

Abstract
Thyroid transcription factor‐1 (TTF‐1), also known as NKX2‐1, plays a role as a lineage‐survival oncogene in lung adenocarcinoma that possesses double‐edged sword characteristics. Although evidence from previous studies has steadily accumulated regarding the roles of TTF‐1 in transcriptional regulation of protein‐coding genes, little is known about its regulatory relationship with microRNAs. Here, we utilized an integrative approach designed to extract maximal information from expression profiles of both patient tumors in vivo and TTF‐1‐inducible cell lines in vitro, which identified microRNA (miR)‐532‐5p as a novel transcriptional target of TTF‐1. We found that miR‐532‐5p is directly regulated by TTF‐1 through its binding to a genomic region located 8 kb upstream of miR‐532‐5p, which appears to impose transcriptional regulation independent of that of CLCN5, a protein‐coding gene harboring miR‐532‐5p in its intron 3. Furthermore, our results identified KRAS and MKL2 as novel direct targets of miR‐532‐5p. Introduction of miR‐532‐5p mimics markedly induced apoptosis in KRAS‐mutant as well as KRAS wild‐type lung adenocarcinoma cell lines. Interestingly, miR‐532‐5p showed effects on MEK‐ERK pathway signaling, specifically in cell lines sensitive to siKRAS treatment, whereas those miR‐532‐5p‐mediated effects were clearly rendered as phenocopies by repressing expression or inhibiting the function of MKL2 regardless of KRAS mutation status. In summary, our findings show that miR‐532‐5p is a novel transcriptional target of TTF‐1 that plays a tumor suppressive role by targeting KRAS and MKL2 in lung adenocarcinoma.
Keywords: Characteristics and pathology of human cancer, microRNA/non‐coding RNA respiratory organ, oncogenes and tumor‐suppressor genes
Lung cancer is the leading cause of cancer mortality, with more than 1.5 million deaths worldwide each year. Lung adenocarcinoma is the most prevalent subtype, accounting for approximately 50% of reported cases.1 KRAS and EGFR are the most frequently and mutually exclusively mutated oncogenes in lung adenocarcinoma. KRAS mutations, thought to be an early event in molecular carcinogenesis, elicit persistent activation of downstream signaling pathways such as the RAF‐MEK‐ERK cascade, conferring increased proliferative capacity.2 Although development of specific tyrosine kinase inhibitors has changed treatment strategies for patients with EGFR mutations,3 KRAS has long been considered to be an “undruggable target”, thus treatment of KRAS‐mutant lung adenocarcinomas is a foremost clinical challenge.4, 5
Thyroid transcription factor 1 (TTF‐1), also known as NKX2‐1, is indispensable for peripheral lung development and physiology, and it is used as a lineage marker for both normal and cancerous cells of the terminal respiratory unit of the lung.6 In previous studies, we and others have found that TTF‐1 shows frequent gene amplification and overexpression, and also plays a crucial role as a lineage‐survival oncogene in lung adenocarcinoma.7, 8, 9, 10 Furthermore, we have reported that ROR1 is a direct transcriptional target of TTF‐1 that sustains a favorable balance between pro‐survival and pro‐apoptotic signaling in lung adenocarcinoma cells.11, 12 Interestingly, subsequent studies also revealed that TTF‐1 possesses not only oncogenic, but also tumor suppressive functions, thus showing double‐edged sword characteristics in cancer cells.13
MicroRNAs (miRNAs) are small RNA molecules of ~22 nt in length that repress gene expression by binding to a 3′‐UTR of the target mRNA.14 Following our discoveries of frequent occurrence of let‐7 downregulation and miR‐17‐92 overexpression in lung cancer,15, 16 evidence for the involvement of various miRNAs in lung cancer pathogenesis has been rapidly accumulating.17 However, little is known regarding TTF‐1‐mediated regulation of miRNAs, as previous studies of TTF‐1 were nearly exclusively focused on transcriptional regulation of protein‐coding genes.
In this study, we attempted to identify TTF‐1‐regulated miRNAs in lung adenocarcinoma specimens. To this end, we used an integrative approach designed to extract information from expression profiles of lung adenocarcinoma patients in vivo as well as of TTF‐1‐inducible cell lines in vitro in a combinatorial fashion. We report here identification of miR‐532‐5p as a novel transcriptional target of TTF‐1, and also show that miR‐532‐5p directly represses KRAS and MKL2 and is capable of inducing apoptosis in lung adenocarcinoma cells.
Materials and Methods
Cell lines
The NCI‐H23, NCI‐H441, NCI‐H1299, and NCI‐H2009 lung adenocarcinoma cell lines were purchased from ATCC (Manassas, VA, USA), whereas PC‐9 was obtained from RIKEN Cell Bank (Tsukuba, Japan). ACC‐LC‐319 and ACC‐LC‐94 lung adenocarcinoma cell lines were established by our group. An immortalized lung epithelial cell line, BEAS‐2B, was a generous gift from Curtis C. Harris (National Cancer Institute, Bethesda, MD, USA). The conditions used to culture these cell lines have been previously reported.18 Verification of all cell lines was carried out by short tandem repeat profiling at the Japanese Collection of Research Bioresources, National Institute of Biomedical Innovation of Japan (Osaka, Japan) in February 2015. All cell lines were confirmed to be absent of mycoplasma contamination (MycoAlert; Lonza, Tokyo, Japan).
DNA constructs
Full‐length TTF‐1 cDNA was PCR‐amplified from a pCMV‐TTF‐1 vector and inserted into a pTRE3G vector (Clontech, Shiga, Japan). A homeodomain deletion mutant of TTF‐1 was generated from pCMV‐TTF‐1 using a KOD Plus Mutagenesis kit (Toyobo, Osaka, Japan). To generate a luciferase reporter construct, a 4549‐bp fragment of the KRAS 3′‐UTR and a 3501‐bp fragment of the MKL2 3′‐UTR were amplified from human genomic DNA (Promega, Tokyo, Japan) and cloned into a modified pGL3 vector (Promega). A KOD Plus Mutagenesis kit was then used to mutate four nucleotides in the miR‐532‐5p binding sites. For luciferase promoter assays, a 3158‐bp fragment of the CLCN5 promoter as well as a 2605‐bp fragment of the MIR532 promoter were amplified from human genomic DNA, and inserted into a pGL4.10 vector (Promega). Potential TTF‐1 binding sites were then deleted using a KOD Plus Mutagenesis kit. The sequences of all primers used are listed in Table S1.
Small interfering RNA and miRNA mimics and inhibitors
The miRNA mimics, including pre‐miR‐532‐5p (PM11553) and pre‐miR‐NC#2 (AM17111), were purchased from Ambion/Invitrogen (Grand Island, NY, USA). Cells were transfected with 5 nm miRNA mimics using RNAiMAX (Invitrogen). The siRNAs against TTF‐1, KRAS, MKL2, and SRF, as well as a negative control siRNA, SIC002, were purchased from Sigma (St. Louis, MO, USA). For transfection experiments, 5 nm (KRAS, MKL2, and SRF) or 20 nm (TTF‐1) was used. CCG‐100602 and CCG‐203971 were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Generation of doxycycline‐inducible TTF‐1 cell lines
Doxycycline‐inducible TTF‐1 BEAS‐2B, NCI‐H23, NCI‐H1299, and PC‐9 cell lines were established by use of a Tet‐On 3G Expression System (Clontech), essentially according to the manufacturer's instructions. In the resultant TTF‐1‐inducible cell lines, TTF‐1 expression was induced in each experiment with 1 μg/mL DOX for various time periods.
Microarray analysis and TaqMan‐based quantitative RT‐PCR
Microarray analysis of 75 lung adenocarcinoma tumor tissues was carried out using a SurePrint G3 Human Gene Expression Microarray Kit (version 2) and GeneSpring (version 12.6), both from Agilent (Santa Clara, CA, USA), as previously described.19 All microarray data obtained in this study are available at the Gene Expression Omnibus under the accession number GSE83839. Analysis of individual mRNA expression was undertaken using a High Capacity cDNA Reverse Transcription Kit, Power SYBR Green PCR Master Mix, and a 7500 Fast Real‐Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) with use of the gene‐specific primers listed in Table S1. Expression of each gene was normalized to 18S and GAPDH expression levels, and calculated using the comparative Ct method.
Global miRNA expression profiling analysis was carried out using a TaqMan MicroRNA Reverse Transcription kit, TaqMan Low Density Array Human MicroRNA Panels (A, version 2.0; B, version 3.0), and a Prism 7900HT Sequence Detection System (Thermo Fisher Scientific), as previously described.18 All TaqMan Low Density Array analysis data obtained in this study are available at the Gene Expression Omnibus under the accession number GSE83838. The expression of individual miRNAs was determined by quantitative (q)RT–PCR analysis using TaqMan MicroRNA Assay and TaqMan MicroRNA RT kits, along with a 7500 Fast Sequence Detection System (Thermo Fisher Scientific). The non‐coding RNA RNU44 was used as an internal control for normalization.
Definition of TTF‐1 module and selection of candidate miRNAs
We defined the TTF‐1 module as a surrogate of the transcriptional activity of TTF‐1 based on two‐color microarray data, essentially as previously described.18 In brief, we selected genes that were persistently up‐ or downregulated within 24 h after TTF‐1 induction in at least two of the four TTF‐1‐inducible cell lines, BEAS‐2B, NCI‐H23, NCI‐H1299, and PC‐9. Consequently, 81 genes were selected as those consisting of the TTF‐1 module. Using our mRNA expression profile dataset comprised of 75 human lung adenocarcinoma tissues, we defined and calculated actTTF‐1 as described below, which was considered to reflect the TTF‐1 module activity in each tumor:
where n is the number of TTF‐1 module genes, z i is the z‐score of the log2 normalized signal ratio in each sample for gene i, s i is the sign function of i, and U and D sets of up‐ and downregulated genes, respectively. We then searched for candidate miRNAs that showed a correlation with the TTF‐1 module activity using both miRNA (GSE51853, Arima et al.)20 and mRNA (GSE83836, this study) expression profile datasets.
Gene ontology enrichment analysis
Gene Ontology (GO) enrichment analysis was performed using the tool available at the Gene Ontology Consortium website (http://geneontology.org/, GO database released 2015‐08‐06).
Western blot analysis
Western blot analysis was carried out using standard procedures with Immobilon‐P filters (Millipore Japan, Tokyo, Japan) and an Enhanced Chemiluminescence system (GE Healthcare, Chicago, IL, USA). The primary antibodies used were anti‐TTF‐1 (8G7G3/1; Thermo Fisher Scientific), anti‐KRAS (N234, Santa Cruz Biotechnology, Dallas, TX, USA), anti‐MKL2) (14613), anti‐ERK1/2 (9102), anti‐pERK1/2 (T202/Y204) (4377), anti‐MEK (9122), anti‐pMEK (S217/S221) (9121) (all Cell Signaling Technology, Danvers, MA, USA), and anti‐α‐tubulin (T9026, Sigma).
Dual luciferase reporter assay
Dual luciferase reporter assays were undertaken to analyze CLCN5 and MIR532 promoter activities using the respective reporter constructs of pGL4 with and without deletion of potential TTF‐1 binding sites and a pRLTK vector, as well as a Dual Luciferase Reporter Assay System (Promega). pGL3 luciferase reporter constructs carrying the 3′‐UTR of either KRAS or MKL2 with wild‐type or mutated sequences of the respective miR‐532‐5p target sites were utilized along with a pRLTK vector to analyze the effects of the miR‐532‐5p mimic or negative control (each 5 nm), as previously described.18 Firefly luciferase activity was normalized to Renilla luciferase activity for each transfected well. Each assay was performed in triplicate.
Chromatin immunoprecipitation–qRT‐PCR assay
NCI‐H441 (2 × 107) cells were crosslinked with a 1% formaldehyde solution for 7 min at room temperature and subsequently quenched with 2.5 m glycine (Sigma) for 5 min. Cells were harvested in ice‐cold PBS containing a protease inhibitor cocktail (Roche, New York, NY, USA) and processed using an M220 Ultrasonicator (Covaris, Woburn, MA, USA) for 10 min at 6°C with a 5% duty factor to obtain chromatin fragment lengths of 200–500 bp, as judged by the Bioanalyzer DNA High‐Sensitivity kit (Agilent). A portion (0.1%) of the lysate was saved as an input sample, while the remainder was incubated with Dynabeads protein G (Thermo Fisher Scientific) coated with either an anti‐TTF‐1 antibody (H‐190; Santa Cruz Biotechnology) or rabbit IgG control antibody (2729; Cell Signaling Technology) overnight at 4°C on a rotating platform. After repeated washes using a magnetic rack (Thermo Fisher Scientific), TTF‐1‐bound genomic DNA was recovered from Dynabeads and reverse‐crosslinked, then digested with RNase A (Invitrogen) and proteinase K (Thermo Fisher Scientific) for 2 h each at 37 and 55°C, respectively. Following purification with a MinElute Reaction Cleanup Kit (Qiagen, Valencia, CA, USA), qRT‐PCR analysis was undertaken using 100 ng DNA from each of the input and ChIP experiments with primers listed in Table S1. For quantification of DNA recovery, Ct values of the ChIP samples were normalized to that of the input and presented as enrichment over background. At least three independent experiments were carried out for the CLCN5 and MIR532 promoters, as well as for two unrelated genomic regions, TAOK3 and CDH10, which served as negative controls.
Flow cytometry analysis
To determine apoptotic cell death, cells were seeded into 6‐well plates and transfected with 5 nm miR‐532‐5p mimics or siRNAs against KRAS, MKL2, or SRF. Cells were harvested after incubation for 96 h and 1 × 104 cells were subjected to flow cytometry analysis using a FACSCalibur Cell Analyzer (BD Biosciences, San Jose, CA, USA). All assays were carried out in triplicate.
Colorimetric and colony formation assays in vitro and tumor growth assays in vivo
Cells were seeded into 12‐well plates and analyzed using a Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan) at 96 h after transfection with 5 nm miR‐532‐5p mimics, or siRNA against KRAS. For colony formation assays, transfected cells were seeded at a density of 1 × 103 cells per 10‐cm dish and cultured for 10 (ACC‐LC‐94, ACC‐LC‐319, NCI‐H1299) or 14 (NCI‐H23) days. Colonies were then stained with 0.05% Crystal Violet (Sigma) and counted using a cell counting pen (AS One, Osaka, Japan). All of the in vitro assays were carried out in triplicate. ACC‐LC‐319, NCI‐H1299, and ACC‐LC‐94 cells were transfected with either miR‐532‐5p or negative control, then 1 × 106 of each cell type in 200 μL RPMI (Sigma) with 25% Matrigel (BD Biosciences) was injected s.c. into the flanks of BALB/cSlc‐nu/nu mice. Ten days after injection, the mice were killed, and tumors were harvested and weighed (n = 10 for ACC‐LC‐319; n = 5 for ACC‐LC‐94 and NCI‐H1299).
Results
Identification of TTF‐1‐regulated miRNAs
Our search for TTF‐1‐regulated miRNAs was carried out according to the protocol depicted in Figure 1(a). In order to identify miRNAs commonly affected by TTF‐1 in multiple cell lines, we generated four TTF‐1‐inducible cell lines, which included three lung adenocarcinoma cell lines, NCI‐H23, NCI‐H1299, and PC‐9, as well as the normal lung epithelial cell line BEAS‐2B. TTF‐1 was markedly induced in response to DOX treatment in all cell lines (Fig. S1a). Next, we analyzed changes in the miRNA expression profiles of TTF‐1‐inducible BEAS‐2B cells in response to DOX treatment using a previously reported TaqMan array dataset,18 which resulted in identification of 40 different miRNAs that showed greater than two‐fold upregulation by TTF‐1. We also attempted to identify miRNAs regulated by TTF‐1 in vivo. As TTF‐1‐mediated regulation varies based on the presence or absence of cellular cofactors, and because TTF‐1 expression itself may not accurately reflect TTF‐1 transcriptional activity, we incorporated the TTF‐1 module for examination of a panel of 75 surgically resected lung adenocarcinoma tissues and undertook global gene expression analysis of TTF‐1‐inducible BEAS‐2B, NCI‐H23, NCI‐H1299, and PC‐9 cells at 24 h after induction of TTF‐1 by DOX. Using those findings, we subsequently selected a panel of 51 upregulated and 30 downregulated mRNAs that were identified in at least two of the four TTF‐1‐inducible cell lines. Those results accordingly defined the TTF‐1 module as a set of 81 genes reflecting TTF‐1 activity (Fig. S1b, Table S2). Subsequently, we determined TTF‐1 module activity in each of the 75 lung adenocarcinoma tissues and searched for miRNAs showing correlation with TTF‐1 module activity by use of our microarray datasets of both mRNA and miRNA expression in the same panel of 75 lung adenocarcinoma tissues. As a result, a set of 81 miRNAs was identified as significantly associated with TTF‐1 module activity (false discovery rate <0.05). Finally, we combined the information derived from analysis of the association between miRNA expression and experimentally defined TTF‐1 module activity in patient tumor tissues in vivo with that from global miRNA expression profiling in TTF‐1‐induced BEAS‐2B cells in vitro. Eventually, 11 miRNAs were experimentally validated and selected as TTF‐1‐inducible miRNAs, and were shown to be correlated with TTF‐1 activity in the present patients (Table 1). Of note, the panel of 11 miRNAs contained members of the miR‐30 family, miR‐195 and let‐7d, which are known to function as tumor suppressor miRNAs. We selected miR‐532‐5p for further analysis, because of a lack of information in regard to its role in lung cancer development as well as a high degree of TTF‐1‐mediated induction shown in a confirmatory experiment (Fig. 1b) and significant correlation with TTF‐1 module activity (Fig. 1c).
Figure 1.
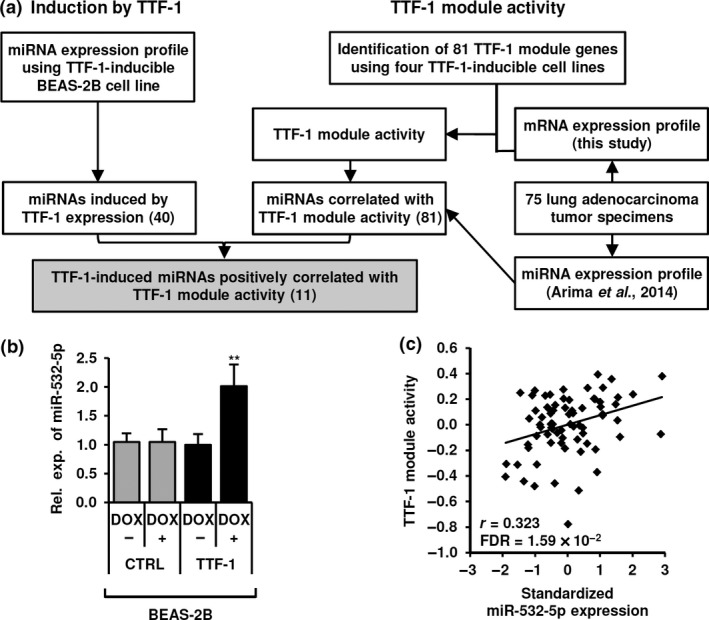
Identification of thyroid transcription factor‐1 (TTF‐1)‐regulated microRNAs (miRNAs). (a) Schematic diagram of strategy used to identify miRNAs regulated by TTF‐1 in vitro and correlated with TTF‐1 activity in vivo. Briefly, 81 genes commonly affected by TTF‐1 in four TTF‐1‐inducible cell lines in vitro were selected to define the TTF‐1 module reflecting TTF‐1 activity. We identified 81 miRNAs that showed a significant correlation with TTF‐1 module activity with the use of both miRNA and mRNA expression profile datasets of 75 lung adenocarcinoma tissues in vivo. Finally, 11 of the 81 miRNAs were found to be present in a panel of 40 miRNAs with 2‐fold upregulation in TTF‐1, but not in control (CTRL) cell line (<1.5‐fold) after doxycycline (DOX) treatment in vitro, which we selected for further analyses. (b) Induction of miR‐532‐5p in response to DOX treatment for 72 h in TTF‐1‐inducible BEAS‐2B cells. Data are shown as mean ± SD of three independent experiments. **P < 0.01, two‐tailed Student's t‐test. Rel. exp., relative expression. (c) Correlation between TTF‐1 module activity and miR‐532‐5p expression in 75 lung adenocarcinoma specimens.
Table 1.
MicroRNAs (miR) showing significant positive correlation with thyroid transcription factor‐1 (TTF‐1) module activity in vivo and more than 2‐fold induction by TTF‐1 in vitro
| Candidate miRNAs | Correlation with TTF‐1 module activity in tumor tissues in vivo | Fold induction in TTF‐1‐inducible BEAS‐2B cells in vitro | |
|---|---|---|---|
| ra | FDR | ||
| hsa‐miR‐532‐5p | 0.323 | 1.59 × 10−2 | 3.95 |
| hsa‐miR‐331 | 0.352 | 8.00 × 10−3 | 3.81 |
| hsa‐miR‐30b | 0.527 | 2.69 × 10−5 | 2.92 |
| hsa‐miR‐30d | 0.574 | 2.18 × 10−6 | 2.88 |
| hsa‐miR‐195 | 0.582 | 2.14 × 10−6 | 2.76 |
| hsa‐miR‐30c | 0.470 | 2.23 × 10−4 | 2.66 |
| hsa‐miR‐26a | 0.573 | 2.18 × 10−6 | 2.62 |
| hsa‐miR‐30a‐5p | 0.498 | 6.88 × 10−5 | 2.50 |
| hsa‐let‐7d | 0.285 | 3.75 × 10−2 | 2.18 |
| hsa‐miR‐328 | 0.486 | 1.25 × 10−4 | 2.05 |
| hsa‐miR‐27b | 0.379 | 3.98 × 10−3 | 2.01 |
Correlation coefficient. FDR, false discovery rate.
MicroRNA‐532‐5p is a transcriptional target of TTF‐1
We then examined how TTF‐1 regulates transcription of miR‐532‐5p, which was found to reside within intron 3 of the CLCN5 gene (Fig. 2a). ENCODE H3K4me3 and H3K27Ac ChIP‐seq data of A549 cells strongly suggested the presence of two potential genomic regions harboring active promoters, which coincided with the putative promoter of the host gene, that is, CLCN5, and a genomic region 8 kb upstream of miR‐532‐5p. Doxycycline treatment of TTF‐1‐inducible NCI‐H1299 and PC‐9 cells showed significant upregulation of both miR‐532‐5p and CLCN5 mRNA (Fig. S2a). Conversely, knockdown of TTF‐1 resulted in markedly reduced miR‐532‐5p expression in both NCI‐H441 and NCI‐H2009 cells, whereas CLCN5 expression was not affected in NCI‐H441, and was rather unexpectedly increased in NCI‐H2009 cells, showing cellular context‐dependent responses (Fig. 2b). Then ChIP–qRT‐PCR analysis was undertaken using NCI‐H2009 cells with an anti‐TTF‐1 antibody and primers amplifying regions within the putative CLCN5 and MIR532 promoters, which contain potential TTF‐1 binding sites. Those results clearly indicated binding of TTF‐1 to both promoters (Fig. 2c), while similar results were obtained in NCI‐H441 cells (Fig. S2b). Furthermore, a dual luciferase assay using reporter constructs of the MIR532 promoter region revealed that deletion of the TTF‐1 binding site significantly impaired promoter activity in NCI‐H2009 (Fig. 2d), as well as in NCI‐H441 cells (Fig. S2c). Following TTF‐1 knockdown in NCI‐H2009 cells, activity of the MIR532 promoter was also reduced (Fig. S2d). In contrast, luciferase reporter activity was significantly increased by deletion of the TTF‐1 binding site within the CLCN5 promoter, a finding consistent with increased CLCN5 mRNA in NCI‐H2009 cells knocked down for TTF‐1. These findings indicated the existence of distinctive regulation characteristics between the MIR532 and CLCN5 promoters.
Figure 2.
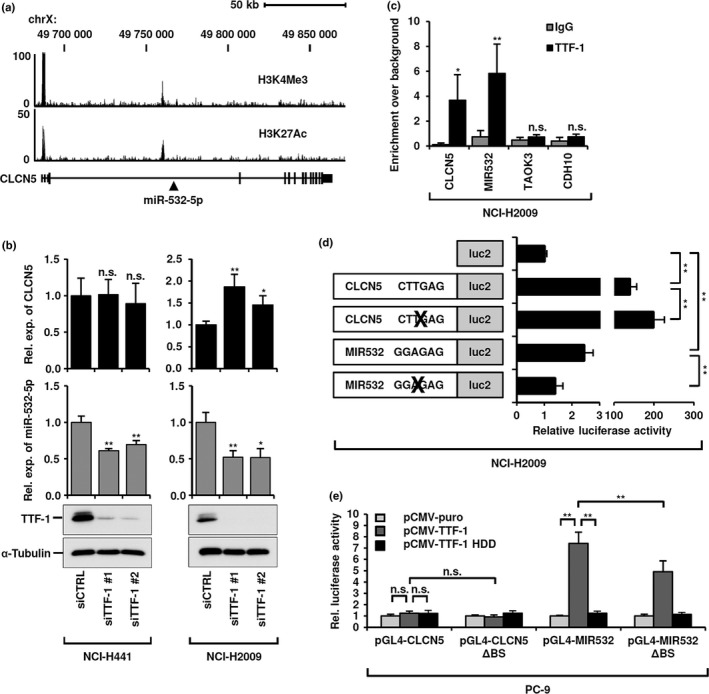
Transcriptional activation of microRNA (miR)‐532‐5p by thyroid transcription factor‐1 (TTF‐1), independent of its host gene CLCN5. (a) Schematic representation of ENCODE ChIP‐seq data of A549 cells. H3K4me3 and H3K27ac data suggested the existence of two distinct promoter regions at the 5′‐UTR of the CLCN5 and MIR532 genes. (b) Quantitative RT‐PCR analysis of CLCN5 mRNA and miR532‐5p expression, as well as Western blot analysis of TTF‐1 in NCI‐H441 and NCI‐H2009 cells knocked down for TTF‐1. (c) ChIP–quantitative RT‐PCR analysis of TTF‐1 binding to CLCN5 and MIR532 promoters in NCI‐H2009 cells. Two unrelated genomic regions, TAOK3 and CDH10, served as negative controls. (d) Dual luciferase assay of CLCN5 and MIR532 promoters carrying wild‐type or TTF‐1 binding site‐deleted sequences in NCI‐H2009 cells. (e) Dual luciferase assay of intact or TTF‐1 binding site‐deleted (ΔBS) CLCN5 and MIR532 promoters in PC‐9 cells introduced with either wild‐type (TTF‐1) or homeodomain deletion mutant (TTF‐1 HDD) TTF‐1 expression constructs. All data shown represent mean ± SD of at least three independent experiments. *P < 0.05; ** P < 0.01, two‐tailed Student's t‐test. n.s., not significant; Rel. exp., relative expression.
To further investigate this issue, PC‐9 cells were co‐transfected with dual luciferase reporter constructs containing the CLCN5 or MIR532 promoter with the TTF‐1 binding site intact or deleted, along with a TTF‐1 expression vector or that carrying deletion of its DNA‐binding homeodomain. MIR532 promoter activity was markedly activated in response to TTF‐1 introduction, whereas it was completely abolished by deletion of the TTF‐1 homeodomain (Fig. 2e). MIR532 promoter activity was also significantly but not completely impaired by deletion of the TTF‐1 binding site, suggesting possible existence of a cryptic TTF‐1 binding site. Interestingly, the CLCN5 reporter construct did not show any increase in luciferase activity in TTF‐1‐transfected PC‐9 cells, suggesting that TTF‐1 either induces or represses transcription of CLCN5, potentially dependent on the presence or absence of cofactor(s). Taken together, our data clearly show that TTF‐1‐mediated miR‐532‐5p induction is independent of regulation of CLCN5.
KRAS is a direct target of miR‐532‐5p
We then sought to identify the target(s) of miR‐532‐5p with potential functional involvement in lung cancer development. Microarray analysis of miR‐532‐5p‐introduced NCI‐H23 and NCI‐H1299 cells was carried out, and the resultant datasets were used to identify a set of possible direct target genes of miR‐532‐5p with the aid of TargetScan Human Release 6.2; http://www.targetscan.org/vert_61/ in a combinatorial fashion, which consequently identified 40 genes as strong candidates (Table S3). This set of 40 genes was then subjected to GO enrichment analysis, which showed significant enrichment of genes with such biological processes as MAPK cascade, cellular response to growth factor stimulus, and the epidermal growth factor receptor signaling pathway (Table 2). We further selected genes commonly shared among biological processes enriched with the potential targets. Seven genes were found to be included in at least five of the seven biological processes (Table S4), with KRAS ranked on top based on context+ score; thus, it attracted much of our subsequent attention.
Table 2.
Enriched processes among candidate microRNA‐532‐5p target genes
| GO biological processes (GO database released 2015‐08‐06) | Number of genes | P‐value |
|---|---|---|
| MAPK cascade | 9 | 4.4 × 10−4 |
| Fc receptor signaling pathway | 8 | 3.1 × 10−3 |
| Positive regulation of macromolecule biosynthetic process | 14 | 9.0 × 10−3 |
| Cellular response to growth factor stimulus | 10 | 1.0 × 10−2 |
| Epidermal growth factor receptor signaling pathway | 7 | 1.8 × 10−2 |
| Positive regulation of cellular biosynthetic process | 14 | 2.0 × 10−2 |
| Regulation of cellular macromolecule biosynthetic process | 21 | 2.5 × 10−2 |
GO, gene ontology.
Quantitative RT‐PCR and Western blot analyses showed a significant reduction of KRAS in miR‐532‐5p‐introduced NCI‐H23 and NCI‐H1299 cells (Fig. 3a). Similar results were also obtained in two additional lung adenocarcinoma cell lines, ACC‐LC‐94 and ACC‐LC‐319 (Fig. S3a). Next, to examine whether KRAS is directly repressed by miR‐532‐5p, NCI‐H23 cells were transfected with miR‐532‐5p as well as dual luciferase reporter constructs carrying the KRAS 3′‐UTR with and without mutations at a potential miR‐532‐5p binding site. Repression of luciferase reporter activity by miR‐532‐5p was strongly impaired by the presence of mutations corresponding to the seed sequence of miR‐532‐5p, indicating that KRAS is a direct target of miR‐532‐5p (Fig. 3b).
Figure 3.
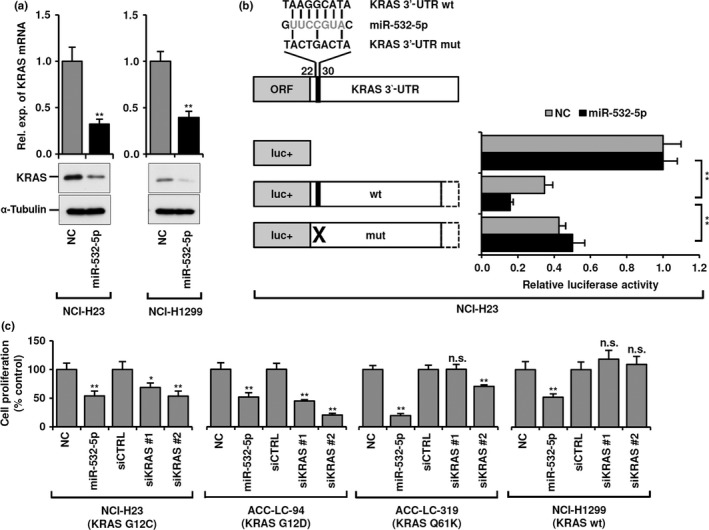
Identification of KRAS as a target of microRNA (miR)‐532‐5p in lung adenocarcinoma cells. (a) Quantitative RT‐PCR and Western blot analyses of KRAS in miR‐532‐5p‐introduced NCI‐H23 and NCI‐H1299 cells. Rel. exp., relative expression. (b) Dual luciferase assay using reporter constructs carrying wild‐type (wt) or mutant (mut) sequences of the KRAS 3′‐UTR in NCI‐H23 cells transiently transfected with miR‐532‐5p mimics or negative control (NC). (c) Colorimetric assay of four lung adenocarcinoma cell lines with and without KRAS mutations. Cells were analyzed at 96 h after transfection with either miR‐532‐5p mimics or two independent siRNAs against KRAS. All data shown represent mean ± SD of three independent experiments. *P < 0.05; **P < 0.01, two‐tailed Student's t‐test.
We further examined whether miR‐532‐5p introduction has an effect on lung adenocarcinoma proliferation, similar to KRAS knockdown, using three lung adenocarcinoma cell lines with mutant KRAS (NCI‐H23, ACC‐LC‐94, and ACC‐LC‐319), as well as NCI‐H1299 with wild‐type KRAS (Fig. 3c). In all of the lung adenocarcinoma cell lines examined, significantly reduced cell proliferation was noted when miR‐532‐5p was introduced. Similarly, KRAS knockdown significantly inhibited cell proliferation of NCI‐H23 and ACC‐LC‐94 cells. In contrast, NCI‐H1299 with wild‐type KRAS showed resistance to siKRAS treatment, whereas ACC‐LC‐319 with mutant KRAS was mostly unaffected. These results suggest that miR‐532‐5p plays a suppressive role in lung adenocarcinoma, in part through direct repression of KRAS, and that miR‐532‐5p may also target another gene that plays a crucial role in lung adenocarcinoma development.
MicroRNA‐532‐5p targets MKL2, induces apoptosis, and inhibits lung adenocarcinoma proliferation
Accordingly, we excluded genes constituting the same biological processes as KRAS from the list of 40 potential targets and searched for additional relevant targets of miR‐532‐5p. As a result, MKL2, a transcriptional co‐activator of SRF and SMAD3,21, 22 was shown to be an intriguing candidate accounting for significant growth inhibition of siKRAS‐resistant ACC‐LC‐319 and NCI‐H1299 cells. Expression of MKL2 was repressed at both the mRNA and protein levels following miR‐532‐5p introduction in NCI‐H23 and NCI‐H1299 cells (Fig. 4a), as well as in ACC‐LC‐94 and ACC‐LC‐319 cells (Fig. S4a). Also, a dual luciferase assay was carried out using reporter constructs containing the MKL2 3′‐UTR with and without mutations at potential miR‐532‐5p binding sites. Mutations corresponding to the seed sequence of miR‐532‐5p at one of the two potential binding sites significantly increased luciferase reporter activity in NCI‐H23 cells treated with miR‐532‐5p (Fig. 4b), whereas abolition of both binding sites markedly restored that activity to nearly the baseline level.
Figure 4.
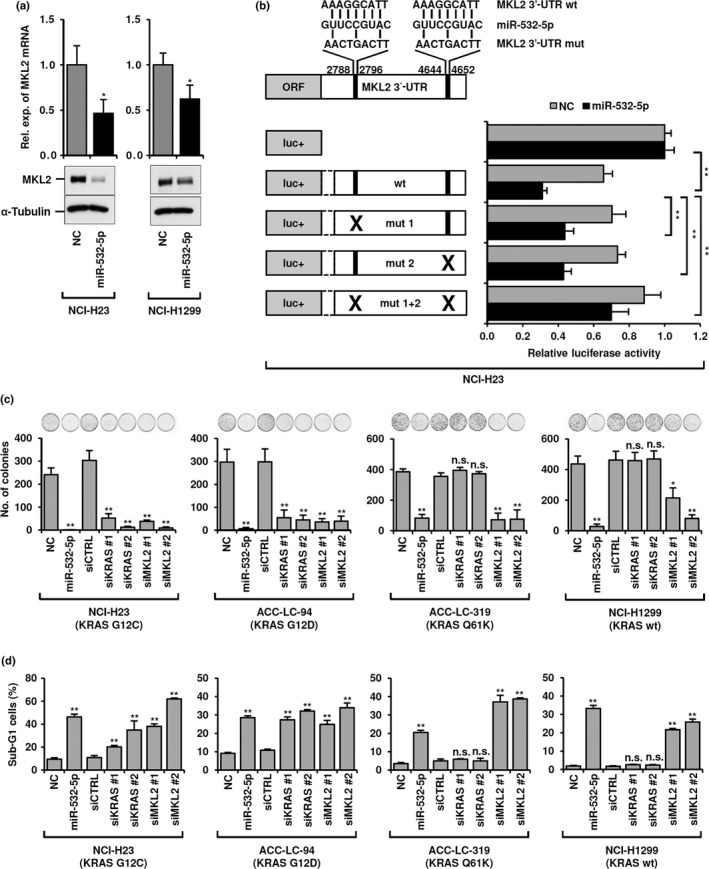
MKL2 as a microRNA (miR)‐532‐5p target and biological effects of miR‐532‐5p introduction compared with KRAS or MKL2 knockdown in lung adenocarcinoma cells. (a) Quantitative RT‐PCR and Western blot analyses of MKL2 in miR‐532‐5p‐introduced NCI‐H23 and NCI‐H1299 cells. Rel. exp., relative expression. (b) Dual luciferase assay using reporter constructs carrying wild‐type (wt) or mutant (mut 1, mut 2, mut 1 + 2) sequences of the MKL2 3′‐UTR in NCI‐H23 cells transiently transfected with miR‐532‐5p mimics or negative control (NC). (c) Colony formation assay for the same four lung adenocarcinoma cell lines with and without KRAS mutations. Treatment with miR‐532‐5p and siMKL2 markedly inhibited colony formation in all four cell lines, whereas siKRAS treatment significantly reduced the number of colonies only in NCI‐H23 and ACC‐LC‐94 cells. (d) Flow cytometry analysis was used to determine sub‐G1 populations in the same panel of four lung adenocarcinoma cell lines. Sub‐G1 populations were markedly increased in all four cell lines when treated with either miR‐532‐5p or siMKL2, whereas siKRAS treatment significantly induced apoptosis only in NCI‐H23 and ACC‐LC‐94 cells. All experiments were carried out in triplicate and data shown represent mean ± SD. *P < 0.05; **P < 0.01, two‐tailed Student's t‐test. n.s., not significant.
Next, we examined the effects of miR‐532‐5p, siKRAS, and siMKL2 treatment on colony formation and found significantly reduced numbers of colonies following miR‐532‐5p and siMKL2 treatment in all four cell lines (Fig. 4c). In contrast, siKRAS specifically inhibited colony formation in NCI‐H23 and ACC‐LC‐94 cells, with a clear dependence on mutant KRAS‐mediated signaling, whereas that was not seen in ACC‐LC‐319 and NCI‐H1299 cells. Consistent findings were obtained in flow cytometry measurements of sub‐G1 cell populations, an indicator of apoptosis induction (Fig. 4d). These results point to the notion that TTF‐1‐induced miR‐532‐5p plays a role as a negative regulator of at least two biologically pertinent and distinct target genes.
We next investigated whether there were distinct responses in terms of MAPK signaling when lung adenocarcinoma cell lines were treated with either miR‐532‐5p, siKRAS, or siMKL2, with special attention given to their dependence on mutant KRAS‐mediated signaling (Fig. 5a). NCI‐H23 and ACC‐LC‐94 cells with apparent KRAS dependency showed clear inhibition of MEK and ERK phosphorylation in response to treatment with miR‐532‐5p and siKRAS, but not with siMKL2. In contrast, such inhibitory effects toward MEK and ERK activation were not observed in NCI‐H1299 and ACC‐LC‐319 cells treated with miR‐532‐5p, siKRAS, or siMKL2. These results suggested that the repressive effect of miR‐532‐5p on lung adenocarcinoma survival is mediated not only by inhibition of the MAPK pathway through targeting of KRAS, but also by inhibition of another vital unidentified molecular pathway, most likely the MKL2‐SRF pathway by targeting MKL2.
Figure 5.
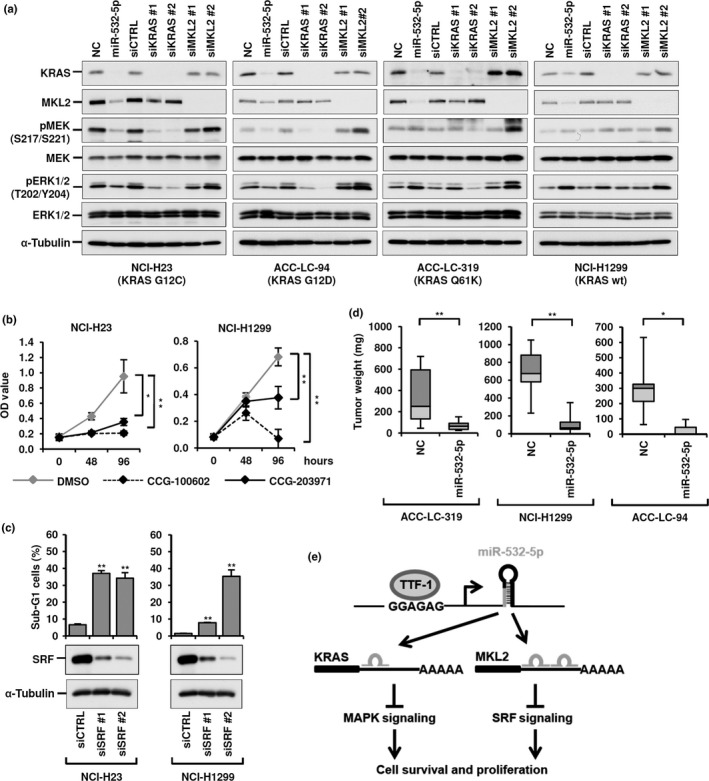
Cancer cell growth decreased by microRNA (miR)‐532‐5p itself and miR‐532‐5p‐affected downstream pathways. (a) Western blot analysis of MEK‐ERK signaling in four lung adenocarcinoma cell lines with and without KRAS mutations. Note that MEK‐ERK signaling was inhibited by both miR‐532‐5p and KRAS knockdown in NCI‐H23 and ACC‐LC‐94, but not ACC‐LC‐319 or NCI‐H1299 cells. (b) Colorimetric assay of effects of treatment with CCG‐100602 and CCG‐203971, MKL‐SRF pathway inhibitors, in NCI‐H23 and NCI‐H1299 cells. (c) Flow cytometry and Western blot analyses of NCI‐H23 and NCI‐H1299 cells treated with siRNA against SRF, for which MKL2 plays a role as a co‐activator. (d) Decreased tumor growth of miR‐532‐5p‐introduced lung adenocarcinoma cell lines in vivo. (e) Schematic diagram summarizing the present findings: thyroid transcription factor‐1 (TTF‐1) induces miR‐532‐5p, which in turn targets KRAS and MKL2, thus inhibiting two crucial downstream pathways, leading to induction of apoptosis in lung adenocarcinoma cells. *P < 0.05; **P < 0.01, two‐tailed Student's t‐test. NC, negative control.
In order to investigate our speculation, we treated NCI‐H23 and NCI‐H1299 cells with CCG‐10060223 and CCG‐203971,24 which inhibit MKL‐SRF‐mediated gene transcription. Colorimetric assays revealed marked inhibition of cell proliferation in response to MKL‐SRF pathway inhibition in both cell lines (Fig. 5b). In addition, we observed significant induction of apoptosis by knockdown of SRF in both NCI‐H23 and NCI‐H1299 cells (Fig. 5c), suggesting that miR‐532‐5p‐induced MKL2 repression elicits apoptosis, at least in part, due to consequential impairment of the transcriptional activating function of the MKL2–SRF complex.
Finally, we investigated the effects of miR‐532‐5p introduction in a xenograft model using ACC‐LC‐319, NCI‐H1299, and ACC‐LC‐94 cells. Decreased expressions of KRAS and MKL2 were observed in miR‐532‐5p‐introduced ACC‐LC‐94 xenografts at 5 days after injection (Fig. S5), whereas the tumor weights in animals with miR‐532‐5p‐introduced cells at 10 days after injection were significantly reduced as compared to those introduced with negative control in all three cell lines (Fig. 5d). Taken together, the present findings clearly indicate that miR‐532‐5p is a novel transcriptional target of TTF‐1 and that miR‐532‐5p plays a role in tumor suppression, at least in part, by targeting KRAS and MKL2 in lung adenocarcinomas (Fig. 5e).
Discussion
In the present study, we applied an integrative approach that combined expression profiles of patient tumor tissues in vivo and experimental data of cell lines in vitro, and our results identified miR‐532‐5p as a functionally relevant transcriptional target of TTF‐1. Thyroid transcription factor‐1 regulates miR‐532‐5p by binding to its own promoter rather than that of CLCN5 harboring miR‐532‐5p in its intron 3. Along this line, it is important to note that, although miR‐532‐5p forms an miRNA cluster together with miR‐188, miR‐500a, miR‐500b, miR‐362, miR‐501, and miR‐660, other miRNAs were filtered out during our search because of their low expression and/or lack of correlation with TTF‐1 in patients.
TTF‐1 was initially identified as a lineage‐survival oncogene in lung adenocarcinoma reports;7, 8, 9, 10 subsequent studies, including our own, showed that it possesses double‐edged sword characteristics,13 including functions deleterious toward cancer.25, 26, 27 The present findings clearly indicate that miR‐532‐5p, a novel target of TTF‐1, possesses apoptosis‐inducing capability, thus transcriptional activation of miR‐532‐5p is considered to reflect a deleterious function of TTF‐1. In addition, multiple additional miRNAs found to be positively associated with TTF‐1 activity are also known to have a tumor‐suppressive function in lung cancer, including miR‐195,28 let‐7d,15 and members of the miR‐30 family,20 providing further supporting evidence of the double‐edged sword characteristics of this enigmatic oncogene. A few reports have been published regarding the reduced expression and tumor‐suppressive functions of miR‐532‐5p in other types of human cancers, including renal cell,29, 30 hepatocellular,31 and ovarian carcinomas.32 MicroRNA‐532‐5p has also been reported to promote tumor proliferation and progression in melanomas33 and gastric cancer cells,34 suggesting its context‐dependent roles in various cancer types. It is important to note that a sizable fraction of TTF‐1‐positive lung adenocarcinomas have low levels of miR‐532‐5p expression, despite the general and significant association of miR‐532‐5p with TTF‐1. In our experiments, we did not find any evidence showing DNA hypermethylation of the MIR532 promoter (data not shown), suggesting involvement of other underlying mechanism(s), such as altered histone modifications and/or lack of co‐activators of TTF‐1 in that cellular context.
The present results clearly revealed that KRAS and MKL2 are direct targets of miR‐532‐5p. KRAS is a well‐established oncogene that is frequently mutated and activated in lung adenocarcinomas. A functional link between Ttf‐1 and Kras mutations was previously reported in a study that used genetically engineered mice, in which Ttf‐1 was shown to inhibit mutant‐Kras‐driven tumorigenesis by repressing a latent gastric differentiation program in lung adenocarcinomas.35 In the present study, we uncovered another link between TTF‐1 and KRAS, as TTF‐1‐regulated miR‐532‐5p was shown to possess a capability to directly repress KRAS by binding to its 3′‐UTR. Along this line, miR‐532‐5p significantly decreased activation of the MEK–ERK axis in lung adenocarcinoma cells dependent on mutant KRAS‐mediated signaling. Interestingly, though Rice et al. previously reported that TTF‐1 transcriptionally activates SREBF2, a host gene of miR‐33a,36 miR‐33a was excluded in the process of our search for TTF‐1‐regulated miRNAs because of its low level of expression.
Together, MKL2 and MKL1 form a family of co‐activators of transcription factors that includes SRF21 and SMAD3,22 that regulates various cellular processes including cell migration, cell growth, and cytoskeleton organization.37 To the best of our knowledge, this is the first study to identify an miRNA targeting MKL2 in cancer. While introduction of miR‐532‐5p elicited apoptosis in lung adenocarcinoma cells regardless of KRAS mutation status, treatment with siRNA against MKL2 clearly rendered a phenocopy of this effect, suggesting crucial involvement of continued MKL2 expression in this devastating cancer. Both siKRAS‐sensitive NCI‐H23 and ‐insensitive NCI‐H1299 cells showed marked sensitivity to two small molecules, CCG‐100602 and CCG‐203971, inhibitors of MKL/SRF‐dependent transcriptional activation. Consistently, SRF knockdown also rendered a phenocopy of the effects of miR‐532‐5p and siMKL2 treatment, indicating a vital role of MKL/SRF‐mediated signaling in lung adenocarcinoma development. Muehlich and colleagues recently reported similar results of nuclear localization of MKL2, reflecting its activated state, and induction of senescence by knocking down MKL1/2 in hepatocellular carcinoma cells.38 A study is needed to address whether miR‐532‐5p or another small molecule can attain sufficient inhibition of MKL‐SRF function without serious side‐effects in lung adenocarcinomas, as well as other types of human cancer.
In conclusion, miR‐532‐5p was revealed as a novel target of TTF‐1 in the present study using an integrative approach that combined both in vitro and in vivo findings. It is conceivable that miR‐532‐5p imposes intricate regulation of TTF‐1 functions together with other identified miRNAs. We also showed that miR‐532‐5p is capable of inducing apoptosis in lung adenocarcinoma and targets two key oncogenes, KRAS and MKL2, suggesting that novel therapeutic strategies using miR‐532‐5p may ultimately prove effective for treating patients with this hard‐to‐cure cancer. Finally, we note that, although Ttf‐1‐knockout mice are born dead with severely impaired lung morphogenesis,39 miR‐532‐5p is upregulated in their lungs from embryonic days 16–18,40 which also warrants future investigation of its potential role in lung development.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- CLCN5
chloride voltage‐gated channel 5
- DOX
doxycycline
- GO
gene ontology
- miR
microRNA
- MKL2
myocardin‐like 2
- qRT‐PCR
quantitative RT‐PCR
- SRF
serum response factor
- TTF‐1
thyroid transcription factor‐1
Supporting information
Fig. S1. Defining thyroid transcription factor‐1 (TTF‐1) module reflecting TTF‐1 activity and correlation between microRNA (miR)‐532‐5p and TTF‐1 expression in vivo.
Fig. S2. Thyroid transcription factor‐1 (TTF‐1) binds to MIR532 promoter and transcriptionally regulates microRNA (miR)‐532‐5p.
Fig. S3. MicroRNA (miR)‐532‐5p‐mediated reduction of both mRNA and protein of KRAS in additional lung adenocarcinoma cell lines.
Fig. S4. MicroRNA (miR)‐532‐5p‐mediated reduction of both mRNA and protein of MKL2 in additional lung adenocarcinoma cell lines.
Fig. S5. Reduction of both KRAS and MKL2 mRNA in microRNA (miR)‐532‐5p‐introduced ACC‐LC‐94 xenografts.
Table S1. Sequences of oligonucleotides used in this study.
Table S2. Genes consisting of the thyroid transcription factor‐1 (TTF‐1) module.
Table S3. List of candidates for microRNA‐532‐5p target genes.
Table S4. Top‐ranked candidates for microRNA‐532‐5p target genes.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (22134005), and Grants‐in‐Aid for Scientific Research (A) from the Japan Society for the Promotion of Science (25250017). S.G. was supported by a Scholarship Program of the MEXT.
Cancer Sci 108 (2017) 1394–1404
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (22134005), Japan Society for the Promotion of Science (25250017).
References
- 1. Detterbeck FC, Decker RH, Tanoue L, Lilenbaum RC. Non‐Small‐Cell Lung Cancer In: DeVita VT, Lawrence TS, Rosenberg SA, eds. DeVita, Hellman, and Rosenberg's Cancer Principles & Practice of Oncology, 10th ed Philadelphia, PA, USA: Wolters Kluwer, 2015; 495–535. [Google Scholar]
- 2. Horn L, de Lima Araujo LH, Nana‐Sinkam P, Otterson GA, Williams TG, Carbone DP. Molecular biology of lung cancer In: DeVita VT, Lawrence TS, Rosenberg SA, eds. DeVita, Hellman, and Rosenberg's Cancer Principles & Practice of Oncology, 10 edn Volume 10. Philadelphia, PA, USA: Wolters Kluwer, 2015; 482–94. [Google Scholar]
- 3. Janne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non‐small‐cell lung cancer: implications for treatment and tumor biology. J Clin Oncol 2005; 23: 3227–34. [DOI] [PubMed] [Google Scholar]
- 4. Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther 2002; 1: 599–606. [DOI] [PubMed] [Google Scholar]
- 5. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell 2014; 25: 272–81. [DOI] [PubMed] [Google Scholar]
- 6. Yatabe Y, Mitsudomi T, Takahashi T. TTF‐1 expression in pulmonary adenocarcinomas. Am J Surg Pathol 2002; 26: 767–73. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka H, Yanagisawa K, Shinjo K et al Lineage‐specific dependency of lung adenocarcinomas on the lung development regulator TTF‐1. Cancer Res 2007; 67: 6007–11. [DOI] [PubMed] [Google Scholar]
- 8. Kendall J, Liu Q, Bakleh A et al Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci USA 2007; 104: 16663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weir BA, Woo MS, Getz G et al Characterizing the cancer genome in lung adenocarcinoma. Nature 2007; 450: 893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwei KA, Kim YH, Girard L et al Genomic profiling identifies TITF1 as a lineage‐specific oncogene amplified in lung cancer. Oncogene 2008; 27: 3635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamaguchi T, Yanagisawa K, Sugiyama R et al NKX2‐1/TITF1/TTF‐1‐Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 2012; 21: 348–61. [DOI] [PubMed] [Google Scholar]
- 12. Yamaguchi T, Lu C, Ida L et al ROR1 sustains caveolae and survival signalling as a scaffold of cavin‐1 and caveolin‐1. Nat Commun 2016; 7: 10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamaguchi T, Hosono Y, Yanagisawa K, Takahashi T. NKX2‐1/TTF‐1: an enigmatic oncogene that functions as a double‐edged sword for cancer cell survival and progression. Cancer Cell 2013; 23: 718–23. [DOI] [PubMed] [Google Scholar]
- 14. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takamizawa J, Konishi H, Yanagisawa K et al Reduced expression of the let‐7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004; 64: 3753–6. [DOI] [PubMed] [Google Scholar]
- 16. Hayashita Y, Osada H, Tatematsu Y et al A polycistronic microRNA cluster, miR‐17‐92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628–32. [DOI] [PubMed] [Google Scholar]
- 17. Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol 2014; 24: R762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tai MC, Kajino T, Nakatochi M et al miR‐342‐3p regulates MYC transcriptional activity via direct repression of E2F1 in human lung cancer. Carcinogenesis 2015; 36: 1464–73. [DOI] [PubMed] [Google Scholar]
- 19. Tomida S, Takeuchi T, Shimada Y et al Relapse‐related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J Clin Oncol 2009; 27: 2793–9. [DOI] [PubMed] [Google Scholar]
- 20. Arima C, Kajino T, Tamada Y et al Lung adenocarcinoma subtypes definable by lung development‐related miRNA expression profiles in association with clinicopathologic features. Carcinogenesis 2014; 35: 2224–31. [DOI] [PubMed] [Google Scholar]
- 21. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003; 113: 329–42. [DOI] [PubMed] [Google Scholar]
- 22. Morita T, Mayanagi T, Sobue K. Dual roles of myocardin‐related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J Cell Biol 2007; 179: 1027–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evelyn CR, Bell JL, Ryu JG et al Design, synthesis and prostate cancer cell‐based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG‐1423. Bioorg Med Chem Lett 2010; 20: 665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bell JL, Haak AJ, Wade SM, Kirchhoff PD, Neubig RR, Larsen SD. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti‐metastasis agents. Bioorg Med Chem Lett 2013; 23: 3826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hosono Y, Yamaguchi T, Mizutani E et al MYBPH, a transcriptional target of TTF‐1, inhibits ROCK1, and reduces cell motility and metastasis. EMBO J 2012; 31: 481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maeda Y, Tsuchiya T, Hao H et al Kras(G12D) and Nk2–1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest 2012; 122: 4388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winslow MM, Dayton TL, Verhaak RG et al Suppression of lung adenocarcinoma progression by Nk2–1. Nature 2011; 473: 101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yongchun Z, Linwei T, Xicai W et al MicroRNA‐195 inhibits non‐small cell lung cancer cell proliferation, migration and invasion by targeting MYB. Cancer Lett 2014; 347: 65–74. [DOI] [PubMed] [Google Scholar]
- 29. Jung M, Mollenkopf HJ, Grimm C et al MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med 2009; 13: 3918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. He H, Wang L, Zhou W et al MicroRNA expression profiling in clear cell renal cell carcinoma: identification and functional validation of key miRNAs. PLoS ONE 2015; 10: e0125672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song X, Wang Z, Jin Y, Wang Y, Duan W. Loss of miR‐532‐5p in vitro promotes cell proliferation and metastasis by influencing CXCL2 expression in HCC. Am J Transl Res 2015; 7: 2254–61. [PMC free article] [PubMed] [Google Scholar]
- 32. Wang F, Chang JT, Kao CJ, Huang RS. High expression of miR‐532‐5p, a tumor suppressor, leads to better prognosis in ovarian cancer both in vivo and in vitro . Mol Cancer Ther 2016; 15: 1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitago M, Martinez SR, Nakamura T, Sim MS, Hoon DS. Regulation of RUNX3 tumor suppressor gene expression in cutaneous melanoma. Clin Cancer Res 2009; 15: 2988–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu X, Zhang Y, Liu Z, Zhang X, Jia J. miRNA‐532‐5p functions as an oncogenic microRNA in human gastric cancer by directly targeting RUNX3. J Cell Mol Med 2016; 20: 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Snyder EL, Watanabe H, Magendantz M et al Nk2–1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell 2013; 50: 185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rice SJ, Lai SC, Wood LW et al MicroRNA‐33a mediates the regulation of high mobility group AT‐hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF‐1/NKX2‐1). J Biol Chem 2013; 288: 16348–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev 2006; 20: 1545–56. [DOI] [PubMed] [Google Scholar]
- 38. Muehlich S, Hampl V, Khalid S et al The transcriptional coactivators megakaryoblastic leukemia 1/2 mediate the effects of loss of the tumor suppressor deleted in liver cancer 1. Oncogene 2012; 31: 3913–23. [DOI] [PubMed] [Google Scholar]
- 39. Kimura S, Hara Y, Pineau T et al The T/ebp null mouse: thyroid‐specific enhancer‐binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 1996; 10: 60–9. [DOI] [PubMed] [Google Scholar]
- 40. Dong J, Jiang G, Asmann YW et al MicroRNA networks in mouse lung organogenesis. PLoS ONE 2010; 5: e10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Defining thyroid transcription factor‐1 (TTF‐1) module reflecting TTF‐1 activity and correlation between microRNA (miR)‐532‐5p and TTF‐1 expression in vivo.
Fig. S2. Thyroid transcription factor‐1 (TTF‐1) binds to MIR532 promoter and transcriptionally regulates microRNA (miR)‐532‐5p.
Fig. S3. MicroRNA (miR)‐532‐5p‐mediated reduction of both mRNA and protein of KRAS in additional lung adenocarcinoma cell lines.
Fig. S4. MicroRNA (miR)‐532‐5p‐mediated reduction of both mRNA and protein of MKL2 in additional lung adenocarcinoma cell lines.
Fig. S5. Reduction of both KRAS and MKL2 mRNA in microRNA (miR)‐532‐5p‐introduced ACC‐LC‐94 xenografts.
Table S1. Sequences of oligonucleotides used in this study.
Table S2. Genes consisting of the thyroid transcription factor‐1 (TTF‐1) module.
Table S3. List of candidates for microRNA‐532‐5p target genes.
Table S4. Top‐ranked candidates for microRNA‐532‐5p target genes.