 4.
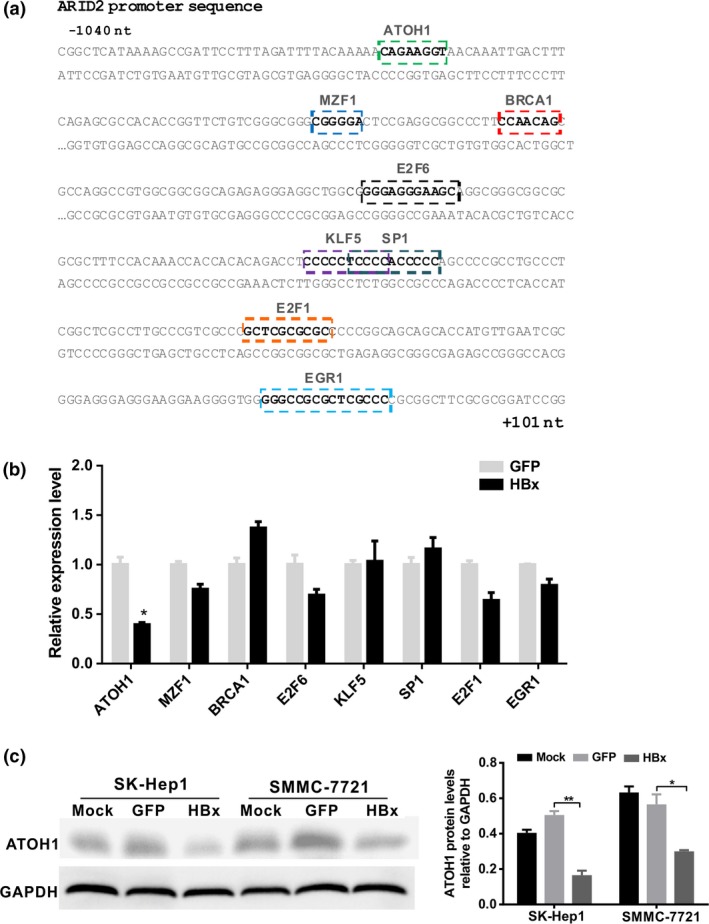
HBx downregulated ATOH1 expression. (a) Illustration of the predicted transcription factor binding sites in the 1‐kb ARID2 promoter region according to the JASPAR database analysis. (b) The mRNA expression levels of transcription factors were detected by real‐time PCR. SK‐Hep1 cells were infected with AdHBx virus or AdGFP control. Total mRNA was isolated at 36 h after infection. Expression of genes encoding the indicated transcription factors was determined by RT‐qPCR (n = 3, *P < 0.05 versus GFP control). (c) Protein expression of ATOH1. SK‐Hep1 cells were treated as described above. At 48 h after infection, cell lysates were used for western blot analysis (n = 3, *P < 0.05; **P < 0.01).
Figure 5.
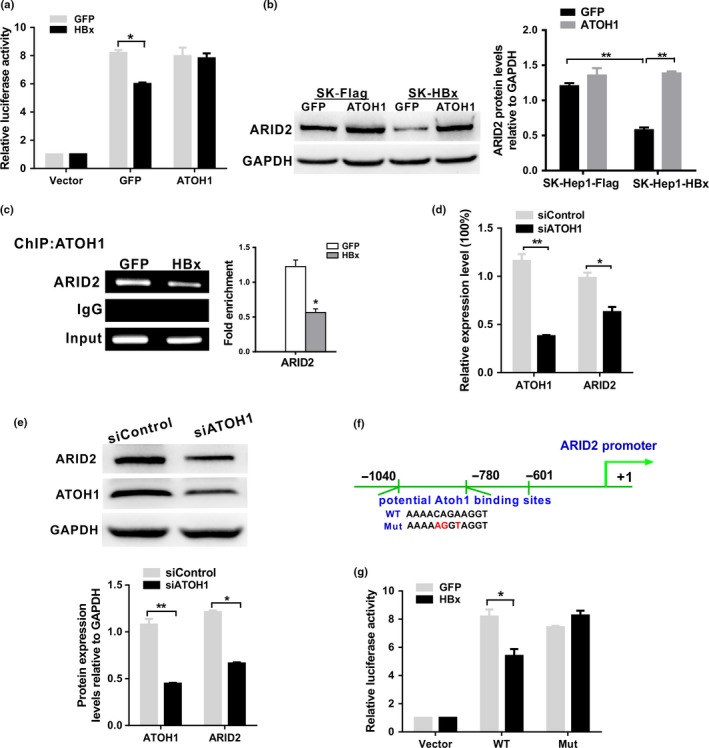
HBx inhibited ARID2 promoter activity via an ATOH1‐dependent pathway. (a) Luciferase activity of the human ARID2 promoter reporter pGL3‐ARID2 in Huh7 cells. Huh7 cells were transfected with pGL3‐ARID2 and then co‐infected with AdGFP control or AdHBx together with AdATOH1. Luciferase activities were measured at 24 h after infection (n = 3, *P < 0.05). (b) ARID2 protein expression was determined by western blotting in Sk‐Hep1/SK‐Hep1‐HBx cells infected with AdATOH1 or AdGFP control (n = 3, **P < 0.01). (c) ChIP assays of cell extracts from Huh7 cells infected with AdHBx or AdGFP using anti‐ATOH1 antibodies. IgG served as a negative control. The relative fold enrichment (bound/input) was measured by qPCR. Data represent the means ± SDs (n = 3, *P < 0.05). (d) and (e) The mRNA and protein expression levels of ARID2 in ATOH1‐depleted SK‐Hep1 cells. SK‐Hep1 cells were infected with lentiviruses carrying ATOH1 shRNA or control shRNA. Cells were performed qRT‐PCR and Western blot assay (n = 3, *P < 0.05; **P < 0.01). (f) Schematic representation of the ARID2 promoter region with the potential ATOH1 binding sites indicated. (g) Luciferase assay of ARID2 promoter constructs with the wild‐type (WT) or mutated ATOH1 binding site in AdGFP‐ or AdHBx‐infected Huh7 cells. Data are shown as means ± SDs (n = 3 independent experiments; *P < 0.05 by Student's t‐test).
To further verify the potential role of ATOH1 in ARID2 transcriptional inhibition induced by HBx, site‐directed mutagenesis targeting ATOH1 binding sites within the ARID2 promoter was performed (Fig. 5f). As expected, HBx failed to inhibit ARID2 promoter activity when the ATOH1 binding site in the ARID2 promoter was mutated (Fig. 5g), indicating that this DNA element was crucial for HBx‐induced ARID2 repression in Huh7 cells. However, co‐IP analysis indicated that anti‐HBx antibodies could not immunoprecipitate ATOH1 in SK‐Hep1 cells, and vice versa (Fig. S2). Thus, HBx may function as a transcriptional repressor of ARID2 expression through inactivation of ATOH1.
ATOH1 was positively correlated with ARID2 expression in HBV‐infected hepatoma cells and HCC liver tissues
To confirm the effects of ATOH1 on ARID2 expression, we examined the levels of ATOH1 and ARID2 in HBV replicative hepatoma cell lines. Western blot analysis showed that both ATOH1 and ARID2 expression levels were significantly decreased in HepG2.2.15 cells and HepAD38 cells stably expressing HBV26 (Fig. S3a). We then evaluated ATOH1 and ARID2 mRNA expression in 24 paired HCC tissues associated with HBV infection. ATOH1 and ARID2 were both downregulated in HCC tissues compared with that in paired adjacent nontumor tissues. Correlative analysis confirmed that ATOH1 expression was positively correlated with ARID2 expression in HCC tissues (P < 0.05; Fig. S3b). Moreover, we examined ATOH1, ARID2 as well as HBx protein expression in 15‐paired HCC tissues. Data showed that ATOH1 and ARID2 were both downregulated in HCC tissues when compared to that in paired adjacent nontumorous tissues (Fig. S3c,d). Taken together, these data provided evidence for synergistic changes in ATOH1 and ARID2 expression in the context of HBV infection both in hepatoma cell lines and human HCC tissues.
ARID2 partially reverses the migration and proliferation of hepatoma cells mediated by HBx in vitro
Previous studies have reported that downregulation of ARID1A, which encodes another ARID protein of the SWI/SNF family, promotes gastric cancer cells or HCC cell migration by repression of epithelial marker E‐cadherin.27, 28 Thus, we further explored the effects of ARID2 on E‐cadherin expression and cell migration in stable HBx‐expressing cell lines. Transwell assays demonstrated that ARID2 significantly inhibited cell migration and enhanced the expression of the epithelial marker E‐cadherin in SK‐HBx and Huh7‐HBx cell lines (Fig. 6a,b). Additionally, EdU assays showed that ARID2 partially abolished the increased proliferation of hepatoma cell lines mediated by HBx (P < 0.05; Fig. 6c). To further verify the direct role of ATOH1 in cell proliferation and migration, we silenced ATOH1 expression by siRNA approach and examined cell proliferation and migration capacity in SK‐Hep1 cells. We found that depletion of ATOH1 largely stimulated cell proliferation, cell migration and suppressed the expression of epithelial marker E‐cadherin in SK‐Hep1 cells. These data indicated that HBx induced ATOH1 inhibition and suppressed ARID2 expression, leading to enhanced hepatoma cell migration and growth (Fig. S4a–c).
Figure 6.
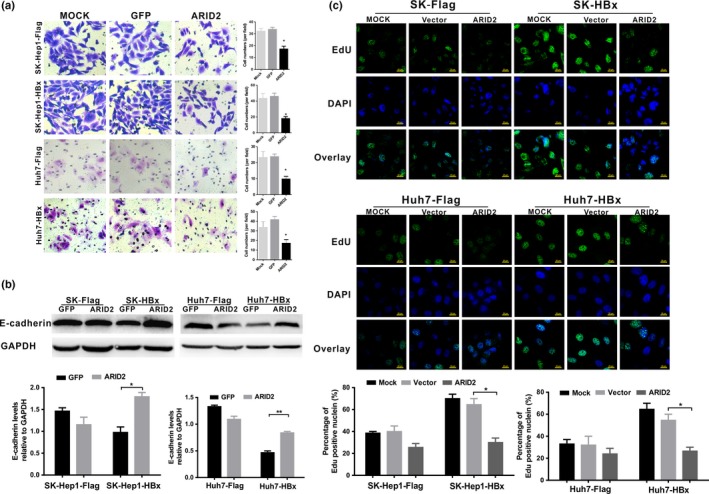
ARID2 partially reversed the enhanced migration and proliferation of hepatoma cells induced by HBx. (a) Transwell assays of cell migration in Sk‐Hep1/Sk‐Hep1‐HBx and Huh7/Huh7‐HBx cells. Cells were infected with AdGFP control or AdARID2 and then subjected to transwell assays. Data represent the results of three independent experiments (means ± SDs; *P < 0.05; **P < 0.01 versus the GFP control; magnification: 200×). (b) E‐cadherin protein expression was determined by western blotting in Sk‐Hep1/SK‐Hep1‐HBx and Huh7/Huh7‐HBx cells infected with AdARID2 or AdGFP control (n = 3, *P < 0.05; **P < 0.01). (c) Cell proliferation was analyzed by EdU incorporation assays. Cells were treated as described above. Data are presented as the means ± SDs (n = 3, *P < 0.05 versus the Vector control).
Discussion
As an oncoprotein, HBx plays a key role in hepatocarcinogenesis.29 HBx has been reported to modulate several cellular processes through direct or indirect regulation of a wide variety of host factors associated with the progression of HCC through various molecular mechanisms.4, 30 Furthermore, natural variants of HBx proteins, including C‐terminal truncated HBx, HBx mutants, and HBV whole‐X protein (HBwx, 56 amino acids longer than HBx), are frequently observed in HBV‐related HCC and are implicated in the development of HCC.31, 32, 33
Recent exome and genome‐wide sequencing studies of human cancers identified significant alterations in the mammalian SWI/SNF complexes. ARID2, which encodes a component of the SWI/SNF chromatin‐remodeling complex, has been identified as a novel tumor‐suppressor gene.34 ARID2 is reported to be truncated mutated in multiple tumors15, 17, 35 and plays important roles in regulating cell differentiation and proliferation. Several groups have reported that ARID2 loss‐of‐function mutations are related to tumor progression and poor survival in patients with HCC and gastric carcinoma.36 A recent study showed that microRNA‐155 accelerates hepatoma growth through targeting of the ARID2‐mediated Akt phosphorylation pathway.37 However, little is known about the functional role of ARID2 in HCC, especially in the context of HBV replication.
Our results here demonstrated that HBV infection, particularly HBx protein, suppressed ARID2 expression in hepatoma cells. We further observed that ARID2 was downregulated in HBV‐related HCC and that ARID2 expression was inversely correlated with HBx protein expression. Furthermore, HBx inhibited ARID2 expression by impairing binding of the transcription factor ATOH1 to the ARID2 promoter. Our results suggested that HBx inhibited the expression of ARID2 and hence may contribute to HCC tumorigenesis.
ATOH1 is a basic helix‐loop‐helix transcription factor. Knockout mice model showed that ATOH1 plays a vital role in the differentiation of neurons and secretory cells in the gut and in postnatal cerebellular development.38 ATOH1 deficiency is frequently observed in many types of tumors and is involved in the development and progression of various types of tumors, such as gastric carcinoma,39 colon cancer,40 medulloblastoma,38 Merkel cell carcinoma,41 and intestinal neuroendocrine tumors.42 In addition, ATOH1 induces the differentiation of gastric cancer stem cells and is correlated with favorable survival in gastrointestinal stromal tumors.39, 43 Although the expressional status of ATOH1 has been reported in many tumor tissues, its role in HCC development has not been investigated.
In this study, we found that HBx inhibited ATOH1 expression and impaired its binding to the ARID2 promoter, leading to downregulation of ARID2 expression. ATOH1 stability is modulated by the ubiquitin‐proteasome degradation system. Sonic hedgehog (SHH) signaling regulates ATOH1 stability via E3 ubiquitin ligase Huwe1.44 Peignon and colleagues reported that the Wnt pathway was involved in downregulation of ATOH1 expression by enhancing the expression of Notch‐related Jagged‐1 and Hes1.45 Furthermore, ATOH1 and β‐catenin proteins are mutually regulated by glycogen synthase kinase (GSK)‐3β degradation in Wnt signaling,46 and HBx and HBx mutants have been found to transactivate Wnt/β‐catenin signaling pathways47 through suppressing GSK‐3β activity or interacting with the suppressor APC to activate Wnt/β‐catenin.32, 48 Further studies are required to determine whether HBx promotes proteasome‐mediated degradation of ATOH1 by activating Wnt/GSK‐3β pathway.
In summary, our findings suggested that HBx downregulated ARID2 through modulation of the transcription factor ATOH1 in hepatoma cells. Moreover, downregulation of ARID2 by HBx contributed to enhanced HCC cell migration and proliferation. Our findings provide new insights into the molecular mechanisms of HCC progression mediated by HBx.
Disclosure statement
The authors declare that they have no conflicts of interest.
Supporting information
Fig S1. (a) The mRNA expression levels of transcription factors. SK‐Hep1 cells and SMMC‐7721 cells were infected with AdHBx or AdGFP control. Total mRNA were isolated at 36 h after infection. The indicated transcription factor genes were detected by Real‐time PCR. (b) SMMC‐7721 cells were treated as shown in Figure S1 (a). The indicated transcription factor genes were determined by qRT‐PCR. (n = 3, *P < 0.05 versus GFP control).
Fig S2. The interaction between HBx and ATOH1 were detected by Co‐IP analysis. SK‐Hep1 cells were co‐transfected with pSEB‐3Flag‐HBx or vector plasmid along with pSEB‐3His‐ATOH1. At 48 h after transfection, cell lysates were subjected to Co‐IP assay with the indicated antibodies.
Fig S3. (a) Protein levels of ARID2 and ATOH1 were detected by western blot analysis in HepG2, HepG2.2.15 and HepAD38 cells with or without tetracycline treatment. (b) Correlation analysis of ATOH1 and ARID2 mRNA expression in 24 paired HCC tissues (*P < 0.05, r = 0.43, Pearson's correlation). (c and d) Protein levels of ARID2 and ATOH1 as well as HBx were detected by immunohistochemical staining and western blot analysis in HBV‐related HCC tissues.
Fig S4. (a) Transwell assays of Sk‐Hep1 cells infected with lentiviruses carrying ATOH1 shRNA or control shRNA. Data represent the results of three independent experiments (means ± SDs; **P < 0.01 versus the siControl; magnification: 200×). (b) E‐cadherin protein expression was determined by western blotting (n = 3, **P < 0.01). (c) Cell proliferation was analyzed by EdU incorporation assays. Data are presented as the means ± SDs (n = 3, **P < 0.01 versus siControl).
Table S1. Primer sequences.
Acknowledgements
We would like to thank Dr. T.‐C He (University of Chicago, USA) for providing the plasmids pSEB‐3Flag and pSEB‐3His, the AdEasy system, and the adenovirus AdGFP. We are also grateful to Professor Ning‐shao Xia (Xiamen University) for providing HBV transgenic mice. This study was supported by research grants from China National Natural Science Foundation (grant nos. 81572683 and 81371827, to NT), Natural Science Foundation Project of CQ CSTC (grant no. cstc2015jcyjBX0011, to NT), the Program for Innovation Team of Higher Education in Chongqing (grant no. CXTDX201601015), and the Leading Talent Program of CQ CSTC (to NT).
Cancer Sci 108 (2017) 1328–1337
Funding Information
The Program for Innovation Team of Higher Education in Chongqing, (Grant / Award Number: ‘CXTDX201601015‘) China National Natural Science Foundation, (Grant / Award Number: ‘81371827‘,’81572683 ‘) Natural Science Foundation Project of CQ CSTC , (Grant / Award Number: ‘cstc2015jcyjBX0011‘)
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Mittal S, El‐Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol 2013; 47(Suppl): S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brechot C, Kremsdorf D, Soussan P et al Hepatitis B virus (HBV)‐related hepatocellular carcinoma (HCC): molecular mechanisms and novel paradigms. Pathol Biol (Paris) 2010; 58: 278–87. [DOI] [PubMed] [Google Scholar]
- 4. Ng SA, Lee C. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol 2011; 46: 974–90. [DOI] [PubMed] [Google Scholar]
- 5. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus‐induced hepatocellular carcinoma. J Gastroenterol Hepatol 2011; 26(Suppl 1): 144–52. [DOI] [PubMed] [Google Scholar]
- 6. Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up‐regulated microRNA‐143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009; 50: 490–9. [DOI] [PubMed] [Google Scholar]
- 7. Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64: 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang XD, Wang Y, Ye LH. Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol Med 2014; 11: 182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim HR, Lee SH, Jung G. The hepatitis B viral X protein activates NF‐kappaB signaling pathway through the up‐regulation of TBK1. FEBS Lett 2010; 584: 525–30. [DOI] [PubMed] [Google Scholar]
- 10. Yan Z, Cui K, Murray DM et al PBAF chromatin‐remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon‐responsive genes. Genes Dev 2005; 19: 1662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mohrmann L, Langenberg K, Krijgsveld J, Kal AJ, Heck AJ, Verrijzer CP. Differential targeting of two distinct SWI/SNF‐related Drosophila chromatin‐remodeling complexes. Mol Cell Biol 2004; 24: 3077–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amaddeo G, Guichard C, Imbeaud S, Zucman‐Rossi J. Next‐generation sequencing identified new oncogenes and tumor suppressor genes in human hepatic tumors. Oncoimmunology 2012; 1: 1612–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guichard C, Amaddeo G, Imbeaud S et al Integrated analysis of somatic mutations and focal copy‐number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012; 44: 694–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kan Z, Zheng H, Liu X et al Whole‐genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 2013; 23: 1422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li M, Zhao H, Zhang X et al Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet 2011; 43: 828–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hodis E, Watson IR, Kryukov GV et al A landscape of driver mutations in melanoma. Cell 2012; 150: 251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manceau G, Letouze E, Guichard C et al Recurrent inactivating mutations of ARID2 in non‐small cell lung carcinoma. Int J Cancer 2013; 132: 2217–21. [DOI] [PubMed] [Google Scholar]
- 18. Lim CH, Cho YK, Kim SW et al The chronological sequence of somatic mutations in early gastric carcinogenesis inferred from multiregion sequencing of gastric adenomas. Oncotarget 2016; 7: 39758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu P, Wu D, You Y et al miR‐208‐3p promotes hepatocellular carcinoma cell proliferation and invasion through regulating ARID2 expression. Exp Cell Res 2015; 336: 232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duan Y, Tian L, Gao Q et al Chromatin remodeling gene ARID2 targets cyclin D1 and cyclin E1 to suppress hepatoma cell progression. Oncotarget 2016; 7: 45863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. You J, Yang H, Lai Y, Simon L, Au J, Burkart AL. ARID2, p110alpha, p53, and beta‐catenin protein expression in hepatocellular carcinoma and clinicopathologic implications. Hum Pathol 2015; 46: 1068–77. [DOI] [PubMed] [Google Scholar]
- 22. Nakazato H, Takeshima H, Kishino T et al Early‐stage induction of SWI/SNF mutations during esophageal squamous cell carcinogenesis. PLoS ONE 2016; 11: e0147372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu MH, Ma WL, Hsu CL et al Androgen receptor promotes hepatitis B virus‐induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med 2010; 2: 32ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie Q, Chen L, Shan X et al Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int J Cancer 2014; 135: 635–46. [DOI] [PubMed] [Google Scholar]
- 25. Rubinson DA, Dillon CP, Kwiatkowski AV et al A lentivirus‐based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet 2003; 33: 401–6. [DOI] [PubMed] [Google Scholar]
- 26. Ladner SK, Otto MJ, Barker CS et al Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 1997; 41: 1715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan HB, Wang XF, Zhang Q et al Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E‐cadherin transcription. Carcinogenesis 2014; 35: 867–76. [DOI] [PubMed] [Google Scholar]
- 28. He F, Li J, Xu J et al Decreased expression of ARID1A associates with poor prognosis and promotes metastases of hepatocellular carcinoma. J Exp Clin Cancer Res 2015; 34: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Zhang H, Ye L. Effects of hepatitis B virus X protein on the development of liver cancer. J Lab Clin Med 2006; 147: 58–66. [DOI] [PubMed] [Google Scholar]
- 30. Sung WK, Lu Y, Lee CW, Zhang D, Ronaghi M, Lee CG. Deregulated direct targets of the hepatitis B virus (HBV) protein, HBx, identified through chromatin immunoprecipitation and expression microarray profiling. J Biol Chem 2009; 284: 21941–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim H, Lee SA, Kim BJ. X region mutations of hepatitis B virus related to clinical severity. World J Gastroenterol 2016; 22: 5467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z, Tang J, Cai X et al HBx mutations promote hepatoma cell migration through the Wnt/beta‐catenin signaling pathway. Cancer Sci 2016; 107: 1380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Liu H, Yi R et al Hepatitis B virus whole‐X and X protein play distinct roles in HBV‐related hepatocellular carcinoma progression. J Exp Clin Cancer Res 2016a; 35: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davoli T, Xu AW, Mengwasser KE et al Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013; 155: 948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cajuso T, Hanninen UA, Kondelin J et al Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int J Cancer 2014; 135: 611–23. [DOI] [PubMed] [Google Scholar]
- 36. Aso T, Uozaki H, Morita S, Kumagai A, Watanabe M. Loss of ARID1A, ARID1B, and ARID2 expression during progression of gastric cancer. Anticancer Res 2015; 35: 6819–27. [PubMed] [Google Scholar]
- 37. Zhang L, Wang W, Li X et al MicroRNA‐155 promotes tumor growth of human hepatocellular carcinoma by targeting ARID2. Int J Oncol 2016b; 48: 2425–34. [DOI] [PubMed] [Google Scholar]
- 38. Flora A, Klisch TJ, Schuster G, Zoghbi HY. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science 2009; 326: 1424–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han ME, Baek SJ, Kim SY, Kang CD, Oh SO. ATOH1 can regulate the tumorigenicity of gastric cancer cells by inducing the differentiation of cancer stem cells. PLoS One 2015; 10: e0126085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kazanjian A, Noah T, Brown D, Burkart J, Shroyer NF. Atonal homolog 1 is required for growth and differentiation effects of notch/gamma‐secretase inhibitors on normal and cancerous intestinal epithelial cells. Gastroenterology 2010; 139: 918–28, 28.e1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leonard JH, Cook AL, Van Gele M et al Proneural and proneuroendocrine transcription factor expression in cutaneous mechanoreceptor (Merkel) cells and Merkel cell carcinoma. Int J Cancer 2002; 101: 103–10. [DOI] [PubMed] [Google Scholar]
- 42. Heiskala K, Arola J, Heiskala M, Andersson LC. Expression of Reg IV and Hath1 in neuroendocrine neoplasms. Histol Histopathol 2010; 25: 63–72. [DOI] [PubMed] [Google Scholar]
- 43. Huang H, Zhai X, Zhu H et al Upregulation of Atoh1 correlates with favorable survival in gastrointestinal stromal tumor. Int J Clin Exp Pathol 2014; 7: 7123–30. [PMC free article] [PubMed] [Google Scholar]
- 44. Forget A, Bihannic L, Cigna SM et al Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev Cell 2014; 29: 649–61. [DOI] [PubMed] [Google Scholar]
- 45. Peignon G, Durand A, Cacheux W et al Complex interplay between beta‐catenin signalling and Notch effectors in intestinal tumorigenesis. Gut 2011; 60: 166–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsuchiya K, Nakamura T, Okamoto R, Kanai T, Watanabe M. Reciprocal targeting of Hath1 and beta‐catenin by Wnt glycogen synthase kinase 3beta in human colon cancer. Gastroenterology 2007; 132: 208–20. [DOI] [PubMed] [Google Scholar]
- 47. Wei Y, Neuveut C, Tiollais P, Buendia MA. Molecular biology of the hepatitis B virus and role of the X gene. Pathol Biol (Paris) 2010; 58: 267–72. [DOI] [PubMed] [Google Scholar]
- 48. Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/beta‐catenin signaling. Cancer Lett 2011; 300: 162–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. (a) The mRNA expression levels of transcription factors. SK‐Hep1 cells and SMMC‐7721 cells were infected with AdHBx or AdGFP control. Total mRNA were isolated at 36 h after infection. The indicated transcription factor genes were detected by Real‐time PCR. (b) SMMC‐7721 cells were treated as shown in Figure S1 (a). The indicated transcription factor genes were determined by qRT‐PCR. (n = 3, *P < 0.05 versus GFP control).
Fig S2. The interaction between HBx and ATOH1 were detected by Co‐IP analysis. SK‐Hep1 cells were co‐transfected with pSEB‐3Flag‐HBx or vector plasmid along with pSEB‐3His‐ATOH1. At 48 h after transfection, cell lysates were subjected to Co‐IP assay with the indicated antibodies.
Fig S3. (a) Protein levels of ARID2 and ATOH1 were detected by western blot analysis in HepG2, HepG2.2.15 and HepAD38 cells with or without tetracycline treatment. (b) Correlation analysis of ATOH1 and ARID2 mRNA expression in 24 paired HCC tissues (*P < 0.05, r = 0.43, Pearson's correlation). (c and d) Protein levels of ARID2 and ATOH1 as well as HBx were detected by immunohistochemical staining and western blot analysis in HBV‐related HCC tissues.
Fig S4. (a) Transwell assays of Sk‐Hep1 cells infected with lentiviruses carrying ATOH1 shRNA or control shRNA. Data represent the results of three independent experiments (means ± SDs; **P < 0.01 versus the siControl; magnification: 200×). (b) E‐cadherin protein expression was determined by western blotting (n = 3, **P < 0.01). (c) Cell proliferation was analyzed by EdU incorporation assays. Data are presented as the means ± SDs (n = 3, **P < 0.01 versus siControl).
Table S1. Primer sequences.