

Abstract
Tumor metastasis occurs naturally in pancreatic cancer, and the efficacy of chemotherapy is usually poor. Precision medicine, combining downregulation of target genes with chemotherapy drugs, is expected to improve therapeutic effects. Therefore, we developed a combined therapy of microRNA‐21 antisense oligonucleotides (ASO‐miR‐21) and gemcitabine (Gem) using a targeted co‐delivery nanoparticle (NP) carrier and investigated the synergistic inhibitory effects on pancreatic cancer cells metastasis and growth. Polyethylene glycol–polyethylenimine–magnetic iron oxide NPs were used to co‐deliver ASO‐miR‐21 and Gem. An anti‐CD44v6 single‐chain variable fragment (scFvCD 44v6) was used to coat the particles to obtain active and targeted delivery. Our results showed that the downregulation of the oncogenic miR‐21 by ASO resulted in upregulation of the tumor‐suppressor genes PDCD4 and PTEN and the suppression of epithelial–mesenchymal transition, which inhibited the proliferation and induced the clonal formation, migration, and invasion of pancreatic cancer cells in vitro. The co‐delivery of ASO‐miR‐21 and Gem induced more cell apoptosis and inhibited the growth of pancreatic cancer cells to a greater extent than single ASO‐miR‐21 or Gem treatment in vitro. In animal tests, more scFvCD 44v6‐PEG‐polyethylenimine/ASO‐magnetic iron oxide NP/Gem accumulated at the tumor site than non‐targeted NPs and induced a potent inhibition of tumor proliferation and metastasis. Magnetic resonance imaging was used to observed tumor homing of NPs. These results imply that the combination of miR‐21 gene silencing and Gem therapy using an scFv‐functionalized NP carrier exerted synergistic antitumor effects on pancreatic cancer cells, which is a promising strategy for pancreatic cancer therapy.
Keywords: Gemcitabine, microRNA‐21, nanomedicine, neoplasm metastasis, pancreatic cancer
Most pancreatic cancer patients die of local relapse or distant metastasis even if they have received adjuvant chemotherapy, including Gem.1 Therefore, the development of novel, effective therapeutic approaches is a critical requirement for improving the prognosis of patients with this lethal disease. Based on the progress of molecular biological research on pancreatic cancer, new therapeutic strategies capable of interrupting aggressive tumor progression seem to hold the greatest promise.
Recently, gene therapy, especially therapy involving miRNAs, has emerged as a new class of therapeutic strategies for various tumors.2, 3, 4 MicroRNA‐21 is one of the more intensively studied miRNAs, and it has been detected in several cancer types, including pancreatic cancer.5 Studies strongly suggest that miR‐21 has oncogenic activity and plays a central role in cell function and survival as well as in cancer initiation and progression.6, 7 When miR‐21 was inhibited by ASO, the development of cancer‐associated phenotypes in cell lines was also inhibited. Therefore, the oncogenic miR‐21 is a potent target for the treatment of pancreatic cancer. It was also reported that the inhibition of miR‐21 increased sensitivity to Gem‐induced apoptosis in pancreatic cancer and to other chemotherapeutic drugs in several types of cancers.8, 9 However, few studies have focused on the effect of Gem therapy on the metastasis of pancreatic cancer in vivo. Moreover, in studies that reported a combination therapy using ASO‐miR‐21 and Gem, most of them gave the two agents separately in different drug delivery systems.10, 11 There were many disadvantages of using two drug carriers, including different biocompatibility and biodistribution, increased drug toxicity, and decreased drug combination activity. The lack of efficient delivery vectors hampered the clinical application of gene therapy. In addition, although Gem is the first‐line chemotherapeutic drug for pancreatic cancer, the lack of a targeted drug delivery system resulted in acute toxicity to normal tissues and multidrug resistance, which limited its clinical effectiveness.
An NP drug‐delivery system is an ideal vector because it can target the delivery of a drug to a tumor through its surface modification. It has been reported that when chemotherapeutic drugs are encapsulated in or attached to nano vectors, their acute toxicities can be reduced, and the therapeutic efficacy can be improved because they are delivered directly into cancer tissues.12, 13 In addition, with the protection of NPs, the loaded RNA can be shielded from degradation and its lifetime in the blood can be prolonged. We previously reported that NPs functionalized with scFvCD44v6 resulted in active tumor‐targeted, sustained siRNA and chemotherapy delivery, efficient gene silencing, and enhanced antitumor effects.14, 15, 16, 17 We previously constructed a dual nano vector, PEG‐PEI‐IONPs, for siRNA and Gem delivery.18 In the current study, we engineered CD44v6‐targeted PEG‐PEI‐IONPs carrying ASO‐miR‐21 and Gem as a combination drug for targeted co‐delivery into CD44v6‐expressing tumor cells. Although a similar dual miR‐21 ASO and Gem delivery approach using PEGylated‐poly(lactide)‐co‐glycolide) NPs was reported recently,19 those NPs were not functionalized with active tumor‐targeting elements, and only in vitro experiments were carried out in hepatocellular carcinoma cell lines. In the present study, the potential of synergistic anticancer activity of the multifunctional nanomedicine was evaluated both in pancreatic cancer cell lines and allograft animal models. In vivo biodistribution of NPs and the carcinoma liver metastasis was also observed by MRI.
Materials and Methods
Materials
Antisense oligonucleotide‐miR‐21, scrambled miRNA ASO (ASO‐NC) and FAM‐labeled control ASO were purchased from GenePharma (Shanghai, China). The MTS was purchased from Promega (Beijing, China). Antibodies against the following proteins were used: PDCD4, Bcl‐2, Bax (Abcam, Cambridge, UK), PTEN, E‐cadherin, and vimentin (Cell Signaling Technology, Danvers, MA, USA), GAPDH (Beyotime, Shanghai, China). Peroxidase‐conjugated secondary antibodies (HRP‐goat anti‐rat and HRP‐goat anti‐rabbit) were purchased from Cell Signaling Technology.
Cell culture
The human pancreatic cancer cell lines PANC‐1 and MIA PaCa‐2 were obtained from the ATCC (Rockefeller, MD, USA). The cells were maintained at 37°C and 5% CO2 in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Biological Industries, Beit Haemek, Israel).
Synthesis and surface modification of PEG‐PEI‐IONPs with scFvCD44v6
Single‐chain variable fragment targeted to human CD44 variant 6 and PEG‐PEI‐IONPs were generated as described in our previous study (details in Data S1).14, 18
Cell treatments
To evaluate the cytotoxicity of the combined therapy, cells were incubated with ASO‐NC‐NPs, Gem‐sol, ASO‐NC‐Gem‐NPs, or ASO‐miR‐21‐Gem‐NPs. The concentration of Gem was varied from 1 to 20 μM, and the amount of PEG‐PEI‐IONPs was calculated based on the amount of Gem. The concentration of ASO used was 100 nmol/L per well,20 and cells treated with PBS were used as a negative control.
To evaluate cellular apoptosis induced by the combined therapy, the concentration of Gem was an equivalent dose (3 μM), and cells incubated with PBS served as a control.
Cell proliferation, colony formation, migration, and invasion assays
We used MTS to undertake cell proliferation assays. Soft agar was used for colony formation assays, and the Boyden chamber system with a polycarbonate membrane (8‐μm pore size; Corning Life Sciences, Corning, MA, USA) was used to carry out cell migration and invasion assays (details in Data S1).
Magnetic resonance imaging experiments
Magnetic resonance imaging scans were used to observe the biodistribution of targeted NPs and non‐targeted NPs in vivo. Mice (n = 3) were injected with scFv functionalized NPs (targeted NPs) and non‐functionalized NPs (non‐targeted NPs) through the tail vein. The amount of nanocomplex was calculated according to the amount of Fe (4.48 μg Fe/g bodyweight of the mice).15 The MRI scans were undertaken pre‐injection and at 2, 4, and 6 h after injection using a volumetric wrist coil.
In vivo antitumor effects
The animal studies were carried out according to the approved protocols of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Animal Care and Use Committee of Sun Yat‐sen University (Guangzhou, China). Female BALB/c nude mice (4–5 weeks old, 13 ± 1 g) were purchased from the Animal Facility Center of Sun Yat‐sen University. When the tumor volume had reached approximately 50 mm3, the mice were randomly divided into seven experimental groups (n = 3 per group) that received the following treatments: (i) PBS; (ii) ASO‐NC‐NPs; (iii) ASO‐miR‐21‐NPs; (iv) Gem‐sol; (v) ASO‐NC‐Gem‐NPs; (vi) ASO‐miR‐21‐Gem‐NPs; and (vii) scFv‐ASO‐miR‐21‐Gem‐NPs, respectively. Two doses of 2 mg/kg Gem and 80 μg ASO per mouse were given through the tail vein 3 days apart, and the PBS treatment group was used as a control. The tumor volumes and bodyweights of the animals were measured and recorded every 3 days for 3 weeks. At the end of the experiment, all of the mice were killed, and the isolated tumors were weighed and then fixed for histological study.
Statistical analysis
The statistical analysis of the data was carried out using one‐way anova, and the Bonferroni method was used to determine the significant difference (SPSS for Windows, Version 13.0; SPSS Inc., Chicago, IL, USA). The results were expressed as the mean ± standard error ( ± SE), and a probability level of P < 0.05 was considered statistically significant.
Results
Downregulation of miR‐21 and expression of targeted proteins
After ASO‐miR‐21‐NPs transfection, the relative expression level of miR‐21 was reduced and the expression of miR‐21 negative controlling PDCD4 and PTEN protein were upregulated in both PANC‐1 and MIA PaCa‐2 cells (P < 0.05) (Fig. 1).
Figure 1.
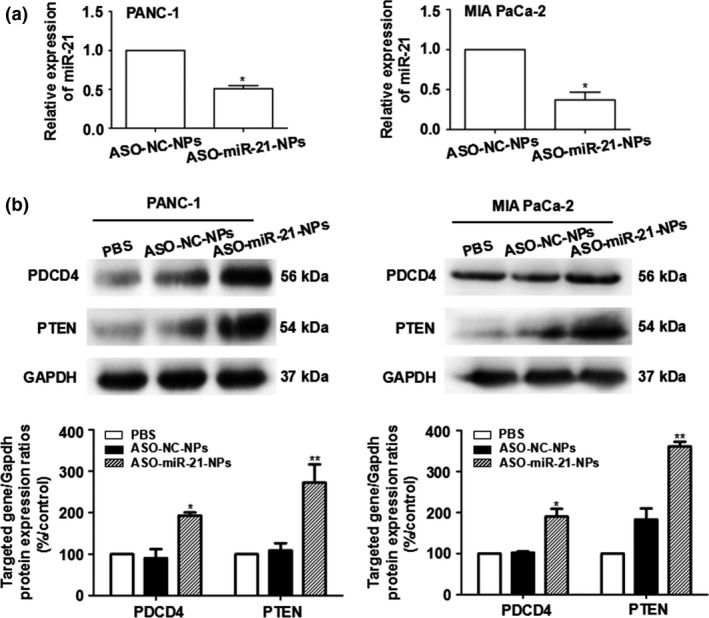
Antisense oligonucleotide nanoparticles (ASO‐NPs) inhibited the expression of microRNA‐21 (miR‐21) in pancreatic cancer cells. MicroRNA‐21 expression levels decreased (a) and the protein expression of miR‐21‐targeted genes PDCD4 and PTEN increased (b) in PANC‐1 and Mia PaCa‐2 cells after transfection with ASO‐miR‐21‐NPs. Concentration of ASO was 100 nmol/L. *P < 0.05, **P < 0.01 versus ASO ‐ NC‐NPs (negative control).
Cell proliferation and colony formation after ASO‐miR‐21‐mediated miR‐21 suppression
Both PANC‐1 and MIA PaCa‐2 cells showed defective proliferation and colony formation after exposure to ASO‐miR‐21‐NPs compared to cells exposed to ASO‐NC‐NPs. As shown in Figure 2(a), the proliferation of PANC‐1 cells decreased to 65.11 ± 1.20% and that of MIA PaCa‐2 cells decreased to 70.78 ± 4.75% (both P < 0.05). The plate colony formation assay was performed to detect the ability of a single cancer cell to reproduce. The soft agar colony formation assay was used to confirm cellular anchorage‐independent growth in vitro. Figure 2(b) shows the colony formation results of the two cell lines. The colony numbers in the plates and in soft agar of PANC‐1 cells were reduced to 47.04 ± 4.16% and 23.79 ± 4.0%, respectively (P < 0.05), and those of the MIA PaCa‐2 cells were reduced to 32.95 ± 11.06% and 28.33 ± 5.0%, respectively (P < 0.05).
Figure 2.
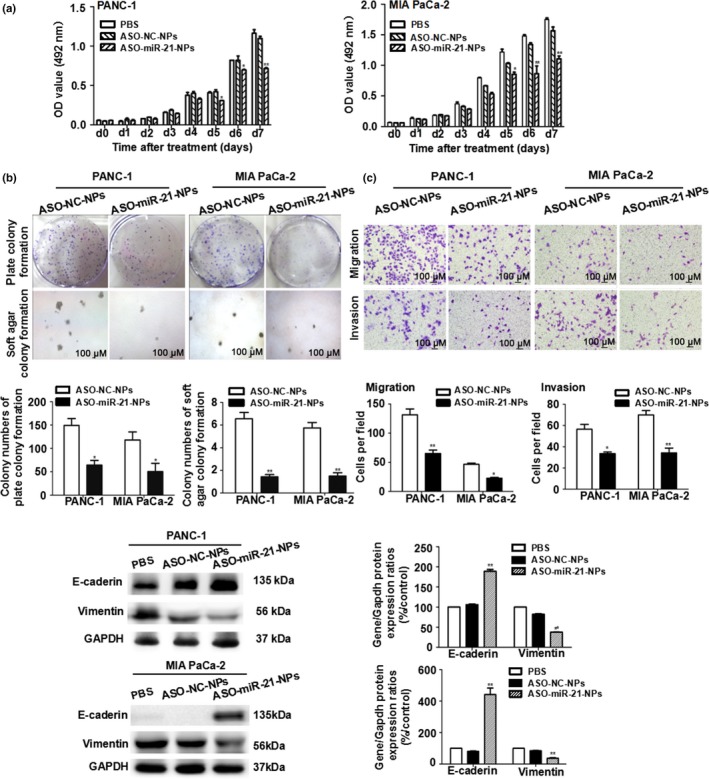
Proliferation, colony formation, migration assay, and epithelial–mesenchymal transition protein expression of PANC‐1 and Mia PaCa‐2 pancreatic cancer cells. Cells were transfected with microRNA‐21 antisense oligonucleotide (ASO‐miR‐21) or ASO‐NC (negative control). (a) Cells were reseeded in 96‐well plates and detected by MTS daily for seven consecutive days. (b) Cells were reseeded in 6‐well plates or soft agar to evaluate colony formation. Magnification, ×40. (c) Cells were reseeded in the upper chamber to evaluate cell migration and invasion. Magnification, ×100. (d) Detection of epithelial–mesenchymal transition protein expression. Data are shown as the mean ± SE (n = 3). *P < 0.05, **P < 0.01 versus ASO ‐ NC nanoparticles (NPs).
Suppression effects on cell migration and invasion
Migration and invasion are two key factors in the spread of pancreatic cancer, and oncogenic miR‐21 is thought to be involved in malignant phenotypes. As shown in Figure 2(c), compared with ASO‐NC‐NPs treatment, miR‐21 knockdown with ASO significantly suppressed migration and invasion in the two cell lines. The E‐cadherin and vimentin proteins that relate to EMT and contribute to cell migration and invasion were also detected by Western blot analysis. Consistent with the results of the biological studies for PANC‐1 and MIA PaCa‐2 cells, the expression levels of the E‐cadherin protein were upregulated to 178.06 ± 4.71% and 545.93 ± 41.21% in cells treated with ASO‐miR‐21‐NPs, whereas vimentin levels were downregulated to 45.73 ± 0.35% and 43.66 ± 3.69% (P < 0.01) (Fig. 2d).
Synergistic effects of ASO‐miR‐21 and Gem on cellular apoptosis and cytotoxicity
The highest apoptosis rate was detected both in PANC‐1 and Mia PaCa‐2 cells receiving ASO‐miR‐21‐Gem‐NPs treatment (P < 0.05 compared with other groups) (Fig. 3a). Consistent with the flow cytometry results, the most significant downregulation of Bcl‐2 protein was observed in the combined therapy groups in PANC‐1 cells (34.39 ± 3.77%) and Mia PaCa‐2 cells (26.30 ± 2.33%) (Fig. 3b). These results verified the synergistic effect of pancreatic cancer cell on apoptosis induced by the combination of ASO‐miR‐21 and Gem. Consequently, the combined ASO‐miR‐21‐Gem‐NPs therapy resulted in the enhanced cytotoxicity of pancreatic cancer cells (Fig. 3c). The IC50 concentration of Gem‐sol and ASO‐miR‐21‐Gem‐NPs was 38.92 μM versus 22.38 μM for PANC‐1 cells (P < 0.05) and 38.29 μM versus 6.65 μM for Mia PaCa‐2 cells (P < 0.01), respectively.
Figure 3.
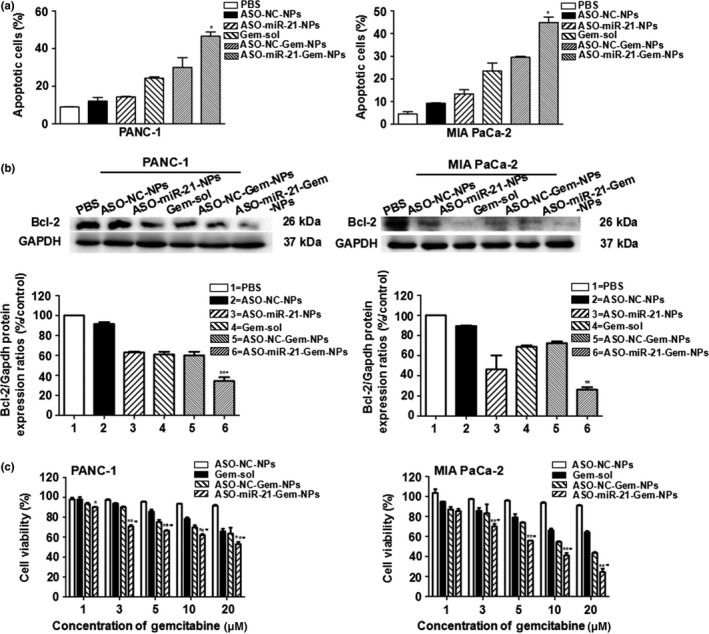
In vitro synergetic antitumor effects of microRNA‐21 antisense oligonucleotide (ASO‐miR‐21) and gemcitabine (Gem) combination therapy. PANC‐1 and Mia PaCa‐2 pancreatic cancer cells were treated with PBS, ASO‐NC (negative control) nanoparticles (NPs), ASO‐miR‐21‐NPs, Gem solution (Gem‐sol), ASO‐NC‐Gem‐NPs, or ASO‐miR‐21‐Gem‐NPs. (a) Cells were harvested and stained with annexin V‐FITC/propidium iodide, and the cellular apoptosis assay was undertaken using flow cytometry analysis. *P < 0.05 versus ASO ‐ NC‐Gem‐NPs. (b) Western blot assay was carried out to detect the apoptotic protein Bcl‐2. **P < 0.01 versus ASO‐NC‐Gem‐NPs; # P < 0.05 versus ASO‐miR‐21‐NPs. (c) MTS assays were undertaken to evaluate cell viability. Concentration of Gem was 1–20 μM, concentration of ASO was 100 nmol/L, and the cells treated with PBS were used as a control. *P < 0.05, **P < 0.01 versus Gem; # P < 0.05 versus ASO ‐ NC‐Gem‐NPs. Data are shown as mean ± SE (n = 3).
Biodistribution and tumor‐targeted effects of NPs detected by MRI
We used MIA PaCa‐2 cells to establish human tumor xenograft models in BALB/c nude mice based on the more significant cytotoxicity results in vitro. In addition, we confirmed that both PANC‐1 cells and MIA PaCa‐2 cells were CD44v6‐positive (Data S1, Fig. S1a). CD44v6 is overexpressed in pancreatic cancer and scFvCD44v6 could bind to CD44v6(+) cells efficiently and selectively.14, 21 Because IONP is an MRI negative contrast agent, the more IONPs that accumulated in the tissues, the lower the T2 value detected. The biodistribution and tumor‐targeted ability of targeted NPs and non‐targeted NPs was continuously observed with MRI after systemic injection. As shown in Figure 4(a), both targeted and non‐targeted NPs circulated in the bloodstream soon after the injection, and the MRI signal decreased in the liver. Two hours after injection, the T2 values of tumor tissues began to decrease and both the targeted NPs and non‐targeted NPs signals were lower at 4 h than the baseline signals obtained pre‐injection, indicating the accumulation of NPs in tumor tissues. However, the MRI signal intensity of the non‐targeted group increased and had returned to almost the original level by 6 h after injection, whereas the targeted NPs group maintained a lower signal, suggesting the selective and long‐lasting accumulation of the targeted NPs in pancreatic tumors (Data S1, Fig. S1b). As shown in Figure 4(b), the MRI signal intensity of liver decreased significantly after injection, and the metastatic nodules were clearly imaged, which led us to observe liver tumor metastasis non‐invasively by MRI during the antitumor studies reported in subsequent sections. Prussian blue staining was carried out on tumor tissue sections to identify the targeted delivery of NPs. Consistent with the MRI imaging results, there were a few Prussian blue‐positive cells in the tumors treated with targeted NPs but few in the tumors treated with non‐targeted NPs.
Figure 4.
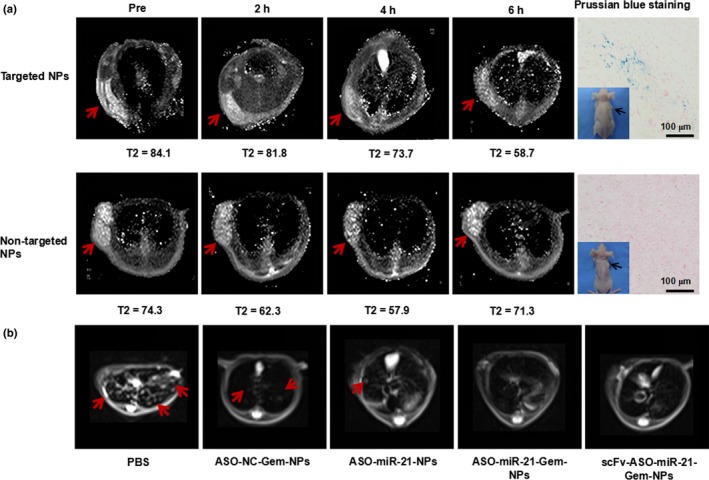
Distribution and tumor‐targeted ability of nanoparticles (NPs) and pancreatic cancer hepatic metastases monitored with MRI. (a) Accumulation of targeted and non‐targeted NPs in tumor tissue confirmed using Prussian blue staining of tumor tissue sections. (b) Metastatic nodules in tumor‐bearing mice were imaged by MRI after different treatments with PBS, negative control antisense oligonucleotide and gemcitabine (ASO‐NC‐Gem‐NPs), ASO‐microRNA‐21 (miR‐21)‐NPs, ASO‐miR‐21‐Gem‐NPs combination therapy, or single‐chain variable fragment (ScFv)‐ASO‐miR‐21‐Gem‐NPs. Treatment with ASO‐miR‐21 efficiently reduced pancreatic carcinoma liver metastasis.
Nanomedicine containing ASO‐miR‐21 and Gem reduced tumor metastasis and growth in vivo
As shown in Figure 5, the most effective tumor suppression was observed in mice treated with the combination. The tumor volumes and weights of mice treated with scFv‐ASO‐miR‐21‐Gem‐NPs were 145.00 ± 7.25 mm3 and 0.11 ± 0.01 g, respectively, 21 days after treatment. These values were much smaller than those for mice treated with PBS, ASO‐NC‐NPs, ASO‐miR‐21‐NPs, Gem‐sol, ASO‐NC‐Gem‐NPs, or ASO‐miR‐21‐Gem‐NPs (P < 0.05). In addition, a change in bodyweight is often used to gauge the toxicity of therapy factors.22 A loss of bodyweight was observed in mice receiving Gem‐sol, whereas the weights of mice receiving the nanomedicine treatments, including ASO‐NC‐NPs, ASO‐miR‐21‐NPs, ASO‐NC‐Gem‐NPs, ASO‐miR‐21‐Gem‐NPs, and scFv‐ASO‐miR‐21‐Gem‐NPs, were similar to the weights of mice in the PBS‐treated control group (Fig. 5b). With increasing duration of treatment, mice receiving Gem‐sol showed obvious weight loss, especially after 2 weeks, indicating the relatively high toxicity of non‐targeted cytotoxic drugs compared to biocompatible nanomedicines.
Figure 5.
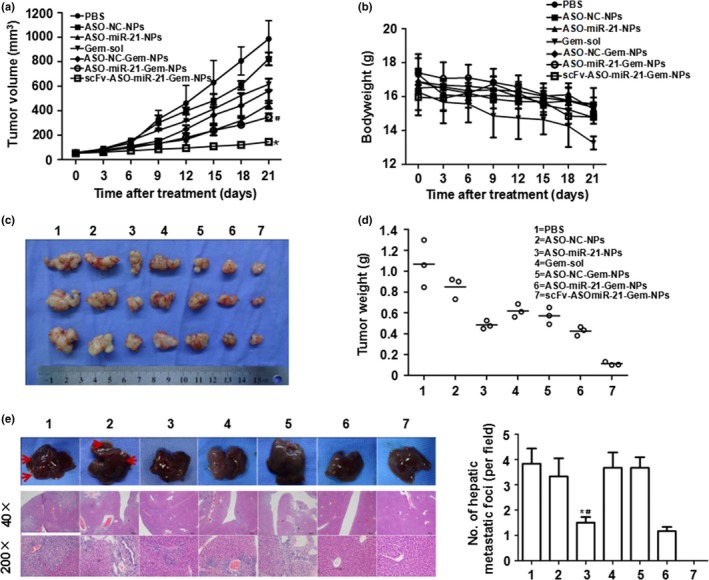
Tumor suppression in animal tests. MIA PaCa‐2 pancreatic cancer cells were inoculated s.c. into the right flank of BALB/c nude mice. When the tumor volume had reached approximately 50 mm3, the mice were randomly divided into seven groups, and received different treatments. Tumor volumes (a) and bodyweights (b) of the animals were measured every 3 days for 3 weeks. At the end of the experiment, the mice were killed and the tumors were isolated (c) and weighed (d). *P < 0.05 versus nanoparticles with microRNA‐21 antisense oligonucleotide and gemcitabine (ASO‐miR‐21‐Gem‐NPs); # P < 0.05 versus ASO ‐ NC‐Gem‐NPs (negative control). Livers were isolated and metastatic lesions were evaluated by tissue section H&E staining (e). *P < 0.05 versus Gem solution (Gem‐sol); # P < 0.05 versus ASO ‐ NC‐Gem‐NPs. (c, e): 1, PBS; 2, ASO‐NC‐NPs; 3, ASO‐miR‐21‐NPs; 4, Gem‐sol; 5, ASO‐NC‐Gem‐NPs; 6, ASO‐miR‐21‐Gem‐NPs; 7, single‐chain variable fragment (scFv)‐ASO‐miR‐21‐Gem‐NPs. Data are shown as mean ± SE (n = 3).
Liver metastasis commonly occurs in advanced pancreatic cancer. We observed liver metastasis using MRI and found that there were still metastatic nodules in the liver after treatment with Gem alone (Fig. 4b). However, when ASO‐miR‐21 was combined with Gem, the metastasis was inhibited. As shown in Figure 5(e), the liver metastatic lesions were confirmed by H&E staining. The liver metastases of mice receiving scFv‐ASO‐miR‐21‐Gem‐NPs, ASO‐miR‐21‐Gem‐NPs, or miR‐21 ASO‐NPs were significantly fewer in number than those of the mice receiving Gem alone, ASO‐NC‐Gem‐NPs, ASO‐NC‐NPs, or the PBS control (P < 0.05). These results indicated that the addition of ASO‐miR‐21 therapy significantly inhibited the liver metastasis of pancreatic cancer.
Histological and immunohistochemical studies of tumor tissues
Immunohistochemical studies and H&E staining were carried out on tumor sections to observe tumor cell density, cellular apoptosis, and protein expression associated with miR‐21 silencing, metastasis, and apoptosis. Decreased cancer cell densities were observed in tumor tissues in mice that received ASO‐miR‐21‐NPs, Gem‐sol, ASO‐NC‐Gem‐NPs, ASO‐miR‐21‐Gem‐NPs, or scFv‐ASO‐miR‐21‐Gem‐NPs treatments (Fig. 6). The most obvious decrease in the density of cancer cells was observed in mice treated with scFv‐ASO‐miR‐21‐Gem‐NPs. The expression of the PDCD4 and PTEN proteins was increased the most in scFv‐ASO‐miR‐21‐Gem‐NPs‐treated mice, indicating that ASO‐miR‐21 was delivered into the tumor tissue by the targeted nanovectors in vivo and suppressed miR‐21 efficiently. In addition, consistent with the results of the in vitro Western blot assays (Fig. 2d), the downregulation of vimentin and the upregulation of E‐cadherin were also detected in mice in which miR‐21 had been inhibited, especially those treated with scFv‐ASO‐miR‐21‐Gem‐NPs. The TUNEL results were consistent with tumor regression and indicated that scFv‐ASO‐miR‐21‐Gem‐NPs induced the highest level of apoptotic nuclei, upregulated Bax protein, and downregulated Bcl‐2 protein. These results suggested that the enhanced anti‐metastasis and anti‐proliferation effects on pancreatic cancer were accomplished by the targeted co‐delivery of ASO and Gem.
Figure 6.
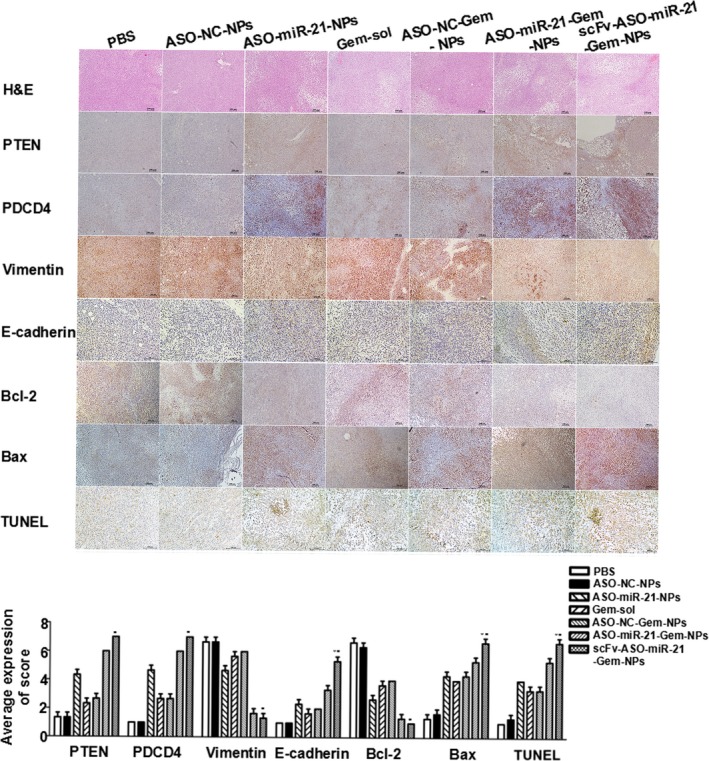
Histological studies of tumor sections from BALB/c nude mice s.c. inoculated with MIA PaCa‐2 pancreatic cancer cells into the right flank. When the tumor volume had reached approximately 50 mm3, the mice were randomly divided into seven groups, and received different treatments: PBS nanoparticles with negative control antisense oligonucleotide (ASO‐NC‐NPs); ASO‐microRNA‐21 (miR‐21)‐NPs; gemcitabine solution (Gem‐sol); ASO‐NC‐Gem‐NPs; ASO‐miR‐21‐Gem‐NPs; and single‐chain variable fragment (scFv)‐ASO‐miR‐21‐Gem‐NPs. Magnification, ×200 for E‐cadherin and TUNEL; ×100 for other images. *P < 0.05 versus ASO‐miR‐21‐Gem‐NPs; # P < 0.05 versus ASO‐miR‐21‐NPs.
Discussion
In this study, we developed an effective targeted therapy and a strategy for the MRI of pancreatic cancer through the combination of ASO‐miR‐21 and Gem using multifunctional NPs, PEG‐PEI‐IONPs. Our previous studies showed that the addition of a small amount of cationic polymer, including PEI and poly(L‐lysine), to the polymer matrix could significantly increase siRNA encapsulation and transfection efficiency and improve the siRNA release and gene silencing effects.23, 24 In the present study, we used PEG‐PEI‐IONPs for the simultaneous delivery of ASO‐miR‐21 and Gem. Polyethylenimine was able to condense the ASO through the positive charges on the polymeric chain and protect them from enzymatic degradation. We characterized PEG‐PEI‐IONPs complexes as a small particle size and suitable surface potential (Data S1, Fig. S2a,b). Gemcitabine was connected to the IONPs through a tetrapeptide (GFLG) linker that was sensitive to a lysosomal cysteine protease, cathepsin B.25 There was an increased level of cathepsin B in pancreatic cancer cells, and the GFLG linker was stable in serum, which enabled the controlled release of Gem inside the tumor cells26, 27 (Data S1, Fig. S2c).
The N/P ratio (N represents the nitrogen atoms of PEI, and P represents the phosphate groups of miRNA) of 20 was confirmed to be appropriate for PEG‐PEI‐IONPs/ASO for further examination by tests for gene encapsulation ability, cytotoxicity, and transfection efficiency (Data S1, Fig. S3). Then we observed the influence of miR‐21 silencing on the biofunction of pancreatic ductal adenocarcinoma. Neoplastic cells were characterized as showing increased growth, decreased apoptosis, and the ability to induce angiogenesis and metastasis to distant organs. In addition, anchorage‐independent growth is one of the hallmarks of EMT.28 It was reported that miR‐21 targeted and inhibited a number of tumor suppressor genes, including PDCD4, PTEN, and TPM1, indicating the oncogenic potential of miR‐21.29 Treatment with PEG‐PEI‐IONPs/ASO‐miR‐21 inhibited cell proliferation and induced colony formation, migration, and invasion, which was consistent with the upregulation of the tumor suppressor genes PTEN and PDCD4 and the inhibition of the EMT‐related genes E‐cadherin and Vimentin after miR‐21 knockdown. In addition, previous studies have identified several other targets of miR‐21 that contribute to reduced apoptosis, increased tumorigenicity, and progression, including TPM1, TIMP3, MMP2, MMP9, VEGF, and Maspin. 30 In summary, all of these results verify that the ASO‐miR‐21‐NPs treatment results in the reversal of the neoplastic cellular phenotype.
Nevertheless, the curative effect of gene therapy alone remains limited. Gemcitabine is considered the first‐line agent for pancreatic cancer therapy. However, primary or secondary drug resistance and metastasis during Gem therapy is still a major problem that remains to be solved.31 Emerging evidence has indicated that overexpressed miR‐21 is strongly associated with Gem resistance in pancreatic cancer and other types of cancer. However, the emphasis has been on clinical retrospective research, statistical analyses of miR‐21 expression, and the prognoses of Gem‐treated pancreatic ductal adenocarcinoma patients. Although the synergistic antitumor effect of ASO‐miR‐21 and Gem has been investigated in vitro and in vivo, these two agents were used in separate drug‐delivery systems, and there have been few studies of the molecular mechanisms affecting metastasis.8, 11 Hence, in the current study, we investigated the synergetic antitumor activities through a “two in one” combination of ASO‐miR‐21 and Gem loaded in a co‐delivery NP carrier, and we studied the molecular mechanisms of the effects on apoptosis and metastasis.
Although PEG‐PEI‐IONPs can co‐deliver Gem and ASO‐miR‐21 into pancreatic cancer cells and exert significant synergetic antitumor effects in vitro, for their use in vivo, stability and tumor‐targeted ability are the NP's most important properties. The IONP core of the NP is an effective MRI contrast enhancement reagent that has been used to detect lymph node metastasis in patients, indicating their biosafety.32, 33 When examining T2‐weighted images of mice bearing MIA PaCa‐2 s.c. xenografts, a remarkable signal decrease was observed in the tumor, indicating that the NPs accumulated in tumor tissues. Prussian blue staining of tumor tissue sections confirmed the presence of IONPs. In addition, the potential metastatic lesions in the liver were clearly delineated owing to the MRI contrast effect of the IONPs, thus we could detect liver metastatic lesions in mice after different treatments. The inhibition of tumor growth and metastasis in mice confirmed the MRI results. Gemcitabine encapsulated in NPs with or without ASO showed stronger antitumor effects than Gem‐sol due to the enhanced permeability and retention effect of the NPs.34 After systemic treatment, the size of the NPs prevented their rapid renal clearance and prolonged their circulation time. The longer that NPs circulate in the blood, the more NPs leak out from the tumor vessels and accumulate in the tumor tissues. The growth of tumors in mice treated with ASO‐miR‐21‐Gem‐NPs or scFv‐ASO‐miR‐21‐Gem‐NPs was slower than in mice treated with ASO‐NC‐Gem‐NPs or ASO‐NC‐NPs, indicating the synergistic antitumor effects of Gem and ASO‐miR‐21. Furthermore, the scFv‐ASO‐miR‐21‐Gem‐NPs showed stronger antitumor activities than the ASO‐miR‐21‐Gem‐NPs. This result might be due to better tumor uptake mediated by the scFv “positive targeting” mechanism. After accumulating in tumor tissues, most non‐targeted NPs circulated back to the bloodstream because of the high interstitial fluid pressure in tumors, whereas targeted NPs could be captured effectively by tumor cells because the scFv recognized and bound to the antigen on the tumor cells.
Previous studies have investigated the repression of cancer metastasis mediated by ASO‐miR‐21 in melanoma, clear‐cell renal cell carcinoma, and colorectal cancer, but less attention has been paid to pancreatic cancer.35, 36, 37 MIA PaCa‐2 is a pancreatic cancer cell line with metastatic potential.38 Results of animal experiments were consistent with the in vitro experiments that showed that ASO‐miR‐21 inhibited the EMT of pancreatic cancer cells, reduced metastasis, and increased the cellular apoptosis induced by Gem. Treatment with scFv‐ASO‐miR‐21‐Gem‐NPs significantly reduced liver metastasis.
In summary, the combination of ASO‐miR‐21 and Gem has synergetic effects on inhibiting pancreatic cancer progression. It has been reported that the targeted suppression of the PIK3R1 gene might be the molecular mechanism in tumor metastasis modulated by miR‐21 in breast cancer.39 Further studies focusing on possible suppressors of miR‐21 and the downstream pathway leading to tumor metastasis in pancreatic cancer are needed. Based on our results, the scFv‐ASO‐miR‐21‐Gem‐loaded NPs have low systemic toxicity and significant theranostic benefits for targeting pancreatic cancer. With this special vector, we can administer combined medicines to treat pancreatic cancer and use MRI to evaluate efficacy and to detect tumor metastasis.
In conclusion, we have revealed the synergetic inhibitory effect of miR‐21 ASO and Gem on human pancreatic cancer cells using a targeted co‐delivery nanomedicine strategy. The downregulation of oncogenic miR‐21 by ASO resulted in the upregulation of tumor‐suppressor genes and the inhibition of EMT, which consequently decreased the proliferation and clonal formation, migration, and invasion of pancreatic cancer cells in vitro, and markedly reduced liver metastasis in vivo. Co‐delivery of ASO and Gem using NPs induced more cell apoptosis and inhibited the growth of pancreatic cancer cells to a greater extent than individual ASO or Gem treatments, both in vitro and in vivo. These results suggest that the combination of ASO‐miR‐21 and chemotherapy using scFv functionalized NPs is promising for pancreatic cancer treatment.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- ASO
antisense oligonucleotide
- ASO‐miR‐21‐Gem‐NPs
PEG‐PEI‐IONPs/ASO‐miR‐21‐Gem
- ASO‐NC‐Gem‐NPs
PEG‐PEI‐IONPs/ASO‐NC‐Gem
- ASO‐NC‐NPs
PEG‐PEI‐IONPs/ASO‐NC
- ASO‐miR‐21‐NPs
PEG‐PEI‐IONPs/ASO‐miR‐21
- EMT
epithelial–mesenchymal transition
- Gem
gemcitabine
- Gem‐sol
gemcitabine solution
- IONP
magnetic iron oxide nanoparticle
- miRNA
microRNA
- MTS
3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐ 2H‐tetrazolium
- NC
negative control
- NP
nanoparticle
- PDCD4
programmed cell death 4
- PEI
polyethylenimine
- PTEN
phosphatase and tensin homolog
- scFv
single‐chain variable fragment antibody
- TPM1
tropomyosin 1
Supporting information
Fig. S1. (a) Expression of CD44v6 in PANC‐1 and MIA PaCa‐2 cells detected by immunofluorescence and Western blot analyses. (b) Percentage of MRI signal changes detected in tumor tissue after injection of targeted nanoparticles (NPs) or non‐targeted NPs in MIA PaCa‐2 bearing mice.
Fig. S2. Particle size and zeta potential (a) and transmission electronic microscopy images (TEM) of nanoparticles (NPs). (b) Controlled release of gemcitabine (Gem) from NPs.
Fig. S3. (a) Gel retardation assay of nanoparticles (NPs). (b) Influence of NPs on cell viability. (c) Flow cytometry assay to observe the transfection efficiency of NPs.
Data S1. Supplementary information.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81502503, 81572396, and 81672408), Natural Science Foundation of Guangdong Province, China (Grant Nos. 2016A030310191 and 2014A030313050), Science and Technology Planning Project of Guangdong Province, China (Grant No.2013B021800233) Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20130171120093), Science and Technology Program of Guangzhou, China (Grant No. 201508020013), Program of Science and Technology star of Zhujiang Guangzhou city, China (Grant No. 201610010078), Key Laboratory of Malignant Tumor Molecular Mechanism and Translational Medicine of Guangzhou Bureau of Science and Information Technology (Grant No. [2013]163), Key Laboratory of Malignant Tumor Gene Regulation and Target Therapy of Guangdong Higher Education Institutes (Grant No. KLB09001), Medical Science Foundation of Guangdong Province (Grant No. A2014262), and Medical Science Foundation of Sun Yat‐sen University (Grant No. 14ykpy29).
Cancer Sci 108 (2017) 1493–1503
Funding information
This work was supported by the National Natural Science Foundation of China (81502503, 81572396, 81672408); Natural Science Foundation of Guangdong Province, China (2016A030310191, 2014A030313050); Science and Technology Planning Project of Guangdong Province, China (Grant No.2013B021800233) Specialized Research Fund for the Doctoral Program of Higher Education (20130171120093); Science and Technology Program of Guangzhou, China (201508020013); Program of Science and Technology star of Zhujiang Guangzhou city, China (201610010078); Key Laboratory of Malignant Tumor Molecular Mechanism and Translational Medicine of Guangzhou Bureau of Science and Information Technology ([2013]163); Key Laboratory of Malignant Tumor Gene Regulation and Target Therapy of Guangdong Higher Education Institutes (KLB09001); Medical Science Foundation of Guangdong Province (A2014262); Medical Science Foundation of Sun Yat‐sen University (14ykpy29).
Contributor Information
Kaihong Huang, Email: huangkh@mail.sysu.edu.cn.
Linjuan Zeng, Email: zenglinj@mail.sysu.edu.cn.
References
- 1. Siegel RL, MillerK D, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29. [DOI] [PubMed] [Google Scholar]
- 2. Wu ZH, Pfeffer LM. MicroRNA regulation of F‐box proteins and its role in cancer. Semin Cancer Biol 2016; 36: 80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonci D, Coppola V, Patrizii M et al A microRNA code for prostate cancer metastasis. Oncogene 2016; 35: 1180–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi DB, Wang YW, Xing AY et al C/EBPα‐induced miR‐100 expression suppresses tumor metastasis and growth by targeting ZBTB7A in gastric cancer. Cancer Lett 2015; 369: 376–85. [DOI] [PubMed] [Google Scholar]
- 5. Wang W, Li J, Zhu W et al MicroRNA‐21 and the clinical outcomes of various carcinomas: a systematic review and meta‐analysis. BMC Cancer 2014; 14: 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009; 10: 704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang Y, Yang YB, Zhang XH, Yu XL, Wang ZB, Cheng XC. MicroRNA‐21 gene and cancer. Med Oncol 2013; 30: 376. [DOI] [PubMed] [Google Scholar]
- 8. Giovannetti E, Funel N, Peters GJ et al MicroRNA‐21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res 2010; 70: 4528–38. [DOI] [PubMed] [Google Scholar]
- 9. Hong L, Han Y, Zhang Y et al MicroRNA‐21: a therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets 2013; 17: 1073–80. [DOI] [PubMed] [Google Scholar]
- 10. Dhayat SA, Abdeen B, Köhler G, Senninger N, Haier J, Mardin WA. MicroRNA‐100 and microRNA‐21 as markers of survival and chemotherapy response in pancreatic ductal adenocarcinoma UICC stage II. Clin Epigenetics 2015; 7: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moriyama T, Ohuchida K, Mizumoto K et al MicroRNA‐21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Mol Cancer Ther 2009; 8: 1067–74. [DOI] [PubMed] [Google Scholar]
- 12. Wang H, Wu Y, Zhao R, Nie G. Engineering the assemblies of biomaterial nanocarriers for delivery of multiple theranostic agents with enhanced antitumor efficacy. Adv Mater 2013; 25: 1616–22. [DOI] [PubMed] [Google Scholar]
- 13. Zhao X, Li F, Li Y et al Co‐delivery of HIF1α siRNA and gemcitabine via biocompatible lipid‐polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 2015; 46: 13–25. [DOI] [PubMed] [Google Scholar]
- 14. Chen Y, Huang K, Li X, Lin X, Zhu Z, Wu Y. Generation of a stable anti‐human CD44v6 scFv and analysis of its cancer‐targeting ability in vitro. Cancer Immunol Immunother 2010; 59: 933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Wang W, Lian G et al Development of an MRI‐visible nonviral vector for siRNA delivery targeting gastric cancer. Int J Nanomedicine 2012; 7: 359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qian C, Wang Y, Chen Y et al Suppression of pancreatic tumor growth by targeted arsenic delivery with anti‐CD44v6 single chain antibody conjugated nanoparticles. Biomaterials 2013; 34: 6175–84. [DOI] [PubMed] [Google Scholar]
- 17. Zeng L, Li J, Wang Y et al Combination of siRNA‐directed Kras oncogene silencing and arsenic‐induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine 2014; 10: 463–72. [DOI] [PubMed] [Google Scholar]
- 18. Li J, Chen Y, Zeng Linjuan et al A nanoparticle carrier for co‐delivery of gemcitabine and small interfering rna in pancreatic cancer therapy. J Biomed Nanotechnol 2016; 12: 1654–66. [DOI] [PubMed] [Google Scholar]
- 19. Devulapally R, Foygel K, Sekar TV, Willmann JK, Paulmurugan R. Gemcitabine and antisense‐microRNA co‐encapsulated PLGA‐PEG polymer nanoparticles for hepatocellular carcinoma therapy. ACS Appl Mater Interfaces 2016; 8: 33412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mace TA, Collins AL, Wojcik SE, Croce CM, Lesinski GB, Bloomston M. Hypoxia induces the overexpression of microRNA‐21 in pancreatic cancer cells. J Surg Res 2013; 184: 855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gansauge F, Gansauge S, Zobywalski A et al Differential expression of CD44 splice variants in human pancreatic adenocarcinoma and in normal pancreas. Cancer Res 1995; 55: 5499–503. [PubMed] [Google Scholar]
- 22. Zhang XD, Wu D, Shen X, Liu PX, Fan FY, Fan SJ. In vivo renal clearance, biodistribution, toxicity of gold nanoclusters. Biomaterials 2012; 33: 4628–38. [DOI] [PubMed] [Google Scholar]
- 23. Zeng L, Li J, Li J et al Effective suppression of the Kirsten rat sarcoma viral oncogene in pancreatic tumor cells via targeted small interfering RNA delivery using nanoparticles. Pancreas 2015; 44: 250–9. [DOI] [PubMed] [Google Scholar]
- 24. Chen Y, Lian G, Liao C et al Characterization of polyethylene glycol‐grafted polyethylenimine and superparamagnetic iron oxide nanoparticles (PEG‐g‐PEI‐SPION) as an MRI‐visible vector for siRNA delivery in gastric cancer in vitro and in vivo. J Gastroenterol 2013; 48: 809–21. [DOI] [PubMed] [Google Scholar]
- 25. Lee GY, Qian WP, Wang L et al Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano 2013; 7: 2078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lock LL, Cheetham AG, Zhang P, Cui H. Design and construction of supramolecular nanobeacons for enzyme detection. ACS Nano 2015; 7: 4924–32. [DOI] [PubMed] [Google Scholar]
- 27. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand‐targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov 2015; 14: 203–19. [DOI] [PubMed] [Google Scholar]
- 28. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest 2009; 119: 1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frampton AE, Castellano L, Colombo T et al MicroRNAs cooperatively inhibit a network of tumor suppressor genes to promote pancreatic tumor growth and progression. Gastroenterology 2014; 146: 268–77. [DOI] [PubMed] [Google Scholar]
- 30. Pan X, Wang ZX, Wang R. MicroRNA‐21: a novel therapeutic target in human cancer. Cancer Biol Ther 2010; 10: 1224–32. [DOI] [PubMed] [Google Scholar]
- 31. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371: 1039–49. [DOI] [PubMed] [Google Scholar]
- 32. Gao Z, Ma T, Zhao E et al Small is smarter: Nano MRI contrast agents – advantages and recent achievements. Small 2016; 12: 556–76. [DOI] [PubMed] [Google Scholar]
- 33. Harisinghani MG, Barentsz J, Hahn PF et al Noninvasive detection of clinically occult lymph‐node metastases in prostate cancer. N Engl J Med 2003; 348: 2491–9. [DOI] [PubMed] [Google Scholar]
- 34. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 2011; 63: 136–51. [DOI] [PubMed] [Google Scholar]
- 35. Yang CH, Yue J, Pfeffer SR, Handorf CR, Pfeffer LM. MicroRNA miR‐21 regulates the metastatic behavior of B16 melanoma cells. J Biol Chem 2011; 286: 39172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiong B, Cheng Y, Ma L, Zhang C. MiR‐21 regulates biological behavior through the PTEN/PI‐3K/Akt signaling pathway in human colorectal cancer cells. Int J Oncol 2013; 42: 219–28. [DOI] [PubMed] [Google Scholar]
- 37. Yu G, Yao W, Gumireddy K et al Pseudogene PTENP1 functions as a competing endogenous RNA to suppress clear‐cell renal cell carcinoma progression. Mol Cancer Ther 2014; 13: 3086–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deer EL, González‐Hernández J, Coursen JD et al Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010; 39: 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan LX, Liu YH, Xiang JW et al PIK3R1 targeting by miR‐21 suppresses tumor cell migration and invasion by reducing PI3K/AKT signaling and reversing EMT, and predicts clinical outcome of breast cancer. Int J Oncol 2016; 48: 471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) Expression of CD44v6 in PANC‐1 and MIA PaCa‐2 cells detected by immunofluorescence and Western blot analyses. (b) Percentage of MRI signal changes detected in tumor tissue after injection of targeted nanoparticles (NPs) or non‐targeted NPs in MIA PaCa‐2 bearing mice.
Fig. S2. Particle size and zeta potential (a) and transmission electronic microscopy images (TEM) of nanoparticles (NPs). (b) Controlled release of gemcitabine (Gem) from NPs.
Fig. S3. (a) Gel retardation assay of nanoparticles (NPs). (b) Influence of NPs on cell viability. (c) Flow cytometry assay to observe the transfection efficiency of NPs.
Data S1. Supplementary information.