

Abstract
Platinum‐based therapeutic strategies have been widely used in ovarian cancer treatment. However, drug resistance has greatly limited therapeutic efficacy. Recently, tolerance to cisplatin has been attributed to other factors unrelated to DNA. p62 (also known as SQSTM1) functions as a multifunctional hub participating in tumorigenesis and may be a therapeutic target. Our previous study showed that p62 was overexpressed in drug‐resistant ovarian epithelial carcinoma and its inhibition increased the sensitivity to cisplatin. In this study, we demonstrate that the activity of the NF‐κB signaling pathway and K63‐linked ubiquitination of RIP1 was higher in cisplatin‐resistant ovarian (SKOV3/DDP) cells compared with parental cells. In addition, cisplatin resistance could be reversed by inhibiting the expression of p62 using siRNA. Furthermore, deletion of the ZZ domain of p62 that interacts with RIP1 in SKOV3 cells markedly decreased K63‐linked ubiquitination of RIP1 and inhibited the activation of the NF‐κB signaling pathway. Moreover, loss of the ZZ domain from p62 led to poor proliferative capacity and high levels of apoptosis in SKOV3 cells and made them more sensitive to cisplatin treatment. Collectively, we provide evidence that p62 is implicated in the activation of NF‐κB signaling that is partly dependent on RIP1. p62 promotes cell proliferation and inhibits apoptosis thus mediating drug resistance in ovarian cancer cells.
Keywords: Drug resistance, NF‐KB pathway, ovarian cancer, p62/SQSTM1, RIP1
Platinum (Pt)‐based chemotherapies are frequently used as an adjuvant for ovarian cancer treatment.1 However, drug resistance is still a major obstacle that limits efficacy of Pt‐based therapeutic regimens.2 Since the 1970s, tolerance to Pt has been blamed on effects unrelated to DNA damage. Recently, investigators have paid more attention to these “non‐nuclear” effects and the molecular pathways that are involved in the cell survival and apoptotic escape.3 Our previous study showed that sequestosome‐1 (p62/SQSTM1) was highly expressed in cisplatin‐resistant ovarian cancer cells. Furthermore, resistance to Pt was removed when we inhibited the p62 expression with the siRNA.4 These results suggested that p62 may play a key role in the mechanism of platinum‐resistance.
p62, also known as SQSTM1, was the first identified autophagy adaptor and is important for cellular detoxification and resistance to nutrient stress.5 However, in contrast to other autophagy adaptors, p62 also functions as a multifunctional hub. It participates in many important cellular events that control proliferation and apoptosis, particularly during tumorigenic conditions.6, 7, 8, 9 The autophagy‐independent role for p62 is reported in various cancers, including ovarian,10 breast,11 liver,12 and kidney.13 Increasing evidence suggests that elevated levels of p62 are correlated with poor prognosis in epithelial ovarian cancer.14 Furthermore, knockout of p62 in mice significantly increases the sensitivity of pulmonary adenocarcinomas to doxorubicin which indicates that p62 may be involved in the regulation of survival signaling.15 Our previous work suggested that p62 accumulation may activate transcription factor Nrf2 to confer cancer cells resistance to chemotherapy.10 Recent advances in the understanding of p62 function suggest that it increases the activity of the nuclear factor kappa B (NF‐κB) pathway in many cell systems.9, 16, 17, 18 High NF‐κB activity may trigger several pro‐survival genes expression that contributes to chemotherapeutic resistance. Thus, identifying the role of p62 in the activation of NF‐κB may improve the understanding of the mechanism of drug resistance. Furthermore, given the complex roles of p62, research that goes beyond simple increases or decreases in levels will be necessary to understand the precise regulation of this molecule in pro‐survival signaling pathways during tumorigenesis.
Receptor interacting serine/threonine kinase 1 (RIP1, also known as RIPK1) has emerged as a key upstream regulator that controls pro‐survival signaling as well as the activation of multiple cell death pathways.19, 20, 21, 22 Furthermore, the ability of RIP1 to modulate these key cellular events is tightly controlled by ubiquitination. The lysine (K) 63‐linked polyubiquitin chains serve as a docking site to mediate the formation of a complex upstream of NF‐κB that promotes the activation of pro‐survival signaling.20, 23, 24, 25 Previous studies have indicated that deletion of the ZZ (Zinc finger) domain of p62 abolished binding of RIP1 to p62 and inhibited the activation of the NF‐κB signaling pathway.26 However, we still know little about the role of these key molecules in ovarian cancer. Here, we explored the effects of cisplatin on ovarian cancer cells and found that p62 could activated the NF‐κB signaling pathway. And this activation was dependent on K63‐linked ubiquitination of RIP1 that subsequently mediated drug resistance.
Materials and Methods
Reagents and antibodies
Cisplatin, trypan blue and 3‐(4, 5‐Dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide (MTT) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). The FITC Annexin V Apoptosis Detection Kit was purchased from BD Biosciences (State of New Jersey, USA). ViaFect Transfection Reagent was purchased from Promega (Madison, MI, USA). Nuclear and cytoplasmic protein extraction kit was purchased from Beyotime (China). The following anti‐body were used: anti‐p62 (Abcam, Cambridge, MA, USA); anti‐p50, anti‐p65, anti‐β actin (Proteintech, Chicago, IL, USA); anti‐RIP1, anti‐K63‐linkage Specific Polyubiquitin (Cell Signaling Technology, Beverly, Massachusetts, USA); anti‐Lamin A/C, anti‐IκB, anti‐pIκB (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Cell lines and cell culture
SKOV3 cells and their cisplatin‐resistant clone SKOV3/DDP were purchased from the Chinese Academy of Medical Sciences and Peking Union Medical College. Both cell lines were cultured in Roswell Park Memorial Institute (RPMI)‐1640 culture medium (Gibco Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2. SKOV3/DDP cells were cultured in the presence of 1 μg/mL cisplatin to maintain the resistance.
Cell transfection
p62‐siRNA and non‐target siRNA (Scramble) were obtained from Genechem (Shanghai, China).The p62‐siRNA (si‐p62) sequence was GAC‐ATC‐TTC‐CGAATC‐TAC‐A and the non‐target siRNA (Scramble) was TTC‐TCC‐GAA‐CGT‐GTC‐ACG‐T. The pcDNA3.1‐p62 and pcDNA3.1‐ΔZZ‐p62 (ZZ domain truncated mutation) and empty pcDNA3.1 vector (NC) were constructed by Sangon Biotech (Shanghai, China). 25 × 104 cells were placed into 6‐well plates. After 36 h cells were transfected with siRNA or plasmids using transfection Reagent (Promega, Madison, MI, USA) according to the manufacturer's instructions.
Cell viability assays
8 × 103 cells were seeded in 96‐well plates in each well. After 36 h cells were treated with different concentrations of cisplatin after transfection. And the MTT assay was used to evaluate cell viability. We measured the absorbance at 570 nm using a Vmax Microplate Reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Trypan blue staining
Cells were harvested and suspended in phosphate‐buffered saline (PBS). For staining, Trypan blue was used at a concentration of 0.4%. Mix 90 μL cells and 10 μL Trypan blue in 1.5 mL tube for 2 min and count cells with the hemocytometer while dead cells have blue cytoplasm. Finally, we examined the percentage of cells that have clear cytoplasm (viable cells) versus total cells.
Luciferase assay
The cells were transfected with the NF‐κB‐Luc reporter plasmid using transfection reagent (Promega, Madison, MI, USA). After drug treatment luciferase activity was measured using the Dual‐Luciferase assay kits (Promega, Madison, WI, USA) and normalized on the basis of Renilla luciferase activity.
Western blot analysis
Proteins were subjected to Western blotting as previously described.27 Then incubated with primary antibodies overnight at 4°C. Horse radish peroxidase (HRP)‐conjugated secondary antibodies were incubated (Proteintech, Chicago, IL, USA) according to the manufacturer's instructions. Then, immunodetection was performed using ECL reagent (Thermo Fisher Scientific) and visualized by a Syngene Bio Imaging (Synoptics, Cambridge, UK).
Immunoprecipitation assay
Cells were lysed in NP40 lysis buffer. Equal amounts of lysates were immunoprecipitated with 2 μg of the RIP1or K63‐linkage Specific Polyubiquitin antibody overnight at 4°C. 25 μL of protein A and G agarose (Beyotime, China) were used each sample. Beads were washed with PBS three times with 1 mL each. The eluted proteins were examined by using western blot.
Immunofluorescence staining and confocal laser microscopy
Cells were washed and fixed in 4% (w/v) paraformaldehyde/PBS for 20 min and subjected to proteinase K digestion for 1 min. Then permeabilized with 0.1% Triton X‐100 for 15 min. After blocking with bovine serum albumen for 30 min cells were incubated with primary antibody overnight at 4°C. The next day, stained with FITC/Texas Red‐conjugated secondary antibodies (1:200 dilution; Proteintech, Chicago, IL, USA) for 30 min in the dark. Finally, Cells were treated with Hoechst 33342/H2O (1 μg/mL) for 2 min, and Images were acquired by an Olympus FV1000 confocal laser microscope.
Flow cytometry
Cells (25 × 104 cells/well) were seeded in 6‐well plates. After exposure to different treatment, cells were collected and performed Annexin‐V FITC/PI staining according to the manufacturer's instructions. Samples were analyzed by Accuri C6 Flow Cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
EDU
Cells were seeded in 96‐well plate and exposed to 50 μM of 5‐ethynyl‐29deoxyuridine (EdU, RiboBio, Guangzhou, China) according to the manufacturer's instructions. Subsequently, the DNA contents were stained with Hoechst 33342 for 30 min and visualized under a microscopy (Olympus, Tokyo, Japan).
Caspase activity assay (Promega)
Cells were collected and Caspase‐Glo 3/7 Assay (Promega, Madison, MI, USA) was used as described in the manufacturer's instructions. Microplate reader (FLUOstar Omega, BMG LABTECH, German) was used for detecting.
RNA extraction and quantitative real‐time PCR
Total RNA extracted and First‐strand cDNAs synthesized were described as before.4 Quantitative real‐time PCR was done by using the MX300P instrument (Agilent, USA) followed by a 2‐step PCR protocol. The primers sequences are as follows: primers for BCL2L1, 5′‐TCCATCGATTCAGTCCCT‐3′.Primers for CCL2 5′‐CAATCAATGCCCCAGTCACC‐3′, 5′‐ CCTGAA CCCACTTCTGCTTG‐3′. Primers for TGFB1 5′‐GGATAACACACTGCAAGTGG‐3′, 5′‐GAGCTGAAGCAAT AGTTGGTG‐3′. Primers for CSF3 5′‐GCTTCCTGCTCAAGTGCTTA ‐3′, 5′‐TTCCCAGTTCTTCCATCTGC‐3′. Primers for IL‐6 5′‐CTGCAGCCACTGGTTCTGT‐3′, 5′‐CCAGAGCTGTGCAGATGAGT‐3′. Primers for ACTB 5′‐ATATCGCGTCGCTGGTCGTC‐3′, 5′‐AGGATGGCGTGAGGGAGAGC‐3′. The relative expression was calculated by ΔCt among different experimental groups normalized to ACTB expression.
Statistical analyses
Statistical analysis was performed using GraphPad Prism 5 (La Jolla, CA, USA). All the data are presented as means ± SD and carried out using the Student's t‐test. P < 0.05 was considered statistically significant.
Results
Cisplatin treatment activates the NF‐κB pathway and an increase in RIP1 K63‐linked ubiquitination in SKOV3/DDP cells
Through previous work, we determined that expression of p62 was different in parental SKOV3 human ovarian epithelial carcinoma cell lines and their established cisplatin‐resistant subline SKOV3/DDP. SKOV3/DDP cells expressed higher levels of p62 and were less sensitive to cisplatin (CDDP) treatment.4 To investigate whether the differential overexpression of p62 is correlated with a pro‐survival mechanism, we evaluated the NF‐κB signaling pathway in both cell lines. As shown in Figure 1a, p50/p65 levels markedly increased in the nucleus and phosphorylation of IκB also increased following cisplatin treatment in SKOV3/DDP cells. This result is in agreement with current knowledge on the activation of the canonical NF‐κB pathway, which results in the phosphorylation of IκB, its subsequent degradation and the release of p50/p65 complex.28, 29, 30 The luciferase reporter assay and immunofluorescence analysis confirmed our speculation that the NF‐κB signaling pathway was activated in SKOV3/DDP cells following cisplatin treatment (Fig. 1b,c).
Figure 1.
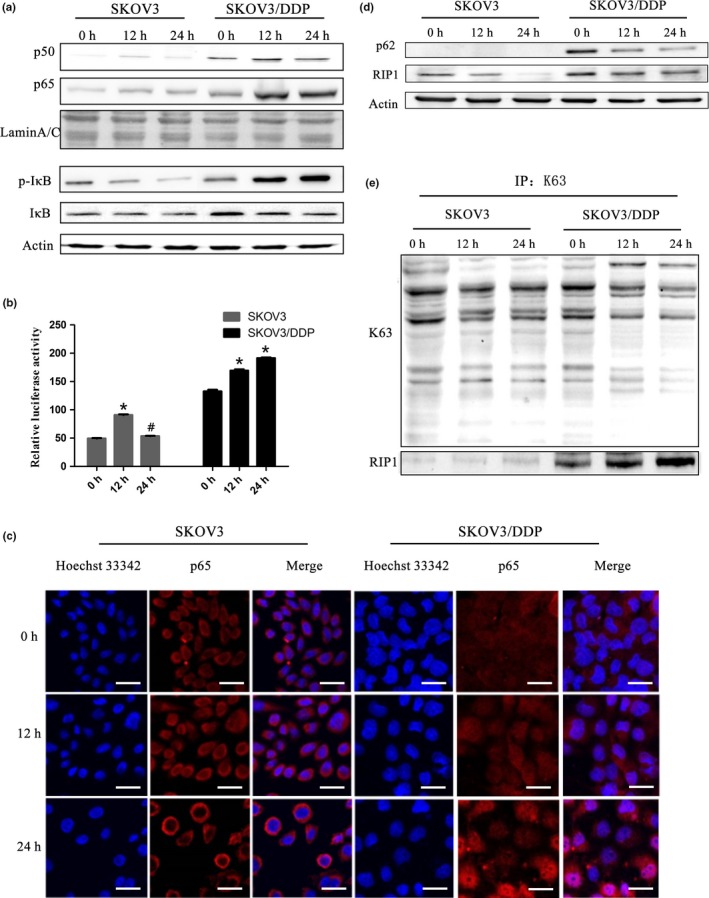
Cisplatin treatment activates the NF‐κB pathway and an increase in RIP1K63‐linked ubiquitination in SKOV3/DDP cells. (a) Cells were treated with cisplatin (6 μg/mL) and expression of NF‐κB p65 and NF‐κB p50 in the nucleus and IκB, p‐IκB in whole cell lysates was analyzed by western blotting. (b) SKOV3 and SKOV3/DDP cells were transiently transfected with a pNF‐κB‐Luc vector. The luciferase activity was assessed and normalized on the basis of Renilla luciferase activity (mean ± SD, n = 3. *P < 0.05, vs control). (c) SKOV3 and SKOV3/DDP cells were stained with Hoechst 33342 and antibodies against p65. They were observed under confocal laser microscopy (scale bar, 25 μm). (d) SKOV3 and SKOV3/DDP cells were immunoblotted (IB) with anti‐p62 and anti‐RIP1. (e) Immunoprecipitation (IP) was performed using the anti‐K63 antibody followed by western blotting using anti‐RIP1 antibody.
As noted above, RIP1 is involved in the canonical activation of NF‐κB. Thus, we investigated the K63‐linked ubiquitination of RIP1. We observed higher basal expression level of RIP1 in SKOV3/DDP cells (Fig. 1d). Furthermore, the co‐immunoprecipitation results showed that cisplatin treatment increased K63‐polyubiquitinated RIP1 over time in SKOV3/DDP cells but not in SKOV3 cells (Fig. 1e). These results prompted us to further explore the relationship between these molecules and cisplatin sensitivity in ovarian cancer cells.
Upregulation of NF‐κB signaling and K63 ubiquitination of RIP1 are dependent on p62/SQSTM1 in SKOV3/DDP cells
To investigate whether p62 affected the activation of NF‐κB or ubiquitination of RIP1, we used siRNA against p62 to inhibit its expression in SKOV3/DDP cells (Fig. 2a). In agreement with our previous study, cell viability decreased following cisplatin treatment when p62 expression was inhibited (Fig. 2b). Furthermore, using a luciferase assay we also observed p62 siRNA markedly induced downregulation of NF‐κB transcriptional activity (Fig. 2d). Interestingly, the amount of co‐immunoprecipitated K63‐polyubiquitinated RIP1 also decreased (Fig. 2c). These data indicated that p62 functioned as a positive regulator for the activation of both the NF‐κB pathway and RIP1.
Figure 2.
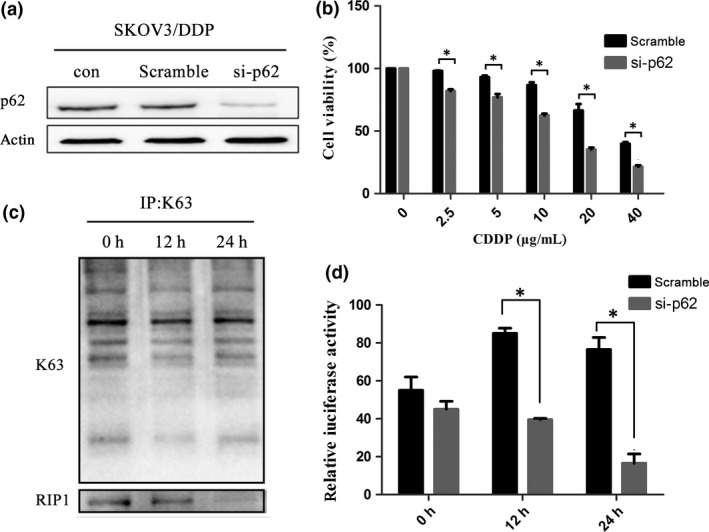
Downregulation of p62/SQSTM1 inhibits the NF‐κB pathway and RIP1K63‐linked ubiquitination in SKOV3/DDP cells. (a) SKOV3/DDP cells were transfected with si‐p62 or control siRNA (Scramble). After 24 h, cells were treated with cisplatin and the expression of p62 was analyzed by western blotting. (b) Cell viability was evaluated by MTT assay (mean ± SD, n = 3. *P < 0.05, vs scramble). (c) SKOV3/DDP cells were transfected with si‐p62. Protein lysates were used for immunoprecipitations with an anti‐K63 antibody and analyzed by western blotting using anti‐RIP1 antibody. (d) SKOV3/DDP cells with si‐p62 or control siRNA were transiently transfected with a pNF‐κB–Luc vector. The luciferase activity was assessed and normalized on the basis of Renilla luciferase activity (mean ± SD, n = 3. *P < 0.05, vs scramble).
p62/SQSTM1 activates the NF‐κB pathway by increasing K63 ubiquitination of RIP1 following cisplatin treatment in SKOV3 cells
A previous study showed that in 293T cells p62 interacted with RIP1 through a specific structure known as the ZZ domain.17 In this study, to investigate whether p62 upregulated NF‐κB through regulating the ubiquitination of RIP1, a ZZ domain truncation mutation (ΔZZ) of p62 and a wild type (wt)‐p62 were transfected into SKOV3 cells (Fig. 3a). Co‐immunoprecipitation revealed that the K63‐linked ubiquitination of RIP1 was downregulated when p62 no longer bound to RIP1 compared with cells overexpressing wt‐p62 (Fig. 3b). These result suggested that p62 can truly affect the ubiquitination of RIP1.
Figure 3.
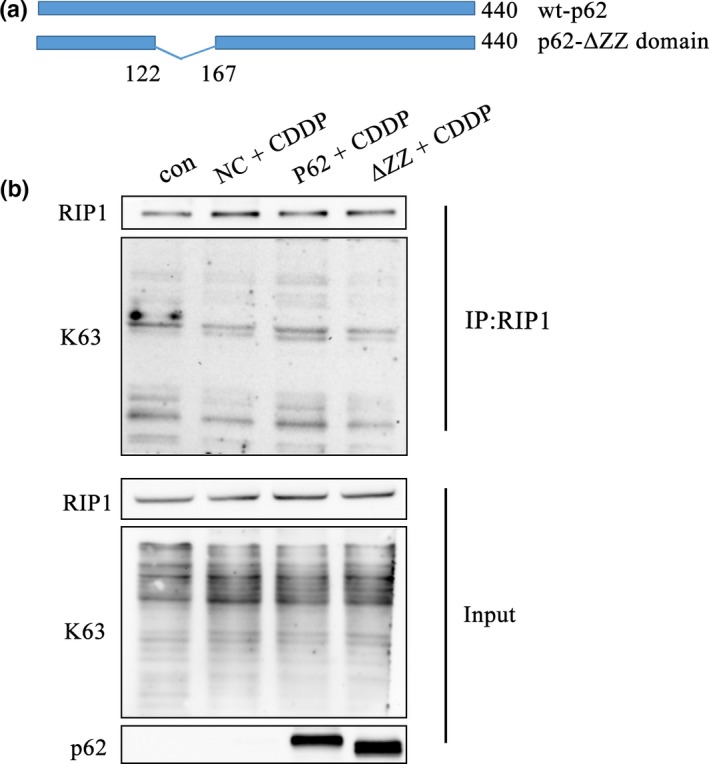
Deleting the ZZ domain of p62 inhibits the K63 ubiquitination of RIP1 following the treatment of cisplatin in SKOV3 cells. (a) Schematic representation of the p62 wild type and mutant ZZ domain constructs. (b) After transfection with wt‐p62 and ΔZZ‐p62, SKOV3 cells were treated with cisplatin (6 μg/mL) for 24 h. Immunoprecipitation was performed using the anti‐RIP1 antibody followed by western blotting using anti‐RIP1 and anti‐K63 antibody.
We then investigated the activity of NF‐κB in ΔZZ‐p62 and wt‐p62 expressing cells. As expected, deleting the ZZ domain decreased the luciferase activity compared with wt‐p62 following the same dose treatment of cisplatin (Fig. 4a). The results of western blotting and immunofluorescence showed that the p50/p65 subunits were barely expressed in the nucleus when the p62 ZZ domain was absent (Fig. 4b–d). These results indicated that p62 was necessary to activate the NF‐κB pathway by regulating the ubiquitination of RIP1.
Figure 4.
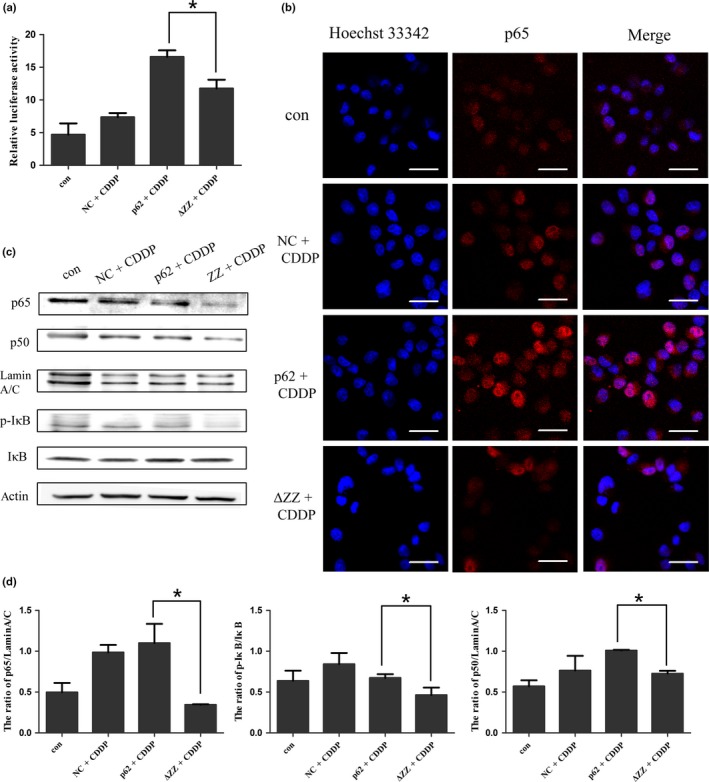
Deleting the ZZ domain of p62 inhibited the activation of NF‐κB signaling pathway in SKOV3 cells. (a) SKOV3 cells were transfected with wt‐p62 and ΔZZ‐p62. The luciferase activity was assessed and normalized on the basis of Renilla luciferase activity (mean ± SD, n = 3. *P < 0.05, vs wt‐p62). (b) Localization of p65 and nuclear staining with Hoechst 33342 were observed with confocal laser microscopy (scale bar, 25 μm). (c) Western blotting was used to analyze the expression of NF‐κB p65, NF‐κB p50 in the nucleus and IκB and p‐IκB in whole cell lysates of SKOV3 cells after transfection of different p62 plasmids. (d) Quantitation of NF‐κB p65, NF‐κB p50, IκB/p‐IκB protein levels. Data were presented as mean ± SD, n = 3. *P < 0.05, vs wt‐p62.
Abolishing the regulation of RIP1 K63‐linked ubiquitination by p62/SQSTM1 increases the sensitivity of SKOV3 cells to cisplatin
We have showed that inhibition of the K63‐linked ubiquitination of RIP1 by mutating the key domain of p62 may downregulate the activity of NF‐κB signaling pathway. Following this point, it is reasonable to speculate that loss of p62 ZZ domain may also affect the cell sensitivity to chemotherapy. In the following part, we examined cell viability using an MTT assay. As shown in (Fig. 5a), overexpression of ΔZZ‐p62 decreased the sensitivity to cisplatin treatment compared with cells transfected with wt‐p62. The trypan blue staining also suggested that transfection of the ΔZZ mutant decreased the number of viable cells (Fig. 5b).
Figure 5.
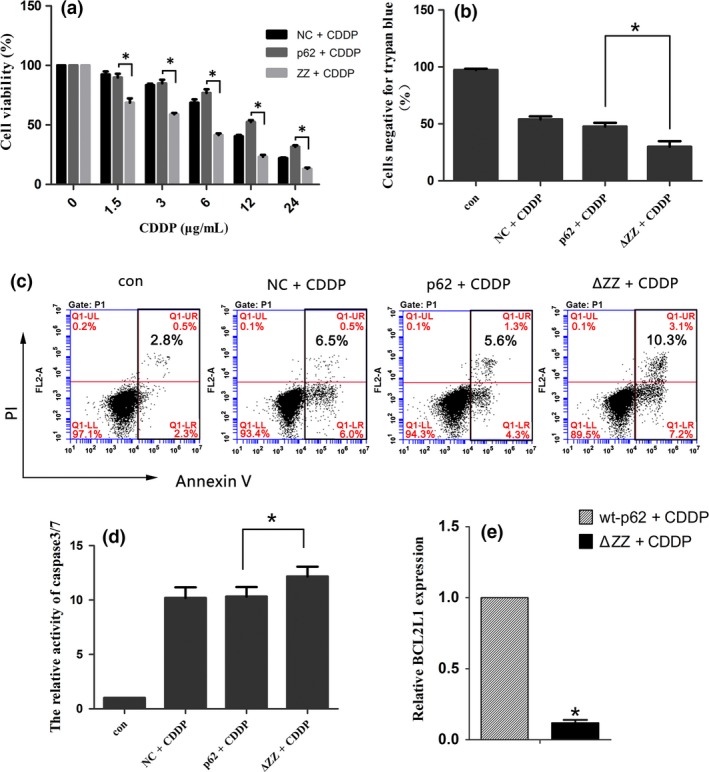
Abolishing the regulation of RIP1 K63‐linked ubiquitination by p62/SQSTM1 increases the sensitivity of SKOV3 cells to cisplatin. (a) The viability of cells was analyzed by an MTT assay. Data were presented as mean ± SD, n = 3. *P < 0.05, vs wt‐p62. (b) SKOV3 cells were treated as before. Cells were stained with Trypan blue and observed under a microscope. Data were presented as mean ± SD, n = 3. *P < 0.05, vs wt‐p62. (c) The apoptosis ratio of SKOV3 cells was detected by flow cytometry analysis. (d) Cells were analyzed using a caspase3/7 activity assay (Promega). Data are presented as mean ± SD, n = 3. *P < 0.05 vs wt‐p62. (e) The mRNA expression of BCL2L1 in SKOV3 cells after transfection of wt‐p62 and ΔZZ‐p62 plasmid. ACTB was used for normalization.Data are presented as mean ± SD, n = 3. *P < 0.05 vs wt‐p62.
We then determined if the reduction in cell number was the result of apoptosis induction or a failure in proliferation. First, we evaluated apoptosis using flow cytometry. As shown in Figure 5c, overexpression of ΔZZ‐p62 in SKOV3 cells increased apoptosis with the treatment of cisplatin. Furthermore, the caspase3/7 activity assay also confirmed this speculation (Fig. 5d).Then we investigated proliferation by examining DNA replication using EdU Cell Proliferation Assay. EdU (5‐Ethynyl‐2′‐ deoxy uridine) is incorporated into cellular DNA during replication and newly synthesized DNA is quantitated by the levels of red stain.31, 32 As shown in Figure 6a, the cells in the replicative phase were inhibited after transfection with ΔZZ‐p62 compared with wt‐p62. Interestingly, staining with Hoechst 33342 also indicated an increase in apoptotic chromatin condensation.
Figure 6.
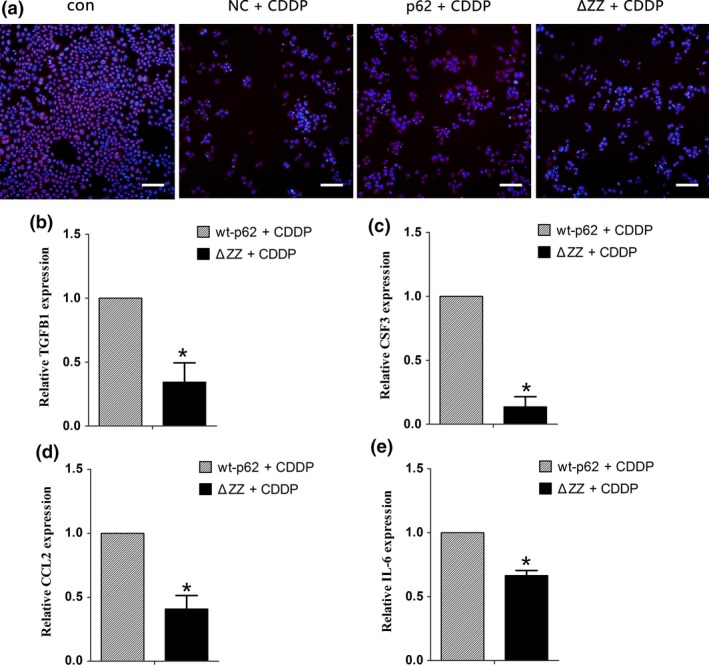
The loss of the ZZ domain decreases cell proliferation in SKOV3 cells. (a) Merged pictures show the overlay of EDU stain and Hoechst33342 (scale bar, 50 μm). (b–e) Quantitative RT‐PCR validation of well‐known NF‐κB regulated genes TGFB1, CSF3, CCL2, and IL6 in SKOV3 cells after transfection of wt‐p62 and ΔZZ‐p62 plasmid. ACTB was used for normalization. Data are presented as mean ± SD, n = 3. *P < 0.05 vs wt‐p62.
To further understand the underlying molecular mechanisms of drug resistance induced by NF‐κB activation, we then investigated several reported NF‐κB target genes33, 34: BCL2 like 1 (BCL2L1, also known as BCLX) is an anti‐apoptotic protein regulated by NF‐κB; Interleukin 6 (IL6) belongs to the cytokine/chemokine family; C‐C motif chemokine ligand 2 (CCL2) is a regulator of cell proliferation and migration; Transforming growth factor beta 1 (TGFB1) and colony stimulating factor 3 (CSF3) are both well‐studied growth factors. Our results indicated that compared with wt‐p62, SKOV3 cells transfected with ΔZZ‐p62 showed lower levels of BCL2L1 expression (Fig. 5e). Consistent with this result deleting the ZZ domain of p62 decreased expression of genes involved in proliferation (Fig. 6b–e). These results indicated that high levels of p62 in cisplatin‐resistant ovarian cancer cells may protect them from death by evading apoptosis and promoting proliferation through activation of NF‐κB signaling.
Discussion
Chemoresistance has been recognized as one of the crucial features of cancer whereby tumor cells escape the toxicity induced by drugs.35 Although the antitumor effects of platinum compounds are related to DNA damage, this chemical feature means that Pt‐based drugs also affect a variety of important cellular events including proliferation and apoptosis.36, 37 Therefore, understanding the role of these pathways may be helpful in combatting resistance. Our study suggests that the p62‐RIP1‐NF‐κB pathway plays an important role in promoting cell survival and downregulating this pathway can significantly increase the sensitivity of ovarian cancer cells to cisplatin (Fig. 7).
Figure 7.
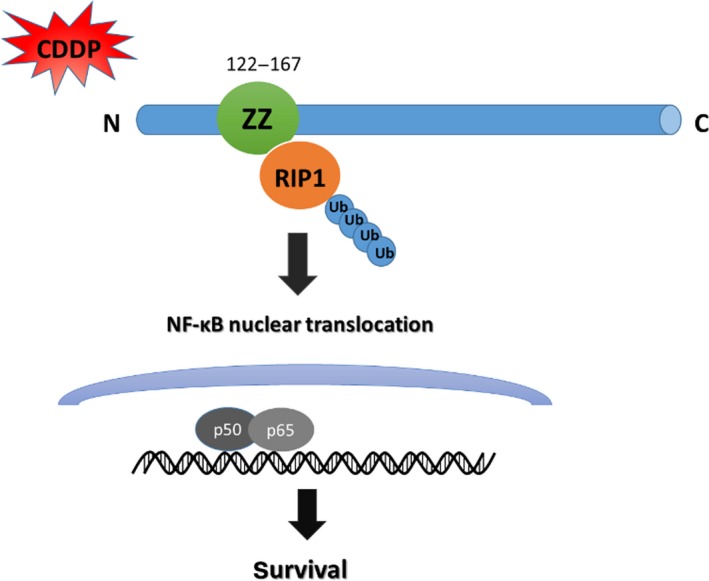
p62‐RIP1‐NF‐κB regulation pathway in ovarian cancer cells. p62 activating the NF‐κB pro‐survival pathway depends on K63‐ubiquitinated RIP1 with cisplatin treatment. Furthermore, the activation of nuclear factor‐ κB (NF‑κB) promotes the transcription of target pro‐survival genes.
We found that basal p62 expression was higher in cisplatin‐resistant ovarian cancer cells (Fig. 1d). And p62 was truly related to resistant mechanism, not only cisplatin but other drug, in SKOV3/DDP cells during our previous study (Fig. S1). Although p62 protein levels decreased with cisplatin treatment in SKOV3/DDP cells, which was the result of autophagy, p62 mRNA transcripts still remained constant.4 Furthermore, downregulating the expression of p62 increased the sensitivity to chemotherapy (Fig. 2b). Thus we believe that the mechanism of drug resistance in ovarian cancer cells is associated with the high level of p62. However, the precise molecular mechanisms of how p62 contributed to drug resistance in ovarian cancer remained poorly understood. p62 is overexpressed in many tumors and functions as a multi‐functional hub that mediates its interactions with many binding partners. These include proteins involved in cellular transformation, proliferation and signaling such as NF‐κB and MAPK.38, 39, 40 We observed an increase in the transcriptional activity of NF‐κB in drug‐resistant SKOV3/DDP ovarian cancer cells when compared with parental SKOV3 cells. This result is in agreement with the previous findings in ovarian cancer cells.41, 42 Therefore, it was reasonable to focus on the p62‐mediated regulation of the pro‐survival signaling complex NF‐κB and explore the mechanism of chemoresistance in ovarian cancer cells.
RIP1 is a member of the RIP serine/threonine kinase family and binds with death receptors such as tumor necrosis factor (TNF) and Fas to promote apoptosis. Interestingly, cancer cells also show greater sensitivity to TNF‐induced death when RIP1 is absent because of inactivation of the NF‐κB survival pathway.23, 43 Recent studies suggested that the K63‐linked polyubiquitin chains of RIP1 can rapidly recruit the IKK complex to promote phosphorylation of NFKBIA and activate NF‐κB signaling, which contributed to cell survival in lymphoma glioblastoma, and colorectal cancer cells.44, 45, 46 Furthermore, Sanz established the relationship between RIP1 and p62 in 293T cells using co‐immunoprecipitation. She found that deletion of the ZZ domain of p62 significantly inhibited the binding of RIP1. This interaction increased the expression of pro‐survival genes through upregulating the NF‐κB signaling pathway.17 In the current study, cisplatin treatment resulted in an increase of K63‐linked ubiquitination of RIP1 in drug‐resistant ovarian cancer cells. When we deleted the ZZ domain that interacts with RIP1, the K63‐ubiquitinated form of RIP1 was reduced. Furthermore, the transcriptional activity of NF‐κB was also inhibited. We then evaluated sensitivity to cisplatin in cells harboring the deleted domain. These cells showed a reduced proliferative capacity and an increase in apoptosis when they lost regulation of RIP1. They are supported by prior studies from Teramachi's laboratory who also observed that cell growth was inhibited by using the inhibitor XRK3F2 that specifically blocks the ZZ domain of p62 in myeloma cells.47 These results confirm our hypothesis that p62 can regulate the NF‐κB signaling pathway through the ubiquitination of RIP1 in ovarian cancer cells.
However, there are still many questions to be answered. We observed that overexpression of wt‐p62 did not completely block apoptosis in ovarian cancer cells. We speculate that in SKOV3/DDP cells the escape from apoptosis was partly dependent on the activation of p62‐RIPK1‐NF‐κB survival pathway under the stress of chemotherapy. However, overexpression of the multi‐functional p62 protein is likely to have pleiotropic effects.48, 49, 50 Furthermore, p62 may also function as a switch that regulates opposing pathways depending on its interaction with different molecules in physiological and pathological conditions.
Collectively, our study provides evidence that, in ovarian cancer cells, p62 participates in the mechanism of cisplatin‐resistance by activating the NF‐κB pro‐survival pathway, and this process depends on K63‐ubiquitinated RIP1. These data contribute to understanding the role of “non‐nuclear” targets in chemotherapy and may aid development of an effective therapeutic strategy to overcome Pt‐based drug resistance in ovarian cancer.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- CDDP/DDP
cisplatin
- NF‐κB
nuclear factor kappa B
- p50
NFKB1 (nuclear factor kappa B subunit 1)
- p62/SQSTM1
sequestosome‐1
- p65
RELA proto‐oncogene
- RIP1
Receptor interacting serine/threonine kinase 1
- ZZ
Zinc finger
Supporting information
Fig. S1 p62 participated in the mechanism of VK3‐resistance in SKOV3/DDP cells (a) SKOV3 and SKOV3/DDP cells were treated with varying doses of VK3 for 8 and 16 h. Cell viability was determined by MTT assay. Data were presented as a mean ± SD, n = 3. (b) Western blot analysis for the nuclear Nrf2 protein in transfected SKOV3/DDP cells.
Acknowledgments
The work was supported by The National Natural Science Foundation of China (No. 81672948, 81202552, 81501982, 81472419) and Jilin Provincial Research Foundation for International Science and Technology Cooperation Projects, China (Nos. 20160414005GH). And we are grateful to State Key Laboratory on Integrated Optoelectronics College of Electronic Science and Engineering Jilin University for providing confocal laser microscope.
Cancer Sci 108 (2017) 1405–1413
Funding Information
The National Natural Science Foundation of China, (‘81672948‘, ‘81202552‘, ‘81501982‘, ‘81472419‘); Jilin Provincial Research Foundation for International Science and Technology Cooperation Projects, China, (20160414005GH).
Contributor Information
Jing Su, Email: sujing@jlu.edu.cn.
Lian‐Kun Sun, Email: sunlk@jlu.edu.cn.
References
- 1. Alkema NG, Wisman GB, van der Zee AG, van Vugt MA, de Jong S. Studying platinum sensitivity and resistance in high‐grade serous ovarian cancer: different models for different questions. Drug Resist Update 2016; 24: 55–69. [DOI] [PubMed] [Google Scholar]
- 2. Ferreira JA, Peixoto A, Neves M et al Mechanisms of cisplatin resistance and targeting of cancer stem cells: adding glycosylation to the equation. Drug Resist Update 2016; 24: 34–54. [DOI] [PubMed] [Google Scholar]
- 3. Gatti L, Cassinelli G, Zaffaroni N, Lanzi C, Perego P. New mechanisms for old drugs: insights into DNA‐unrelated effects of platinum compounds and drug resistance determinants. Drug Resist Update 2015; 20: 1–11. [DOI] [PubMed] [Google Scholar]
- 4. Yu H, Su J, Xu Y et al p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer 2011; 47: 1585–94. [DOI] [PubMed] [Google Scholar]
- 5. Moscat J, Karin M, Diaz‐Meco M. p62 in cancer: signaling adaptor beyond autophagy. Cell 2016; 167: 606–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 2015; 282: 4672–8. [DOI] [PubMed] [Google Scholar]
- 7. Moscat J, Diaz‐Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137: 1001–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moscat J, Diaz‐Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci 2012; 37: 230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J, Yang Z, Dong J. P62: an emerging oncotarget for osteolytic metastasis. J Bone Oncol 2016; 5: 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia M, Yu H, Gu S et al p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1‐Nrf2‐ARE system. Int J Oncol 2014; 45: 2341. [DOI] [PubMed] [Google Scholar]
- 11. Puvirajesinghe TM, François B, Ashish J et al Identification of p62/SQSTM1 as a component of non‐canonical Wnt VANGL2–JNK signalling in breast cancer. Nat Comm 2016; 7: 10318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saito T, Ichimura Y, Taguchi K et al p62/Sqstm1 promotes malignancy of HCV‐positive hepatocellular carcinoma through Nrf2‐dependent metabolic reprogramming. Nat Comm 2016; 7: 12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen K, Zeng J, Xiao H et al Regulation of glucose metabolism by p62/SQSTM1 through HIF1α. J Cell Sci 2016; 129: 817–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwadate R, Inoue J, Tsuda H et al High expression of SQSTM1/p62 protein is associated with poor prognosis in epithelial ovarian cancer. Acta Histochem Cytochem 2014; 47: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duran A, Linares JF, Galvez AS et al The signaling adaptor p62 is an important NF‐kappaB mediator in tumorigenesis. Cancer Cell 2008; 13: 343–54. [DOI] [PubMed] [Google Scholar]
- 16. Trocoli A, Bensadoun P, Richard E et al p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ 2014; 21: 1852–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manley S, Williams JA, Ding WX. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med 2013; 238: 525–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xi G, Shen X, Wai C, Vilas CK, Clemmons DR. Hyperglycemia stimulates p62/PKCζ interaction, which mediates NF‐κB activation, increased Nox4 expression, and inflammatory cytokine activation in vascular smooth muscle. FASEB J 2015; 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 2013; 14: 727–36. [DOI] [PubMed] [Google Scholar]
- 20. Liu XY, Lai F, Yan XG et al RIP1 kinase is an oncogenic driver in melanoma. Cancer Res 2015; 75: 1736–48. [DOI] [PubMed] [Google Scholar]
- 21. Lin Y. RIP1‐mediated signaling pathways in cell survival and death. Control 2014; 23–43. [Google Scholar]
- 22. de Almagro MC, Goncharov T, Izraeltomasevic A et al Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Diff 2017; 24: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha‐induced NF‐kappaB activation. J Biol Chem 2006; 281: 13636–43. [DOI] [PubMed] [Google Scholar]
- 24. Yang Y, Xia F, Hermance N et al A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF‐kappaB and p38 mitogen‐activated protein kinase (MAPK)/MAPK‐activated protein 2 responses to DNA damage. Mol Cell Biol 2011; 31: 2774–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emmerich CH, Ordureau A, Strickson S et al Activation of the canonical IKK complex by K63/M1‐linked hybrid ubiquitin chains. Proc Natl Acad Sci USA 2013; 110: 15247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanz L, Sanchez P, Lallena MJ, Diaz‐Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF‐κB activation. EMBO J 1999; 18: 3044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma L, Xu Y, Su J et al Autophagic flux promotes cisplatin resistance in human ovarian carcinoma cells through ATP‐mediated lysosomal function. Int J Oncol 2015; 47: 1890. [DOI] [PubMed] [Google Scholar]
- 28. Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell 2008; 132: 344–62. [DOI] [PubMed] [Google Scholar]
- 29. Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF‐kappaB pathway activation in multiple myeloma. Blood 2010; 115: 3541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF‐kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 2005; 30: 43–52. [DOI] [PubMed] [Google Scholar]
- 31. Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo . Proc Natl Acad Sci USA 2008; 105: 2415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flomerfelt FA, Gress RE. Analysis of cell proliferation and homeostasis using EdU labeling. Methods Mol Biol 2016; 1323: 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang L, Shao L, Creighton CJ et al Function of phosphorylation of NF‐kB p65 ser536 in prostate cancer oncogenesis. Oncotarget 2015; 6: 6281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galluzzi L, Vitale I, Michels J et al Systems biology of cisplatin resistance: past, present and future. Cell Death Dis 2014; 5: e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gottesman MM. Mechanisms of cancer drug resistance. Medicine 2002; 53: 615–27. [DOI] [PubMed] [Google Scholar]
- 36. Safaei R, Larson BJ, Cheng TC et al Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug‐resistant human ovarian carcinoma cells. Mol Cancer Ther 2005; 4: 1595–604. [DOI] [PubMed] [Google Scholar]
- 37. Kelland L. The resurgence of platinum‐based cancer chemotherapy. Nat Rev Cancer 2007; 7: 573–84. [DOI] [PubMed] [Google Scholar]
- 38. Wei H, Guan JL. Blocking tumor growth by targeting autophagy and SQSTM1 in vivo . Autophagy 2015; 11: 854–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura K, Kimple AJ, Siderovski DP, Johnson GL. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF‐kappaB activation. J Biol Chem 2010; 285: 2077–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moscat J, Diaz‐Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci 2007; 32: 95–100. [DOI] [PubMed] [Google Scholar]
- 41. Yang YI, Ahn JH, Lee KT, Shih Ie M, Choi JH. RSF1 is a positive regulator of NF‐kappaB‐induced gene expression required for ovarian cancer chemoresistance. Cancer Res 2014; 74: 2258–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Annunziata CM, Stavnes HT, Kleinberg L et al Nuclear factor kappaB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 2010; 116: 3276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Puliyappadamba VT, Chakraborty S, Chauncey SS et al Opposing effect of EGFRWT on EGFRvIII‐mediated NF‐kappaB activation with RIP1 as a cell death switch. Cell Rep 2013; 4: 764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu P, Shi KJ, An JJ et al The LEF1/CYLD axis and cIAPs regulate RIP1 deubiquitination and trigger apoptosis in selenite‐treated colorectal cancer cells. Cell Death Dis 2014; 5: e1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosebeck S, Rehman AO, Apel IJ et al The API2‐MALT1 fusion exploits TNFR pathway‐associated RIP1 ubiquitination to promote oncogenic NF‐kappaB signaling. Oncogene 2014; 33: 2520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellail AC, Olson JJ, Yang X, Chen ZJ, Hao C. A20 ubiquitin ligase‐mediated polyubiquitination of RIP1 inhibits caspase‐8 cleavage and TRAIL‐induced apoptosis in glioblastoma. Cancer Discov 2012; 2: 140–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Teramachi J, Silbermann R, Yang P et al Blocking the ZZ domain of sequestosome1/p62 suppresses myeloma growth and osteoclast formation in vitro and induces dramatic bone formation in myeloma‐bearing bones in vivo . Leukemia 2016; 30: 390–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang S, Okamoto K, Yu C, Sinicrope FA. p62/sequestosome‐1 up‐regulation promotes ABT‐263‐induced caspase‐8 aggregation/activation on the autophagosome. J Biol Chem 2013; 288: 33654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zeng RX, Zhang YB, Fan Y, Wu GL. p62/SQSTM1 is involved in caspase‐8 associated cell death induced by proteasome inhibitor MG132 in U87MG cells. Cell Biol Int 2014; 38: 1221–6. [DOI] [PubMed] [Google Scholar]
- 50. Wong WW, Silke J. Another facet of ubiquitylation: death. J Mol Cell Biol 2009; 1: 80–1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 p62 participated in the mechanism of VK3‐resistance in SKOV3/DDP cells (a) SKOV3 and SKOV3/DDP cells were treated with varying doses of VK3 for 8 and 16 h. Cell viability was determined by MTT assay. Data were presented as a mean ± SD, n = 3. (b) Western blot analysis for the nuclear Nrf2 protein in transfected SKOV3/DDP cells.