

Abstract
Small‐cell lung cancer (SCLC) accounts for approximately 15% of all lung cancers, and is characterized as extremely aggressive, often displaying rapid tumor growth and multiple organ metastases. In addition, the clinical outcome of SCLC patients is poor due to early relapse and acquired resistance to standard chemotherapy treatments. Hence, novel therapeutic strategies for the treatment of SCLC are urgently required. Accordingly, several molecular targeted therapies were evaluated in SCLC; however, they failed to improve the clinical outcome. The receptor tyrosine kinase MET is a receptor for hepatocyte growth factor (HGF), and aberrant activation of HGF/MET signaling is known as one of the crucial mechanisms enabling cancer progression and invasion. Here, we found that the HGF/MET signaling was aberrantly activated in chemoresistant or chemorelapsed SCLC cell lines (SBC‐5, DMS273, and DMS273‐G3H) by the secretion of HGF and/or MET copy number gain. A cell‐based in vitro assay revealed that HGF/MET inhibition, induced either by MET inhibitors (crizotinib and golvatinib), or by siRNA‐mediated knockdown of HGF or MET, constrained growth of chemoresistant SCLC cells through the inhibition of ERK and AKT signals. Furthermore, treatment with either crizotinib or golvatinib suppressed the systemic metastasis of SBC‐5 cell tumors in natural killer cell‐depleted SCID mice, predominantly through cell cycle arrest. These findings reveal the therapeutic potential of targeting the HGF/MET pathway for inhibition, to constrain tumor progression of SCLC cells showing aberrant activation of HGF/MET signaling. We suggest that it would be clinically valuable to further investigate HGF/MET‐mediated signaling in SCLC cells.
Keywords: Animal model, HGF, MET, small‐cell lung cancer, targeted therapy
Lung cancer is the leading cause of cancer‐related deaths worldwide.1 Approximately 15% of lung cancer cases are classified as being small‐cell lung cancer (SCLC), an extremely aggressive cancer type characterized by rapid tumor growth leading to multiple organ metastases, such as to the brain, bone, liver, and/or lymph nodes.2 In fact, approximately 70% of SCLC patients are categorized as suffering extensive disease with distant metastases at the time of diagnosis. For the past two decades, the standard treatment for these advanced SCLC patients has been combined chemotherapy, comprising treatment with cisplatin and either etoposide or irinotecan. The response rate for this therapy is approximately 70–80%; however, the median survival time is only 9–12 months, and the overall survival rate of SCLC patients with extensive disease at 5 years is only 5–10%, largely due to early relapse and acquired resistance.2, 3, 4, 5, 6. Thus, novel therapeutic strategies for the treatment of SCLC are urgently required to improve patient clinical outcomes. Several molecularly targeted therapies have been evaluated and trialed; however, these failed to improve the clinical outcome for this disease 7.
The receptor tyrosine kinase MET acts as the receptor for hepatocyte growth factor (HGF), such that MET activated by HGF‐binding forms a homodimer, and transduces downstream signals to various pathways.8, 9, 10 In several cancers, aberrant activation of HGF/MET signaling is reported as a crucial mechanism enabling cancer progression and invasion.10, 11 Recently, a MET exon 14 skipping mutation was reported to be a novel driver mutation in non‐small‐cell lung cancer (NSCLC) patients.12 Similarly, MET‐activating mutations and dysregulated autocrine/paracrine HGF/MET signaling have both been reported in SCLC patients; furthermore, MET phosphorylation predicts poor clinical outcome in SCLC.13, 14, 15, 16 Canadas et al. recently reported that HGF stimulation in SCLC cells promotes epithelial–mesenchymal transition and chemoresistance, whereas treatment with a MET inhibitor restored chemosensitivity.17
Although activated MET is reported as a prognostic factor for poor clinical outcome in SCLC, the mechanisms underlying the deleterious effects of aberrant activation of the HGF/MET pathway in SCLC have not been clearly elucidated.16 We previously established a mouse model of multiple‐organ SCLC metastasis, by utilizing natural killer (NK) cell‐depleted SCID mice, to produce an animal model whose tumor distribution resembles that of advanced SCLC patients.18, 19, 20 The aim of the present study was to evaluate the effect of inhibiting the HGF/MET pathway on tumor progression in SCLC with multi‐organ metastasis, using this in vivo mouse xenograft model, as well as various cell‐based in vitro assays.
Materials and Methods
Cell cultures and reagents
The human SCLC cell lines SBC‐3 and SBC‐5, were kindly provided by Dr. K. Kiura (Okayama University, Okayama, Japan). DMS237 and DMS273‐G3H were obtained as reported previously.21 Briefly, DMS273‐G3H was established using tumor cells from a bone metastasis after we had implanted DMS273 orthotopically into the left lung of nude mice. H196 and H1048 were purchased from ATCC (Manassas, VA, USA). DMS114 and DMS454 were purchased from the European Collection of Authenticated Cell Cultures (Porton Down, Hampshire, UK). All cells were maintained in RPMI‐1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (10 μg/mL) in a humidified, 5% CO2 incubator at 37°C. All cells were passaged for a total of less than 3 months before being replaced by frozen early‐passage stocks. Cells were regularly screened for mycoplasma using a MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA). Golvatinib was obtained from Selleck Chemicals (Houston, TX, USA), and crizotinib was obtained from Active Biochem (Kowloon, Hong Kong).
Antibodies and Western blot analysis
Protein aliquots (25 μg) were separated by SDS‐PAGE (Bio‐Rad, Hercules, CA, USA) and transferred to PVDF membranes (Bio‐Rad). The membranes were washed three times and then incubated with Blocking One solution (Nacalai Tesque, Kyoto, Japan) for 1 h at room temperature. The membranes were then incubated overnight at 4°C with primary antibodies against anti‐MET (25H2), anti‐phospho‐MET (pMET) (Tyr1234/1235), anti‐protein kinase B (AKT), anti‐phospho‐AKT (Ser473), anti‐cleaved poly(ADP‐ribose) polymerase (PARP) (Asp214), anti‐cleaved caspase‐3 (Asp175) (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti‐human HGF (200 μg/mL), anti‐human/mouse/rat ERK (Erk1/Erk2; 0.2 μg/mL), anti‐phospho‐Erk1/Erk2 (T202/Y204; 0.1 μg/mL), GAPDH antibodies (1:1000; R&D Systems, Minneapolis, MN, USA), anti‐cyclin A (H432), anti‐cyclin B1 (GNS1), anti‐cyclin D1 (A12), or anti‐cyclin E (HE12) antibodies (1:200; Santa Cruz Biotechnology, Dallas, Texas, USA). The membranes were then washed three times and incubated for 1 h at room temperature with species‐specific HRP‐conjugated secondary antibodies. Immunoreactive bands were visualized with SuperSignal West Dura Extended Duration Substrate (an enhanced chemiluminescent substrate) (Pierce Biotechnology, Rockford, IL, USA). Each experiment was carried out independently at least three times.
Cell viability assay
Cell viability was measured using the MTT dye reduction method.22 Tumor cells (1–3 × 103 cells/100 μL/well) in RPMI‐1640 medium with 10% FBS, were plated onto 96‐well plates and cultured with the indicated compound for 72 h. Afterwards, 50 μg MTT solution (2 mg/mL; Sigma‐Aldrich, St. Louis, MO, USA) was added to each well. Plates were incubated for 2 h, the medium was removed, and the dark blue crystals in each well were dissolved in 100 μL DMSO. Absorbance was measured with a microplate reader at a test wavelength of 550 nm and a reference wavelength of 630 nm. Percentage growth was determined relative to untreated controls.
Knockdown of siRNA
Duplexed Stealth RNAi against MET (RSS351362; Invitrogen, Carlsbad, CA, USA) and HGF (HSS179213; Invitrogen), and Silencer Select siRNA for Negative Control no. 1 (Invitrogen), were transfected with Lipofectamine RNAiMAX (Invitrogen) in accordance with the manufacturer's instructions.
Cytokine production
Cells (2 × 105) were cultured in RPMI‐1640 medium with 10% FBS for 24 h, then washed with PBS and incubated for 48 h in 2 mL of the same medium. The culture medium was then harvested and centrifuged, and the level of HGF determined using a Quantikine ELISA kit (R&D Systems) for targeted cytokines in accordance with the manufacturer's protocols. All samples were run in triplicate. Color intensity was measured at 450 nm using a spectrophotometric plate reader. Concentrations of growth factors were determined in comparison to standard curves.
Fluorescent in situ hybridization
The MET 7q31.2 chromosomal locus was labeled with LSI MET Spectrum Red Probe (Abbott, Abbott Park, IL, USA). Centromere 7, labeled with Spectrum Green Probe (CEP7 (D7Z1), Abbott), was paired to a control for copy number. Then FISH was carried out using standard methods.23 Only nuclei with unambiguous CEP7 signals were scored for MET signal number.
Cell apoptosis
Cells (3 × 103) were seeded into each well of a 96‐well, white‐walled plate, incubated overnight, and treated with the indicated compounds or vehicle (DMSO) for 8 h. Cellular apoptosis was analyzed by measuring caspase‐3/7 activity with a Caspase‐Glo 3/7 assay kit (Promega, Madison, WI, USA) in accordance with the manufacturers’ protocols. Apoptotic cells in tumor xenografts were detected by TUNEL staining, using the In Situ Cell Death Detection Kit, POD (Sigma‐Aldrich) in accordance with the manufacturer's protocols.
Tumor cell inoculation in mice with SCID
Five‐week‐old male SCID mice were obtained from Clea Japan (Tokyo, Japan). Mice were pretreated with anti‐mouse interleukin‐2b antibody to deplete NK cells and thereby facilitate the formation of metastatic sites.24 Two days later, 1.2 × 106 SBC‐5/EGFP‐Eluc cells were introduced into mice by tail vein injection. After 10 days, mice were randomized (n = 6 per group) and drugs were given once daily by oral gavage. Tumor luminescence and body weight were measured twice weekly. Mice were monitored daily for general condition.
This study was carried out in strict accordance with the recommendations stipulated in the Guide for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science, and Technology, Japan. The utilized protocol was approved by the Ethics Committee on the Use of Laboratory Animals and the Advanced Science Research Center, Kanazawa University, Kanazawa, Japan (approval no. AP‐153499). Surgery was carried out once animals were anesthetized with sodium pentobarbital, and all efforts were made to minimize animal suffering.
Luciferase expression and radiographic analyses with an IVIS imaging system
After inoculation, the development of tumors was tracked in live mice by repeated non‐invasive optical imaging of tumor‐specific luciferase activity using the IVIS Lumina XR Imaging System (PerkinElmer, Alameda, CA, USA) as described.20 The intensity of the bioluminescence signal was analyzed using Living Image 4.0 software (PerkinElmer) by serially quantifying the peak photon flux in a selected region of interest within a given tumor. The intensity of the bioluminescence signal was corrected to take into account the total area of the region of interest, and the elapsed time for which bioluminescence signals were read by the charge coupled device camera. The corrected value was then expressed as photons/s.
Histological analyses of tumors
Formalin‐fixed, paraffin‐embedded tissue sections (4‐μm thick) were deparaffinized. Proliferating cells were detected by incubating tissue sections with Ki‐67 antibody (clone MIB‐1; Dako, Glostrup, Denmark). Antigens were retrieved by microwaving tissue sections in 10 mM citrate buffer (pH 6.0). After incubation with a secondary antibody and treatment with the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA), peroxidase activity was visualized by a DAB reaction. The sections were counterstained with hematoxylin.
Quantification of immunohistochemistry results
The five areas containing the highest numbers of stained cells within each section were selected for histological quantification by light or fluorescence microscopy at 400‐fold magnification.
Statistical analysis
Differences between groups were analyzed using one‐way anova. All statistical analyses were undertaken using GraphPad StatMate 4 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
Expression of HGF and MET in SCLC cell lines
To evaluate the effects of HGF/MET activation in SCLC, we first measured the expression of HGF with Western blot analysis, and the levels of both total MET and pMET proteins, in eight SCLC cell lines (Fig. 1a). Both HGF and pMET proteins were expressed in SBC‐5, DMS273, and DMS273‐G3H cells at varying levels, whereas DMS114 cells did not express either pMET or total MET proteins, despite high levels of HGF expression. To confirm HGF production, we measured the levels of HGF protein in the cell culture supernatant by ELISA assay. Hepatocyte growth factor was highly expressed in SBC‐5, DMS114, DMS273, and DMS273‐G3H cells, consistent with the results of the Western blot analysis (Fig. 1b). These results indicate that HGF/MET signaling is activated in some SCLC cell lines.
Figure 1.
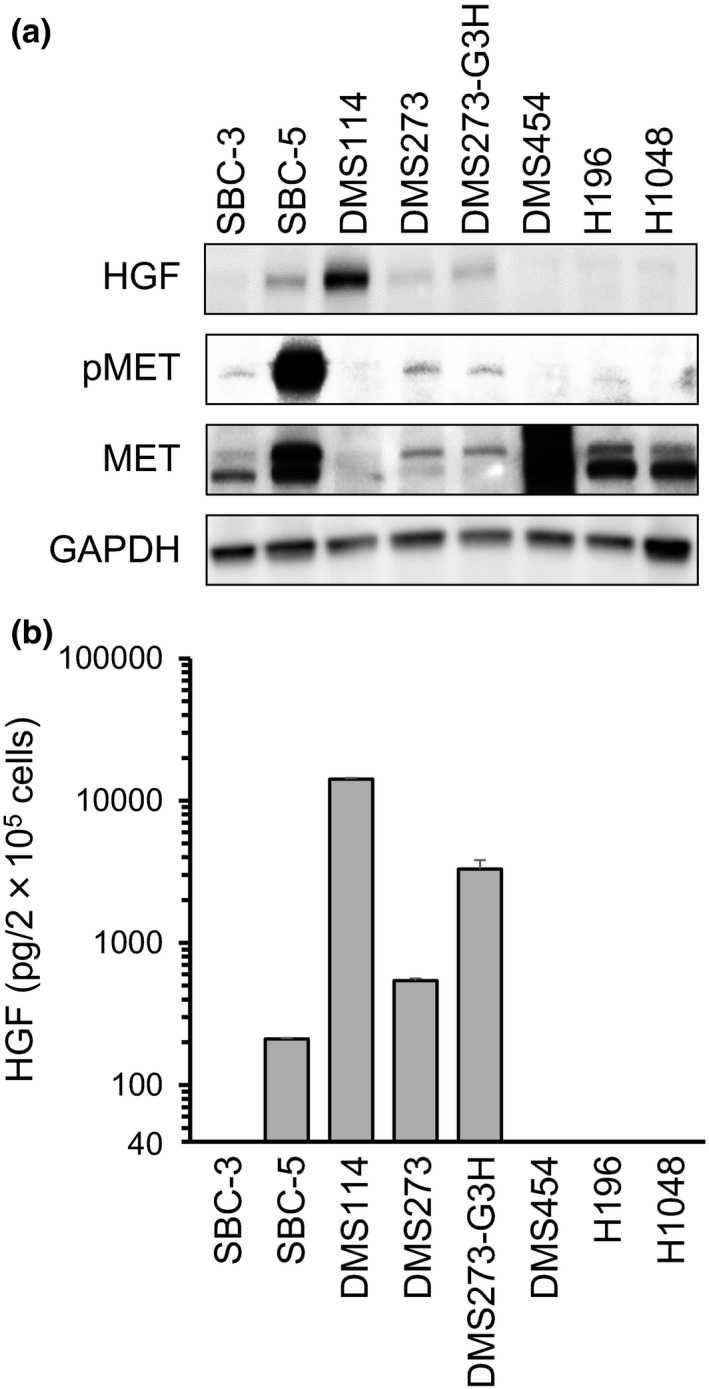
Expression of hepatocyte growth factor (HGF), phosphorylated MET (pMET), and total MET protein in small‐cell lung cancer cells. (a) Protein expression in cell lysates was evaluated by Western blot analysis. Three independent experiments were carried out, and a representative result is shown. (b) HGF production in small‐cell lung cancer cells. Cells were incubated in RPMI‐1640 medium for 48 h before culture supernatants were harvested. Levels of ligand expression in the collected supernatants was determined by ELISA (sensitivity ˃40 pg/mL). Error bars indicate SD of triplicate cultures.
Growth and viability of SCLC cells effectively suppressed by MET inhibitors
To examine the effect of MET inhibitors on the viability of SCLC cells, we used crizotinib and golvatinib. Treatment with either crizotinib or golvatinib alone significantly inhibited both the viability and growth of SBC‐5, DMS273, and DMS273‐G3H cells, each of which was identified to show high levels of both HGF and pMET expression (Fig. 2). The IC50 values of crizotinib or golvatinib in these SCLC cell lines were as follows. SBC‐5: crizotinib, 0.112 μM; golvatinib, 0.094 μM. DMS273: crizotinib, 0.208 μM; golvatinib, 0.613 μM. DMS273‐G3H: crizotinib, 0.432 μM; golvatinib, 0.535 μM. In contrast, the MET inhibitors had only a minimal effect on the other SCLC cell lines (Fig. S1). In addition, exogenous HGF did not affect the cell viability of these cells without HGF expression, although Western blot analyses revealed that the phosphorylation of MET, AKT, and ERK was increased in the presence of HGF (Fig. S2). To investigate the downstream effects of the inhibitors, we examined the phosphorylation statuses of MET, AKT, and ERK proteins using Western blot analysis. In the presence of either crizotinib or golvatinib, the phosphorylation of MET, AKT, and ERK was markedly inhibited in SCLC cells (Fig. 2c). Together, these results clearly show that the MET inhibitors crizotinib and golvatinib were able to inhibit the viability and growth of SCLC cells showing high expression of both HGF and pMET by inhibition of both MAPK and phosphoinositide‐3 kinase signaling.
Figure 2.
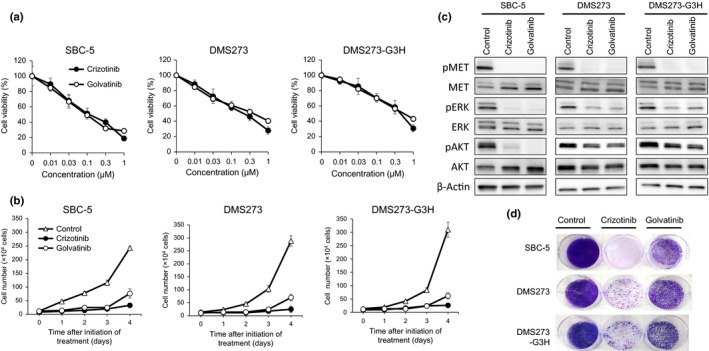
MET inhibitors effectively suppress the growth of small‐cell lung cancer cells showing aberrant activation of the hepatocyte growth factor (HGF)/MET pathway, through inhibition of ERK and protein kinase B (AKT) signals. (a) SBC‐5, DMS273, and DMS273‐G3H cells (1–2 × 103 per well) were incubated with various concentrations of either crizotinib or golvatinib for 72 h. Cell viability was determined by MTT assay. Data are shown as the mean ± SD of three independent experiments. (b) SBC‐5, DMS273, and DMS273‐G3H cells (1 × 104 per well) were incubated with 1 μM crizotinib or golvatinib, then harvested and counted daily. Data are shown as the mean ± SD of three independent experiments. (c) SBC‐5, DMS273, and DMS273‐G3H cells were treated with either 1 μM crizotinib or golvatinib for 2 h. Cell lysates were evaluated for protein expression by Western blot analysis. Three independent experiments were carried out, and a representative result is shown. p, phosphorylated. (d) SBC‐5, DMS273, and DMS273‐G3H cells (1 × 104 per well) were treated with 1 μM crizotinib or golvatinib every 72 h for 14 days. Cell plates were stained with crystal violet and imaged. Two independent experiments were carried out, and a representative plate is shown.
To confirm whether the viability of the SCLC cells was mediated by the HGF/MET pathway, we next knocked down MET and HGF using specific siRNAs. The viability of all three SCLC cell lines was inhibited by knockdown of either MET or HGF gene expression, and this effect was most pronounced in the SBC‐5 cell line (Fig. 3a). Western blot analysis revealed that the phosphorylation of AKT and ERK was inhibited by MET knockdown, whereas HGF knockdown inhibited ERK phosphorylation to a lesser degree than AKT phosphorylation (Fig. 3b). These results indicated that the viability of these SCLC cells was dependent on the HGF/MET pathway, and suggested that MET may be a promising molecular therapeutic target in SCLC cells that show aberrant activation of the HGF/MET pathway.
Figure 3.
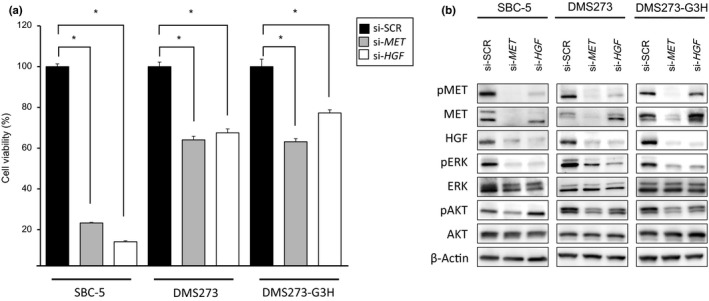
Viability of small‐cell lung cancer cells showing aberrant activation of hepatocyte growth factor (HGF)/MET is dependent on the HGF/MET pathway. SBC‐5, DMS273, and DMS273‐G3H cells were treated with siRNAs (si‐) specific to either MET or HGF, or scrambled controls (SCR). (a) Cell viability was determined by MTT assay 72 h after siRNA treatment. Data are shown as the mean ± SD of three independent experiments. *P < 0.05 by Student's t‐test, si‐SCR versus si‐MET or si‐HGF. (b) Cell lysates were evaluated for protein expression by Western blot analysis 48 h after siRNA treatment. Three independent experiments were carried out, and a representative result is shown. AKT, protein kinase B; p, phosphorylated.
Small‐cell lung cancer cells showing aberrant activation of the HGF/MET pathway show MET copy number gain
In general, MET activation is induced not only by HGF‐mediated stimulation, but also by altered MET gene expression, for example, as caused by activating mutations or gene amplification. Thus, to identify underlying mechanisms for the aberrant activation of the HGF/MET pathway, we next investigated possible sources of abnormal MET gene expression in SCLC cells. We found that the MET gene copy number was remarkably increased according to FISH analysis of SBC‐5, DMS273, and DMS273‐G3H cells; however, remarkable copy number gain were not detected in DMS114 cells, which had the highest secretion of HGF in the eight cell lines (Table 1). In contrast, neither MET amplification (Table 1) nor previously reported SCLC‐associated MET mutation (for example, E168D, R988C, T1010I, and R1166Q) (data not shown), was detected in the SCLC cell lines. These results suggest that the MET copy number gain, in addition to the secretion of HGF, may contribute to the aberrant activation of the HGF/MET pathway in SCLC cells.
Table 1.
MET gene copy number significantly increased in small‐cell lung cancer cell lines
| Cell line | MET : CEP7 ratio | Gene copy number | Positive cells (%) |
|---|---|---|---|
| SBC‐5 | 1.3 | <2 | 0 |
| 3–4 | 20 | ||
| >5 | 80 | ||
| DMS273 | 1.0 | <2 | 5 |
| 3–4 | 65 | ||
| >5 | 30 | ||
| DMS273‐G3H | 1.0 | <2 | 3 |
| 3–4 | 8 | ||
| >5 | 89 | ||
| DMS‐114 | 0.8 | <2 | 12 |
| 3–4 | 82 | ||
| >5 | 6 |
MET copy number determined by FISH.
MET inhibitors promote cell cycle arrest of SCLC cells showing aberrant activation of the HGF/MET pathway
To further assess the underlying mechanism by which crizotinib and golvatinib inhibit MET activity, we investigated the effect of these compounds on cell cycle and apoptosis. Western blot analysis revealed that the level of cyclin A, a known key modulator of the S–G2 cell cycle phase, was remarkably decreased in SCLC cells by treatment with either crizotinib or golvatinib (Fig. 4a). Cyclin A was also decreased by knockdown of either MET or HGF using specific siRNAs in SBC‐5 cells (Fig. S3). In contrast, MET inhibitor treatment did not affect the expression of cleaved PARP or cleaved caspase‐3, and furthermore did not inhibit caspase‐3/7 activity, all of which are associated with apoptosis (Fig. 4). These results suggest that crizotinib and golvatinib inhibit cell viability predominantly through cell cycle arrest in the S–G2 phase, but do not induce apoptosis.
Figure 4.
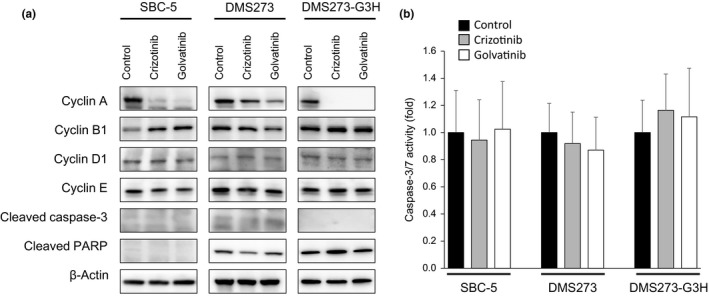
MET inhibitors induce cell cycle arrest, but not apoptosis, in small‐cell lung cancer cells. (a) SBC‐5, DMS273, and DMS273‐G3H cells were treated with 1 μM crizotinib or golvatinib for 72 h. Cell lysates were then evaluated for protein expression by Western blot analysis. Three independent experiments were carried out, and a representative result is shown. PARP, poly(ADP‐ribose) polymerase. (b) SBC‐5, DMS273, and DMS273‐G3H cells were treated with 1 μM crizotinib or golvatinib for 8 h, and the activity of caspase‐3/7 was then measured using a Caspase‐Glo3/7 assay kit. Error bars represent mean ± SD.
MET inhibitors significantly inhibited tumor progression of SBC‐5 cells in NK cell‐depleted SCID mice
We next examined the efficacy of the MET inhibitors crizotinib and golvatinib on tumor progression using our previously established in vivo imaging model with SBC‐5 EGFP‐Eluc‐transfected (SBC‐5/EGFP‐Eluc) cells.20 Bioluminescent signals were detected in the SBC‐5/EGFP‐Eluc cells, and observed to increase in a time‐dependent manner, consistent with the time‐course of metastasis formation.20 Having confirmed that EGFP‐Eluc expression had no effect on the sensitivity of the SBC‐5 cells to either crizotinib or golvatinib in an in vitro assay (Fig. S4), the SBC‐5/EGFP‐Eluc cells were injected i.v. into NK‐cell depleted SCID mice. We initiated treatment with 100 mg/kg crizotinib or 50 mg/kg golvatinib prior to detection of bioluminescent signals of systemic metastasis. The results of our analysis show that the expected increase in systemic metastasis was significantly inhibited by daily oral treatment with 100 mg/kg crizotinib or 50 mg/kg golvatinib compared to the control group (Fig. 5a,b). Furthermore, the phosphorylation of MET, ERK, and AKT, as well as the expression of cyclin A, was inhibited by treatment with either crizotinib or golvatinib, consistent with the results of our in vitro analyses (Fig. 5c). None of the groups of mice showed significant weight loss (Fig. S5a). We next assayed cell proliferation and apoptosis by immunohistochemical staining of liver tumors. Crizotinib and golvatinib treatment significantly reduced the number of proliferating cells in liver tumors as compared to controls (Fig. 5d,e); in contrast, the expression of cleaved PARP and cleaved caspase 3 were not altered by MET inhibitor treatment (Fig. S5b,c). These results show that, in the mouse model used here, the MET inhibitors crizotinib and golvatinib were able to constrain the progression of systemic metastasis by SCLC cells by cell cycle arrest, but not apoptosis.
Figure 5.
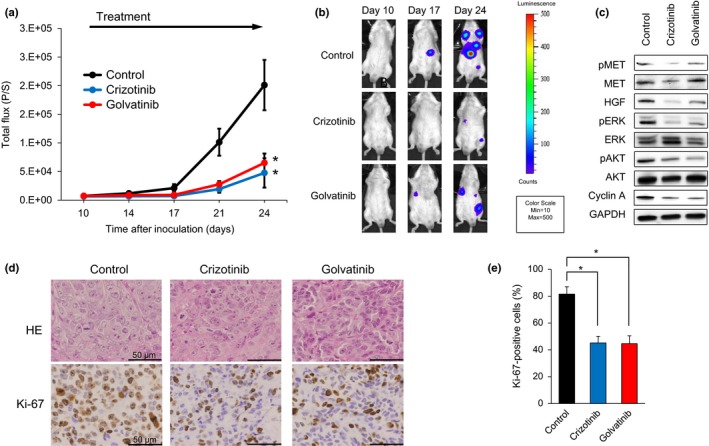
MET inhibitors significantly inhibit tumor progression of SBC‐5 small‐cell lung cancer cells in an in vivo multi‐organ metastasis mouse model. (a) EGFP‐Eluc‐transfected SBC‐5 (SBC‐5/EGFP‐Eluc) cells were injected i.v. into natural killer cell‐depleted SCID mice. At 10 days after inoculation, the mice were randomized into vehicle (control), crizotinib (100 mg/kg), or golvatinib (50 mg/kg) treatment groups (n = 6 per group), and began treatment once daily by oral gavage. Luminescence was evaluated twice weekly. Bars indicate standard error. *P < 0.05 by Mann–Whitney test, versus control group. (b) Representative images of mice showing merged bioluminescence and photograph. (c) Liver tumors were resected from mice 3–4 h after treatment on day 35. Relative levels of proteins observed in each tumor were determined by Western blot analysis. AKT, protein kinase B; HGF, hepatocyte growth factor; p, phosphorylated. (d) Representative images of liver tumors immunohistochemically stained with antibodies to human Ki‐67. Bar = 50 μm. (e) Quantification of proliferating cells in liver tumors, determined as the observed percentage of Ki‐67‐positive cells. The data shown represent the mean of five areas ± SD. *P < 0.05 by Student's t‐test, versus control group.
Discussion
In the present study, we showed that the aberrant activation of the HGF/MET pathway mediates the growth and survival of SCLC cells showing HGF secretion and/or MET copy number gain. Furthermore, the MET inhibitors crizotinib and golvatinib were able to inhibit cell growth and prevent systemic metastasis of SCLC cells in vivo, predominantly by cell cycle arrest. These findings indicate that inhibition of HGF/MET signaling may constrain tumor progression of SCLC cells showing aberrant activation of the HGF/MET pathway.
It is well known that aberrant activation of the HGF/MET pathway promotes cancer progression, invasion, and metastasis. We and other researchers have reported that the alternative bypass signaling by HGF/MET (e.g., by HGF overexpression22 or MET amplification25) induces resistance to both epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitors (TKIs) in EGFR‐mutated NSCLC cells, and anaplastic lymphoma kinase (ALK)‐TKIs in ALK‐rearranged NSCLC cells.22, 26, 27 In addition, we recently reported that MET copy number gain induced EGFR‐TKI gefitinib resistance in leptomeningeal carcinomatosis of EGFR‐mutant lung cancer cells.28 Thus, HGF/MET pathway activation in lung cancer is associated not only with malignant transformation, but also with drug resistance that enables cell survival. Furthermore, it has been shown that HGF/MET is the pivotal pathway disrupted in the pathophysiology of SCLC tumors. Ozasa et al. reported that the MET activation caused by an increase of MET gene loci promotes resistance to cytotoxic anticancer drugs in SCLC cells.29 Sakamoto et al. showed that treatment with MET inhibitors constrained the formation of distant metastases in SCLC cells showing high HGF production in an orthotopic model.21 Maulik et al. reported that HGF stimulation resulted in phosphorylation of multiple cytoskeletal proteins such as p125FAK or PYK2 in H69 SCLC cells.30 Wang et al. showed that siRNA could inhibit c‐MET expression reducing both SCLC cell growth and invasion in vitro and in vivo.31 We have shown here that the survival of the SBC‐5, DMS273, and DMS273‐G3H SCLC cell lines, identified to show aberrant activation of the HGF/MET pathway, was dependent on HGF/MET signaling. Each of these cell lines was found to overexpress HGF as a result of autocrine loops, and to show activation of pMET by MET gene copy number gain. Notably, each of these SCLC cell lines were derived from tumors of chemoresistant or chemorelapsed SCLC patients. This suggests that the therapeutic targeting of HGF/MET signaling may be effective in treating SCLC even in patients showing resistance to conventional cytotoxic anticancer drugs.
To determine the best subsequent treatment option, lung cancer patients who show resistance to chemotherapy or targeted therapy often undergo rebiopsy. This procedure may provide an opportunity to identify underlying sources of drug resistance, such as histological or genetic changes.32, 33 For instance, EGFR‐T790M mutations are often observed in tumors from EGFR‐mutation NSCLC patients that are rebiopsied after acquiring resistance to first‐generation EGFR‐TKIs.34 For resistant or relapsed SCLC patients, it may be beneficial to diagnose recurrent tumors by rebiopsy, to ascertain whether they show aberrant activation of the HGF/MET pathway. Identification of the genetic mechanisms underlying acquired drug resistance may lead to early intervention treatment with MET inhibitors, potentially prolonging the duration before tumor recurrence. Thus, our findings suggest that it may be beneficial, even in SCLC patients who are resistant to cytotoxic chemotherapy, to re‐evaluate the overexpression of HGF and/or the phosphorylation of MET proteins, so as to identify those patients that are likely to respond positively to treatment with MET inhibitors.
A major impediment to this process is the difficulty of identifying tumors that show aberrant activation of the HGF/MET pathway within the clinical setting. It may be critical to detect not only increased levels of MET phosphorylation, but also to identify MET copy number gain, MET gene activating mutations, and MET gene amplifications. In addition, having identified these genetic changes, the most effective therapeutic strategy for each patient needs to be determined, for example, designing combinations of chemotherapy with MET inhibitor, sequential treatment regimes, maintenance or therapy. In this study, crizotinib did not show antitumor effects after systemic metastasis had already spread (Fig. S5d). Other factors might affect tumor progression in the large number of tumor cells with systemic metastasis. According to our results, early treatment with MET inhibitors, while the number of tumor cells is still low, could suppress systemic metastasis. Continued research in this area is essential to improve both the genetic diagnosis and design of tailored therapy regimes for SCLC patients.
In the present study, we showed that treatment with MET inhibitors suppressed the spread of systemic metastasis induced by SCLC cells in NK cell‐depleted SCID mice, predominantly through cell cycle arrest, but not apoptosis. Interestingly, we also showed that siRNA‐induced knockdown of MET reduced the expression of HGF proteins, suggesting that HGF‐induced autocrine systems may be dependent on MET activation. However, we could not clarify the exact mechanisms. Elucidation of the mechanisms underlying the reduction in HGF protein expression induced by MET knockdown will require further investigation.
Crizotinib is an ATP‐competitive, small‐molecule inhibitor of MET kinase that is already approved for the treatment of patients with ALK fusion‐positive lung cancer in several countries including Japan, and for NSCLC patients with ALK fusion or ROS1 fusions in the USA.35, 36 Golvatinib is a dual MET and vascular endothelial growth factor receptor‐2 TKI under clinical development.37, 38 It is clinically valuable to assess the impacts of these compounds on the inhibition of systemic metastatic tumors in mouse xenograft models, as has been shown here.
In summary, we showed in the present study that HGF/MET signaling was aberrantly activated in chemoresistant or chemorelapsed SCLC cell lines (SBC‐5, DMS273, and DMS273‐G3H) by the secretion of HGF and/or MET copy number gain. The MET inhibitors exerted antitumor effects on these SCLC cell lines in both cell‐based in vitro assays and in vivo xenograft models. These findings indicate the therapeutic potential of inhibiting HGF/MET signaling to constrain tumor progression in SCLC cells showing aberrant activation of the HGF/MET pathway. Therefore, we suggest that it would be clinically valuable to further investigate HGF/MET‐mediated signaling in SCLC cells.
Disclosure Statement
Seiji Yano obtained research grants from Chugai Pharmaceutical Co. Ltd. The other authors have no conflict of interest.
Supporting information
Fig. S1. MET inhibitors did not suppress the growth of small‐cell lung cancer SBC‐3, DMS114, DMS454, H1048, or H196 cells.
Fig. S2. Effect of exogenous hepatocyte growth factor (HGF) in small‐cell lung cancer cells without HGF expression.
Fig. S3. Inhibition of MET or hepatocyte growth factor (HGF) induced cell cycle arrest.
Fig. S4. EGFP‐Eluc expression did not affect the sensitivity of SBC‐5 cells to MET inhibitors.
Fig. S5. Analysis of additional data in vivo systemic metastasis mouse model.
Acknowledgments
The authors thank Drs. Katsuyuki Kiura and Mitsune Tanimoto (Okayama University) for kindly providing SBC‐3 and SBC‐5 cells, and Dr. Masayuki Miyasaka (Osaka University) for providing the TM‐β1 hybridoma. This work was supported by the Japan Society from the Promotion of Science (KAKENHI Grant No. JP16H05308, to S.Y.).
Cancer Sci 108 (2017) 1378–1385
Funding Information
Japan Society for the Promotion of Science, (Grant/Award Number: ‘JP16H05308‘)
References
- 1. Bethesda M. SEER Cancer Stat Facts: Lung and Bronchus Cancer. National Cancer Institute. http://seer.cancer.gov/statfacts/html/lungb.html.
- 2. van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small‐cell lung cancer. Lancet 2011; 378(9804): 1741–55. [DOI] [PubMed] [Google Scholar]
- 3. Noda K, Nishiwaki Y, Kawahara M et al Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small‐cell lung cancer. N Engl J Med 2002; 346(2): 85–91. [DOI] [PubMed] [Google Scholar]
- 4. Amarasena IU, Walters JA, Wood‐Baker R, Fong K. Platinum versus non‐platinum chemotherapy regimens for small cell lung cancer. Cochrane Database Syst Rev 2015; (8): CD006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roth BJ, Johnson DH, Einhorn LH et al Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small‐cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992; 10(2): 282–91. [DOI] [PubMed] [Google Scholar]
- 6. Hann CL, Rudin CM. Fast, hungry and unstable: finding the Achilles’ heel of small‐cell lung cancer. Trends Mol Med 2007; 13(4): 150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arcaro A. Targeted therapies for small cell lung cancer: Where do we stand? Crit Rev Oncol Hematol 2015; 95(2): 154–64. [DOI] [PubMed] [Google Scholar]
- 8. Bottaro DP, Rubin JS, Faletto DL et al Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 1991; 251(4995): 802–4. [DOI] [PubMed] [Google Scholar]
- 9. Naldini L, Vigna E, Narsimhan RP et al Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto‐oncogene c‐MET. Oncogene 1991; 6(4): 501–4. [PubMed] [Google Scholar]
- 10. Boccaccio C, Comoglio PM. Invasive growth: a MET‐driven genetic programme for cancer and stem cells. Nat Rev Cancer 2006; 6(8): 637–45. [DOI] [PubMed] [Google Scholar]
- 11. Danilkovitch‐Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Investig 2002; 109(7): 863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol 2009; 4(1): 5–11. [DOI] [PubMed] [Google Scholar]
- 13. Ma PC, Kijima T, Maulik G et al c‐MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Can Res 2003; 63(19): 6272–81. [PubMed] [Google Scholar]
- 14. Voortman J, Harada T, Chang RP et al Detection and therapeutic implications of c‐Met mutations in small cell lung cancer and neuroendocrine tumors. Curr Pharm Des 2013; 19(5): 833–40. [PMC free article] [PubMed] [Google Scholar]
- 15. Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signalling and specific inhibition of c‐MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer 2007; 97(3): 368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arriola E, Canadas I, Arumi‐Uria M et al MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF‐induced effects in MET mutant cell lines. Br J Cancer 2011; 105(6): 814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Canadas I, Rojo F, Taus A et al Targeting epithelial‐to‐mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res 2014; 20(4): 938–50. [DOI] [PubMed] [Google Scholar]
- 18. Yano S, Sone S. Novel metastasis model of human lung cancer cells representing different histological types in SCID mice depleted of NK cells. Gan To Kagaku Ryoho 1997; 24(4): 489–94. [PubMed] [Google Scholar]
- 19. Yano S, Muguruma H, Matsumori Y et al Antitumor vascular strategy for controlling experimental metastatic spread of human small‐cell lung cancer cells with ZD6474 in natural killer cell‐depleted severe combined immunodeficient mice. Clin Cancer Res 2005; 11(24 Pt 1): 8789–98. [DOI] [PubMed] [Google Scholar]
- 20. Takeuchi S, Fukuda K, Arai S et al Organ‐specific efficacy of HSP90 inhibitor in multiple‐organ metastasis model of chemorefractory small cell lung cancer. Int J Cancer 2016; 138(5): 1281–9. [DOI] [PubMed] [Google Scholar]
- 21. Sakamoto S, Inoue H, Ohba S et al New metastatic model of human small‐cell lung cancer by orthotopic transplantation in mice. Cancer Sci 2015; 106(4): 367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Can Res 2008; 68(22): 9479–87. [DOI] [PubMed] [Google Scholar]
- 23. Suda K, Murakami I, Katayama T et al Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res 2010; 16(22): 5489–98. [DOI] [PubMed] [Google Scholar]
- 24. Yano S, Nishioka Y, Izumi K et al Novel metastasis model of human lung cancer in SCID mice depleted of NK cells. Int J Cancer 1996; 67(2): 211–7. [DOI] [PubMed] [Google Scholar]
- 25. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316(5827): 1039–43. [DOI] [PubMed] [Google Scholar]
- 26. Tanimoto A, Yamada T, Nanjo S et al Receptor ligand‐triggered resistance to alectinib and its circumvention by Hsp90 inhibition in EML4‐ALK lung cancer cells. Oncotarget. 2014; 5(13): 4920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang W, Li Q, Yamada T et al Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res 2009; 15(21): 6630–8. [DOI] [PubMed] [Google Scholar]
- 28. Nanjo S, Arai S, Wang W et al MET copy number gain is associated with gefitinib resistance in leptomeningeal carcinomatosis of EGFR‐mutant lung cancer. Mol Cancer Ther 2017; 16(3): 506–15. [DOI] [PubMed] [Google Scholar]
- 29. Ozasa H, Oguri T, Maeno K et al Significance of c‐MET overexpression in cytotoxic anticancer drug‐resistant small‐cell lung cancer cells. Cancer Sci 2014; 105(8): 1032–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maulik G, Kijima T, Ma PC et al Modulation of the c‐Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res 2002; 8(2): 620–7. [PubMed] [Google Scholar]
- 31. Wang ZX, Lu BB, Yang JS, Wang KM, De W. Adenovirus‐mediated siRNA targeting c‐Met inhibits proliferation and invasion of small‐cell lung cancer (SCLC) cells. J Surg Res 2011; 171(1): 127–35. [DOI] [PubMed] [Google Scholar]
- 32. Suda K, Murakami I, Sakai K et al Small cell lung cancer transformation and T790M mutation: complimentary roles in acquired resistance to kinase inhibitors in lung cancer. Sci Rep 2015; 5: 14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nosaki K, Satouchi M, Kurata T et al Re‐biopsy status among non‐small cell lung cancer patients in Japan: a retrospective study. Lung Cancer 2016; 101: 1–8. [DOI] [PubMed] [Google Scholar]
- 34. Janne PA, Yang JC, Kim DW et al AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N Engl J Med 2015; 372(18): 1689–99. [DOI] [PubMed] [Google Scholar]
- 35. Zou HY, Li Q, Lee JH et al An orally available small‐molecule inhibitor of c‐Met, PF‐2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Can Res 2007; 67(9): 4408–17. [DOI] [PubMed] [Google Scholar]
- 36. Solomon BJ, Mok T, Kim DW et al First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med 2014; 371(23): 2167–77. [DOI] [PubMed] [Google Scholar]
- 37. Nakagawa T, Tohyama O, Yamaguchi A et al E7050: a dual c‐Met and VEGFR‐2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci 2010; 101(1): 210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang W, Li Q, Takeuchi S et al Met kinase inhibitor E7050 reverses three different mechanisms of hepatocyte growth factor‐induced tyrosine kinase inhibitor resistance in EGFR mutant lung cancer. Clin Cancer Res 2012; 18(6): 1663–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. MET inhibitors did not suppress the growth of small‐cell lung cancer SBC‐3, DMS114, DMS454, H1048, or H196 cells.
Fig. S2. Effect of exogenous hepatocyte growth factor (HGF) in small‐cell lung cancer cells without HGF expression.
Fig. S3. Inhibition of MET or hepatocyte growth factor (HGF) induced cell cycle arrest.
Fig. S4. EGFP‐Eluc expression did not affect the sensitivity of SBC‐5 cells to MET inhibitors.
Fig. S5. Analysis of additional data in vivo systemic metastasis mouse model.