

Abstract
Advances in next‐generation sequencing (NGS) technologies have enabled physicians to test for genomic alterations in multiple cancer‐related genes at once in daily clinical practice. In April 2015, we introduced clinical sequencing using an NGS‐based multiplex gene assay (OncoPrime) certified by the Clinical Laboratory Improvement Amendment. This assay covers the entire coding regions of 215 genes and the rearrangement of 17 frequently rearranged genes with clinical relevance in human cancers. The principal indications for the assay were cancers of unknown primary site, rare tumors, and any solid tumors that were refractory to standard chemotherapy. A total of 85 patients underwent testing with multiplex gene assay between April 2015 and July 2016. The most common solid tumor types tested were pancreatic (n = 19; 22.4%), followed by biliary tract (n = 14; 16.5%), and tumors of unknown primary site (n = 13; 15.3%). Samples from 80 patients (94.1%) were successfully sequenced. The median turnaround time was 40 days (range, 18–70 days). Potentially actionable mutations were identified in 69 of 80 patients (86.3%) and were most commonly found in TP53 (46.3%), KRAS (23.8%), APC (18.8%), STK11 (7.5%), and ATR (7.5%). Nine patients (13.0%) received a subsequent therapy based on the NGS assay results. Implementation of clinical sequencing using an NGS‐based multiplex gene assay was feasible in the clinical setting and identified potentially actionable mutations in more than 80% of patients. Current challenges are to incorporate this genomic information into better therapeutic decision making.
Keywords: Actionable mutation, genotype‐directed therapy, multiplex gene assay, next‐generation sequencing, precision cancer medicine
With the paradigm shift to precision cancer medicine, there is a growing recognition that understanding of genomic architecture enables the adoption of better therapeutic strategies and that genotype‐directed therapy can improve the clinical outcomes of cancer patients.1 In current clinical practice, hotspot‐based, single‐gene testing approaches, such as those testing for EGFR mutations in non‐small cell lung cancer or RAS mutations in colorectal cancer, have commonly been used.2, 3 However, several recent studies have reported that more comprehensive characterization of genomic alterations is necessary for successful identification of patients who may benefit from molecularly targeted therapies, and could provide more clinical benefits for individual patients.4, 5, 6, 7, 8
Recent technological innovations in next‐generation sequencing (NGS) have facilitated comprehensive genomic profiling of human cancers through whole‐genome, whole‐exome, and whole‐transcriptome sequencing, making it possible to deliver genomically informed personalized cancer therapy to individual patients.9, 10, 11, 12 In particular, NGS‐based multiplex gene assays can analyze a large number of pre‐selected genes with clinical relevance to human cancers at once and are powerful tools for the simultaneous screening of numerous cancer‐related genes in the clinical setting.
Clinical sequencing generally refers to sequencing of a genome or exome by NGS technologies for clinical applications.13 To date, this approach has been adopted not only in oncology but also in a variety of medical fields such as genetic analysis of neurologic disorders or genetic phenotyping of infectious diseases. In this study, we have defined clinical sequencing as the characterization of the tumor genomic variants that may confer sensitivity to a specific molecularly targeted therapy using an NGS‐based multiplex gene assay.
In April 2015, we introduced clinical sequencing using an NGS‐based multiplex gene assay (OncoPrime) into daily clinical practice.14 We describe here the feasibility and diagnostic yield of the NGS assay in an initial cohort of patients with advanced solid tumors.
Patients and Methods
Patient population
Between April 2015 and July 2016, 85 patients with histopathologically confirmed solid tumors underwent an NGS‐based multiplex gene assays (OncoPrime) at Kyoto University Hospital. The principal indications for the assay were cancers of unknown primary site, rare tumors, and any solid tumors refractory to standard chemotherapy. This study was approved by the Ethics Committee of the Kyoto University Graduate School of Medicine (G692) and all patients provided written informed consent for the use of genomic and clinical data for research purposes.
NGS‐based multiplex gene assay (OncoPrime)
OncoPrime is an NGS‐based multiplex gene assay designed for clinical tumor genomic analyses. This NGS assay can sequence the entire coding region of 215 genes and concurrently examine the rearrangement of 17 frequently rearranged genes with clinical or preclinical relevance in human solid tumors (Table S1).
After the NGS assay was ordered by the treating physician, 5–10 slices of 10 μm sections of archival formalin‐fixed paraffin‐embedded (FFPE) tumor tissue (tumor content ≥20%) or DNA extracted from fresh frozen tumor tissue at our institution were shipped to a Clinical Laboratory Improvement Amendment (CLIA)‐certified laboratory of EA Genomics (Morrisville, NC, USA). DNA extraction was performed by EA Genomics. Solution hybridization targeted 3861 exons of 215 cancer‐related genes and 59 introns of 17 genes commonly rearranged in human cancers. Sequencing was performed on an Illumina HiSeq 2500 machines (San Diego, CA, USA). Variant calling was done using variant calling software (VarPROWL) in a CLIA‐certified laboratory of EA Genomics.
The turnaround time (TAT) was defined as the period between the date of ordering OncoPrime and that of receiving NGS assay results by the treating physician.
Definition of actionability
Actionability implies that a protein product of the mutated gene can impact clinical decision making for patient treatment. The NGS‐based multiplex gene assay provides an enormous amount of information about genomic alterations within tumors; however, sometimes it is challenging to determine whether identified genomic alterations are actionable or not.15 In addition, the evidence behind actionability ranges from sufficient clinical data to only preclinical evidence, and several actionability classification schemes have been proposed.12, 16, 17, 18
In this study, we defined a genomic alteration as actionable if the identified alterations met any of several criteria:
It can be directly targeted by a United States Food and Drug Administration (FDA)‐approved drug (such as a BRAF inhibitor targeting a BRAF mutation, an EGFR inhibitor targeting an EGFR mutation, or an HER2 kinase inhibitor targeting an HER2 mutation).
It is a signaling pathway component that can be targeted by an FDA‐approved drug (such as a mammalian target of rapamycin inhibitor for PIK3CA mutation, a smoothened homolog inhibitor for the PTCH1 mutation, or a mitogen‐activated protein kinase kinase inhibitor for RAS mutation).
It predicts treatment response to an FDA‐approved drug (such as a poly [ADP‐ribose] polymerase inhibitor for BRCA1/2 mutation).
It can be targeted directly or indirectly by an investigational agent that is available in early clinical trials. For instance, this category includes such genes as CTNNB1 or TP53.
It is a biomarker for which only preclinical data is available. For instance, this category includes such genes as IGF2R or SMAD4.
Variant filtering and reporting
The procedure for variant prioritization and filtering was as follows Fig. (S1). First, all silent mutations in non‐reference alleles were removed, keeping mutations that were missense, nonsense, or involved splicing junctions. Second, all non‐reference alleles that appeared in >1% of the population were removed, as these were likely germline events. Third, all non‐reference alleles with allele frequencies <4% and >95% were removed, as these were below the specified limit of detection of the assay and likely germline events, respectively. Finally, the importance of variants was prioritized based on membership in the following databases: Online Mendelian Inheritance in Man (https://www.omim.org/),19 ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/),20 Clinical Trial.gov (https://clinicaltrials.gov/), Drug Bank (https://www.drugbank.ca/), Catalogue of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic),21 and the Cancer Genome Atlas (https://cancergenome.nih.gov/). NGS data were annotated by N‐of‐One, Inc. (Concord, MA, USA). Genomic alterations that were potentially targetable with an FDA‐approved drug or an investigational agent tested in early clinical trials were reported as potentially actionable mutations. Including variants of unknown significance, a maximum of 14 variants was listed in a final report and returned to the treating physician.
Molecular tumor board
Our institutional molecular tumor board comprises medical and surgical oncologists, pathologists, bioinformaticians, and medical geneticists. The molecular tumor board meetings are held for an hour every one or 2 weeks, and in general, 15–20 board members and clinicians attend the meeting. The molecular tumor board discusses genetically informed treatment options and other issues such as the possibility of germline variants in two to three patients and supports the treating physician for appropriate use of sequencing data. T.K, M.K, Y.Y, M.K, M.N, E.N, H.M, S.M, K.T, S.M, H.H, H.S, S.K, Y.O and M.M were the board members.
Results
Patient characteristics
The patient characteristics are summarized in Table 1. The median age was 58 years (range, 8–82 years). Most patients (80.0%) had solid tumors refractory to standard chemotherapy, and the remainder had cancers of unknown primary site (15.3%) or rare tumors (4.8%). The most common solid tumor types tested were pancreatic (n = 19; 22.4%), followed by biliary tract (n = 14; 16.5%).
Table 1.
Patient demographics and clinical characteristics
| Characteristics | Number of patients (%) |
|---|---|
| Sex, no. (%) | |
| Female | 47 (55.3) |
| Male | 38 (44.7) |
| Age, years | |
| Median | 58 |
| Range | 8–82 |
| Indication, no. (%) | |
| Cancers of unknown primary site | 13 (15.3) |
| Rare tumors | |
| Liposarcoma | 1 (1.2) |
| Malignant schwannoma | 1 (1.2) |
| Calcifying fibrous tumor | 1 (1.2) |
| Thymic | 1 (1.2) |
| Solid tumors refractory to standard chemotherapy | |
| Pancreatic | 19 (22.4) |
| Biliary tract | 14 (16.5) |
| Colorectal | 10 (11.8) |
| Gastric | 6 (7.1) |
| Lung | 4 (4.7) |
| Esophageal | 3 (3.5) |
| Liver | 3 (3.5) |
| Breast | 2 (2.4) |
| Ovarian | 2 (2.4) |
| Brain | 1 (1.2) |
| Melanoma | 1 (1.2) |
| Neuroendocrine tumor | 1 (1.2) |
| Peritoneum | 1 (1.2) |
| Uterine body | 1 (1.2) |
Feasibility of the NGS‐based multiplex gene assay
Archival FFPE tumor tissue was available for the NGS assay in 62 patients (Fig. 1). Because the remaining patients did not have appropriate archival FFPE tumor tissue, they underwent endoscopic biopsy (n = 7), fine needle biopsy (n = 12), or excisional biopsy (n = 4) of their primary or metastatic lesions. DNA extracted from the archival FFPE tumor tissue or fresh frozen tumor tissue was used for the NGS assay. The success rate of the NGS assay was 82.3% (51 of 62 patients) when using DNA extracted from archival FFPE tumor tissue, whereas it was 95.7% (22 of 23 patients) when using DNA extracted from fresh frozen tumor tissue. In 12 of 85 patients (14.1%), the initial NGS assay failed because of insufficient tissue quantity (n = 3), failed library preparation (n = 4), or high duplicate read rate (>80%) and/or low insert size (<120 bp) (n = 5). Among these 12 patients, for six patients from whom archival FFPE tumor tissue was used and one patient from whom fresh frozen tumor tissue was used in the first assay, the second NGS assay was successful when alternative DNA extracted from fresh frozen tumor tissue was used. As a result, the NGS assay was successful for 80 patients (94.1%) and the final report could be completed. In this group of 80 patients, the median of DNA yield and concentration of library DNA were 1853 ng (range, 125–31850) and 5.40 nM (range, 0.20–36.93), respectively (Fig. S2). The mean depth for sequencing reactions were analyzed in 58 patients, and its median figure was 4397 (range, 2123–6674) (Fig. S3). The median TAT was 40 days (range, 18–70) (Fig. S4).
Figure 1.
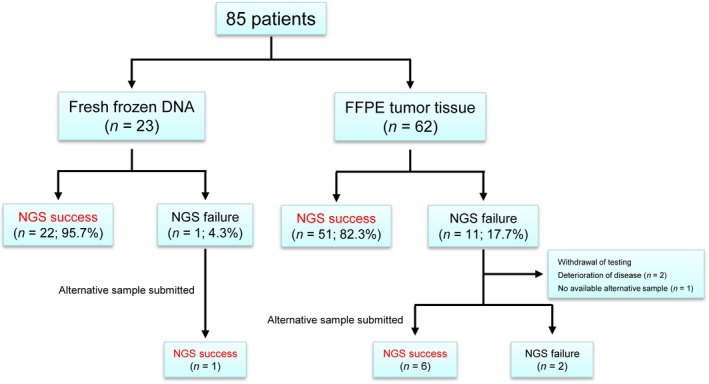
Feasibility of the next‐generation sequencing (NGS)‐based multiplex gene assay. NGS success means that NGS was successfully completed, and the treating physician could receive the NGS assay results from the laboratory. Also, NGS failure means that NGS was not successfully completed, and the treating physician could not receive the NGS assay results.
Identification of potentially actionable mutations
Of the 80 patients with NGS success, at least one potentially actionable mutation was identified in 69 patients (86.3%). The median number of actionable mutations per patient was 2 (range, 0–6). Potentially actionable mutations were identified throughout different tumor types and were most commonly found in TP53 (46.3%), KRAS (23.8%), APC (18.8%), STK11 (7.5%), and ATR (7.5%) genes (Fig. 2, Table S2).
Figure 2.
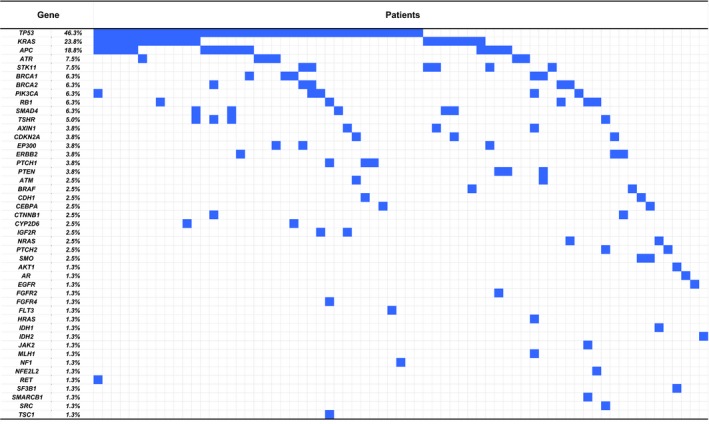
Heat map describing potentially actionable mutations identified in each patient. Each column represents one patient, and each row represents one gene. Potentially actionable mutations are shown in blue.
Therapeutic implications of actionable mutations
To explore the impact of the NGS assay results on subsequent treatment decision making, we classified the patients based on the availability of drugs suggested by genomic testing (Fig. 3). Forty‐two patients (60.9%) had genomic alterations that were potentially targetable with an approved or off‐label drug in Japan. Nineteen patients (27.5%) had genomic alterations that were not targetable with an approved or off‐label drug in Japan, but were targetable with an FDA‐approved drug. Eight patients (11.6%) had genomic alterations that were potentially targetable with an investigational agent only available in early clinical trials.
Figure 3.
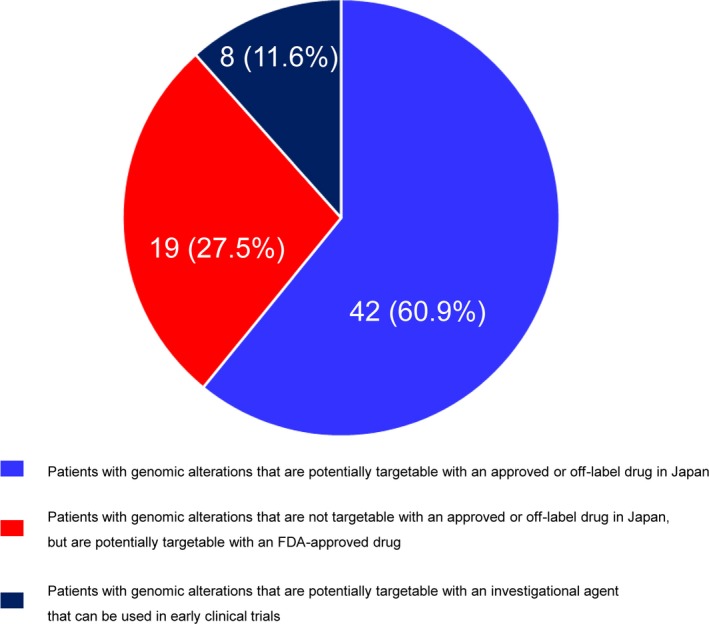
Distribution of patients according to the availability of drugs suggested by next‐generation sequencing (NGS) assay results.
After the NGS assay, nine patients (13.0% of those with actionable mutations) received subsequent therapy based on the NGS assay results (Table 2). At the start of treatment based on the NGS assay results, we discussed the therapeutic indications and possible treatment options at our molecular tumor board meeting. The doses and regimen schedules were adjusted at the discretion of the treating physicians according to the general condition of each patient. Six patients received genotyped‐directed therapy with molecularly targeted agents as suggested by the NGS assay results. Notably, among these patients, a patient with cancer of unknown primary site was found to harbor an EGFR mutation, which had not been identified by conventional hotspot‐based gene assays, most likely because of low allele frequency. This patient was treated with an EGFR tyrosine kinase inhibitor (erlotinib) and consequently experienced a remarkable tumor response with improvement in symptoms. Three patients with BRCA1 or BRCA2 mutations did not receive genotype‐directed therapy with molecularly targeted agents, such as olaparib, because they were not approved in Japan at that time. Instead, they received platinum‐based therapies because these have been reported to elicit better responses in these patients.22, 23, 24
Table 2.
Patients who received subsequent therapy based on next‐generation sequencing (NGS) assay results
| Tumor type | Gene | Mutation | Drugs | Treatment response | References | |
|---|---|---|---|---|---|---|
| Genotype‐directed therapy | Lung | PIK3CA | Splice site 814‐1G>A | AZD5363 | Discontinuation due to adverse effects | Li et al.35 |
| Gastric | PIK3CA | p.Glu542Lys | Everolimus | Progressive disease | Janku et al.36, Loi et al.37, Deming et al.38 | |
| Unknown primary site | EGFR | p.Leu858Arg | Erlotinib | Remarkable response | Rosell et al.39, Mok et al.40, Tsao et al.41 | |
| Pancreatic | PTEN |
p.Asp92Glu p.Cys130Phe |
Everolimus | Termination of treatment before evaluation of response due to poor general condition | Wu et al.42 | |
| Biliary tract | ERBB2 | p.Ser310Phe p.Gly660Asp | Afatinib | Stable disease | Sequist et al.43, Suzawa et al.44 | |
| Biliary tract | ERBB2 | p.Gly776Arg | Afatinib | Discontinuation due to adverse effects | Sequist et al.43, Suzawa et al.44 | |
| Genotype‐relevant therapy | Unknown primary site | BRCA2 | p.Ser76* | Gemcitabine plus cisplatin | Termination of treatment before evaluation of response due to poor general condition | Lowery et al.22, Maxwell et al.23, Golan et al.24 |
| Pancreatic | BRCA2 | p.Gln3026* | S‐1 plus oxaliplatin | Partial response | Lowery et al.22, Maxwell et al.23, Golan et al.24 | |
| Liver | BRCA1 | p.Leu52Phe | 5‐FU plus cisplatin | Termination of treatment before evaluation of response due to poor general condition | Lowery et al.22, Maxwell et al.23, Golan et al.24 |
5‐FU, 5‐fluorouracil.
Incidental findings
It is possible that the clinical NGS assay incidentally revealed germline variants. With regard to these incidental findings, the American College of Medical Genetics and Genomics (ACMG) has published their recommendations on the management of incidental germline findings from somatic mutation profiling in the clinical setting.25 Because our gene panel contained 20 of 56 genes for which the ACMG recommends return of pathogenic germline variants, we carefully reviewed whether the mutations found in these 20 genes were derived from germline variants based on the patients’ personal and family histories of cancer and reported allele frequencies. Pathogenicity was determined based on ClinVar and gene‐specific databases, such as Insight (https://www.insight-group.org/), LOVD (http://www.lovd.nl/3.0/home), or the database of the University of Utah Department of Pathology and ARUP Laboratories (http://arup.utah.edu/database/BRCA/). Consequently, among 80 patients with NGS success, five patients (6.3%) had a suspected pathogenic germline variant in at least one of the 20 genes: BRCA1 (n = 1), BRCA2 (n = 1), TP53 (n = 2), and BRCA1 and TP53 (n = 1) (Table 3). Patients 1 and 2 did not desire to undergo germline testing because they considered that the result did not influence their subsequent cancer treatment. By Sager sequencing using genomic DNA extracted from peripheral lymphocytes, we confirmed that TP53 p.Gly245Asp in patient 3 and TP53 p.Arg175His in patient 5 were somatic variants and BRCA1 p.Gln934* in patient 5 was a germline variant. Patient 4 had already been diagnosed with hereditary breast and ovarian cancer (HBOC) elsewhere.
Table 3.
Patients with suspected pathogenic germline variants
| Patient | Age | Sex | Tumor type | Family history | Gene | Mutation | Germline testing† |
|---|---|---|---|---|---|---|---|
| 1 | 44 | M | Pancreatic | − | BRCA2 | p.Gln3026* | – |
| 2 | 78 | F | Colorectal | − | TP53 | p.Arg273His | – |
| 3 | 57 | M | Colorectal | − | TP53 | p.Gly245Asp | Somatic |
| 4 | 39 | F | Breast (HBOC) | + | BRCA1 | p.Leu63* | Germline |
| 5 | 82 | M | Gastric | − | BRCA1 | p.Gln934* | Germline |
| TP53 | p.Arg175His | Somatic |
HBOC, hereditary breast and ovarian cancer. †Patients 1 and 2 did not desire to undergo germline testing.
Discussion
We reviewed our experience with the first consecutive 85 patients who underwent an NGS‐based multiplex gene assay in a CLIA‐certified laboratory at our institution. The success rate of the NGS assay using clinical samples such as archival FFPE or fresh frozen tumor tissue was 94.1%, supporting the feasibility of clinical sequencing in the daily clinical practice.
In this study, most (86.3%) of the patients were found to harbor at least one potentially actionable mutation. However, the median TAT was 40 days (range, 18–70), which was not satisfactory for a clinical setting because it caused delay in treatment initiation. One of the main reasons for the long TAT was long transport time because the NGS assay had to be performed in a CLIA‐certified laboratory in the United States. A shorter TAT is an essential to promptly initiate cancer treatment, especially for patients with advanced tumors refractory to standard chemotherapy. Indeed, some patients unfortunately missed the chance to receive a treatment based on the NGS assay results because of the deterioration of their disease. Therefore, shortening the TAT is very much required.
Several studies have reported that genomic information facilitates the adoption of better therapeutic strategies and that genotype‐directed therapy can improve the clinical outcomes of cancer patients.26 In fact, we observed notable treatment response in a few patients. In this regard, rigorous studies are needed to investigate whether genotype‐directed therapy proposed by comprehensive genomic profiling can result in better clinical outcome in cancer patients. Several prospective studies are now underway for verifying the clinical utility of comprehensive genomic profiling.27, 28
The number of patients who could receive a therapy according to NGS assay results was relatively limited (13.0% of those with actionable mutations). In general, to receive genotype‐directed therapy based on NGS assay results, it is necessary to use an off‐label drug or an investigational agent that is available only in early clinical trials. However, in Japan, there are several barriers, such as a high costs and statutory regulations, to accessing these drugs in the clinic. Moreover, available clinical trials are open in limited institutions and for selected patients, thus limiting patient enrollment. In accordance with our experience, previous studies have also pointed out these problems as obstacles to promoting precision medicine in the field of cancer treatment.29, 30, 31 To overcome these problems, amendments of social rules and regulations would be needed, to push the application of NGS assay from the research setting into daily clinical practice.
With regard to incidental germline variants, we observed five patients (6.3%) who had a suspected germline variant. Sanger sequencing using genomic DNA showed that one patient (patient 3) had a somatic variant of TP53 and one (patient 5) had somatic and germline variants of TP53 and BRCA1, respectively. One patient with HBOC (patient 4) had a confirmed germline variant of BRCA1. As for the remaining two patients (patients 1 and 2), we provided detailed information about the results to the patients and their families. Although they understood the significance of germline testing, they did not desire further testing because they considered the results would not influence any subsequent cancer treatment. Meric‐Bernstam et al. recently reported that 4.3% patients who underwent clinical NGS assays for advanced cancers probably had pathogenic germline variants in one of 19 genes for which the ACMG recommends return of pathogenic germline variants, with BRCA1, BRCA2, and TP53 being the most common.32 Although there were a few limitations to the present study such as the limited sample size and bias in the types of tumor tested, we estimate that the frequency of incidental germline variants in the Japanese population may be comparable to or lower than in the series of Meric‐Bernstam et al.
The analysis of the cancer genome using NGS provides extensive information about the genomic alterations within tumors. In this regard, molecular tumor boards play a pivotal role in the appropriate understanding and clinical use of NGS assay results.33, 34 In our institution, we regularly hold an institutional multi‐disciplinary molecular tumor board meetings comprising medical oncologists, surgical oncologists, pathologists, bioinformaticians, and medical geneticists, and discuss possible treatment options based on NGS assay results and other issues such as incidental findings.
In conclusion, the implementation of clinical sequencing using an NGS‐based multiplex gene assay is feasible in the clinical setting. Although the clinical utility of comprehensive genomic profiling has not yet been completely evaluated, we believe that implementation of clinical sequencing using an NGS‐based multiplex gene assay would facilitate the rapid molecular classification of tumors and has a great potential for promoting precision cancer medicine.
Disclosure Statement
The authors have declared no conflicts of interest.
Supporting information
Fig. S1. A flow of variant filtering process in OncoPrimeTM.
{kind=link}
Fig. S2. DNA yield and concentration of library DNA of each patient.
{kind=link}
Fig. S3. Mean depth for sequencing reactions of each patient.
{kind=link}
Fig. S4. Histogram of the turnaround time between the date of ordering OncoPrimeTM and that of receiving NGS assay results by the treating physician.
{kind=link}
Table S1. Genes sequenced in OncoPrimeTM.
Table S2. Potentially actionable mutations identified in OncoPrimeTM.
Acknowledgments
We gratefully thank M. Imano, E. Sasaki, K. Ashida, and M. Funakoshi for their excellent technical assistance and secretarial help. This study was supported by the Practical Research for Innovative Cancer Control and Program for Integrated Database of Clinical and Genomic Information from Japan Agency for Medical Research and Development, AMED.
Cancer Sci 108 (2017) 1440–1446
Funding information
Japan Agency for Medical Research and Development, AMED.
References
- 1. Jameson JL, Longo DL. Precision medicine‐personalized, problematic, and promising. N Engl J Med 2015; 372: 2229–34. [DOI] [PubMed] [Google Scholar]
- 2. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 3. Douillard JY, Oliner KS, Siena S et al Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369: 1023–34. [DOI] [PubMed] [Google Scholar]
- 4. Von Hoff DD, Stephenson JJ Jr, Rosen P et al Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010; 28: 4877–83. [DOI] [PubMed] [Google Scholar]
- 5. Frampton GM, Fichtenholtz A, Otto GA et al Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013; 31: 1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsimberidou AM, Wen S, Hong DS et al Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res 2014; 20: 4827–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takeda M, Sakai K, Terashima M et al Clinical application of amplicon‐based next‐generation sequencing to therapeutic decision making in lung cancer. Ann Oncol 2015; 26: 2477–82. [DOI] [PubMed] [Google Scholar]
- 8. Kuboki Y, Yamashita S, Niwa T et al Comprehensive analyses using next‐generation sequencing and immunohistochemistry enable precise treatment in advanced gastric cancer. Ann Oncol 2016; 27: 127–33. [DOI] [PubMed] [Google Scholar]
- 9. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second‐generation sequencing. Nat Rev Genet 2010; 11: 685–96. [DOI] [PubMed] [Google Scholar]
- 10. Mardis ER. A decade's perspective on DNA sequencing technology. Nature 2011; 470: 198–203. [DOI] [PubMed] [Google Scholar]
- 11. MacConaill LE. Existing and emerging technologies for tumor genomic profiling. J Clin Oncol 2013; 31: 1815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Allen EM, Wagle N, Stojanov P et al Whole‐exome sequencing and clinical interpretation of formalin‐fixed, paraffin‐embedded tumor samples to guide precision cancer medicine. Nat Med 2014; 20: 682–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014; 370: 2418–25. [DOI] [PubMed] [Google Scholar]
- 14. Kou T, Kanai M, Matsumoto S, Okuno Y, Muto M. The possibility of clinical sequencing in the management of cancer. Jpn J Clin Oncol 2016; 46: 399–406. [DOI] [PubMed] [Google Scholar]
- 15. Carr TH, McEwen R, Dougherty B et al Defining actionable mutations for oncology therapeutic development. Nat Rev Cancer 2016; 16: 319–29. [DOI] [PubMed] [Google Scholar]
- 16. Andre F, Mardis E, Salm M, Soria JC, Siu LL, Swanton C. Prioritizing targets for precision cancer medicine. Ann Oncol 2014; 25: 2295–303. [DOI] [PubMed] [Google Scholar]
- 17. Vidwans SJ, Turski ML, Janku F et al A framework for genomic biomarker actionability and its use in clinical decision making. Oncoscience 2014; 1: 614–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meric‐Bernstam F, Johnson A, Holla V et al A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst 2015; 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res 2015; 43: D789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landrum MJ, Lee JM, Riley GR et al ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; 42: D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forbes SA, Beare D, Gunasekaran P et al COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015; 43: D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lowery MA, Kelsen DP, Stadler ZK et al An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011; 16: 1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maxwell KN, Domchek SM. Cancer treatment according to BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol 2012; 9: 520–8. [DOI] [PubMed] [Google Scholar]
- 24. Golan T, Kanji ZS, Epelbaum R et al Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014; 111: 1132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Green RC, Berg JS, Grody WW et al ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013; 15: 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwaederle M, Zhao M, Lee JJ et al Association of biomarker‐based treatment strategies with response rates and progression‐free survival in refractory malignant neoplasms: a meta‐analysis. JAMA Oncol 2016; 2: 1452–9. [DOI] [PubMed] [Google Scholar]
- 27. Brower V. NCI‐MATCH pairs tumor mutations with matching drugs. Nat Biotechnol 2015; 33: 790–1. [DOI] [PubMed] [Google Scholar]
- 28. McNeil C. NCI‐MATCH launch highlights new trial design in precision‐medicine era. J Natl Cancer Inst 2015; 107. [DOI] [PubMed] [Google Scholar]
- 29. Meric‐Bernstam F, Brusco L, Shaw K et al Feasibility of large‐scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 2015; 33: 2753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodon J, Soria JC, Berger R et al Challenges in initiating and conducting personalized cancer therapy trials: perspectives from WINTHER, a Worldwide Innovative Network (WIN) Consortium trial. Ann Oncol 2015; 26: 1791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med 2016; 375: 1289–94. [DOI] [PubMed] [Google Scholar]
- 32. Meric‐Bernstam F, Brusco L, Daniels M et al Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol 2016; 27: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tafe LJ, Gorlov IP, de Abreu FB et al Implementation of a molecular tumor board: the impact on treatment decisions for 35 patients evaluated at Dartmouth‐Hitchcock medical center. Oncologist 2015; 20: 1011–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwaederle M, Parker BA, Schwab RB et al Molecular tumor board: the University of California‐San Diego Moores Cancer Center experience. Oncologist 2014; 19: 631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li J, Davies BR, Han S et al The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient‐derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med 2013; 11: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Janku F, Tsimberidou AM, Garrido‐Laguna I et al PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 2011; 10: 558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loi S, Michiels S, Baselga J et al PIK3CA genotype and a PIK3CA mutation‐related gene signature and response to everolimus and letrozole in estrogen receptor positive breast cancer. PLoS ONE 2013; 8: e53292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deming DA, Leystra AA, Farhoud M et al mTOR inhibition elicits a dramatic response in PI3K‐dependent colon cancers. PLoS ONE 2013; 8: e60709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosell R, Carcereny E, Gervais R et al Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–46. [DOI] [PubMed] [Google Scholar]
- 40. Mok TS, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–57. [DOI] [PubMed] [Google Scholar]
- 41. Tsao MS, Sakurada A, Cutz JC et al Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 42. Wu R, Hu TC, Rehemtulla A, Fearon ER, Cho KR. Preclinical testing of PI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioid adenocarcinoma. Clin Cancer Res 2011; 17: 7359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sequist LV, Yang JC, Yamamoto N et al Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013; 31: 3327–34. [DOI] [PubMed] [Google Scholar]
- 44. Suzawa K, Toyooka S, Sakaguchi M et al Antitumor effect of afatinib, as a human epidermal growth factor receptor 2‐targeted therapy, in lung cancers harboring HER2 oncogene alterations. Cancer Sci 2016; 107: 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. A flow of variant filtering process in OncoPrimeTM.
Fig. S2. DNA yield and concentration of library DNA of each patient.
Fig. S3. Mean depth for sequencing reactions of each patient.
Fig. S4. Histogram of the turnaround time between the date of ordering OncoPrimeTM and that of receiving NGS assay results by the treating physician.
Table S1. Genes sequenced in OncoPrimeTM.
Table S2. Potentially actionable mutations identified in OncoPrimeTM.