

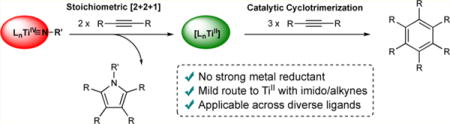
Abstract
Low-valent TiII species have typically been synthesized by the reaction of TiIV halides with strong metal reductants. Herein we report that TiII species can be generated simply by reacting TiIV imido complexes with 2 equiv of alkyne, yielding a metallacycle that can reductively eliminate pyrrole while liberating TiII. In order to probe the generality of this process, TiII-catalyzed alkyne trimerization reactions were carried out with a diverse range of TiIV precatalysts.
Graphical abstract
INTRODUCTION
Low-valent Ti reagents have played an important role in many molecular transformations over the past 50 years.1 In particular, TiII intermediates have been invoked in a rich and varied range of stoichiometric and catalytic reactions: N2 fixation,2 McMurry coupling,1b,3 alkyne cyclotrimerization,1c,4 Pauson–Khand cycloaddition,5 cyclopropanation,6 and oxidative addition reactions with many other electrophiles.7 Formally, TiII coordination complexes are fairly uncommon because of the extremely high thermodynamic stability of the TiIV oxidation state. Nevertheless, discrete TiII complexes have been isolated and characterized across diverse ligand sets: cyclopentadienyl,2c,8 calixarene,4,9 isocyanide,10 porphyrin,7a,11 pyridine,7b and 1,2-bis(dimethylphosphino)ethane.12 Similarly, “masked” TiII complexes have also been reported in which the low-valent state is stabilized by strong π-acceptors such as alkyne,13 alkene,14a–c and carbonyl.14d
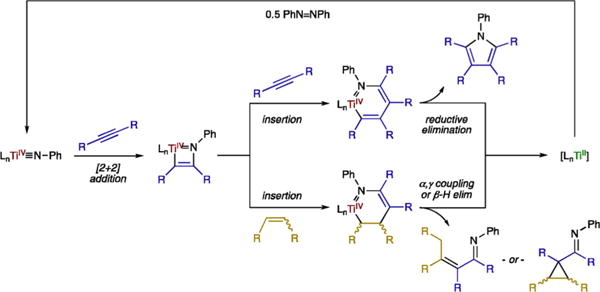
Given that they are highly reducing, TiII reagents have typically been formed from the reaction of TiIV halide precursors with powerful reductants: KC8,2,7b LiAlH4,11 Na/Hg,15 and Mg.16 Recently, we have reported several catalytic oxidative C–N bond forming reactions that proceed through a formal TiII/TiIV redox couple (Scheme 1). These reactions presumably generate a TiII intermediate in the absence of a strong metal reductant by coupling a TiIV imido unit with two alkynes (or an alkyne and an alkene).17
Scheme 1. Previously Reported TiII/TiIV Redox Catalytic Reactions.
Intrigued by this unusual and mild route to form TiII species in situ, we have set out to demonstrate the generality of forming TiII species from the reaction of alkynes with TiIV imido complexes. These transient TiII complexes were then examined for their competency as catalysts for alkyne trimerization. Herein we report that many diverse Ti imido precatalyst structures are capable of generating TiII intermediates and subsequently trimerizing alkynes. By comparing structurally similar catalyst classes, we have drawn several qualitative conclusions and empirical trends.
RESULTS AND DISCUSSION
We began our investigation by synthesizing a diverse set of Ti imido complexes based mostly on ligand architectures that are well-established in titanium-catalyzed hydroamination and polymerization reactions (Figure 1). These ligands can be divided into several categories: first, complexes varying in simple monoanionic X-type ligands: halides (1–3),18 pyrrolides (4 and 5),19 and aryloxides (6 and 7);20 second, polyhapto ligands: 1,4-bis(trimethylsilyl)cyclooctatetraene (8),21 cyclo-pentadienyl Cp (9 and 10),22 and pentamethylcyclopentadienyl Cp* (11);22 third, LX-type bidentate ligands: amidate (12),23 phenoxyimino (13),24 and amidinate (14 and 15)25 that may be hemilabile; and last, a Zr analogue (16) included in the series to allow for reactivity comparison within the group 4 triad. In contrast to titanium, ZrII is considerably harder to access.26
Figure 1.
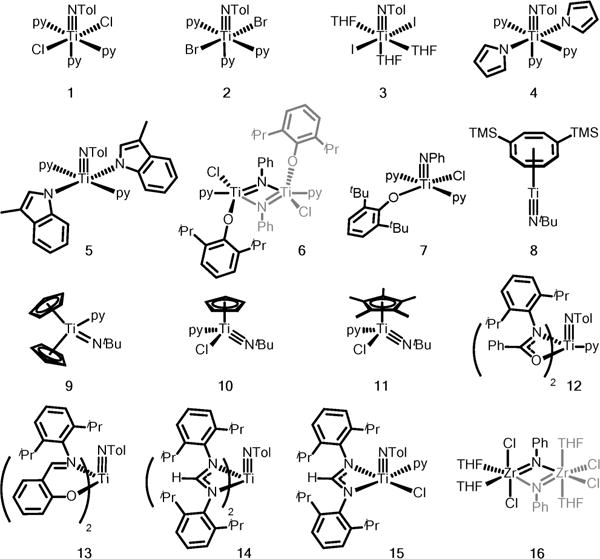
Ti and Zr imido precatalysts investigated for the generality of forming MII intermediate.
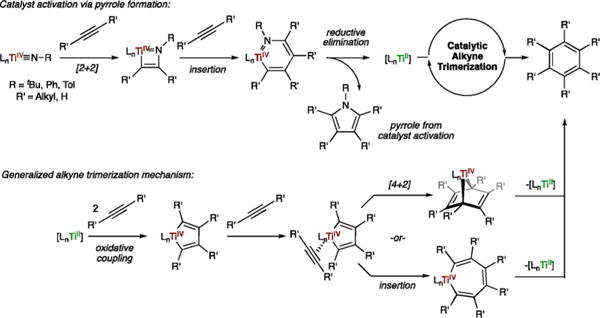
Based on our previous mechanism for TiII/TiIV redox catalysis, TiII species capable of alkyne trimerization can be generated by the coupling of 2 alkynes with a TiIV imido and subsequent elimination of one equivalent of pyrrole per Ti (Scheme 2, top).17 This activation process occurs through a [2 + 2] cycloaddition27 of the TiIV imido with an alkyne, followed by insertion of the second alkyne into the metallacycle28 and finally reductive elimination of pyrrole to yield the TiII trimerization catalyst.29 While there may be catalyst-to-catalyst variation in the mechanistic details of trimerization catalysis, TiII is generally understood30 to trimerize alkynes by first oxidatively coupling two alkynes to yield a metallacyclopentadiene that can then undergo [4 + 2] cycloaddition with a third alkyne to yield a titananorbornadiene intermediate, which is then displaced by alkyne to liberate the trimerized product (Scheme 2, bottom). Alternately, the metallacyclopentadiene could insert the third equivalent of alkyne to produce a metallacycloheptatriene, which upon reductive elimination yields the trimerized product. In general, the [4 + 2] mechanism appears to be more broadly invoked,4a although a universal mechanism for trimerization is unlikely given the diverse range of molecular structures capable of catalyzing this reaction.
Scheme 2. Catalytic Alkyne Trimerization with TiIV Imido Catalysts.
Catalytic alkyne trimerization reactions were carried out with 5 mol % of each Ti imido precatalyst and either 1-hexyne or 3-hexyne in C6D5Br at 115 °C for 16 h. Unsymmetrical 1-hexyne can yield two alkyne trimer regioisomers (1,3,5- and 1,2,4-tri-n-butylbenzene A and B, respectively) and three pyrrole regioisomers (2,4-, 2,5-, and 3,4-di-n-butylpyrrole, C–E, respectively) (Table 1). The expected statistical distribution between the two alkyne trimer products is 1:3 (A/B),30c while the distribution of pyrrole products is 2:1:1 (C/D/E). The 2,4-and 2,5-disubstituted pyrroles were the major products in the reactions reported herein, and no significant additional 1H NMR peaks that could plausibly be assigned as the 3,4-regioisomers were observed in any of the catalytic experiments. Where possible, the pyrrole regioisomers were independently synthesized via alternate routes to confirm their characterization, although some are inaccessible using modern synthetic techniques (See Table 1 and Supporting Information for details). 3-Hexyne can only yield hexaethylbenzene F and 2,3,4,5-tetraethylpyrrole G (Table 2).
Table 1.
1-Hexyne Trimerization Dataa
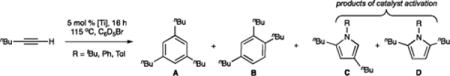
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| [Ti] | % trimer yield | A/B | % [Ti] act.b | C/D | % conversion | TONc |
| 1 | quant. | 27:73 | 37 ± 4 | 86:14 | 100 ± 0 | 18 ± 2 |
| 2 | 87 ± 1 | 35:65 | 6 ± 2 | 78:23 | 89 ± 3 | 94 ± 31 |
| 3d | 94 ± 4 | 39:61 | 5 ± 1 | 100:0 | 98 ± 2 | 132 ± 38 |
| 4 | 61 ± 5 | 24:76 | 32 ± 3 | 75:25 | 93 ± 4 | 12 ± 2 |
| 5 | 85 ± 2 | 39:61 | 20 ± 10 | 60:40 | 100 ± 0 | 28 ± 14 |
| 6 | quant. | 31:69 | 30 ± 4 | 86:14 | 100 ± 0 | 21 ± 4 |
| 7 | 41 ± 8 | 37:63 | 31 ± 8 | 65:35 | 64 ± 16 | 9 ± 4 |
| 8 | 62 ± 4 | 33:67 | 36 ± 9 | 100:0e | 100 ± 0 | 11 ± 4 |
| 9 | 53 ± 2 | 34:66 | 11 ± 1 | 100:0e | 91 ± 3 | 32 ± 4 |
| 10 | 71 ± 2 | 27:73 | 61 ± 3 | 100:0e | 100 ± 0 | 7 ± 0 |
| 11 | 50 ± 4 | 37:62 | 33 ± 4 | 100:0e | 75 ± 4 | 10 ± 1 |
| 12 | 34 ± 9 | 15:85 | 57 ± 19 | 37:63 | 64 ± 7 | 4 ± 2 |
| 13 | 40 ± 5 | 17:83 | 15 ± 2 | 69:31 | 55 ± 14 | 17 ± 3 |
| 14 | 18 ± 3 | 39:61 | 7 ± 1 | 31:69 | 56 ± 13 | 17 ± 4 |
| 15 | 65 ± 5 | 21:79 | 38 ± 1 | 65:35 | 76 ± 12 | 11 ± 1 |
| 16 | 24 ± 2 | 42:58 | 0 ± 0 | 53 ± 5 | ||
Conditions: 5 mol % [Ti], 0.4 M 1-hexyne, C6D5Br, 16 h, 115 °C, average of 2ȓ4 runs. Quantitation determined by in situ 1H NMR.
% Ti activated = (yield of C + D)/[Ti]tot.
TON = (yield of A + B)/Ti activated.
<5 min, room temperature |
D (R = NtBu) could not be independently synthesized/characterized, although raw spectra indicate only formation of C.
Table 2.
3-Hexyne Trimerization Dataa
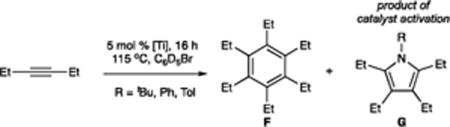
| ||||
|---|---|---|---|---|
|
| ||||
| [Ti] | % trimer yield | % [Ti] act.b | % conversion | TONc |
| 1 | 62 ± 13 | 80 ± 12 | 73 ± 17 | 4 ± 2 |
| 2 | 55 ± 21 | 57 ± 12 | 77 ± 12 | 5 ± 2 |
| 3d | 83 ± 6 | 17 ± 7 | 81 ± 3 | 35 ± 14 |
| 4 | 24 ± 8 | 33 ± 18 | 54 ± 16 | 5 ± 3 |
| 5 | 6 ± 5 | 42 ± 6 | 15 ± 0 | 1 ± 1 |
| 6 | 38 ± 2 | 48 ± 11 | 72 ± 18 | 5 ± 1 |
| 7 | 3 ± 3 | 70 ± 9 | 49 ± 11 | 0 ± 0 |
| 8 | 68 ± 7 | 74 ± 14 | 87 ± 2 | 6 ± 1 |
| 9 | 1 ± 0 | 8 ± 2 | 12 ± 0 | 1 ± 0 |
| 10 | 0 ± 0 | 1 ± 0 | 7 ± 2 | 0 ± 0 |
| 11 | 3 ± 0 | 41 ± 7 | 49 ± 8 | 1 ± 0 |
| 12 | 0 ± 0 | 7 ± 4 | 42 ± 13 | 0 ± 0 |
| 13 | 0 ± 0 | 0 ± 0 | 42 ± 11 | |
| 14 | 2 ± 2 | 6 ± 4 | 20 ± 15 | 2 ± 2 |
| 15 | 1 ± 0 | 25 ± 1 | 22 ± 6 | 0 ± 0 |
| 16 | 1 ± 0 | 0 ± 0 | 35 ± 4 | |
Conditions: 5 mol % [Ti], 0.4 M 3-hexyne, C6D5Br, 16 h, 115 °C, average of 2–4 runs. Quantitation determined by in situ 1H NMR.
% Ti activated = (yield of G)/[Ti]tot.
TON = (yield of F)/Ti activated.
Room temperature.
Given that the pyrrole byproduct formation is stoichiometric with respect to TiII formation, the amount of TiIV activated toward catalysis can be determined by quantifying the amount of pyrrole formed in a given reaction. Additionally, control experiments with structurally analogous TiIV halide precatalysts yield no trimerization (see Table S2), indicating that a Lewis acid mechanism for trimerization can be ruled out and that all productive catalysis most likely occurs through TiII. As a result, one can gain further insight into catalyst activity by calculating a “real” TON for each catalyst: the amount of trimer generated per activated Ti center. While this number may not truly reflect actual turnover given that catalyst dis-/comproportionation or ligand redistribution may occur, it is nonetheless instructive in qualitatively comparing catalyst systems.
All precatalysts examined were active for trimerization catalysis with 1-hexyne, albeit with starkly different degrees of activation and rates of catalysis (Table 1), demonstrating the generality of accessing a TiII intermediate from a TiIV imido unit. Most of the catalysts examined did not deviate significantly from the statistical distribution of alkyne trimers, although the ratio of pyrrole byproduct regioisomers varied widely. In general, precatalysts with poor to moderate trimerization yields were observed to have poor mass balances that may be a result of alkyne oligomerization,31 catalyst decomposition, or off-cycle/arrested alkyne-bound Ti complexes (metallacyclopropene, metallacyclopentadiene, and η6-arene)32 that were either incapable of or slower at catalytic turnover.
Results with catalysts bearing simple monoanionic ligands (1–3) indicate that electron-poor metal centers with weaker donor ligands (as measured by Odom via ligand donor parameterization) are better for 1-hexyne trimerization: I (3) > Br (2) > Cl (1).33 The apparent TON also increases as the metal center becomes more electron-poor. Most strikingly, even though only 5% of 3 was activated, it yielded 94% trimerization at room temperature in less than 5 min. In contrast, 2 slowly trimerized at room temperature, while 1 (and other catalysts) required high temperatures for productive catalysis to occur. The regioselectivity of both 1-hexyne trimerization and pyrrole formation trends toward sterically favored products A and C as the electrophilicity of the catalyst increases.
Catalysts 6 and 7 allow for a direct comparison of steric effects on activation and catalysis. While the amount of precatalyst activated is approximately the same for both, catalysis with more sterically encumbered 7 is incomplete under standard conditions, yielding significantly less trimerization product than that with 6. Intriguingly, although there are marginal differences in the regioselectivity of trimerization between both, catalyst 6 is significantly more selective than 7 for 2,4-disubstituted pyrrole activation product A. In fact, the overall activation and catalytic profile of 6 are very similar to those of 1. This similarity may indicate that some ligand redistribution/disproportionation occurs in these monoaryloxide complexes.
Varying the substituents on cyclopentadienyl-supported Ti catalysts 9–11 significantly affected both the amount of catalyst activated and the degree of alkyne trimerization. Cp2-substituted 9 exhibits the lowest amount of catalyst activation but has the highest apparent TON of all three Cp-substituted catalysts, indicating that the bulkier and more electron-rich Cp2 TiII active species may be more long-lived and/or more reactive than the monoligated analogues. More sterically encumbered Cp* derivative 11 has a lower degree of activation than Cp counterpart 10, although the apparent TONs for both are similar, indicating that in these systems the initial activation of the CpTi(≡NtBu) fragment by incoming alkynes is more sensitive to sterics than alkyne trimerization by a putative CpTiII species.
Catalysts 12–15, supported by bidentate potentially hemi-labile ligands, did not react to full conversion under standard catalytic conditions. Similar to Cp derivatives 9–11, bis-ligated bidentate ligands undergo lower catalyst activation but have higher apparent TONs, indicating that although mono ligation may aid in catalyst activation due to the steric sensitivity of [2+2+1] pyrrole formation it may also lead to less stable and/or less active trimerization catalysts. Unfortunately, there is no correlation between selectivity in pyrrole activation and 1-hexyne trimerization in any of these systems.
Remarkably, catalysts 12 and 14 show preference for the selective formation of 2,5-disubstituted pyrrole activation product D, which results from a Markovnikov [2 + 2] addition of 1-hexyne to the Ti≡NTol fragment followed by 2,1-insertion of 1-hexyne into the resulting azametallacycle. This regioselectivity is surprising given that hydroamination of terminal aliphatic alkynes with aniline23b,34 by catalyst 12 favors the opposite [2 + 2] products, in a ratio of approximately 1.6:1 anti-Markovnikov to Markovnikov. While these results are apparently contradictory, they are likely a result of different rate-determining steps of catalysis (for example, in Schafer’s hydroamination report, [2 + 2] addition is reversible and protonolysis by aniline is rate determining) or a function of a change in catalyst speciation. In hydroamination catalysis, there is a large excess of Lewis basic amine present that may coordinate to Ti throughout the catalytic cycle;34 however, in these trimerization experiments, no such strong Lewis base exists.
Given successful catalysis with a diverse range of Ti catalysts, we synthesized and tested a Zr imido analogue, 16. Interestingly, 16 trimerized 1-hexyne in poor yield in the absence of any detectable pyrrole activation byproduct. This result leads to one of two possible conclusions: (1) Trimerization by Zr occurred through a Lewis acid mechanism such as that reported by Floriani et al.35 for ZrCl4. (2) Small amounts of Zr≡NPh were activated in a quantity undetectable by 1H NMR and GC/MS, and catalysis occurred in a manner similar to Ti. While ZrCl4 is known to trimerize alkynes through the Lewis acid pathway, control experiments with ZrCl4 under our specific reaction conditions yielded no alkyne trimerization. (Arene coordination to ZrCl4, both from C6D5Br and 1,3,5-trimethoxybenzene, inhibits Lewis acid catalysis. See Table S2.) Thus, neither pathway can be fully ruled out.
With an internal alkyne such as 3-hexyne, alkyne trimerization becomes more challenging (Table 2). In all cases examined, the apparent TON and yield of hexaethylbenzene were lower than those in the 1-hexyne reactions. Interestingly, the amount of pyrrole formed via [2+2+1] of 3-hexyne was typically higher than the reactions with 1-hexyne. This is likely the result of the relative rates of activation versus trimerization: In most 1-hexyne reactions, trimerization rapidly depletes the amount of alkyne available for further catalyst activation, whereas with 3-hexyne, catalyst activation can effectively compete with slower alkyne trimerization.
Trends in catalyst activity similar to those observed in 1-hexyne reactions can also be observed in the 3-hexyne reactions. For example, the most electron-deficient simple halide 3 substantially outperforms other monoanionic analogues 1, 2, 4, and 5, despite a lower degree of catalyst activation. Increasing the steric profile of monoligated complexes also suppresses productive catalysis, as 6 gives moderate yields of hexaethylbenzene while the bulkier 7 only yields trace product. Disappointingly, bidentate ligands 9–15 yielded no productive catalysis despite some pyrrole production and instead led to poor mass balances.
CONCLUSIONS
In summary, we have demonstrated the generality of obtaining a reduced “TiII” intermediate by the coupling of a TiIV imido and 2 equiv of alkyne, generating a stoichiometric amount of pyrrole as a byproduct. Remarkably, a very diverse range of catalyst structures are reasonably efficient at generating TiII intermediates via [2+2+1]. The degrees of catalyst activation and catalyst activity are highly dependent on the structure of the Ti complex. In general, electron-poor Ti complexes, such as those derived from Ti(NTol)(THF)3I2 precatalyst 3, are far superior for alkyne trimerization compared to other electron-rich and/or multidentate ligands. Additionally, while most precatalysts predominantly generated 2,4-disusbstituted pyrroles on activation with 1-hexyne, hemilabile ligand scaffolds such as 12 and 14 demonstrated selectivity toward 2,5-disubstituted pyrroles.
More generally, these trimerization reactions illustrate an important design principle for early transition metal catalysis involving redox at the metal: stabilization of low-valent states. While there have been significant recent advances in the use of redox noninnocent ancillary ligands36 to modulate similar transformations, one may also consider that redox noninnocent reactants or products can play a similar role; in this case, π-backdonation from TiII into arenes and alkynes is certainly critical to accessing low-valent states and for catalysis. Similarly, π-backdonation into CO in Pauson–Khand reactions,5 alkenes in the Kulinkovich reaction,6 and azobenzene in [2+2+1] pyrrole syntheses17 is integral for productive reactivity and catalyst stability. This research will provide new potential access points to carry out various other stoichiometric and catalytic reactions with low-valent Ti under mild conditions and a future platform for new catalyst development toward selective pyrrole syntheses.
EXPERIMENTAL SECTION
General Considerations
All air- and moisture-sensitive reactions were carried out in a nitrogen-filled glovebox. Standard solvents for air-and moisture-sensitive reactions were either deoxygenated by sparging with N2 and dried by passing through activated alumina columns of a Pure Process Technology solvent purification system (benzene, ether, pentanes, hexanes, THF, or CH2Cl2) or vacuum-transferred from Na/Ph2CO (C6D6) or CaH2 (CDCl3). C6D5Br was synthesized following a literature procedure,37 degassed, dried over CaH2, and filtered through basic alumina prior to use. Commercial PhCF3 was vacuum transferred from CaH2 and filtered through basic alumina prior to use.
Ti(NtBu)Cl2py3,18 precatalysts 1,18 8,21 and 9–1122 were synthesized according to a literature procedure. Dimeric [Ti(NPh)-Cl2py2]2 was prepared by extended heating of Ti(NPh)Cl2py318 under vacuum. Liquid alkynes and other reagents were freeze–pump–thaw degassed three times and passed through activated basic alumina prior to use.
1H and 13C NMR spectra were recorded on Bruker Avance III HD 400 and 500 MHz spectrometers. Chemical shifts were referenced to the residual protio-solvent impurity for 1H (s, 7.16 ppm for C6D5H; s, 7.26 for CHCl3; s 7.30, 7.02, and 6.94 ppm for C6 D4 HBr)38 and solvent carbons for 13C (t, 128.1 ppm for C6D6; t, 77.2 ppm for CDCl3). X-ray data were collected using a Bruker Photon 100 CMOS diffractometer for data collection at 123(2) K using Cu Kα radiation (normal parabolic mirrors). The data intensity was corrected for absorption and decay (SADABS). Final cell constants were obtained from least-squares fits of all measured reflections and the structure was solved and refined using SHELXL-2014/7.39 Details regarding refined data and cell parameters are available in Table S3. CCDC entries 1524349–1524352 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, United Kingdom, fax: (+44) 1223-336-033, or e-mail: deposit@ccdc.cam.ac.uk
Synthesis of Ti(N(p-tolyl))(C5H5N)3Br2 (2)
TiBr4 (604 mg, 1.64 mmol, 1.0 equiv), N-(p-tolyl)-N,N-bis(trimethylsilyl)amine (404 mg, 1.61 mmol, 1.0 equiv), and 8 mL of CH2Cl2 were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was then sealed with a Teflon screw cap, heated to 60 °C, and stirred for 0.5 h. After cooling to room temperature the mixture was left to stir for 1.5 h before diluting with 5 mL of hexanes. The reaction mixture was then filtered through a medium frit and washed with hexanes (3 × 3 mL). The resulting solid was then dissolved with 4 mL of pyridine and 2 mL of CH2Cl2. After stirring for 15 min, the solution was further diluted with 10 mL of CH2Cl2, filtered through a plug of Celite, layered with hexanes, and placed in a −35 °C freezer overnight. The resulting tan/green solid was collected and washed with hexanes to give 2 (356 mg, 40% yield). Elemental analysis was not attempted as complex decomposition would occur under prolonged drying on the vacuum line. 1H NMR (400 MHz, CDCl3): δ 9.14 (br s, 4H, o-py-H), 8.86 (br s, 2H, axial o-py-H), 7.84 (t, 3JHH = 7.6 Hz, 2H, p-py-H), 7.72 (t, 3JHH = 7.6 Hz, 1H, axial p-py-H), 7.36 (t, 3JHH = 6.7 Hz, 4H, m-py-H), 7.26 (br s, 2H, axial m-py-H), 6.99 (d, 3JHH = 7.8 Hz, 2H, m-NTol-H), 6.88 (d, 3JHH = 8.0 Hz, 2H, o-NTol-H), 2.25 (s, 3H, NC6H4-CH3). 13C NMR (101 MHz, CDCl3): δ 152.1, 151.3 (br), 138.8, 137.1 (br), 132.3, 128.8, 124.2, 124.1, 123.9 (br, 2C), 21.2 (NC6H4-CH3).
Synthesis of Ti(N(p-tolyl))THF3I2 (3)
TiI4 (442 mg, 0.796 mmol, 1.0 equiv), N-(p-tolyl)-N,N-bis(trimethylsilyl)amine (200 mg, 0.795 mmol, 1.0 equiv), and 5 mL of toluene were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was then sealed with a Teflon screw cap, heated to 75 °C, and stirred for 2 h. After cooling to room temperature, the mixture was diluted with 5 mL of hexanes. The reaction mixture was then filtered through a medium frit and washed with hexanes (3 × 2 mL). The solid was collected, treated with 5 mL of THF, and heated to 60 °C until all the solids dissolved to give a red solution. The solution was then layered with 5 mL of hexanes and placed in a −35 °C freezer overnight to give 3 as X-ray quality red block crystals which were washed with cold hexanes (165 mg, 33% yield). Elemental analysis was not attempted as complex decomposition would occur under prolonged drying on the vacuum line. 1H NMR (400 MHz, CDCl3): δ 6.93 (d, 3JHH = 8.3 Hz, 2H, m-NTol-H), 6.84 (d, 3JHH = 8.1 Hz, 2H, o-NTol-H), 4.54 (br s, 8H, 2,5-THF-H), 3.77 (br s, 4H, axial 2,5-THF-H), 2.25 (s, 3H, NC6H4-CH3), 2.13 (br s, 8H, 3,4-THF-H), 1.85 (br s, 4H, axial 3,4-THF-H). 13C NMR (101 MHz, CDCl3): δ 160.3, 133.7, 128.8, 123.6, 76.6 (br-s, 1C), 68.4 (br-s, 1C), 25.5 (br-s, 1C), 21.2 (NC6H4-CH3).
Synthesis of Ti(N(p-tolyl))(C5H5N)3(C4H4N)2 (4)
First, Li-(C4H4N) was prepared. Pyrrole (2.50 g, 37.3 mmol, 1.0 equiv) and 10 mL of toluene were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was then cooled in the glovebox coldwell to −75 °C. nBuLi (2.5 M, 18 mL, 44.7 mmol, 1.2 equiv) was added dropwise to the vial over 15 min. The reaction was allowed to stir while warming to room temperature. Afterward, excess hexanes were added to precipitate out the lithium pyrrolide salt, which was collected on a medium frit, washed with more hexanes, and dried overnight under vacuum to ensure removal of toluene.
Li(C4H4N) (200 mg, 2.77 mmol, 4.0 equiv), 1 (318 mg, 0.69 mmol 1.0 equiv), and 2 mL of THF were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was then sealed with a Teflon screw cap and stirred overnight at room temperature. The reaction mixture changed to a dark red color over this period of time. Solvent was removed in vacuo, and the remaining solid was dissolved in benzene and filtered through Celite. The filtrate was lyophilized to give 4 (350 mg, 92% yield). Elemental analysis for C30H30N6Ti (calcd, found): C (68.97, 68.88), H (5.79, 5.79), N (16.09, 16.02). 1H NMR (400 MHz, C6D6): δ 8.34 (br s, 2H, axial o-py-H), 8.13 (br s, 4H, o-py-H), 7.53 (br s, 4H, o-NC5H4-H), 6.96 (d, 3JHH = 7.8 Hz, 2H, m-NTol-H), 6.88 (br s, 1H, axial p-py-H), 6.82 (d, 3JHH = 7.8 Hz, 2H, o-NTol-H), 6.76 (br s, 4H, m-NC5H4-H), 6.59–6.56 (m, 4H, axial m-py-H and p-py-H), 6.31 (br s, 4H, m-py-H), 2.06 (s, 3H, NC6H4-CH3). 13C NMR (101 MHz, C6D6): δ 151.0, 138.1, 130.6, 129.2, 128.6, 127.2, 124.4, 123.4, 108.8, 21.0 (NC6H4-CH3).
Synthesis of Ti(N(p-tolyl))(C5H5N)2(skatolide)2 (5)
First, Li skatolide was synthesized. Skatole (2.50 g, 19.1 mmol, 1.0 equiv) and 10 mL of toluene were added to a 50 mL round-bottomed flask equipped with a stirbar in a N2-filled glovebox. This was then cooled in the glovebox coldwell to −75 °C. nBuLi (2.5 M, 9 mL, 22.9 mmol, 1.2 equiv) was added dropwise to the round-bottomed flask over 15 min. The reaction was allowed to stir while warming to room temperature. Afterward, excess hexanes was added to precipitate out the lithium skatolide salt which was collected on a medium frit, washed with more hexanes and dried overnight under vacuum to ensure removal of toluene.
Li skatolide (100 mg, 0.729 mmol, 2.2 equiv), 1 (150 mg, 0.325 mmol, 1.0 equiv) and 2 mL of THF were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was then sealed with a Teflon screw cap and stirred overnight at room temperature. The reaction mixture changed to a dark red color over this period of time. The solvent was removed in vacuo and the resulting solid was dissolved in benzene, filtered through Celite and the filtrate was lyophilized to give 5 as an oily red solid (130 mg, 70% yield). 1H NMR (400 MHz, C6D6): δ 8.15 (d, 3JHH = 5.0 Hz, 4H, o-py-H), 7.84–7.82 (m, 2H, Ar-H), 7.32–7.31 (m, 4H, Ar-H), 7.13 (d, 3JHH = 8.2 Hz, 2H, m-NTol-H), 6.89 (d, 3JHH = 8.1 Hz, 2H, o-NTol-H), 6.39 (t, 3JHH = 7.6 Hz, 2H, p-py-H), 5.99 (t, 3JHH = 6.7 Hz, 4H, m-py-H), 2.53(s, 6H, −CH3), 2.09 (s, 3H, NC6H4-CH3). 13C NMR (126 MHz, C6D6): δ 161.1, 150.4, 138.3, 130.9, 130.5, 129.4, 128.4, 124.5, 122.9, 121.6, 119.6, 118.6, 111.7, 21.0 (NC6H4-CH3), 10.5 (−CH3).
Synthesis of [Ti(μ-NPh)(C5 H5 N)(2,6-iPr2PhO)Cl]2 (6)
This procedure was adapted from that used for synthesis of a similar compound with a different imido substituent.20a 2,6-Diisopropylphe-nol (17.4 g, 97.6 mmol, 1.0 equiv) and 40 mL of THF were added to a 100 mL round-bottomed flask equipped with a stirbar in a N2-filled glovebox and cooled in the glovebox freezer to −35 °C. Solid NaH (2.66 g, 111 mmol, 1.1 equiv) was added slowly to the stirring cooled solution. Caution: This reaction will exotherm. The mixture turned deep green in color. Upon full addition, the mixture was warmed to room temperature and stirred for 2 h. The mixture was then filtered through a Celite plug, washed with THF, and the filtrate solvents were removed in vacuo to give 2,6-iPr2PhONa as a white solid.
Next, 2,6-iPr2PhONa (700 mg, 3.50 mmol, 2.5 equiv) was dissolved in 6 mL of THF in a 20 mL scintillation vial in a N2-filled glovebox. The solution was then added dropwise to a suspension of [Ti(NPh)Cl2py2]2 (1.03 g, 1.40 mmol, 1.0 equiv) in 2 mL of THF in a separate 20 mL scintillation vial equipped with a stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 16 h before removal of solvents in vacuo. The solids were extracted into 5 mL of CH2Cl2, filtered through a Celite plug to remove NaCl, layered with 5 mL of hexanes, and cooled in a −35 °C freezer. The dark red/black crystalline material was collected and washed with hexanes to give 6. (470 mg, 48% yield). 1H NMR (400 MHz, CDCl3): δ 8.57 (d, 3JHH = 4.9 Hz, 4H, o-py-H), 7.47 (t, 3JHH = 7.6 Hz, 2H, p-py-H), 7.10 (d, 3JHH = 7.6 Hz, 4H, Ar-H), 7.01–6.95 (m, 6H, Ar-H), 6.77 (t, 3JHH = 7.8 Hz, 4H, Ar-H), 6.54–6.49 (m, 6H, Ar-H), 3.76 (br s, 4H, iPr2C-H), 1.17 (d, 3JHH = 6.8 Hz, 24H, iPr-H). 13C NMR (101 MHz, CDCl3): δ 163.3, 161.4, 149.8, 138.3, 138.1, 127.6, 124.2, 123.2, 122.7, 122.0, 118.8, 26.8, 23.9.
Synthesis of Ti(NPh)(C5H5N)2 (2,6-tBu2PhO)Cl (7)
This procedure was adapted from that used for synthesis of a similar compound with a different imido substituent.20a 2,6-Di-tert-butylphenol (1.00 g, 4.85 mmol, 1.0 equiv) and 10 mL of THF were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. Caution: This reaction will exotherm. Solid NaH (150 mg, 6.25 mmol, 1.3 equiv) was added slowly to the solution, and the resulting mixture was left to stir uncapped for 30 min to allow for the evolution of H2. This was then sealed with a Teflon screw cap and stirred at room temperature for 16 h. Afterward, the mixture was filtered through a Celite plug to remove residual NaH and dried in vacuo to give 2,6-tBu2PhONa as a white solid.
2,6-tBu2PhONa (227 mg, 0.994 mmol mmol, 2.4 equiv, 1.2 equiv per Ti center) was dissolved in 6 mL of THF in a 20 mL scintillation vial in a N2-filled glovebox. This solution was added dropwise to a suspension of [Ti(NPh)Cl2py2]2 (305 mg, 0.414 mmol, 1.0 equiv) in 2 mL of THF in a separate 20 mL scintillation vial equipped with a small stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 16 h followed by removal of solvents in vacuo. The solids were extracted into 5 mL of CH2Cl2, filtered through a Celite plug to remove NaCl, layered with 5 mL of hexanes, and cooled in a −35 °C freezer. The globular solids were collected, crushed, and dried in vacuo to yield 7 (150 mg, 48% yield). 1H NMR (400 MHz, CDCl3): δ 9.27 (br s, 4H, o-py-H), 7.86 (br s, 2H, p-py-H), 7.44 (br s, 4H, m-py-H), 7.32 (d, 3JHH = 7.7 Hz, 1H, m-C6H t3Bu2-H), 6.92 (t, 3JHH = 7.6 Hz, 2H, o-NPh-H), 6.85 (t, 3JHH = 7.8 Hz, 1H, p-C6H t3Bu2-H), 6.64 (t, 3JHH = 7.2 Hz, 1H, p-NPh-H), 6.36 (d, 3JHH = 8.0 Hz, 2H, m-NPh-H), 1.53–1.21 (m, 18H, tBu-H).
Synthesis of Ti(N(p-tolyl))(C5H5N)((N-2′,6′-iPr2Ph)-phenylamidate)2 (12)
N-(2′,6′-Diisopropylphenyl) (phenyl)-(amide)40 (186 mg, 0.661 mmol, 2.2 equiv), KBn (86 mg, 0.660 mmol, 2.2 equiv), and 2 mL of benzene were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was stirred at room temperature for 10 min until a colorless solution formed. The colorless solution was added directly into a suspension of 1 (139 mg, 0.301 mmol, 1.0 equiv) and 8 mL of benzene in a separate 20 mL scintillation vial equipped with a small stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 1 h before passing through a plug of Celite and drying the filtrate in vacuo to give a brown solid. The crude product was dissolved in 10 mL of ether and filtered through a glass fiber filter paper fitted in a pipet. Then, 10 mL of hexanes was layered onto the ether solution, and the mixture was placed in a −35 °C freezer to yield 12 as brown crystals. (132 mg, 55% yield). 1H NMR (400 MHz, C6D6): δ 9.34 (d, 3JHH = 4.4 Hz, 2H, o-py-H), 7.86 (d, 3JHH = 7.2 Hz, 4H, Ar-H), 7.23 (br s, 4H, Ar-H), 7.19 (br s, 2H, Ar-H), 6.96–6.87 (m, 6H, Ar-H), 6.74 (d, 3JHH = 8.1 Hz, 2H, m-NTol-H), 6.70 (t, 3JHH = 7.6 Hz, 1H, p-py-H), 6.59 (d, 3JHH = 8.2 Hz, 2H, o-NTol-H), 6.41 (t, 3JHH = 6.4 Hz, 2H, m-py-H), 4.19 (br s, 2H, iPr2C-H), 3.59 (br s, 2H, iPr2C-H), 2.02 (s, 3H, NC6H4-CH3), 1.29–1.19 (m, 12H, iPr-H), 1.12–1.10 (m, 6H, iPr-H), 0.78 (br s, 6H, iPr-H). 13C NMR (101 MHz, C6D6): δ 158.0, 151.6, 143.1, 142.1, 139.1, 133.5, 131.4, 130.3, 129.7, 128.9, 128.0, 125.8, 125.0, 124.2, 124.1, 28.6, 28.2, 24.6, 23.9, 21.0 (NC6H4-CH3).
Synthesis of Bis(2,6-iPr2Ph-salycilaldimino)Ti(N(p-tolyl)) (13)
First, 2,6-iPr2Ph-salycilaldimine41 (170 mg, 0.604 mmol, 1.2 equiv), KBn (79 mg, 0.607 mmol, 1.2 equiv), and 4 mL of benzene were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was stirred at room temperature for 15 min until a yellow solution formed. The yellow solution was added dropwise to a suspension of 1 (234 mg, 0.507 mmol, 1.0 equiv) and 10 mL of benzene in a separate 20 mL scintillation vial equipped with a small stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 3 h before filtering through a plug of Celite and drying the filtrate in vacuo. The orange-red solid was dissolved in 15 mL of ether and filtered through a glass fiber filter paper fitted in a pipet. The ether filtrate was concentrated to 2.5 mL before layering with 2.5 mL of hexanes. The solution was placed in a −35 °C freezer to yield 13 as a mixture of fine X-ray quality orange crystals and orange powder (160 mg from three crops of recrystallization, 45% yield). Elemental analysis for C45H51N3O2Ti (calcd, found): C (75.72, 74.12), H (7.20, 6.96), N (5.89, 5.69). 1H NMR (400 MHz, C6D6): δ 8.15 (s, 2H, H-C‗N), 7.34–7.27 (m, 4H, Ar-H), 7.18 (d, 3JHH = 2.1 Hz, 1H, Ar-H), 7.16 (br s, 1H, Ar-H), 7.11 (dd, 3JHH = 7.0 Hz, 4JHH = 1.7 Hz, 1H, Ar-H, 7.09 (dd, 3J = 6.9 Hz, 4JHH = 1.6 Hz, 1H, Ar-H), 7.04 (dd, HH 3JHH = 7.8 Hz, 4JHH = 1.7 Hz, 2H, Ar-H), 6.56 (d, 3JHH = 7.9 Hz, 4H, Ar-H), 6.46 (d, 3JHH = 8.3 Hz, 2H, m-NTol-H), 6.22 (d, 3JHH = 8.1 Hz, 2H, o-NTol-H), 3.89 (hept, 3JHH = 6.7 Hz, 2H, iPr2C-H), 2.60 (hept, 3JHH = 6.8 Hz, 2H, iPr2C-H), 1.95 (s, 3H, NC6H4-CH3), 1.15 (d, 3JHH= 6.9 Hz, 6H, iPr), 1.11 and 1.10 (d, 3JHH = 6.9 Hz, 12H, iPr), 0.95 (d, 3JHH = 6.9 Hz, 6H, iPr). 13C NMR (101 MHz, C6D6): δ 168.9, 167.5, 160.1, 149.6, 142.4, 141.5, 136.0, 134.3, 129.7, 126.9, 124.5, 124.0, 123.3, 122.3, 120.3, 117.7, 29.5, 28.3, 25.3, 24.9, 23.8, 22.9, 21.0 (NC6H4-CH3).
Synthesis of Ti(N(p-tolyl))(N,N′-(2,6-iPr2Ph)2formamidinate)2 (14)
First, N,N′-(2,6-iPr2Ph)2formamidine42 (454 mg, 1.25 mmol, 2.1 equiv), KBn (162 mg, 1.25 mmol, 2.1 equiv), and 2 mL of THF were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was stirred at room temperature for 10 min until a colorless solution formed. The colorless solution was added directly into a suspension of 1 (280 mg, 0.607 mmol, 1.0 equiv) and 4 mL of THF in a separate 20 mL scintillation vial equipped with a small stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 2 h before filtering through a plug of Celite and drying the filtrate under vacuum to give a brown solid. Then, 10 mL of ether was added to the solid, and the suspension was filtered through a medium frit. The powder residue was dried in vacuo for 3 h to give 14. The ether filtrate was further concentrated in vacuo to 5 mL, layered with 5 mL of hexanes, and left to stand at room temperature to yield more of 14 as a brown solid (300 mg, 56% yield from both the powder and solid). X-ray quality crystals were grown from a 2:1 ether/hexanes layering mixture. 1H NMR (400 MHz, C6D6): δ 8.29 (s, 2H, C-H), 7.07 (s, 12H, Ar-H), 6.94 (d, 3JHH = 8.2 Hz, 2H, m-NTol-H), 6.79 (d, 3JHH = 8.1 Hz, 2H, o-NTol-H), 4.58 (br s, 4H, iPr2C-H), 3.09 (br s, 4H, iPr2C-H), 2.09 (s, 3H, NC6H4-CH3), 1.01 (br s, 48H, iPr). 13C NMR (101 MHz, C6D6): δ 164.9, 162.0, 143.6, 131.3, 128.6, 126.0, 124.7, 124.0, 28.2 (iPr), 25.7 (br, iPr-CH), 23.4 (br, iPr-CH), 21.0 (NC6H4-CH3).
Synthesis of Ti (N (p - toly l)) (C5 H5 N) (N, N′ -(2,6-iPr2 Ph)2 formamidinate)Cl (15)
First, N,N ′-(2,6-iPr2Ph)2formamidine (201 mg, 0.551 mmol, 1.2 equiv), KBn (72 mg, 0.551 mmol, 1.2 equiv), and 4 mL of THF were added to a 20 mL scintillation vial equipped with a small stirbar in a N2-filled glovebox. This was stirred at room temperature for 10 min until a colorless solution formed. The colorless solution was added directly into a suspension of 1 (211 mg, 0.457 mmol, 1.0 equiv) and 10 mL of THF in a separate 20 mL scintillation vial equipped with a small stirbar. This was then sealed with a Teflon screw cap and stirred at room temperature for 3 h before filtering through a plug of Celite and drying in vacuo to give a brown solid. The solid was dissolved in 15 mL of ether, and insoluble material was removed via filtration through a glass fiber filter paper fitted in a pipet. Then, 5 mL of hexanes were layered upon the ether solution, and the mixture was left to stand at room temperature to yield X-ray quality brown crystals of 15 (133 mg, 46% yield). 1H NMR (400 MHz, C6D6): δ 8.73 (d, 3JHH = 4.9 Hz, 2H, o-py-H), 8.11 (s, 1H, C-H), 7.10 (br s, 5H, Ar-H), 7.08 (d, 3JHH = 8.4Hz, 2H, m-NTol-H), 6.82 (d, 3JHH = 8.1 Hz, 2H, o-NTol-H), 6.53 (t, 3JHH = 7.7 Hz, 1H, p-py-H), 6.23 (t, 3JHH = 8.0 Hz, 2H, m-py-H), 4.03 (br s, 4H, iPr2C-H), 2.05 (s, 3H, NC6H4-CH3), 1.25 (d, 3JHH = 6.0 Hz, 24H, iPr-H). 13C NMR (101 MHz, C6D6): δ 164.7, 160.7, 149.9, 144.5, 143.3, 138.6, 131.5, 128.9, 128.6, 126.0, 124.3, 124.1, 123.7, 28.1 (iPr), 24.8 (br, iPr-CH), 21.0 (NC6H4-CH3).
Synthesis of [Zr(μ-NPh)THF2Cl2]2 (16)
16 was synthesized via a modification of the literature procedure, starting from ZrBn4 instead of Zr(CH2TMS)4.43 ZrCl4(THF)2 (4.47 g, 11.8 mmol, 1.0 equiv) and 100 mL of THF were added to a 250 mL round-bottomed flask equipped with a stirbar in a N2-filled glovebox. Separately, ZrBn4 (5.40 g, 11.8 mmol, 1.0 equiv) was dissolved in 25 mL of THF in a 50 mL round-bottomed flask. The ZrBn4 solution was added in dropwise to the THF solution of ZrCl4 with rapid stirring. The flask was sealed, covered in aluminum foil, and stirred at room temperature for 5 h to in situ generate ZrCl2Bn2. Afterward, aniline (2.21 g, 23.7 mmol, 2.0 equiv) in 10 mL of THF was added dropwise to the reaction mixture. The reaction was stirred for 13 h at room temperature while covered in aluminum foil. Volatiles were then removed under vacuum, and the residual brown-yellow solid was dissolved in a minimal amount of 5:1 CH2Cl2/THF, transferred into two 20 mL vials, and layered with an equal volume of pentane. The solutions were placed in a −35 °C freezer for 3 days to afford 16 as a yellow crystalline solid (8.68 g, 92% yield). 1H NMR (400 MHz, CDCl3): δ 7.18 (t, 3JHH = 7.8 Hz, 4H, m-NPh-H), 7.10 (d, 3JHH = 7.2 Hz, 4H, o-NPh-H), 6.76 (t, 3JHH = 7.2 Hz, 2H, p-NPh-H), 3.99 (br s, 16H, 2,5-THF-H), 1.71 (br s, 16H, 3,4-THF-H). 13C NMR (101 MHz, CDCl3): δ 152.9, 128.5, 121.7, 121.0, 72.8 (br s), 25.5.
General Procedure for Catalytic Alkyne Trimerization
Precatalyst (5 mol % Ti, 0.01 mmol, 0.02 M) and 0.5 mL of stock solution were added to a Teflon-tape-lined screw-cap NMR tube in a N2-filled glovebox. This was then sealed with a Teflon screw cap and heated at 115 °C for 16 h. The stock solution was prepared by adding either 3-hexyne or 1-hexyne (0.4 M) to C6D5Br with 1,3,5-trimethoxybenzene (0.04 M) acting as an internal standard. Quantitative 1H NMR spectra of the catalytic mixture were recorded before and after heating on the Bruker Avance III HD 400 MHz spectrometers (Acquisition time = 5 s; delay time = 30 s; dummy scans = 0; number of scans = 8). The initial precatalyst quantity of 0.01 mmol was used for catalyst activation calculations. Ti(NTol)-THF3I2 (3) was an exception to the general procedure: Trimerization 3-hexyne was completed at room temperature over 16 h.
Supplementary Material
Acknowledgments
Financial support was provided by the University of Minnesota (start-up funds, Heisig/Gleysteen fellowship for T.A.W.), the ACS Petroleum Research Fund (ACS-PRF 54225-DNI3) and the National Institutes of Health (1R35GM119457). Departmental equipment was purchased via grants from the NSF (MRI 1224900) (Bruker-AXS D8 Venture diffractometer) and the NIH (S10OD011952) (NMR Facility) with matching funds from the University of Minnesota.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organo-met.7b00096.
Full NMR and XRD data, as well as experimental controls and characterization of all products of catalysis-(PDF)
Crystallographic information file for 3 and 13–15 (CIF)
ORCID
Ian A. Tonks: 0000-0001-8451-8875
Notes
The authors declare no competing financial interest.
References
- 1.(a) McMurry JE. Acc Chem Res. 1974;7:281–286. [Google Scholar]; (b) Fürstner A, Bogdanović B. Angew Chem, Int Ed Engl. 1996;35:2442–2469. [Google Scholar]; (c) Okamoto S. Chem Rec. 2016;16:857–872. doi: 10.1002/tcr.201500277. [DOI] [PubMed] [Google Scholar]; (d) Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835–2886. doi: 10.1021/cr990277l. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hanna TE, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2004;126:14688–14689. doi: 10.1021/ja045884r. [DOI] [PubMed] [Google Scholar]; (b) Vol’Pin ME, Shur VB. Nature. 1966;209:1236–1236. [Google Scholar]; (c) Bercaw JE, Marvich RH, Bell LG, Brintzinger HH. J Am Chem Soc. 1972;94:1219–1238. [Google Scholar]
- 3.(a) Fuerstner A, Hupperts A, Ptock A, Janssen E. J Org Chem. 1994;59:5215–5229. [Google Scholar]; (b) Balu N, Nayak SK, Banerji A. J Am Chem Soc. 1996;118:5932–5937. [Google Scholar]
- 4.(a) Yamamoto K, Nagae H, Tsurugi H, Mashima K. Dalton Trans. 2016;45:17072–17081. doi: 10.1039/c6dt03389j. [DOI] [PubMed] [Google Scholar]; (b) Morohashi N, Yokomakura K, Hattori T, Miyano S. Tetrahedron Lett. 2006;47:1157–1161. [Google Scholar]; (c) Calderazzo F, Marchetti F, Pampaloni G, Hiller W, Antropiusová H, Mach K. Chem Ber. 1989;122:2229–2238. [Google Scholar]
- 5.(a) Kablaoui NM, Hicks FA, Buchwald SL. J Am Chem Soc. 1997;119:4424–4431. [Google Scholar]; (b) Hicks FA, Kablaoui NM, Buchwald SL. J Am Chem Soc. 1999;121:5881–5898. [Google Scholar]; (c) Sturla SJ, Buchwald SL. Organometallics. 2002;21:739–748. [Google Scholar]
- 6.(a) Kulinkovich OG. Pure Appl Chem. 2000;72:1715. [Google Scholar]; (b) Kulinkovich OG, Sviridov SV, Vasilevski DA. Synthesis. 1991;1991:234–234. [Google Scholar]
- 7.(a) Wang X, Woo LK. J Org Chem. 1998;63:356–360. [Google Scholar]; (b) Wijeratne GB, Zolnhofer EM, Fortier S, Grant LN, Carroll PJ, Chen CH, Meyer K, Krzystek J, Ozarowski A, Jackson TA, Mindiola DJ, Telser J. Inorg Chem. 2015;54:10380–10397. doi: 10.1021/acs.inorgchem.5b01796. [DOI] [PubMed] [Google Scholar]; (c) Kool LB, Rausch MD, Alt HG, Herberhold M, Honold B, Thewalt U. J Organomet Chem. 1987;320:37–45. [Google Scholar]
- 8.Lukešová L, Horáček M, Štěpnička P, Fejfarová K, Gyepes R, Císařová I, Kubišta J, Mach K. J Organomet Chem. 2002;663:134–144. [Google Scholar]
- 9.Ladipo FT, Sarveswaran V, Kingston JV, Huyck RA, Bylikin SY, Carr SD, Watts R, Parkin S. J Organomet Chem. 2004;689:502–514. [Google Scholar]
- 10.Allen JM, Ellis JE. J Organomet Chem. 2008;693:1536–1542. [Google Scholar]
- 11.Woo LK, Hays JA, Young VG, Day CL, Caron C, D’Souza F, Kadish KM. Inorg Chem. 1993;32:4186–4192. [Google Scholar]
- 12.(a) Jensen JA, Wilson SR, Schultz AJ, Girolami GS. J Am Chem Soc. 1987;109:8094–8096. [Google Scholar]; (b) Morris RJ, Girolami GS. Inorg Chem. 1990;29:4167–4169. [Google Scholar]
- 13.(a) Rosenthal U. Russ Chem Bull. 2014;63:2577–2582. [Google Scholar]; (b) Haehnel M, Ruhmann M, Theilmann O, Roy S, Beweries T, Arndt P, Spannenberg A, Villinger A, Jemmis ED, Schulz A, Rosenthal U. J Am Chem Soc. 2012;134:15979–15991. doi: 10.1021/ja3070649. [DOI] [PubMed] [Google Scholar]; (c) Rosenthal U, Pellny PM, Kirchbauer FG, Burlakov VV. Acc Chem Res. 2000;33:119–129. doi: 10.1021/ar9900109. [DOI] [PubMed] [Google Scholar]
- 14.(a) Hill JE, Fanwick PE, Rothwell IP. Organometallics. 1992;11:1771–1773. [Google Scholar]; (b) Cohen SA, Auburn PR, Bercaw JE. J Am Chem Soc. 1983;105:1136–1143. [Google Scholar]; (c) Mullins SM, Duncan AP, Bergman RG, Arnold J. Inorg Chem. 2001;40:6952–6963. doi: 10.1021/ic010631+. [DOI] [PubMed] [Google Scholar]; (d) Graham TW, Kickham J, Courtenay S, Wei P, Stephan DW. Organometallics. 2004;23:3309–3318. [Google Scholar]
- 15.Frazier BA, Wolczanski PT, Keresztes I, DeBeer S, Lobkovsky EB, Pierpont AW, Cundari TR. Inorg Chem. 2012;51:8177–8186. doi: 10.1021/ic300590t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ohkubo M, Mochizuki S, Sano T, Kawaguchi Y, Okamoto S. Org Lett. 2007;9:773–776. doi: 10.1021/ol062963u. [DOI] [PubMed] [Google Scholar]; (b) Okamoto S, He J-Q, Ohno C, Oh-iwa Y, Kawaguchi Y. Tetrahedron Lett. 2010;51:387–390. [Google Scholar]
- 17.(a) Davis-Gilbert ZW, Yao LJ, Tonks IA. J Am Chem Soc. 2016;138:14570–14573. doi: 10.1021/jacs.6b09939. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gilbert ZW, Hue RJ, Tonks IA. Nat Chem. 2015;8:63–68. doi: 10.1038/nchem.2386. [DOI] [PubMed] [Google Scholar]
- 18.Blake AJ, Collier PE, Dunn SC, Li WS, Mountford P, Shishkin OV. J Chem Soc, Dalton Trans. 1997:1549–1558. [Google Scholar]
- 19.(a) Swartz DL, II, Staples RJ, Odom AL. Dalton Trans. 2011;40:7762–7768. doi: 10.1039/c1dt10127g. [DOI] [PubMed] [Google Scholar]; (b) Odom AL, McDaniel TJ. Acc Chem Res. 2015;48:2822. doi: 10.1021/acs.accounts.5b00280. [DOI] [PubMed] [Google Scholar]
- 20.(b) Tillack A, Khedkar V, Jiao H, Beller M. Eur J Org Chem. 2005;2005:5001–5012. [Google Scholar]; (a) Collier PE, Blake AJ, Mountford P. J Chem Soc, Dalton Trans. 1997:2911–2920. [Google Scholar]
- 21.Dunn SC, Hazari N, Jones NM, Moody AG, Blake AJ, Cowley AR, Green JC, Mountford P. Chem - Eur J. 2005;11:2111–2124. doi: 10.1002/chem.200401104. [DOI] [PubMed] [Google Scholar]
- 22.Dunn SC, Mountford P, Robson A. J Chem Soc, Dalton Trans. 1997:293–304. [Google Scholar]
- 23.(a) Zhang Z, Leitch DC, Lu M, Patrick BO, Schafer LL. Chem - Eur J. 2007;13:2012–2022. doi: 10.1002/chem.200600735. [DOI] [PubMed] [Google Scholar]; (b) Yim JCH, Bexrud JA, Ayinla RO, Leitch DC, Schafer LL. J Org Chem. 2014;79:2015–2028. doi: 10.1021/jo402668q. [DOI] [PubMed] [Google Scholar]
- 24.(a) Oakes DCH, Gibson VC, White AJP, Williams DJ. Inorg Chem. 2006;45:3476–3477. doi: 10.1021/ic060146k. [DOI] [PubMed] [Google Scholar]; (b) Strauch J, Warren TH, Erker G, Fröhlich R, Saarenketo P. Inorg Chim Acta. 2000;300–302:810–821. [Google Scholar]
- 25.(a) Groom LR, Schwarz AD, Nova A, Clot E, Mountford P. Organometallics. 2013;32:7520–7539. [Google Scholar]; (b) Elkin T, Botoshansky M, Waymouth RM, Eisen MS. Organometallics. 2014;33:840–843. [Google Scholar]
- 26.(a) Negishi E, Holmes SJ, Tour JM, Miller JA, Cederbaum FE, Swanson DR, Takahashi T. J Am Chem Soc. 1989;111:3336–3346. [Google Scholar]; (b) Cotton FA, Kibala P, Shang M, Wojtczak WA. Organometallics. 1991;10:2626–2630. [Google Scholar]; (c) Plundrich GT, Wadepohl H, Clot E, Gade LH. Chem - Eur J. 2016;22:9283–9292. doi: 10.1002/chem.201601213. [DOI] [PubMed] [Google Scholar]; (d) Pun D, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2008;130:6047–6054. doi: 10.1021/ja801021w. [DOI] [PubMed] [Google Scholar]
- 27.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 28.Vujkovic N, Ward BD, Maisse-François A, Wadepohl H, Mountford P, Gade LH. Organometallics. 2007;26:5522–5534. [Google Scholar]
- 29.Tonks IA, Meier JC, Bercaw JE. Organometallics. 2013;32:3451–3457. [Google Scholar]
- 30.(a) Ladipo FT. Comments Inorg Chem. 2006;27:73–102. [Google Scholar]; (b) Ozerov OV, Ladipo FT, Patrick BO. J Am Chem Soc. 1999;121:7941–7942. [Google Scholar]; (c) Broere DLJ, Ruijter E. Synthesis. 2012;44:2639–2672. [Google Scholar]
- 31.(a) Diercks R, Dieck tom H. Chem Ber. 1985;118:428–435. [Google Scholar]; (b) Pal S, Uyeda C. J Am Chem Soc. 2015;137:8042–8045. doi: 10.1021/jacs.5b04990. [DOI] [PubMed] [Google Scholar]; (c) Roland CD, Li H, Abboud KA, Wagener KB, Veige AS. Nat Chem. 2016;8:791–796. doi: 10.1038/nchem.2516. [DOI] [PubMed] [Google Scholar]
- 32.(a) Ozerov OV, Patrick BO, Ladipo FT. J Am Chem Soc. 2000;122:6423–6431. [Google Scholar]; (b) Hill JE, Balaich G, Fanwick PE, Rothwell IP. Organometallics. 1993;12:2911–2924. [Google Scholar]; (c) Troyanov S. J Organomet Chem. 1994;475:139–147. [Google Scholar]
- 33.(a) Bemowski RD, Singh AK, Bajorek BJ, DePorre Y, Odom AL. Dalton Trans. 2014;43:12299–12305. doi: 10.1039/c4dt01314j. [DOI] [PubMed] [Google Scholar]; (b) DiFranco SA, Maciulis NA, Staples RJ, Batrice RJ, Odom AL. Inorg Chem. 2012;51:1187–1200. doi: 10.1021/ic202524r. [DOI] [PubMed] [Google Scholar]
- 34.Yim JCH, Schafer LL. Eur J Org Chem. 2014;2014:6825–6840. [Google Scholar]
- 35.Musso F, Solari E, Floriani C, Schenk K. Organometallics. 1997;16:4889–4895. [Google Scholar]
- 36.(a) Nguyen AI, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF. Chem Sci. 2011;2:166–169. [Google Scholar]; (b) Munha RF, Zarkesh RA, Heyduk AF. Dalton Trans. 2013;42:3751–3766. doi: 10.1039/c2dt32063k. [DOI] [PubMed] [Google Scholar]; (c) Heins SP, Wolczanski PT, Cundari TR, MacMillan SN. Chem Sci. 2017 doi: 10.1039/C6SC05610E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kozhushkov SI, Yufit DS, Ackermann L. Org Lett. 2008;10:3409–3412. doi: 10.1021/ol8011875. [DOI] [PubMed] [Google Scholar]
- 38.Hayes PG, Piers WE, Parvez M. Chem - Eur J. 2007;13:2632–2640. doi: 10.1002/chem.200601087. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick GM. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 40.Bakthavachalam K, Reddy ND. Organometallics. 2013;32:3174–3184. [Google Scholar]
- 41.Chang S, Jones L, Wang C, Henling LM, Grubbs RH. Organometallics. 1998;17:3460–3465. [Google Scholar]
- 42.Elkin T, Kulkarni NV, Tumanskii B, Botoshansky M, Shimon LJW, Eisen MS. Organometallics. 2013;32:6337–6352. [Google Scholar]
- 43.Dubberley SR, Evans S, Boyd CL, Mountford P. Dalton Trans. 2005:1448–1458. doi: 10.1039/b500507h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.