

Phytoplanktonic eukaryotes form huge panmictic populations and have evolved original hypervariability mechanisms to resist viral attack.
Keywords: picophytoplankton, sex evolution, mating type locus, population genomics, chromothripsis, linkage disequilbrium, GC content evolution, prasinovirus, evolutionary genomics, multiple nucleotide mutation events
Abstract
Tiny photosynthetic microorganisms that form the picoplankton (between 0.3 and 3 μm in diameter) are at the base of the food web in many marine ecosystems, and their adaptability to environmental change hinges on standing genetic variation. Although the genomic and phenotypic diversity of the bacterial component of the oceans has been intensively studied, little is known about the genomic and phenotypic diversity within each of the diverse eukaryotic species present. We report the level of genomic diversity in a natural population of Ostreococcus tauri (Chlorophyta, Mamiellophyceae), the smallest photosynthetic eukaryote. Contrary to the expectations of clonal evolution or cryptic species, the spectrum of genomic polymorphism observed suggests a large panmictic population (an effective population size of 1.2 × 107) with pervasive evidence of sexual reproduction. De novo assemblies of low-coverage chromosomes reveal two large candidate mating-type loci with suppressed recombination, whose origin may pre-date the speciation events in the class Mamiellophyceae. This high genetic diversity is associated with large phenotypic differences between strains. Strikingly, resistance of isolates to large double-stranded DNA viruses, which abound in their natural environment, is positively correlated with the size of a single hypervariable chromosome, which contains 44 to 156 kb of strain-specific sequences. Our findings highlight the role of viruses in shaping genome diversity in marine picoeukaryotes.
INTRODUCTION
Bacterial-sized photosynthetic eukaryotes were first detected by flow cytometry in a Mediterranean lagoon, where they may form blooms with densities up to 105 cells per milliliter (1). Culture-independent metabarcoding of samples of filtered seawater, using the eukaryotic 18S rDNA gene, revealed their large phylogenetical spread over the eukaryotic tree of life and their ubiquitous distribution in the sunlit surface of the ocean (2–4). Mamiellophyceae are the most prevalent picoeukaryotes of the green lineage and are particularly abundant in coastal areas, where picoeukaryotes make up 80% of biomass (5). Within this class, the first genome sequence analysis of two morphologically indistinguishable members of the Ostreococcus genus revealed an unexpectedly high level of protein divergence, suggesting ancient niche differentiation, providing novel insights about the paradox of the plankton (6, 7). Both experimental evolution (8) and environmental studies in the Arctic Ocean (9) suggest rapid adaptation of picophytoplankton to climate-induced environmental changes. The mechanisms involved in adaptation may hinge on preexisting genetic variation, also known as standing genomic variation, or on new mutations (10). Populations that adapt from standing genomic variation have a higher potential for adaptation if the rate of environmental change is fast (11). However, information on standing genomic variation in populations of picophytoplanktonic eukaryotes is scarce and relies on a few marker sequences (12), because most current research efforts focus on the description of interspecific diversity (4).
Here, we analyze the complete genomes of 13 haploid Ostreococcus tauri isolates sampled in the Mediterranean Sea to provide the first complete picture of the level and variation of standing genomic variation within natural populations of picophytoplankton.
RESULTS
Single-nucleotide polymorphism landscape
The high genome coverage obtained for each strain enabled accurate estimation of the nucleotide variation across >94% of the complete genome sequence for a total number of 117,600 single-nucleotide polymorphisms (SNPs; table S1). Synonymous diversity, πS, is 0.01, slightly higher than intergenic diversity, πi = 0.009, and almost five times the nonsynonymous diversity, πNS = 0.002 (table S2). Assuming a mutation rate of 4 × 10−10 mutations per nucleotide per generation in coding regions (13), the effective population size, Ne, in O. tauri can be estimated to be 1.2 × 107. The effective population size of O. tauri is higher than the values that have been reported in other unicellular organisms, such as Saccharomyces cerevisiae (14), Saccharomyces paradoxus (15), or Neurospora crassa (16), although these other species were collected over larger geographical areas. Nonsynonymous SNPs have a skewed site frequency spectrum toward low frequencies, as expected if most mutations changing the amino acid are deleterious (Fig. 1A). Nonsense SNPs, which change an amino acid into a stop codon, have the highest bias toward low frequencies and occur at an average of 14 nonsense mutations per strain. Strikingly, 10% of SNPs occur within multiple-nucleotide polymorphisms (MNPs) (Fig. 1B). MNPs suggest that at least 5% of mutation events affect multiple nucleotides (17), which is consistent with a recent estimation of multinucleotide spontaneous mutations in O. tauri (13), and other species such as S. cerevisiae, Drosophila melanogaster, Arabidopsis thaliana, Caenorhabditis elegans (17), and humans (18).
Fig. 1. Polymorphism features on 18 standard chromosomes.
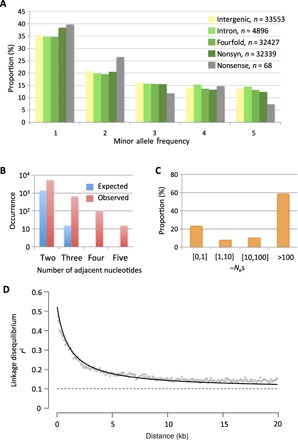
(A) Site frequency spectrum of total SNPs. (B) Number of observed and expected mutation events involved in MNPs. (C) Distribution of fitness effects on nonsynonymous sites. (D) LD between pairs of SNPs, measured as r2, with distance on chromosome 10. Gray line, expected LD for unlinked sites estimated from site frequency spectrum on intergenic sites.
Inference of genome-wide selection and mutation processes
The distribution of fitness effects on nonsynonymous sites can be inferred by comparing the site frequency spectrum of SNPs segregating at nonsynonymous and synonymous sites, where synonymous mutations are assumed to be neutral (19). The distribution of fitness effects at nonsynonymous sites is highly leptokurtic, with relatively few mutations inferred to be slightly deleterious or effectively neutral (Nes < 1, 23%), whereas most mutations are strongly deleterious (70% with Nes > 10; Fig. 1C). There is an excess of GC→AT as compared to AT→GC singletons (binomial test, P < 10−6). The site frequency spectrum on AT↔GC segregating polymorphisms is compatible with a model including GC-biased gene conversion (20) and a nonstationary GC composition (table S3), suggesting that GC content is decreasing in O. tauri. Decreasing GC content seems to be a recurrent trend in GC-rich genomes and has been reported in several vertebrate (21, 22) species. Consistent with GC-biased gene conversion, the GC base composition of chromosomes increases with average recombination rate per chromosome (Spearman ρ = 0.53, P = 0.02).
Pervasive evidence of recombination
The analysis of the polymorphism spectrum along the chromosomes provides pervasive evidence of recombination along 94% of the nuclear genome, with a genome-wide average population recombination rate per nucleotide ρ = 2Ner = 0.0013. However, the average recombination rate is negatively correlated with chromosome size (Spearman ρ = −0.82, P < 0.0001), as expected if the number of crossing-over events is typically one or two per chromosome (23). This inverse relationship between chromosome size and recombination rate is thus expected and was first described in S. cerevisiae (24). On chromosome 10, a medium-size chromosome, linkage disequilibrium (LD), measured as r2, is halved within 2 kb and reaches its minimum at 10-kb distance between SNPs (Fig. 1D). This is higher than in A. thaliana (25) and S. paradoxus (24), where the association between alleles becomes random beyond 25 kb, and close to Lachancea kluyveri (26), where minimum LD is reached within 5 kb. The number of recombination and mutation events in the genealogy of the sample can be used to estimate the minimum meiosis-to-mitosis ratio (12, 27). Calculating this ratio using the 176-kb-long chromosome 20 gave a minimum of 212 recombination events and 469 synonymous mutation events over the genealogy of the sample. Assuming 4 × 10−10 mutations per nucleotide per generation per mitotic cell division (13) and one recombination per chromosome per meiosis gives 212 meioses for 469/((4 × 10−10) × (176/3 × 103)) = 2 × 107 mitoses, that is, a minimum of 1 meiosis every 94,000 mitoses (a frequency of 1 × 10−5 true outcrossing events per generation). This is 10 times larger than our previous estimate based on sequencing 2 kilo–base pair (kbp) (12) but still indicates a high prevalence of asexual division in Ostreococcus. This is similar to the rates of true outcrossing events observed in the yeasts S. cerevisiae (2 × 10−5) (27) and the European population of S. paradoxus (1 × 10−5) (15) but somewhat higher than the frequency observed in Far East Asian population of S. paradoxus (3 × 10−6) (15).
Gene content variation within the population
On standard chromosomes, coverage variation analysis followed by Sanger resequencing enabled three insertions (table S4) and 10 gene deletions (table S5) to be validated. Although the insertions did not contain known genes, the largest deleted gene encodes a type I polyketide synthase (PKS) in strain RCC1559 (7282 amino acids) and the second largest encodes a different type I PKS gene (4766 amino acids) in RCC1108 and RCC1114. Polyketides constitute a large, structurally diverse class of natural products that include a myriad of important pharmaceuticals that exhibit antibacterial, antifungal, immunosuppressive, and antitumor properties. Natural null alleles of PKS genes may help elucidate the polyketide structures, which may unravel novel bioactive compounds from phytoplanktonic microalgae.
Structural variation on the large outlier chromosome supports hypothesis of two ancient mating-type loci in Mamiellophyceae
The coverage of the low-GC region on outlier chromosome 2 of the reference strain was insufficient to call polymorphism data (fig. S1). PacBio sequencing enabled the resolution of the large structural diversity in these regions, revealing two highly divergent loci. Whereas one 650-kb locus, M−, is present in only one strain (RCC4221), the second locus, M+, is 450 kb and is present in the other strains. The spectrum of polymorphism on M+ reveals recombination suppression in the low-GC region (Fig. 2), as suggested for sex-determining regions in eukaryotes (31 to 34). Chromosome 2 was suspected to be a “sex chromosome” upon its first description (28) because of its lower GC content and higher density of transposable elements, a signature of inefficient selection and low-biased gene conversion in nonrecombining regions (29). Using sequence similarities with genes involved in sex in Chlamydomonas and Mamiellophyceae (30), the candidate master sex determination mid gene, a transcription factor of the RWP-RK family, has been previously annotated on O. tauri’s RCC4221 chromosome 2 as well as in three of the four other Mamiellophyceae genomes sequenced at that time (31). This transcription factor controls the expression of genes involved in the minus gamete phenotype in Chlamydomonas (32), and RCC4221 has thus been proposed to be the minus type. The strains without the sex-determining transcription factor thus correspond to the plus mating types. Eighty-one orthologous genes, including housekeeping genes such as an arginine–transfer RNA ligase, are trapped within this region (Fig. 3A), and the molecular evolution of these genes enables us to investigate the age of recombination suppression in the candidate MAT locus. The evolution of a mating-type locus is closely linked with recombination suppression if the alleles encoding for one sex have a deleterious effect on the other sex (antagonistic mutations) (33). The phylogeny of five genes, which are located in the candidate MAT locus in seven Mamiellophyceae species, reveals that the candidate plus and minus haplotypes of O. tauri group together within different species (Fig. 3B). This demonstrates that they have remained genetically isolated during speciation of Ostreococcus and thus have evolved without recombining, as previously reported in the Volvox species complex (34) or in the brown alga Ectocarpus (35). The average amino acid identity between these five orthologous genes between the two Ostreococcus haplotypes is 53%, which is lower than the average amino acid identity between Micromonas and Ostreococcus orthologous genes (58%). The divergence of these two haplotypes could thus pre-date the divergence of Mamiellophyceae, estimated at 600 million years ago (36).
Fig. 2. Variation of the population recombination rate (ρ), nucleotide diversity (π), and GC content (GC) along chromosomes of O. tauri.
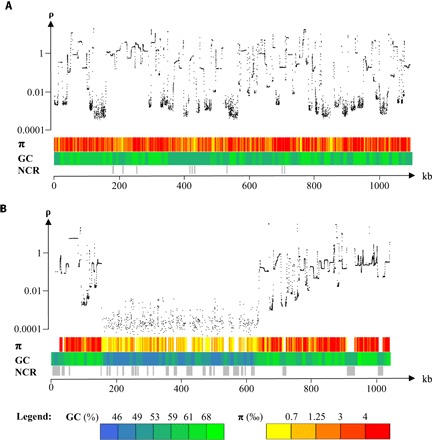
(A) Chromosome 1, a standard chromosome. (B) Chromosome 2. ρ, population-scaled recombination rate (per kilobase) inferred from SNP using the program interval of LDhat package; π, nucleotide diversity per site averaged across 2.5-kb windows; GC, averaged GC percent across 10-kb windows; NCR, noncallable regions of at least 1 kb, where polymorphisms could not be called because of insufficient coverage (<10×) or because of too high coverage suggestive of regions in multiple copies, which are not represented in the reference sequence. ρ and π are computed using SNPs segregating among the 13 strains for chromosome 1 and 12 strains (RCC4221 is excluded) for chromosome 2. For chromosome 2, the reference strain RCC4221 was excluded because of its high divergence with the other strains on the candidate MAT locus region; the sequence of chromosome 2 of strain RCC1115 was used instead.
Fig. 3. Genetic structure of the two candidate MAT loci on chromosome 2 in O. tauri.
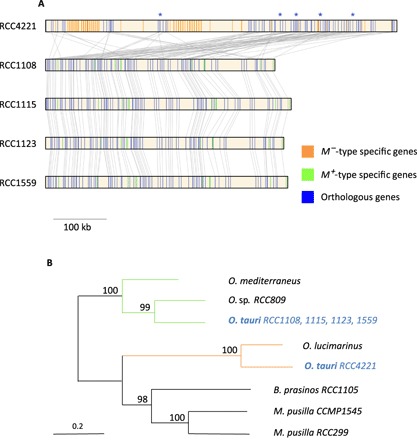
(A) Genes are indicated by vertical lines. Blue lines, orthologous genes between plus and minus type strains; green lines, plus specific genes; orange lines, minus specific genes; gray lines, orthologous relationship between genes on different strains. “*” indicates the position of the five orthologous genes used to build the phylogeny from the different strains. (B) Phylogeny of five orthologous housekeeping genes trapped inside the MAT locus region in seven species of Mamiellophyceae (O. tauri, Ostreococcus mediterraneus, Ostreococcus RCC809, Ostreococcus lucimarinus, Bathycoccus prasinos RCC1105, Micromonas pusilla RCC299, and M. pusilla CCMP1545). Bootstrap maximum likelihood (ML) percentages are indicated on nodes.
Hypervariability on the small outlier chromosome
Like chromosome 2, chromosome 19 is also known as an outlier chromosome because of its lower GC content, its high proportion of species-specific genes, and the fast evolution rates of the few of orthologous genes that it bears (7, 37). Initially, only 7% of the chromosome was callable for SNP analysis. PacBio sequencing confirmed important size variation of this chromosome between strains (38) with sizes from 260 to 415 kb (Fig. 4 and fig. S2). A large part of the size variation is the consequence of duplications and translocations within chromosome 19, except for 44- to 156-kb region that appears to be strain-specific (Fig. 4). One synteny block is composed of 6 to 13 tandem duplications of a 2.5-kb region that is specific to O. tauri and encodes an open reading frame with Pfam domain PFAM04572 (α1,4-glycosyltransferase conserved region). Phylogenetic analysis of these repeats reveals several recent tandem duplications within each strain (fig. S3). Most strain-specific regions have no hit against GenBank, with two exceptions. First, a few genes show close identity to genes located on standard chromosomes, suggesting recent translocations onto chromosome 19. Gene ontology analysis of strain-specific sequences suggests an overrepresentation of genes involved in carbohydrate transport (GO: 0008643, P < 10−4), which are generally overrepresented on this chromosome in Mamiellophyceae (39). Second, two short stretches of DNA (50 to 70 bp) have high identity to O. tauri prasinovirus sequences [OtV1 (40) and OtV5 (41)] in RCC1115 and RCC1108. Prasinoviruses are nucleocytoplasmic large DNA viruses related to giant viruses (42), which infect these microalgae and outnumber them by one order of magnitude in their natural environment (43). The viral sequences correspond to the end of a gene encoding for a viral DNA helicase (OTV1_064 for RCC1108) and a gene with unknown function (OtV1_225 for RCC1115). They are not inserted between direct repeats on chromosome 19, outruling an analogy to the prokaryotic CRISPR (clustered regularly interspaced short palindromic repeats) system (44). The mechanisms generating this hypervariable chromosome thus involve translocations from standard chromosomes and massive internal rearrangements, including successive rounds of tandem duplications.
Fig. 4. Chromosome 19 sequence conservation in six O. tauri strains.
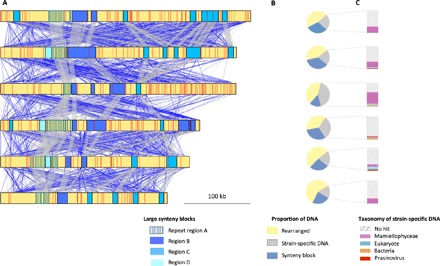
(A) Large synteny blocks along chromosome 19 are represented as rectangles (fig. S6), and red lines indicate location of strain-specific DNA. Pairwise local alignments between strains are represented in gray (sense) or blue (antisense) (blastn with scores between 50 and 500 and percentage alignment identities >95%). (B) Proportion of DNA in chromosome in syntenic regions, in rearranged regions, and in strain-specific regions. (C) Taxonomic affiliation of strain-specific sequences using sequence homology against GenBank (tblastx) (69).
Phenotypic assays
There is significant growth rate variation between strains, with a maximum growth rate variation of ±28% at 20°C (fig. S4) with faster- and slower-growing strains. The susceptibility spectrum to prasinoviruses, isolated from the same sampling locations (45), discriminates among 12 of the 13 strains, because only 2 strains have an identical viral susceptibility spectrum (table S6). Two strains are highly susceptible, whereas one strain is resistant to all 32 viruses tested. Previous results suggested a cost of viral resistance on growth rates, with susceptible strains outcompeting resistant strains (46). The size of chromosome 19 decreases with increasing viral susceptibility (Fig. 5; Pearson ρ = −0.74, P < 0.005), suggesting that the cost of resistance on the division rate may be the direct consequence of the increased metabolic cost associated with the replication and expression of a larger chromosome. Consistent with this metabolic cost hypothesis, experimental evolution of viral resistance in strain RCC4221 was significantly associated with increased gene expression on chromosome 19, which has low levels of expression under virus-free conditions (39).
Fig. 5. Length of chromosome 19 versus susceptibility to prasinoviruses.
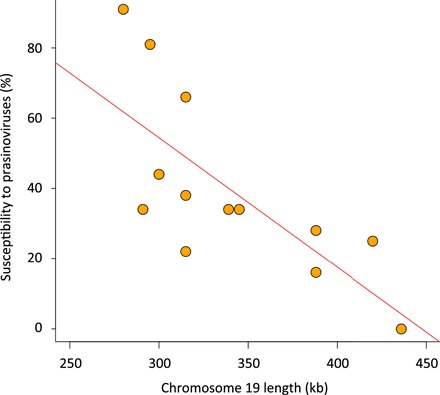
Susceptibility of each strain is estimated by the percent of prasinoviruses lysing a strain and varies from 0% (strain is resistant to all 32 viruses) to 100% (strain is lysed by all 32 viruses). Pearson correlation coefficient ρ = −0.74, P < 0.005.
DISCUSSION
Planktonic marine eukaryotes represent a yet untapped reservoir of genetic biodiversity, and new genera are still being discovered regularly. It is, therefore, not surprising that intraspecific levels of diversity are thus far underexplored in these enigmatic organisms. We previously reported a high level of intergenic nucleotide diversity (πi = 0.01) and occurrence of sexual reproduction based on an analysis of height short intergenic markers in 17 O. tauri strains (13 of them being analyzed here). Here, we confirm these initial findings using full-genome sequences, and we also provide evidence of ancient divergence between two large chromosomal regions with recombination suppression, consistent with a mating-type locus. Future experimentation will be needed to show that genes located in these two candidate mating-type loci are involved in gamete fusion.
This study supports the fact that these species have a very high adaptation potential, in particular to viral pressure. The initial response to selection is predicted to depend almost entirely on standing variation, with de novo mutations becoming gradually more important (11). The time scale of this transition strongly depends on the rate of environmental change, but for slow or moderately fast change, it typically occurs over at least hundreds of generations (11). The chromosome hypervariability reported here is different from previously reported mechanisms such as the genomic islands reported in the cyanobacteria Prochlorococcus (47), the pathogenicity chromosomes in fungi (48), or adaptive immunity genes in mammals. Comparative sequence analysis shows that this level of variation results from massive rearrangements of small repeats, duplications, and translocations from the standard chromosomes, and less frequently from the genome of prasinoviruses. These massive rearrangements recall chromothripsis, in which chromosomes are broken into many pieces, randomly duplicated, and stitched back together in some cancer genomes (49). We recently provided evidence that size variation of chromosome 19 can be induced upon viral infection in O. tauri (39). The significant correlation between the size of the hypervariable chromosome and viral resistance in the population suggests that molecular toolkit involved in this chromosome tinkering has likely evolved during adaptation of cell immunity to infection by large double-stranded DNA viruses.
MATERIALS AND METHODS
Sequencing and assembly
Genome sequencing of 12 O. tauri strains isolated in the northwestern Mediterranean Sea (RCC1108, RCC1110, RCC1112, RCC1114, RCC1115, RCC1116, RCC1117, RCC1118, RCC1123, RCC1558, RCC1559, and RCC1561; fig. S5 and table S7) was performed using Illumina technology at the Joint Genome Institute (JGI), San Francisco. Strains RCC1110, RCC1114, RCC1115, RCC1116, RCC1558, RCC1559, and RCC745 were sequenced with a protocol yielding 76-bp-long paired-end (PE) reads and an expected sequencing depth ranging between 163X and 336X. Strains RCC1108, RCC1112, RCC1117, RCC1118, RCC1123, and RCC1561 were sequenced 6 months later with another protocol yielding 101-bp-long PE reads and an expected sequencing depth ranging from 778X to 1129X. Large noncovered regions on chromosomes 2 and 19 were resolved by PacBio resequencing of seven strains at the Get-PlaGe sequencing platform in Toulouse, France. The libraries were prepared from 10 μg of DNA according to PacBio’s protocols “20-kb Template Preparation Using BluePippin Size-Selection System (15-kb size cut-off)”. The sample quantity and quality controls were validated on Qubit, NanoDrop, Pippin Pulse, and Fragment Analyser. At the end, the quantity of libraries was from 169 to 350 ng. Libraries (0.25 nM) were loaded per SMRT (Single Molecule Real-Time sequencing) on the PacBio RS II System. The run was performed with protocol OneCellPerWell on C4P6 chemistry and 360-min movies. The raw data were assembled using HGAP3 (50). The assemblies of chromosomes 2 and 19 were checked manually with assembly and visualization tools available from the Geneious software (51) and could be assembled from telomere to telomere for the six resequenced strains. These assemblies revealed substantial levels of variation on chromosomes 2 and 19, with sizes ranging from 965 kb (RCC1123) to 1217 kb (RCC4221) for chromosome 2 and 261 kb (RCC1123) to 414 kb (RCC1561) for chromosome 19 (fig. S2). Chromosome 2 and 19 sizes of other strains could be inferred by applying a linear regression between pulsed-field gel electrophoresis–inferred sizes, L, from a previous study (38) and PacBio resequenced sizes: L02 = 1.52*L − 504,781 (R2 = 0.91) and L19 = 0.84*L + 48,062 (R2 = 0.81). Chromosome 19 sequences from each strain were aligned using the dottup program as implemented in Geneious (51) (fig. S6) to define four large syntenic regions, noted A, B, C, and D. Region A is composed of 6 to 13 tandem replications of a 2.5-kb sequence; in RCC1108 and RCC1123 strains, this repeated region has also copies on the reverse strand.
SNP calling
Trimmed reads (52, 53) were mapped onto O. tauri’s RCC4221 genome version 2 (54) with Burrows-Wheeler Aligner (55) (fig. S1). Single-nucleotide variant calling was performed with three different algorithms: (i) GATK UnifiedGenotyper version 2.4.9 (UG), (ii) GATK HaplotypeCaller version 3.3.0 (HC) (56), and (iii) an in-house python script (GENO) developed for organellar data (57). Methods UG and GENO work on a per-locus basis, whereas HC is a haplotype-based method. Before calling variants with UG and HC, reads were locally realigned around indel variants using GATK IndelRealigner. We called SNPs and indel variants from the 13 .bam files with tools UnifiedGenotyper and HaplotypeCaller for haploid organisms, setting option “dcov” to 2000, “stand_call_conf” to 30, and “stand_emit_conf” to 10. This means that sites with a Phred-scaled confidence lower than 30 will be annotated, has “LowQual” in the “FILTER” section, and will be filtered out. Our in-house python scripts (GENO) called variants for the 13 strains in two steps: It first read into the 13 pile-up files obtained with “samtools mpileup –E” from SAMtools version 1.1 (58) to obtain all variants at every position along the genome sequence for each strain. This provided the number of reads supporting each variant. In a second step, our in-house script read into the variant files to keep variants supported by at least 80% of reads covering a given position for a given strain.
For all three methods, variants (vcf format) were filtered (using an in-house python script) according to the following three criteria: First, sites with a Phred-scaled confidence lower than 30 were filtered out. Second, to be callable, a position had to be “well covered” for all 13 strains, that is, covered by at least 10 reads and by less than 1.5 times the average read depth along the genome sequence for this given strain. Third, a variant in a callable position had to be supported by more than 80% of reads covering this position to be considered as a polymorphism. For the GATK UG and HC approaches, we further filtered out positions with genotype quality less than 30.
The number of SNPs and indels called by the different tools varied from ~105,000 (GENO) to ~141,000 (HC) (table S8), with ~92,000 [corresponding to 90% (GENO) and 70% (HC)] of polymorphisms called by all three methods (fig. S7). This substantial level of variation between SNP calling procedures led us to resequence four polymorphic regions containing 278 polymorphic sites by Sanger sequencing (table S9) to assess the sensitivity and specificity of each calling method (table S10). Details on the number of reads, average read mapping depth, coverage along the genome sequence, and number of SNPs and indels called for each strain are presented in table S1.
Comparison of predictions with Sanger resequencing led to the choice of UG version 2.4.9 for SNV calling (table S1) (56). Notably, of the 278 resequenced SNPs, 29 were MNPs (13 dinucleotide polymorphisms and 1 trinucleotide polymorphism). These 29 MNPs were confirmed at all loci.
Large indel (>100 bp) identification and validation
We developed a method to detect insertions, which are not present in the reference genome, and deletions, which are absent from the reference genome. These particular events cause specific profiles in the mapping result (.bam file) that we detected using homemade scripts. First, we aligned reads on the reference genome with a method that allows partial mapping (59). In this context, we defined three types of mapping events (fig. S8A). These mapping types were screened to detect profiles specific to large insertions or deletions. An insertion corresponds to a sequence that is absent from the reference genome and thus to a particular profile that can be detected by screening the mapping result of the reads (fig. S8B). The algorithmic workflow for detecting novel insertions is composed of four steps (table S11). Second, we started from upstream and downstream regions independently to elongate the sequence with unmapped reads to infer the genomic sequence of the insertion. The assembly algorithm is computed on upstream and downstream sets independently. At the end, we compared the sequences obtained from the upstream and downstream set (table S12). Third, we detected deletions by screening the mapping profile of the reads (fig. S8C). Reads from the strain were split into upstream and downstream reads, and defined two initial breakpoints on the reference genome (table S13). Predicted deletions (table S5) and insertions (table S4) were checked by Sanger sequencing (tables S14 and S15).
Population genomic analysis
Population genomic analysis was performed using SnpEff (60) and LDhat for recombination rate (61). To infer short insertions and deletions, we screened the mapping profiles of each strain and validated each candidate insertion and deletion by polymerase chain reaction. SNPGenie (version snpgenie-1.2.2.pl) (62) was used to infer the number of nonsynonymous and synonymous sites in the coding regions of the O. tauri RCC4221 genome (table S2). VCFtools (63) and a set of in-house python codes were used to parse the vcf files containing polymorphisms annotated with SnpEff to compute classical diversity statistics, such as allele frequency spectrum, nucleotide diversity, and Tajima’s D. The LD for pairs of SNPs was measured as the squared allele frequency correlation (r2) using vcftools --hap-r2 (63), with a maximum distance of 100 kb and a minimum distance of 10 bp between a pair of sites. We used biallelic SNPs only; singletons, chromosomes 2 and 19, as well as strains RCC1116, RCC1117, and RCC4221 were excluded from this analysis (fig. S9). r2 values were binned into 1-kb bins. LD decay with genomic distance between sites was evaluated by a nonlinear model (nls function in R). The expected values of r2 between adjacent sites under drift recombination equilibrium with a low level of mutation and adjusted for sample size are (64)
The least square estimate of C = 2Ner is the population-scale recombination rate, where Ne is the effective population size and r is the recombination rate per site per generation. The expected r2 value for sites that are unlinked was estimated numerically from the site frequency spectrum at intergenic sites; it equals 0.094 (0.001).
We then used more recently developed models to estimate the variation of the population-scaled recombination rate along chromosomes. A composite likelihood estimation of the population scale recombination rate (ρ) was obtained using the program interval from the package LDhat (61). LDhat input files were generated from the vcf files using vcftools -ldhat-geno. The interval program was run for each chromosome using 10,000,000 iterations for the rjMCMC procedure, a block penalty of 10, and 10,000 updates between samples. The coalescent method LDhat estimates the parameter ρ = 2Ner, where Ne is the effective population size and r is the genetic map distance across the region analyzed.
Phylogenetic analysis
Chromosome 2 of six strains shares more than 99% nucleotide identity with RCC4221 but on the low-GC candidate MAT locus region. The low-GC region was extracted from each resequenced strain, and manual annotation of this region was performed by homology with genes from the annotation of RCC4221 and RCC1115. Orthologous gene families of five gene sequences of predicted housekeeping genes trapped in the MAT locus region of O. tauri were retrieved from pico-PLAZA (65), and genes that were located in the low-GC region of the big outlier chromosomes in all seven sequenced Mamiellophyceae species (O. tauri, O. lucimarinus, Micromonas RCC299 and CCMP1545, Ostreococcus spp. RCC809, and Bathycoccus RCC1105) were used for phylogenetic reconstruction. Sequence alignment on the concatenated five genes (ostta02g00240, ostta02g01140, ostta02g01460, ostta02g02010, and ostta02g02420; total alignment length, 2130 amino acids) was performed with MAFFT version 7. Phylogenetic reconstructions were based on amino acid sequences using Bayesian inference (BI) and ML. For ML, the best-fit evolutionary model was selected via Akaike Information Criterion using ProtTest version 3.4.2 (66), resulting here in an LG + I + G model. BI was carried out with MrBayes 3.2.6 (67) using a mixed model, with four chains of 106 generations, trees sampled every 1000 generations, and burnin value set to 20% of the sampled trees. We checked that the SD of the split frequencies fell below 0.01 to ensure convergence in tree search. ML reconstructions were carried out using PhyML (68) and validated with 100 bootstrap replicates. Trees obtained with BI and ML were congruent. Phylogenetic analysis of the 2.5-kb repeat region on chromosome 19 suggests recent duplication events within each chromosome and fast evolution of this repeat (fig. S3).
Phenotypic assays
Before starting growth assays, we transferred cultures in L1 medium flasks and let them grow for 2 weeks at 14° and 20°C under 12-hour light/12-hour dark cycles to have enough cells to inoculate cultures. Growth assays were performed in 24-well microtiter plates, with a starting population of ~50,000 cells per well. The number of biological replicates was four for each strain. Cell growth was estimated by optical density at 640 nm 6 days after plate inoculation and normalized by the absorption of the L1 medium at T0. There is a significant variation of growth rates between strains at both temperatures (fig. S4; analysis of variance, P < 10−4 for 20° and 14°C). Significant paired comparisons (Tukey test) are provided in table S16. There is a significant correlation between growth rates at 14° and 20°C (Pearson ρ = 0.72, P = 0.006), consistent with overall faster-growing (for example, RCC1108, RCC1110, and RCC4221) and slower-growing (for example, RCC1123 and RCC1116) strains.
A subset of 32 viruses from 50 viruses was isolated between January 2006 and March 2009 from the northwestern Mediterranean Sea from seven different O. tauri strains (45). A fresh lysate of each virus was prepared (about 108 particles/ml) to test for viral specificity on many different strains, including 12 of the O. tauri strains sequenced in this study (45). To check that viral specificity had remained stable and to obtain viral specificity for one additional strain, RCC1108, RCC1110, RCC1117, RCC4221, RCC1123, and RCC1558 cultures were grown in 24 microtiter wells until green and inoculated with fresh lysates of the viruses. Infection was tested in triplicate, and the result was recorded 3 days later according to the color of the infected culture as compared to the control as follows: no lysis or lysis. The complete lysis spectrum of the sequenced O. tauri population is presented in table S6. Strain virus susceptibility had remained stable for all checked strains since the first assessment (45). All statistical analyses were performed with R (www.r-project.org).
Supplementary Material
Acknowledgments
We would like to thank the GenoToul bioinformatics platform for access to computing cluster facilities and the O. mediterraneus genome consortium for access to the genome annotation. Funding: The work conducted by M.H. and E.R. was funded by LabEx NUMEV and supported by the ATGC bioinformatics platform. The work conducted by the U.S. Department of Energy (DOE) JGI, a DOE Office of Science User Facility, was supported by the Office of Science of the U.S. DOE under contract no. DE-AC02-05CH11231. This work was funded by the European Community’s 7th Framework program FP7 under grant agreement no. 254619 to G.P. and A.E.-W. and by the Agence Nationale de la Recherche under grant agreement ANR-13-JSV6-0005 to G.P. and S.S.-F. Author contributions: S.S.-F., H.M., and N.G. designed phenotypic assays. E.D. and M.K. performed phenotypic assays. R.B.-M., S.Y., M.H., E.R., W.S., J.M., A.K., I.V.G., A.E.-W., and G.P. contributed to bioinformatic analysis of the Illumina data set. M.K., C.D., and G.S. contributed to PacBio sequencing. G.P. performed PacBio assembly and analysis. Y.D. and S.S.-F. performed phylogenetic analysis. A.E.-W. performed distribution of fitness effects analysis. A.K., W.S., J.M., and I.V.G. contributed to RCC1115 annotation and set up the web portal. G.P. and R.B.-M. wrote the manuscript. All authors participated in manuscript revisions. G.P. conceived the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All PE read Illumina data sets have been submitted to GenBank under the following Sequence Read Archive accession nos.: RCC1108: SRP081304, RCC1110: SRP081303, RCC1112: SRP081306, RCC1114: SRX2018736, RCC1115: SRX2018766, RCC1116: SRX2018764, RCC1117: SRP081308, RCC1118: SRP081305, RCC1123: SRP081307, RCC1558: SRX2018765, RCC1559: SRX2018767, RCC1561: SRP081309, and RCC4221: SRX030853. Genome browsers of strains RCC1115 (GenBank accession, NERT01000000) and RCC4221 (GenBank accession, CAID01000001.1 to CAID01000020.1) are available via the JGI genome portals (http://genome.jgi.doe.gov/Ostta4221_3 and http://genome.jgi.doe.gov/Ostta1115_2). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700239/DC1
fig. S1. Read coverage map of reference strain RCC4221 per chromosome for each strain.
fig. S2. Size distribution of chromosomes 2 and 19 in 13 O. tauri strains.
fig. S3. Phylogeny of the 2.5-kb repeat of chromosome 19’s syntenic block A.
fig. S4. Growth rates of 13 strains with 95% confidence interval at 14° and 20°C.
fig. S5. Sampling area and sampling sites of water samples used to isolate the 13 strains of O. tauri.
fig. S6. Dotplot of chromosome 19 between RCC1115 and RCC1559, RCC4221, RCC1108, and RCC1123.
fig. S7. Venn diagram depicting the number of shared or specific polymorphic sites called by each calling method.
fig. S8. Large (>100 bp) indel read mapping features.
fig. S9. Phylogenetic distance tree of 13 O. tauri strains based on 117,600 biallelic SNPs.
table S1. Read mapping and polymorphism statistics for each strain.
table S2. Nucleotide diversity and number of segregating and nonsegregating sites for five types of polymorphisms (synonymous, fourfold degenerate, nonsynonymous, intergenic, and intronic).
table S3. Fitting the models to the site frequency spectrum on fourfold degenerate sites.
table S4. Analysis of gene content of predicted and validated insertions using sequence homology (blastx).
table S5. Functional description of 10 predicted and validated deletions.
table S6. Viral susceptibility and resistance spectrum of O. tauri strains and a collection of 32 prasinoviruses.
table S7. Geographic origin and sampling date of the 13 O. tauri strains.
table S8. Variants, SNPs, and indel polymorphisms for each calling method.
table S9. Features of the four resequenced regions.
table S10. SNP calling validation for GATK UG, GENO, and HC by Sanger resequencing.
table S11. Insertion detection steps.
table S12. Insertion assembly steps.
table S13. Deletion detection steps.
table S14. Position and validation status of 18 predicted insertions.
table S15. Position and validation status of 12 predicted deletions (>500 bp).
table S16. Pairwise growth rates by Tukey test between strains at 20° and 14°C.
REFERENCES AND NOTES
- 1.Courties C., Vaquer A., Troussellier M., Lautier J., Chrétiennot-Dinet M. J., Neveux J., Machado C., Claustre H., Smallest eukaryotic organism. Nature 370, 255 (1994). [Google Scholar]
- 2.Díez B., Pedrós-Alió C., Massana R., Study of genetic diversity of eukaryotic picoplankton in different oceanic regions by small-subunit rRNA gene cloning and sequencing. Appl. Environ. Microbiol. 67, 2932–2941 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moon-van der Staay S. Y., De Wachter R., Vaulot D., Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409, 607–610 (2001). [DOI] [PubMed] [Google Scholar]
- 4.de Vargas C., Audic S., Henry N., Decelle J., Mahé F., Logares R., Lara E., Berney C., Le Bescot N., Probert I., Carmichael M., Poulain J., Romac S., Colin S., Aury J.-M., Bittner L., Chaffron S., Dunthorn M., Engelen S., Flegontova O., Guidi L., Horák A., Jaillon O., Lima-Mendez G., Lukeš J., Malviya S., Morard R., Mulot M., Scalco E., Siano R., Vincent F., Zingone A., Dimier C., Picheral M., Searson S., Kandels-Lewis S.; Oceans Coordinators T., Acinas S. G., Bork P., Bowler C., Gorsky G., Grimsley N., Hingamp P., Iudicone D., Not F., Ogata H., Pesant S., Raes J., Sieracki M. E., Speich S., Stemmann L., Sunagawa S., Weissenbach J., Wincker P., Karsenti E., Eukaryotic plankton diversity in the sunlit ocean. Science 348, 1261605 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Worden A. Z., Nolan J. K., Palenik B., Assessing the dynamics and ecology of marine picophytoplankton: The importance of the eukaryotic component. Limnol. Oceanogr. 49, 168–179 (2004). [Google Scholar]
- 6.Palenik B., Grimwood J., Aerts A., Rouzé P., Salamov A., Putnam N., Dupont C., Jorgensen R., Derelle E., Rombauts S., Zhou K., Otillar R., Merchant S. S., Podell S., Gaasterland T., Napoli C., Gendler K., Manuell A., Tai V., Vallon O., Piganeau G., Jancek S., Heijde M., Jabbari K., Bowler C., Lohr M., Robbens S., Werner G., Dubchak I., Pazour G. J., Ren Q., Paulsen I., Delwiche C., Schmutz J., Rokhsar D., Van de Peer Y., Moreau H., Grigoriev I. V., The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. U.S.A. 104, 7705–7710 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jancek S., Gourbière S., Moreau H., Piganeau G., Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae). Mol. Biol. Evol. 25, 2293–2300 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Schaum C.-E., Rost B., Collins S., Environmental stability affects phenotypic evolution in a globally distributed marine picoplankton. ISME J. 10, 75–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W. K. W., McLaughlin F. A., Lovejoy C., Carmack E. C., Smallest algae thrive as the Arctic Ocean freshens. Science 326, 539 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Barrett R. D. H., Schluter D., Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Matuszewski S., Hermisson J., Kopp M., Catch me if you can: Adaptation from standing genetic variation to a moving phenotypic optimum. Genetics 200, 1255–1274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimsley N., Péquin B., Bachy C., Moreau H., Piganeau G., Cryptic sex in the smallest eukaryotic marine green alga. Mol. Biol. Evol. 27, 47–54 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Krasovec M., Eyre-Walker A. C., Sanchez-Ferandin S., Piganeau G., Spontaneous mutation rate in the smallest photosynthetic eukaryotes. Mol. Biol. Evol. 34, 1770–1779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liti G., Carter D. M., Moses A. M., Warringer J., Parts L., James S. A., Davey R. P., Roberts I. N., Burt A., Koufopanou V., Tsai I. J., Bergman C. M., Bensasson D., O’Kelly M. J. T., van Oudenaarden A., Barton D. B. H., Bailes E., Nguyen A. N., Jones M., Quail M. A., Goodhead I., Sims S., Smith F., Blomberg A., Durbin R., Louis E. J., Population genomics of domestic and wild yeasts. Nature 458, 337–341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai I. J., Bensasson D., Burt A., Koufopanou V., Population genomics of the wild yeast Saccharomyces paradoxus: Quantifying the life cycle. Proc. Natl. Acad. Sci. U.S.A. 105, 4957–4962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellison C. E., Hall C., Kowbel D., Welch J., Brem R. B., Glass N. L., Taylor J. W., Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc. Natl. Acad. Sci. U.S.A. 108, 2831–2836 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrider D. R., Hourmozdi J. N., Hahn M. W., Pervasive multinucleotide mutational events in eukaryotes. Curr. Biol. 21, 1051–1054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Besenbacher S., Sulem P., Helgason A., Helgason H., Kristjansson H., Jonasdottir A., Jonasdottir A., Magnusson O. T., Thorsteinsdottir U., Masson G., Kong A., Gudbjartsson D. F., Stefansson K., Multi-nucleotide de novo mutations in humans. PLOS Genet. 12, e1006315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eyre-Walker A., Woolfit M., Phelps T., The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173, 891–900 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galtier N., Piganeau G., Mouchiroud D., Duret L., GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics 159, 907–911 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duret L., Semon M., Piganeau G., Mouchiroud D., Galtier N., Vanishing GC-rich isochores in mammalian genomes. Genetics 162, 1837–1847 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romiguier J., Ranwez V., Douzery E. J. P., Galtier N., Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res. 20, 1001–1009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller H. J., The mechanism of crossing-over. Am. Nat. 50, 192–313 (1916). [Google Scholar]
- 24.Kaback D. B., Guacci V., Barber D., Mahon J. W., Chromosome size-dependent control of meiotic recombination. Science 256, 228–232 (1992). [DOI] [PubMed] [Google Scholar]
- 25.Nordborg M., Hu T. T., Ishino Y., Jhaveri J., Toomajian C., Zheng H., Bakker E., Calabrese P., Gladstone J., Goyal R., Jakobsson M., Kim S., Morozov Y., Padhukasahasram B., Plagnol V., Rosenberg N. A., Shah C., Wall J. D., Wang J., Zhao K., Kalbfleisch T., Schulz V., Kreitman M., Bergelson J., The pattern of polymorphism in Arabidopsis thaliana. PLOS Biol. 3, e196 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedrich A., Jung P., Reisser C., Fischer G., Schacherer J., Population genomics reveals chromosome-scale heterogeneous evolution in a protoploid yeast. Mol. Biol. Evol. 32, 184–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruderfer D. M., Pratt S. C., Seidel H. S., Kruglyak L., Population genomic analysis of outcrossing and recombination in yeast. Nat. Genet. 38, 1077–1081 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Derelle E., Ferraz C., Rombauts S., Rouzé P., Worden A. Z., Robbens S., Partensky F., Degroeve S., Echeynié S., Cooke R., Saeys Y., Wuyts J., Jabbari K., Bowler C., Panaud O., Piégu B., Ball S. G., Ral J.-P., Bouget F.-Y., Piganeau G., De Baets B., Picard A., Delseny M., Demaille J., Van de Peer Y., Moreau H., Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. U.S.A. 103, 11647–11652 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charlesworth B., Charlesworth D., The degeneration of Y chromosomes. Philos. Trans. R. Soc. B Biol. Sci. 355, 1563–1572 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J.-H., Lin H., Joo S., Goodenough U., Early sexual origins of homeoprotein heterodimerization and evolution of the plant KNOX/BELL family. Cell 133, 829–840 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Worden A. Z., Lee J.-H., Mock T., Rouzé P., Simmons M. P., Aerts A. L., Allen A. E., Cuvelier M. L., Derelle E., Everett M. V., Foulon E., Grimwood J., Gundlach H., Henrissat B., Napoli C., McDonald S. M., Parker M. S., Rombauts S., Salamov A., Von Dassow P., Badger J. H., Coutinho P. M., Demir E., Dubchak I., Gentemann C., Eikrem W., Gready J. E., John U., Lanier W., Lindquist E. A., Lucas S., Mayer K. F. X., Moreau H., Not F., Otillar R., Panaud O., Pangilinan J., Paulsen I., Piegu B., Poliakov A., Robbens S., Schmutz J., Toulza E., Wyss T., Zelensky A., Zhou K., Armbrust E. V., Bhattacharya D., Goodenough U. W., Van de Peer Y., Grigoriev I. V., Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324, 268–272 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Ferris P. J., Goodenough U. W., Mating type in Chlamydomonas is specified by mid, the minus-dominance gene. Genetics 146, 859–869 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charlesworth D., The status of supergenes in the 21st century: Recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evol. Appl. 9, 74–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferris P., Olson B. J. S. C., De Hoff P. L., Douglass S., Casero D., Prochnik S., Geng S., Rai R., Grimwood J., Schmutz J., Nishii I., Hamaji T., Nozaki H., Pellegrini M., Umen J. G., Evolution of an expanded sex-determining locus in Volvox. Science 328, 351–354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed S., Cock J. M., Pessia E., Luthringer R., Cormier A., Robuchon M., Sterck L., Peters A. F., Dittami S. M., Corre E., Valero M., Aury J.-M., Roze D., Van de Peer Y., Bothwell J., Marais G. A. B., Coelho S. M., A haploid system of sex determination in the brown alga Ectocarpus sp. Curr. Biol. 24, 1945–1957 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Parfrey L. W., Lahr D. J. G., Knoll A. H., Katz L. A., Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. U.S.A. 108, 13624–13629 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreau H., Verhelst B., Couloux A., Derelle E., Rombauts S., Grimsley N., Van Bel M., Poulain J., Katinka M., Hohmann-Marriott M. F., Piganeau G., Rouzé P., Da Silva C., Wincker P., Van de Peer Y., Vandepoele K., Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 13, R74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subirana L., Péquin B., Michely S., Escande M.-L., Meilland J., Derelle E., Marin B., Piganeau G., Desdevises Y., Moreau H., Grimsley N. H., Morphology, genome plasticity, and phylogeny in the genus Ostreococcus reveal a cryptic species, O. mediterraneus sp. nov. (Mamiellales, Mamiellophyceae). Protist 164, 643–659 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Yau S., Hemon C., Derelle E., Moreau H., Piganeau G., Grimsley N., A viral immunity chromosome in the marine picoeukaryote, Ostreococcus tauri. PLOS Pathog. 12, e1005965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weynberg K. D., Allen M. J., Ashelford K., Scanlan D. J., Wilson W. H., From small hosts come big viruses: The complete genome of a second Ostreococcus tauri virus, OtV-1. Environ. Microbiol. 11, 2821–2839 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Derelle E., Ferraz C., Escande M.-L., Eychenié S., Cooke R., Piganeau G., Desdevises Y., Bellec L., Moreau H., Grimsley N., Life-cycle and genome of OtV5, a large DNA virus of the pelagic marine unicellular green alga Ostreococcus tauri. PLOS ONE 3, e2250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iyer L. M., Balaji S., Koonin E. V., Aravind L., Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 117, 156–184 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Hingamp P., Grimsley N., Acinas S. G., Clerissi C., Subirana L., Poulain J., Ferrera I., Sarmento H., Villar E., Lima-Mendez G., Faust K., Sunagawa S., Claverie J.-M., Moreau H., Desdevises Y., Bork P., Raes J., de Vargas C., Karsenti E., Kandels-Lewis S., Jaillon O., Not F., Pesant S., Wincker P., Ogata H., Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 7, 1678–1695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., Horvath P., CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Clerissi C., Desdevises Y., Grimsley N., Prasinoviruses of the marine green alga Ostreococcus tauri are mainly species specific. J. Virol. 86, 4611–4619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas R., Grimsley N., Escande M.-l., Subirana L., Derelle E., Moreau H., Acquisition and maintenance of resistance to viruses in eukaryotic phytoplankton populations. Environ. Microbiol. 13, 1412–1420 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Coleman M. L., Sullivan M. B., Martiny A. C., Steglich C., Barry K., DeLong E. F., Chisholm S. W., Genomic islands and the ecology and evolution of Prochlorococcus. Science 311, 1768–1770 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Ma L.-J., van der Does H. C., Borkovich K. A., Coleman J. J., Daboussi M.-J., Di Pietro A., Dufresne M., Freitag M., Grabherr M., Henrissat B., Houterman P. M., Kang S., Shim W.-B., Woloshuk C., Xie X., Xu J.-R., Antoniw J., Baker S. E., Bluhm B. H., Breakspear A., Brown D. W., Butchko R. A. E., Chapman S., Coulson R., Coutinho P. M., Danchin E. G. J., Diener A., Gale L. R., Gardiner D. M., Goff S., Hammond-Kosack K. E., Hilburn K., Hua-Van A., Jonkers W., Kazan K., Kodira C. D., Koehrsen M., Kumar L., Lee Y.-H., Li L., Manners J. M., Miranda-Saavedra D., Mukherjee M., Park G., Park J., Park S.-Y., Proctor R. H., Regev A., Ruiz-Roldan M. C., Sain D., Sakthikumar S., Sykes S., Schwartz D. C., Turgeon B. G., Wapinski I., Yoder O., Young S., Zeng Q., Zhou S., Galagan J., Cuomo C. A., Kistler H. C., Rep M., Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephens P. J., Greenman C. D., Fu B., Yang F., Bignell G. R., Mudie L. J., Pleasance E. D., Lau K. W., Beare D., Stebbings L. A., McLaren S., Lin M.-L., McBride D. J., Varela I., Nik-Zainal S., Leroy C., Jia M., Menzies A., Butler A. P., Teague J. W., Quail M. A., Burton J., Swerdlow H., Carter N. P., Morsberger L. A., Iacobuzio-Donahue C., Follows G. A., Green A. R., Flanagan A. M., Stratton M. R., Futreal P. A., Campbell P. J., Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E. E., Turner S. W., Korlach J., Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., Thierer T., Ashton B., Meintjes P., Drummond A., Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H., Luo X., Qian J., Pang X., Song J., Qian G., Chen J., Chen S., FastUniq: A fast de novo duplicates removal tool for paired short reads. PLOS ONE 7, e52249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanc-Mathieu R., Verhelst B., Derelle E., Rombauts S., Bouget F.-Y., Carré I., Château A., Eyre-Walker A., Grimsley N., Moreau H., Piégu B., Rivals E., Schackwitz W., Van de Peer Y., Piganeau G., An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics 15, 1103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Durbin R., Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blanc-Mathieu R., Sanchez-Ferandin S., Eyre-Walker A., Piganeau G., Organellar inheritance in the green lineage: Insights from Ostreococcus tauri. Genome Biol. Evol. 5, 1503–1511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philippe N., Salson M., Commes T., Rivals E., CRAC: An integrated approach to the analysis of RNA-seq reads. Genome Biol. 14, R30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cingolani P., Platts A., Wang L. L., Coon M., Nguyen T., Wang L., Land S. J., Lu X., Ruden D. M., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McVean G., Awadalla P., Fearnhead P., A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160, 1231–1241 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nelson C. W., Moncla L. H., Hughes A. L., SNPGenie: Estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 31, 3709–3711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R. 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Remington D. L., Thornsberry J. M., Matsuoka Y., Wilson L. M., Whitt S. R., Doebley J., Kresovich S., Goodman M. M., Buckler E. S. IV, Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 98, 11479–11484 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vandepoele K., Van Bel M., Richard G., Van Landeghem S., Verhelst B., Moreau H., Van de Peer Y., Grimsley N., Piganeau G., pico-PLAZA, a genome database of microbial photosynthetic eukaryotes. Environ. Microbiol. 15, 2147–2153 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Darriba D., Taboada G. L., Doallo R., Posada D., jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., Larget B., Liu L., Suchard M. A., Huelsenbeck J. P., MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guindon S., Gascuel O., A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 70.Felsenstein J., Churchill G. A., A Hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 13, 93–104 (1996). [DOI] [PubMed] [Google Scholar]
- 71.McGuire G., Prentice M. J., Wright F., Improved error bounds for genetic distances from DNA sequences. Biometrics 55, 1064–1070 (1999). [DOI] [PubMed] [Google Scholar]
- 72.Paradis E., Claude J., Strimmer K., APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Zhang H., Gao S., Lercher M. J., Hu S., Chen W.-H., EvolView, an online tool for visualizing, annotating and managing phylogenetic trees. Nucleic Acids Res. 40, W569–W572 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lercher M. J., Smith N. G. C., Eyre-Walker A., Hurst L. D., The evolution of isochores: Evidence from SNP frequency distributions. Genetics 162, 1805–1810 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/7/e1700239/DC1
fig. S1. Read coverage map of reference strain RCC4221 per chromosome for each strain.
fig. S2. Size distribution of chromosomes 2 and 19 in 13 O. tauri strains.
fig. S3. Phylogeny of the 2.5-kb repeat of chromosome 19’s syntenic block A.
fig. S4. Growth rates of 13 strains with 95% confidence interval at 14° and 20°C.
fig. S5. Sampling area and sampling sites of water samples used to isolate the 13 strains of O. tauri.
fig. S6. Dotplot of chromosome 19 between RCC1115 and RCC1559, RCC4221, RCC1108, and RCC1123.
fig. S7. Venn diagram depicting the number of shared or specific polymorphic sites called by each calling method.
fig. S8. Large (>100 bp) indel read mapping features.
fig. S9. Phylogenetic distance tree of 13 O. tauri strains based on 117,600 biallelic SNPs.
table S1. Read mapping and polymorphism statistics for each strain.
table S2. Nucleotide diversity and number of segregating and nonsegregating sites for five types of polymorphisms (synonymous, fourfold degenerate, nonsynonymous, intergenic, and intronic).
table S3. Fitting the models to the site frequency spectrum on fourfold degenerate sites.
table S4. Analysis of gene content of predicted and validated insertions using sequence homology (blastx).
table S5. Functional description of 10 predicted and validated deletions.
table S6. Viral susceptibility and resistance spectrum of O. tauri strains and a collection of 32 prasinoviruses.
table S7. Geographic origin and sampling date of the 13 O. tauri strains.
table S8. Variants, SNPs, and indel polymorphisms for each calling method.
table S9. Features of the four resequenced regions.
table S10. SNP calling validation for GATK UG, GENO, and HC by Sanger resequencing.
table S11. Insertion detection steps.
table S12. Insertion assembly steps.
table S13. Deletion detection steps.
table S14. Position and validation status of 18 predicted insertions.
table S15. Position and validation status of 12 predicted deletions (>500 bp).
table S16. Pairwise growth rates by Tukey test between strains at 20° and 14°C.